

Abstract
The optimization of compounds with multiple targets is a difficult multidimensional problem in the drug discovery cycle. Here, we present a systematic, multidisciplinary approach to the development of selective antiparasitic compounds. Computational fragment-based design of novel pteridine derivatives along with iterations of crystallographic structure determination allowed for the derivation of a structure–activity relationship for multitarget inhibition. The approach yielded compounds showing apparent picomolar inhibition of T. brucei pteridine reductase 1 (PTR1), nanomolar inhibition of L. major PTR1, and selective submicromolar inhibition of parasite dihydrofolate reductase (DHFR) versus human DHFR. Moreover, by combining design for polypharmacology with a property-based on-parasite optimization, we found three compounds that exhibited micromolar EC50 values against T. brucei brucei while retaining their target inhibition. Our results provide a basis for the further development of pteridine-based compounds, and we expect our multitarget approach to be generally applicable to the design and optimization of anti-infective agents.
Introduction
The World Health Organization has identified 17 neglected tropical diseases (NTDs) that pose a health burden to over 1.4 billion people.1,2 Parasites of the trypanosomatid family are responsible for two potentially lethal insect-vector borne NTDs: human African trypanosomiasis (HAT, sleeping sickness), caused by Trypanosoma brucei, and leishmaniasis, caused by the intracellular parasite Leishmania spp.(3−7) Current therapeutics are limited by toxicity, poor efficacy, and parasite resistance, thus underlining the need for new chemotherapies.8,9
New antiparasitic agents can be identified by target-based drug design strategies.10−12 The folate pathway enzyme dihydrofolate reductase (DHFR) is a known anticancer, antibacterial, and antimalarial target.13−16 It provides reduced folates, which are crucial to biological processes like DNA, protein, and amino acid synthesis or one-carbon transfer.14,17,18 In trypanosomatids, DHFR inhibition, for example by methotrexate (MTX, 1a), is ineffective due to a metabolic bypass via the biopterin-reducing pteridine reductase 1 (PTR1, Figure 1): when DHFR is inhibited, PTR1 is overexpressed and sustains sufficient reduced folate levels to ensure parasite survival. Thus, when targeting the folate pathway in Leishmania, both DHFR and PTR1 need to be considered.19−21 In T. brucei, RNA interference studies have suggested PTR1 to be a potential antiparasitic target in its own right.22,23 Nonetheless, even nanomolar PTR1 inhibitors have so far shown limited antiparasitic activity in vitro,24,25 suggesting that targeting the T. brucei folate pathway may also benefit from the consideration of both PTR1 and DHFR.
Figure 1.

Overview of pterin activation in the trypanosomatidic folate pathway when DHFR is inhibited and PTR1 provides a metabolic bypass. Under normal conditions (indicated by dashed lines), the DHFR domain of the bifunctional DHFR-TS reduces biological folates to tetrahydrofolate (THF). Serine hydroxymethyl transferase (SHMT) converts THF to 5,10-methylene THF, which has a central role in amino acid synthesis, protein biosynthesis, and one-carbon transfer. It is also required by the TS domain of DHFR-TS to convert deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP), which is necessary for DNA synthesis. PTR1 catalyzes the reduction of unconjugated pterins, like biopterin, and takes over folate reduction when DHFR is inhibited (continuous lines), thus acting as a metabolic bypass and an important additional target for shutting down the trypanosomatidic folate pathway. Both proteins are shown in cartoon representation (DHFR domain of DHFR-TS: purple, PTR1 monomer of the functional tetramer: light pink) with the NADPH/NADP+ cofactor in a stick representation with black carbons and the folate substrate in yellow spheres. In PTR1, an arginine residue from a neighboring subunit that points into the active site is shown in a magenta stick representation.
Screening a set of folate-related compounds against parasitic folate pathway targets previously led to the identification of compounds 1b (methyl-1-(4-(((2,4-diaminopteridin-6-yl)methyl)(methyl)amino)benzoyl)piperidine-4-carboxylate) and 1c (methyl-1-(4-(((2,4-diaminopteridin-6-yl)methyl)amino)benzoyl)piperidine-4-carboxylate) as submicromolar inhibitors of Leishmania major PTR1 (LmPTR1) with Ki values of 0.04 and 0.10 μM, respectively.261c was additionally a micromolar inhibitor of L. major DHFR (LmDHFR) with a weak selectivity for the parasite enzyme over the human DHFR (hDHFR) (Ki of 4 vs 10 μM). In contrast to the parasite DHFR, which is covalently coupled with thymidylate synthase (TS) in a bifunctional DHFR-TS, the hDHFR off-target is monofunctional and shares only about 30% sequence identity with parasite DHFR domains, indicating potential for further optimization of selectivity.27−29
The current study focuses on optimizing pteridine-based compounds for their inhibition of T. brucei PTR1 (TbPTR1) and TbDHFR, in addition to the corresponding Leishmania targets, while ensuring selectivity against the off-target hDHFR. The enzymatic evaluation of reference pteridines reported earlier,26,30 our comparative study of trypanosomatid folate pathway proteins,31 and computational docking studies were first employed for the design of novel pteridine derivatives. Three new crystal structures of complexes of pteridines with TbPTR1 and a complex with LmPTR1 were determined and confirmed the predicted bound orientation of the novel pteridines. A systematic analysis of correlations between computed physicochemical molecular descriptors and observed antiparasitic effects was then performed and allowed us to prioritize promising compounds for synthesis. In total, we identified 26 new pteridine-based multitarget inhibitors showing improved target inhibitory profiles for PTR1 and DHFR of both L. major and T. brucei. Among these inhibitors, we report the first, to the best of our knowledge, apparent picomolar inhibitors of TbPTR1 and several new low nanomolar inhibitors of LmPTR1, which mostly also show selective micromolar to submicromolar inhibition of the parasite DHFR variants. In vitro evaluations of the designed multitarget inhibitors against bloodstream forms of T. brucei brucei revealed low micromolar to submicromolar EC50 values for three of these pteridines. Taken together, we here report a successful application of a systematic multitarget design approach to yield selective pteridine-based antiparasitic compounds affecting multiple trypanosomatid enzymes.
Results and Discussion
Reference Compounds Inhibit both PTR1 and DHFR
To systematically assess multitarget inhibition, we measured the inhibition of TbPTR1, TbDHFR, LmPTR1, LmDHFR, and the off-targets hDHFR and hTS by the folate-related anticancer agent methotrexate (MTX, 1a) and seven further pteridine-based reference compounds (1b–1h, Figure 2 and Table S1, SI).26,30,32 Although 1b–1h were primarily designed as LmPTR1 inhibitors, we found all to be more potent against TbPTR1 than LmPTR1 with 1b being the strongest inhibitor of TbPTR1 with an IC50 of 50 nM against TbPTR1 and 1 μM against LmPTR1 (Figure 2A). Notably, these compounds exhibited micromolar to submicromolar inhibition of LmDHFR and TbDHFR (IC50LmDHFR 0.3–1.4 μM; TbDHFR 0.3–20.05 μM). While MTX (1a) was more potent against the parasite DHFRs, it was not selective (selectivity index SI: TbDHFR/hDHFR = 3 and LmDHFR/hDHFR = 1, Figure 2A). For compounds 1b–1h, higher SI values were observed, ranging up to about 165 for 1b for both TbDHFR and LmDHFR (Figure 2A).
Figure 2.

Inhibitory activities, selectivities, and structures of reference pteridines. (A) Heatmaps show activities given by IC50 values (top) and selectivity indices (SI) (bottom) for the targets and the off-target hDHFR. All values, as well as data for hTS, are given in Table S1. NI: no inhibition; NA: not applicable. (B) Previously published compounds shown were used as reference compounds: 1a is methotrexate; 1b, 1c, and 1h are 6b, 6a, and 6c from Cavazzuti et al.;26 and 1d–1g correspond to 5d, 5b, 6a, and 5a from Corona et al.30
Substrate-like and Methotrexate-Inhibitor-like Binding Modes of the Reference Compounds
Despite the hydrogen-bonding network stabilizing the pteridine ring in the PTR1 active site, for the PTR1 complexes with MTX/1a derivatives, there are two alternative binding modes. Previously determined crystal structures show that compounds 1b and 1c share a substrate-like pterin orientation in the complex with LmPTR1.26 In the same crystal structure, compound 1b also adopts a second, so-called inhibitor-like (or MTX-like) orientation, with the bicyclic ring system flipped by 180° and rotated by 30° (Figures 3A,B and S1).26 Dual binding modes have also been observed in crystallographic complexes of TbPTR1 with small pteridine-based inhibitors.32
Figure 3.

Orientations of reference pteridine compound 1b in crystal structures of LmPTR1 and TbPTR1. (A,B) Compound 1b (cyan carbons) in complex with LmPTR1 (PDB-ID 2qhx) has a substrate-like (A) and an inhibitor-like or MTX-like (B) binding mode. 1b is shown with (A) folate (yellow carbons) superimposed from a TbPTR1 structure (PDB-ID 3bmc) and with (B) MTX (1a, yellow carbons) superimposed from an LmPTR1 structure (PDB-ID 1e7w). The pteridine nitrogens are labeled according to the ring nomenclature. (C) Binding site in the crystal structure determined in this work (PDB-ID 6rx5) of TbPTR1 (gray cartoon, His267′ from the neighboring subunit in lavender) in complex with NADPH/NADP+ and compound 1b, which has the MTX-like binding mode. Interacting residues (in A, B: only Phe113) and the NADPH/NADP+ cofactor are shown in sticks (carbons colored according to protein and black, respectively). In (C), water molecules are shown as red spheres, and the inhibitor is surrounded by the omit map (green wire) contoured at the 2.5 σ level. Hydrogen bonds are represented by brown dashed lines.
We here determined the crystal structure of the ternary complex of TbPTR1 with NADPH/NADP+ and the reference compound 1b (PDB-ID 6rx5, resolution 1.42 Å, experimental details: Tables S2 and S3). It shows that the diaminopteridinyl moiety of 1b adopts only the MTX-like orientation (Figure 3C), resembling its MTX-like binding mode in LmPTR1 (Figures 3B and S2). Consistently, docking studies indicated that all reference pteridines adopt MTX-like binding modes in the different targets and the off-target hDHFR (Table S4 and Figure S5). Therefore, we concluded that the MTX-like binding mode is likely the dominant one, and we focused on the analysis of this binding mode in the subsequent compound design.
Comparative Target/Off-Target Mapping and Docking Studies Support Design Focused on Selective Multitarget Inhibition
To develop enhanced selective inhibitors of the parasite targets, we employed a multitarget-based design approach to improve inhibition of TbPTR1, TbDHFR, LmPTR1, and LmDHFR while retaining low hDHFR off-target inhibition. The next generation of pteridine-like compounds was created by dissecting the part of 1b attached to the pteridine core into three modules. These were N10, the substitution to the N10 position; PABA, the para-amino benzoic acid (PABA) moiety; and Tail, the cyclic glutamate tail (Figure 4). We separately modified each of these modules to obtain three new series of compounds. The modifications of each module were based on binding mode predictions from docking in the different targets and the off-target hDHFR and our previously published optimization guidelines for MTX-like scaffolds.31 The key concepts adopted in the compound design are summarized in Figure 4.
Figure 4.

Structural features of PTR1 and DHFR considered in the multitarget design of selective compounds illustrated for reference compound 1b. (A) Selected residues within 5 Å of the three modules—N10, PABA and Tail—modified in the design procedure. Residues were selected for the complexes of 1b with TbPTR1 (pale gray), TbDHFR (dark gray), LmPTR1 (pale pink), and LmDHFR (dark pink). Residues are colored according to their properties: basic: blue, polar: green, and nonpolar: yellow. The ligand interaction plot is based on Panecka-Hofman et al.31 and provides an overview of residues with similar properties that surround the ligand modules in the different targets (showing only those applied for the design; for full maps, see Figures S3 and S4). In some positions, the amino acid type of the off-target hDHFR is different from parasite DHFR. Differing hDHFR residues are labeled in the top right corner of the corresponding parasite DHFR residue. These positions highlight suitable substitution points to improve selectivity. (B) Surface representations of complexes of 1b with TbPTR1 (left, PDB-ID 6rx5) and TbDHFR (right, MTX-like top-ranked docking pose in PDB-ID 3rg9). The compound tail moiety is fully solvent-exposed in PTR1, whereas it is well-enclosed in DHFR. (C) Surface representations of complexes of 1b with TbPTR1 (left, PDB-ID 6rx5) and LmPTR1 (right, PDB-ID 2qhx, state A). The ligand is more enclosed in the narrow pocket entrance of TbPTR1, while the LmPTR1 pocket has an elongated, widened funnel that can accommodate larger compound tails. In (B,C), 1b is shown in sticks with cyan carbons.
Rationale for N10 Modifications
The binding pockets of the different target proteins were found to share a number of aliphatic residues in the proximity of the N10 substituent of a bound ligand, e.g., Leu209 of TbPTR1; Ile47 and Leu90 of TbDHFR; Leu226 and Leu229 of LmPTR1; Ile20 and Val62 of LmDHFR (Figure 4A).31 Bulkier nonpolar groups in comparison to the methyl of 1b, like the ethyl and propargyl substituents of 2a and 2b, allow for interactions with those hydrophobic moieties. Docking studies suggested that even substituents of the size of benzyl, as in 2c, can be accommodated in the PTR1 and DHFR pockets (Figure 5A,B). Furthermore, such bulky substituents may improve selectivity for the on-targets: The hDHFR pocket has a lower volume compared to the parasite DHFR pockets (pocket volume TbDHFR 353 Å3, LmDHFR 384 Å3, and hDHFR 347 Å3).
Figure 5.

Views of the binding sites showing docked poses of selected pteridine-based inhibitors in the target proteins: TbPTR1 (pale gray) (A,E), TbDHFR (dark gray) (B,F), LmPTR1 (pale pink) (C), and LmDHFR (dark pink) (D). (A) Induced fit (IF) MTX-like docking pose for compound 2c (cyan carbons) in TbPTR1 in the presence of a conserved water molecule (ball-and-stick representation): Trp221 moves (indicated by a brown arrow) to make room for the phenyl of 2c. (B–F) Rigid-body docking poses of 2c in TbDHFR (B), 3c (lime carbons) in LmPTR1 and LmDHFR (C,D), and 4e (purple carbons) in TbPTR1 and TbDHFR (E,F); see text for discussion. Docked poses are shown for N1-deprotonated compounds, but similar orientations were observed for the N1-protonated forms (see Figure S6). For PTR1, all docking poses shown were obtained in the presence of conserved structural water molecules. Generally, similar poses were observed for docking without water. In all panels, proteins are shown in cartoon representation with the important interacting residues (compare Figure 4A) and the NADPH/NADP+ cofactor shown in sticks (carbons colored according to protein and black, respectively). Residues His267′ and Arg287′ from the neighboring subunit are shown in lavender and magenta in TbPTR1 and LmPTR1, respectively. Hydrogen bonds are represented by brown dashed lines. Further IF docking poses are shown in Figures S7 and S8.
Furthermore, as previously demonstrated,31 hDHFR favors hydrogen bond donors in the proximity of N10 and the PABA ring system, whereas the parasite DHFRs allow for favorable interactions with hydrogen bond acceptors. To improve off-target selectivity, we thus replaced N10 by sulfur and the PABA benzene ring by pyridine in 2d.
Although Corona et al.30 found improved selectivity for PTR1 over hDHFR by hydrophilic N10 substitutions, our data for reference compound 1d with a hydroxyethyl substituent did not support this observation (Figure 2A). Docking simulations indicated that interactions with a highly conserved structural water might induce an unfavorable conformation of the substituent’s aliphatic chain (Figure S5A, SI). To relax the geometry while allowing interactions between the substituent and water, we elongated the aliphatic linkage to a hydroxypropyl in 2e.
Thus, in total, the N10 series consists of five novel pteridines (2a–e, Figure 6) modified to improve interactions with PTR1 and parasite DHFR and to exploit the differences in pocket sizes and residues between the parasitic targets and the hDHFR off-target.
Figure 6.

Overview of the modifications in the N10, PABA, and Tail modules explored in the designed compound series with respect to the reference compound 1b. Synthesized members of each designed series are shown in the framed boxes along with the key objectives addressed with the respective modifications. See text for details.
Rationale for PABA Modifications
As a first modification to the PABA moiety, in 3a, we replaced the PABA phenyl group with benzyl (Figure 6). The additional hydrophobic spacer can interact with hydrophobic target residues while resulting in a shifted position of the hydrophilic linker amide. The positioning of hydrophobic and hydrophilic residues surrounding the PABA moiety and the amide linker in the human off-target is different from that of the parasite targets PTR1 and DHFR, which can be exploited to improve selectivity. For the same reason, the amide linker position was also shifted in 3b by substituting the PABA moiety with meta-aminobenzoic acid.
A second key feature of the targets vs off-targets that was used to inform the design of the PABA series relates to the compound tail: Tail regions are solvent-exposed in PTR1 and thus have poorly defined interactions (Figure 4B). In contrast, in DHFR, the tail region is enclosed, and strong interactions occur with the hDHFR off-target.31 We therefore shortened the tail region to achieve full enclosure in the PTR1 binding pocket by replacing PABA by naphthalene (3c) or benzene moieties (nonsubstituted, 3d; or substituted with −CF3, 3e). Docking results showed that the smaller tail fully resides in the PTR1 binding pocket (Figure 5C) and is stabilized by surrounding hydrophobic residues, not only in PTR1 but also in parasite DHFR (Figure 5C,D). Rigid-body docking studies suggested that the bulky naphthalene of 3c may be particularly beneficial in LmPTR1, since this target has a more elongated, open pocket compared to TbPTR1 (Figure 4C). The PABA moiety modifications are therefore suitable for modulating the compound interaction profile in a species-specific manner.
In summary, the PABA series contains five new pteridine derivatives (3a–3e, Figure 6) designed to improve selectivity by exploiting the different surroundings of bound PABA moieties in hDHFR in comparison to the parasite target proteins.
Rationale for Tail Modifications
The surrounding of the compound tail features several hydrophobic residues, particularly in the two T. brucei targets (Figure 4A). Directional interactions with the tail moiety may have limited benefit for the binding affinity in PTR1, since the flexibility of the solvent-exposed tail likely has an entropic contribution. Hydrophobic interactions are geometrically less restrained than, for instance, hydrogen bonds. Therefore, anticipating less pronounced entropic penalties on binding in our designed derivatives, we replaced the methyl ester in the tail of 1b by the more flexible ethyl and propyl in 4a and 4b, respectively. Two aspects may result in selectivity benefits from this approach: The tail region is enclosed by more hydrophobic moieties in parasite DHFR than in the hDHFR off-target (Figure 4A), and residues surrounding the tail have previously been demonstrated to show differing conformational variability in the crystal structures when comparing parasitic targets with the off-target.31
To combine exploitation of the differing patterns of hydrophobic residues in the tail environments of targets and off-targets with improved enclosure in PTR1 (Figure 4A,B), we further modified the tail to an unsubstituted piperidine (4c) or replaced piperidine with an unsubstituted benzene (4d). Compound 4e, with benzene attached via a flexible ethyl linkage to an MTX-like amide, can benefit from nonpolar and aromatic residues surrounding the tail in PTR1 and parasite DHFR and, according to docking predictions, readily adapt to their differing placements in the on-targets; see Figure 5E,F. The docking studies additionally suggest that the flexible aromatic tail can form cation−π interactions with positively charged residues in the entrance of the DHFR pocket (e.g., Arg59 of TbDHFR, Figures 4A and 5F). Additional hydrophobic residues in the target pocket entrance regions, like Pro99 of TbPTR1 (Figure 4A), can be targeted with an altered geometry in combination with methoxylations: 4f and 4g combine a one-carbon spacer between N10 and PABA and amide-linked methoxylated tail portions. In addition, an etheryl linkage to a nonsubstituted (4h), methoxylated (4i), or trimethoxylated (4j) benzyl group was explored. Compounds 4f–4j were collectively designed to interact with the different hydrophobic, aromatic, and positively charged surrounding residues found around the tail region in the various targets (Figure 4A).
Taken together, the tail series comprises 10 new pteridines (4a–4j, Figure 6) with modified tails to target residue patterns distinguishing on- and off-targets and the distinct surroundings of the tail in PTR1 vs DHFR.
Synthesis of Pteridine Derivatives with High Yield
A total of 26 new 2,4-diaminopteridine derivatives and the reference compounds 1b and 1c were (re)synthesized as reported in Schemes 1–8. We applied our methodology for an improved reaction yield of the chemical pteroid step to provide a key intermediate for most of the designed compounds.33 Displacement of the chloride of 6-(chloromethyl)pteridine-2,4-diamine hydrochloride (29, Scheme 1) by the appropriate substituted anilines and aliphatic amino derivatives was carried out in N,N-dimethylacetamide (DMA) at 60 °C microwave (MW) to provide 1b,c, 2a–e, 3a–c, 4a–j, and 5a–f in high yields of 70–90% with reduced reaction time (Schemes 2–7).33
Scheme 1. Synthesis of Derivatives of Compound 29.

Reagents and conditions: (i) SOCl2, reflux, 12 h, 70% yield; (ii) 29 (1.2 equiv), corresponding amine derivative (1 equiv), K2CO3 (3 equiv), KI (0.1 equiv), DMA, 60°C, 20′–30′ MW.
Scheme 8. Synthesis of Compounds 3d,e.

Reagents and conditions: (i) KMnO4, acetone/0.5 M phosphate buffer at pH 7 (1:1 V/V); (ii) EDC·HCl (1.1 equiv), HOBt (0.1 equiv), TEA (2–3 equiv), DMF, rt, overnight.
Scheme 2. Synthesis of Compounds 1b,c, 2a–c, 2e, 4a–c, and 5c, and Intermediates 32–35, 37, 38, and 40–49.

Reagents and conditions: compounds 30, 31, 36, and 39 were purchased from Sigma; (i) acetonitrile or 3-hydroxypropanenitrile, 10% Pd/C, NH4OAc (1 equiv), CH3OH, H2, rt, 24–36 h (32, 33); (ii) alkyl halide (propargyl bromide, (bromomethyl)benzene) (0.5 equiv), K2CO3 (2 equiv), DMF dry, rt, 24 h (34, 35); (iii) SOCl2 (4 equiv), propanol (for 37), EtOH (for 38), reflux, 7–12 h (89 and 96% yield); (iv) EDC·HCl (1.1 equiv), HOBt (0.1 equiv), TEA (2–3 equiv), DMF, rt, overnight (40–49); (v) 29 (1.2 equiv), corresponding amine derivative (1 equiv), K2CO3 (3 equiv), KI (0.1 equiv), DMA, 20′ MW (1b,c, 2a–c, 2e, 4a–c, 5c).
Scheme 7. Synthesis of Compounds 3c, 4d,e, and 5d.

Reagents and conditions: (i) 29 (1.2 equiv), corresponding amine derivative (1 equiv), K2CO3 (3 equiv), KI (0.1 equiv), DMA, 30′ MW (3c, 4d,e, and 5d); (ii) acetonitrile, 10% Pd/C, NH4OAc (1 equiv), CH3OH, H2, rt, 24–36 h (74); (iii) EDC·HCl (1.1 equiv), HOBt (0.1 equiv), TEA (2–3 equiv), DMF, rt, overnight (75).
The PABA amine functionalization was achieved by selective alkylation of primary amines to secondary amines using nitriles as alkylating reagents with Pd/C for intermediates 32 and 33.34,35 Conventional alkylation of the latter with propargyl bromide or (bromomethyl)benzene resulted in derivatives 34 and 35, respectively (Scheme 2).
The reductive alkylation of amines using nitriles was also used to obtain 51 and 74 in Schemes 3 and 7. The isonipecotic acid derivatization was achieved via Fischer esterification using the reagent solvents propanol (37) and EtOH (38), respectively; methyl isonipecotate (36) and piperidine (39) were purchased from Sigma (Scheme 2).
Scheme 3. Synthesis of Compounds 3a and 5a,b.

Reagents and conditions: (i) 3-hydroxypropanenitrile, 10% Pd/C, NH4OAc (1 equiv), CH3OH, H2, rt, 24 h (51); (ii) EDC·HCl (1.1 equiv), HOBt (0.1 equiv), TEA (2–3 equiv), DMF, rt, overnight (52–54); (iii) 29 (1.2 equiv), corresponding amine derivative (1 equiv), K2CO3 (3 equiv), KI (0.1 equiv), DMA, 20′ MW (3a, 5a,b).
The intermediate acid derivatives 30–35 and d and e were condensed to amides through a coupling reaction with the respective amines 36–39 and g using EDC·HCl in dimethylformamide (DMF) as the coupling agent to provide the intermediate products 40–49, 71, 72, and 75, which were then made to react with 29 to obtain the final compounds (1b,c, 2a–d, 3b, 4a–c, 4e, 5c; Schemes 2, 6, 7).
Scheme 6. Synthesis of Compounds 3b and 2d.

Reagents and conditions: (i) EDC·HCl (1.1 equiv), HOBt (0.1 equiv), TEA (2–3 equiv), DMF, rt, overnight (71 and 72); (ii) 29 (1.2 equiv), corresponding amine derivative (1 equiv), K2CO3 (3 equiv), KI (0.1 equiv), DMA, 20′ MW (3b, 2d).
Using the same method, we synthesized the elongated compounds 3a, 4f–g, and 5a,b, characterized by a carbon spacer in the PABA moiety (Schemes 3 and 4).
Scheme 4. Synthesis of Compounds 4f,g.

Reagents and conditions: (i) di-tert-butyl pyrocarbonate (1.05 equiv), dioxane/H2O/1 N NaOH 1/1/1 V/V/V, rt, 6 h (55); (ii) EDC·HCl (1.1 equiv), HOBt (0.1 equiv), TEA (2–3 equiv), DMF, rt, overnight (56 and 57); (iii) TFA, DCM, rt (58 and 59); (iv) 29 (1.2 equiv), corresponding amine derivative (1 equiv), K2CO3 (3 equiv), KI (0.1 equiv), DMA, 20′ MW (4f,g).
To obtain 4f,g (Scheme 4), an additional protection step reaction to guide selective amide functionalization was necessary. The selectivity was achieved via Boc protection in the first step of the reaction of b to obtain 55, which was then coupled with the respective aliphatic amine to give 56 and 57. The target amines were finally obtained by a deprotection step carried out in 30–40% trifluoroacetic acid/dichloromethane (TFA/DCM) in quantitative yield.
The phenoxyphenyl–methanamine derivative intermediates (Scheme 5) were synthesized starting from 4-fluorobenzaldehyde and the respective phenol derivates 60–62 by an SNAr reaction. Subsequently, the primary amines 66–68,36 or functionalized amines 69 and 70 (obtained via a one-pot reductive step), were reacted with 29 to obtain 4h–j and 5e,f.
Scheme 5. Synthesis of Compounds 4h–j and 5e,f.

Reagents and conditions: (i) K2CO3 (3 equiv), DMF, reflux, 16–18 h (63–65); (ii) NH2OH·HCl (1.2 equiv), EtOH, rt, >1 h followed by Zn dust (2.5 equiv) in 12 M HCl (4 equiv), rt, 15′ (66–68); (iii) 29 (1.2 equiv), corresponding amine derivative (1 equiv), K2CO3 (3 equiv), KI (0.1 equiv), DMA, 20′ MW (4h–j, 5e,f); (iv) methylamine (for 69) or benzylamine (for 70), EtOH dry, 60°C, 3 h, then NaBH4 (1.5 equiv), rt, 2 h.
Compounds 3c, 4d, and 5d, with a higher steric hindrance, were obtained with a slightly increased reaction time in a good yield. Finally, to obtain 3d,e, it was necessary to first perform an oxidation reaction. Treatment of Pt–OH in acetone/0.5 M phosphate buffer at pH 7 (1:1 v/v) with KMnO4 gave the oxidized analogue 76, which was subsequently coupled with the selected aliphatic amine to obtain the desired amides (Scheme 8).
Crystal Structures for the PTR1 Targets Confirm the Predicted Interactions and That the Pteridine Derivatives Adopt a Methotrexate-Inhibitor-Like Orientation
The structures of TbPTR1 with two new pteridines, 2a and 2e, and that of LmPTR1 with 2e, were determined to 1.20, 1.11, and 2.10 Å resolution, respectively (see Tables S2 and S3). The structures contain functional enzyme tetramers in the crystallographic asymmetric unit with a similar structure to those previously determined.37,38 In all complexes, the compounds adopt MTX-like binding modes (Figure 7A,B).
Figure 7.

Views of the binding sites of crystal structures of complexes of pteridine-based inhibitors in TbPTR1 and LmPTR1 determined in this work, which confirm the predicted MTX-like binding modes. (A) 2a (green carbons) in TbPTR1 (gray cartoon, His267′ from the neighboring subunit in lavender) and (B) 2e (yellow carbons) in LmPTR1 (pink cartoon, Arg287′ from the neighboring subunit in magenta). Water molecules are shown as red spheres, and the inhibitors are surrounded by the omit map (green wire) contoured at the 2.5 σ level. Interacting residues and the NADPH/NADP+ cofactor are shown in sticks (carbons colored according to protein and black, respectively). Hydrogen bonds are represented by brown dashed lines.
In line with the docking predictions, the overall structure of the TbPTR1 complexes resembles the complex with 1b (compare Figure 7A with 3C). In agreement with the design objective, the N-ethyl moiety of 2a was found to form van der Waals interactions with Val206 and Trp221 on the hydrophobic side of the pocket (Figure 7A). The bulkier N-propylhydroxyl moiety of 2e forms direct and water-mediated hydrogen bonds with Asp161 and receives an intramolecular hydrogen bond from the amine in position 4 on the pteridine system (Figure S2C). The structure of LmPTR1 in complex with 2e (Figure 7B) closely resembles that observed in TbPTR1, except for the terminal piperidine moiety (Figure S2C,D). The latter moiety is highly flexible—a possible orientation is reported in the crystal structure, but further orientations cannot be excluded.
Designed Pteridine Derivatives Have Improved Target and Off-Target Enzyme Inhibitory Activities
The measured inhibitory activities of compounds 2a–e, 3a–e, and 4a–j against the targets TbPTR1, TbDHFR, LmPTR1, and LmDHFR and the off-targets hDHFR and hTS are given in Figure 8 and Table S1. All inhibitory activities are reported as IC50 values, which are commonly used to characterize and rank compounds when screening for enzyme inhibition in drug discovery projects.39 Overall, the inhibitory activities against the PTR1 targets for the designed compounds are improved, as are PTR1 vs off-target selectivities. Indeed, for a small number of compounds (2c, 2d, 4c, 4d, 4e, 5c, and 5d), the IC50 values for inhibition were determined to be either 1 nM or less than 0.1 nM against TbPTR1. As the TbPTR1 assay makes use of low nanomolar concentrations of enzyme, for these very potent compounds, the tight binding limit was approached, and therefore, accurate values of the IC50 values could not be determined.40 Representative dose–response curves are shown in the Supporting Information, showing that only part of the response range could be measured for these compounds for which the IC50 value could also be rather sensitive to any possible errors in dilution or determination of inhibitor or enzyme active site concentration.
Figure 8.
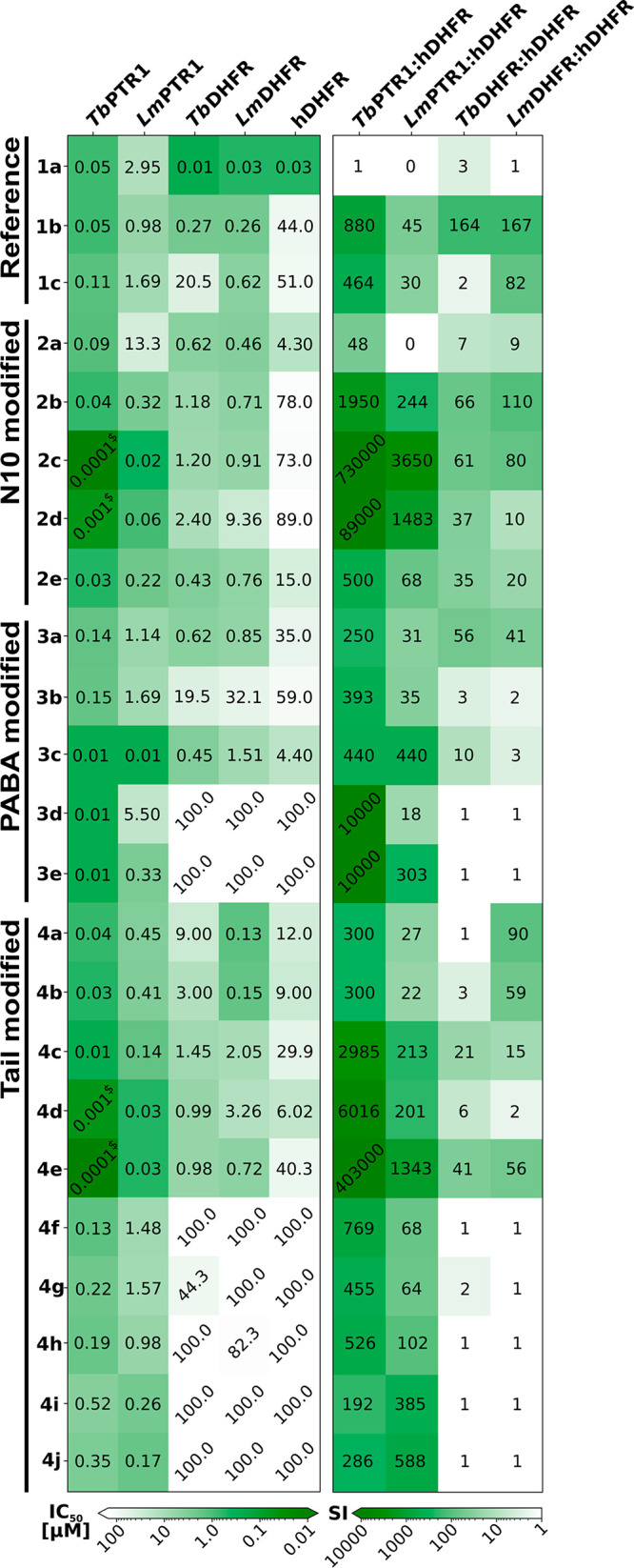
Inhibitory activities (IC50 values, left) and selectivities (selectivity indices (SI), right) of compounds of the designed N10-, PABA-, and Tail-modified series and selected reference compounds against the targets TbPTR1, LmPTR1, TbDHFR, and LmDHFR and the off-target hDHFR. All values, as well as data for hTS, are reported in Table S1. Greener boxes show higher inhibition and selectivity. $ indicates that a precise activity value could not be determined as the tight binding limit was approached.
N10 Modifications Yield Improved PTR1 Inhibitors with Similar Selectivity Trends for Parasite DHFRs
The N10-modified compounds (2a–e; Figure 6) are improved PTR1 inhibitors in comparison to 1b, except for 2a (1b IC50TbPTR1 50 nM, LmPTR1 1 μM; N10 series IC50TbPTR1 < 0.1–90 nM; LmPTR1 0.02–13.3 μM; Figure 8). 2c is the best in the series with IC50 < 0.1 nM against TbPTR1 and an IC50 of 20 nM against LmPTR1.
All compounds are roughly similar to 1b in parasite DHFR inhibition (1b IC50TbDHFR and LmDHFR 0.3 μM; N10 series IC50TbDHFR 0.4–2.4 μM, LmDHFR 0.5–9.4 μM), and selectivities over hDHFR range from 7- to 66-fold for TbDHFR and 9- to 110-fold for LmDHFR, which are somewhat lower than for 1b (SI TbDHFR/hDHFR = 164 and LmDHFR/hDHFR = 167). Thus, mainly PTR1 inhibition benefits from the selected N10 modifications.
PABA Modifications Lead to Strong Variations in the Target Inhibition Profile
The modifications of the PABA moiety in the PABA series (compounds 3a–e; Figure 6) distinctly affect the inhibitory activities against the targets. Smaller compounds with well-enclosed binding poses show varying improvements in inhibitory activity for different PTR1 variants: 3c, in contrast to most of the studied pteridines, is equipotent toward LmPTR1 and TbPTR1 (IC50 10 nM). This notable improvement of LmPTR1 activity is in line with its predicted good steric fit to the LmPTR1 binding pocket shape (compare Figures 4C and 5C). Full enclosure and stabilizing interactions with hydrophobic residues lining the pocket entrance likewise probably contribute to an around 10-fold higher potency of 3d and 3e against TbPTR1 than the most similar reference compound 1c (lacking an N10 substitution) (IC503d, 3e: 10 nM; 1c: 110 nM).
Whereas 3d and 3e do not show inhibitory activity against the parasite DHFR targets, 3c shows similar activity against LmDHFR to 1c (IC50 1.5 and 0.6 μM, respectively) and displays higher activities against both TbDHFR (IC501c: 20.5 μM; 3c: 0.5 μM) and hDHFR (IC501c: 51 μM; 3c: 4 μM). A one-carbon spacer to shift the position of the PABA carbonyl in 3a with respect to 1c improves inhibition of TbDHFR (IC50 0.6 μM) while not significantly affecting inhibition of LmDHFR and hDHFR. Thus, 3a is more selective against TbDHFR than the reference 1c (SI: 56 vs 2).
Taken together, alterations to the PABA moiety, due to its central location in the compound scaffold, different pocket sizes, and surrounding residue patterns in targets (Figure 4A) display highly variable effects on the activity profiles.
Alterations in Tail Geometry Boost PTR1 Inhibition but Can Reduce DHFR Inhibition
In the Tail-modified series (compounds 4a–j; Figure 6), hydrophobic and aromatic residues lining the pocket entrance region of PTR1 were exploited by either tail elongation or shortening. The interactions of these residues with the flexible aromatic tail of 4e (see Figure 5E) likely contribute to the boost of the IC50 against TbPTR1 to the subnanomolar range and to 30 nM against LmPTR1; these are 1000-fold and 57-fold improvements, respectively, in PTR1-inhibitory potencies compared to reference compound 1c. The shortened tails of 4c (unsubstituted piperidine) and 4d (benzene) are stabilized by the same residues and likely benefit from a better enclosure in the PTR1 pocket. Both compounds show improved TbPTR1 and LmPTR1 inhibition compared to 1b (IC50TbPTR1 4c: 10 nM, 4d: 1 nM vs 1b: 50 nM; LmPTR1 4c: 0.1 μM, 4d: 0.03 μM vs 1b: 1.0 μM).
However, shortening of the tail diminishes the inhibition of parasite DHFR, whereas it either does not affect or increases inhibition of the off-target hDHFR. Revisiting the docking predictions provides a possible explanation for this: The piperidine/benzene groups in the tails of 4c and 4d can form more extended hydrophobic interactions with Phe31 of hDHFR than with the corresponding methionine in the parasite DHFR variants (Figure 9). In the parasite protein, moreover, Asn64 in the pocket entrance of hDHFR is replaced by phenylalanine, which, upon interaction with the compound tail, becomes solvent-exposed.
Figure 9.

Docking poses for compound 4c from the Tail series (magenta carbons) in (A,B) TbDHFR and (C) hDHFR, showing differences in exposure and interactions of the PABA and Tail moieties in the two DHFRs. (A) TbDHFR pocket accommodates 4c with its tail enclosed by surrounding residues. hDHFR has a similar shape. TbDHFR is shown in a gray surface representation. (B,C) Views of the binding sites of TbDHFR and hDHFR, which are shown in cartoon representation in gray and green, respectively. Important interacting residues and the NADPH/NADP+ cofactor (black carbons) are shown as sticks. Hydrogen bonds are indicated by brown dotted lines. While the orientations of 4c are rather similar in both DHFR variants, the tail moiety is more solvent-exposed in TbDHFR: the PABA benzene and piperidine of 4c compete for interactions with Phe94 of TbDHFR, which thereby becomes exposed to the solvent. In hDHFR, the corresponding exposed residue is the polar Asn64, and the tail of 4c can interact with Phe31 deeper in the pocket, rendering the mode of binding more favorable in hDHFR. The results are presented for N1-deprotonated compounds, but similar observations were made with N1-protonated compounds (Figure S6).
Pocket size and interaction pattern differences between LmDHFR and other DHFR variants, as also discussed for the PABA series, also affect the Tail-modified compounds: for instance, 4d is more active against both TbDHFR and hDHFR than 1c (IC50TbDHFR 1 vs 21 μM, hDHFR 6 vs 51 μM), while both compounds show similar activity for LmDHFR.
Summary of the Compound Activity Profiles for the N10, PABA, and Tail-Modified Series
Taken together, most of the new pteridine derivatives display 1–2-fold greater inhibition of TbPTR1 than LmPTR1 and are more or equally active against PTR1 than the reference compound 1b. The nanomolar to subnanomolar PTR1 inhibitors show improved selectivity for PTR1 over the off-target hDHFR by up to about 3 orders of magnitude (2c, 4e: TbPTR1 IC50 < 0.1 nM; SI > 400 000) (Figure 8). The IC50 against hDHFR is typically greater than 100 μM, whereas inhibitory activities against TbDHFR and LmDHFR are higher. For parasite DHFR, the compounds with the best inhibitory activities have similar IC50 values to 1b (e.g., LmDHFR IC504a: 0.13 μM, 4b: 0.15 μM, and 1b: 0.26 μM). Thus, the newly designed compounds show improved target inhibitory profiles, particularly for the PTR1 targets, and overall good selectivity for the parasitic proteins.
Inhibitory Activity against T. brucei Is Related to the Hydrophobicity of the Compounds
Following the assessment of the improvement on the target inhibition level, we next determined the antiparasitic effect on T. brucei brucei Lister 427 bloodstream forms and L. infantum intramacrophage amastigotes (Figure 10A and Table S7). The LmPTR1 and LmDHFR proteins are highly similar to the corresponding L. infantum proteins (91 and 96% sequence identity, respectively), but in spite of the improved effect on both target proteins, the designed pteridines are mostly inactive against L. infantum. In contrast, the compounds show activity against T. brucei.
Figure 10.

Antiparasitic activity expressed as percentage of inhibition against T. brucei brucei for reference compounds and members of the N10-, PABA-, and Tail-modified series (A) and the selected representatives of the merged in silico library (B). The average of at least three independent determinations is shown with the standard deviation. The inactive compounds in the Tail-modified series, 4f, 4h, and 4j were omitted. Activities can be found in Table S7.
The multiple correlation coefficient between the TbPTR1 and TbDHFR IC50 values and the T. brucei bloodstream form inhibition is R = 0.35 (eq 3, SI), indicating that the levels of target enzyme inhibition are, for the current compounds, only weakly correlated with the exhibited antiparasitic effect when assuming a linear correlation. PTR1 inhibition alone shows a Pearson correlation R = 0.34 with T. brucei inhibition, whereas R is only 0.24 for DHFR inhibition, possibly because all studied compounds are much stronger inhibitors of PTR1 than DHFR. The low correlation for DHFR inhibition might also arise because of the competition from the high folate concentration in the medium in the parasite assays.
Another reason for the low correlations between parasite and target protein inhibition could be transport issues. For example, the charged compound tail and possible polyglutamylation of the parent MTX (1a) have previously been suggested to influence compound transport.41,42 All the newly designed pteridines lack the glutamate tail, which may affect their in vivo activities, but there might be other structural or physicochemical features that render them more or less active against parasites despite similar target inhibition. For example, for T. brucei, we noticed that while 2c and 4e have similar effects on the targets TbPTR1 (0.1 nM) and TbDHFR (approximately 1 μM), they differ notably in their inhibitory effect on the parasite bloodstream forms (75 vs 40%).
Therefore, we investigated the correlations of physicochemical properties and ADMET predictors with the measured effect on T. brucei; see Table 1. Our aim was to identify which properties were indicative of a better antiparasitic effect, possibly related to better uptake. Overall, only weak correlations of the individual properties with T. brucei inhibition were observed (Pearson R: 0.47–0.55 and −0.41 – −0.54; computed as defined in the SI). The strongest correlation was found for the predicted skin permeability, QPlogKp, as a descriptor linked to lipophilicity, (R: 0.55). The logPo/w and the binding to human serum albumin had slightly weaker correlations with the antiparasitic effect (R: 0.49 and 0.47, respectively). For these properties, an increase in the value corresponds with higher anti-T. brucei activity. In contrast, some properties showed anticorrelation, for instance, the aqueous solubility and the cohesive index44 (R: −0.54 and −0.41, respectively). Taken together, the data indicate an improved antiparasitic effect with increased lipophilicity of the studied compounds.
Table 1. Descriptors with Significant Correlations with the Observed Inhibitory Effect on T. brucei for the Reference Compounds and Pteridines of the N10-, PABA-, and Tail-Modified Series Calculated with QikProp43a.
| predicted property | QPlogKp | QPlogPo/w | QPlogKhsa | cohesive index | CIQPlogS |
|---|---|---|---|---|---|
| R | 0.55 | 0.49 | 0.47 | –0.41 | –0.54 |
| R2 | 0.30 | 0.24 | 0.22 | 0.17 | 0.29 |
| P-value | 0.003 | 0.01 | 0.01 | 0.04 | 0.004 |
| resampling recovery rate (%) | 100 | 96 | 96 | 56 | 96 |
| optimization direction | ↑ | ↑ | ↑ | ↓ | ↓ |
| covered range | –6.62 – −3.60 | –1.02–2.92 | –0.85–0.35 | 0.02–0.04 | –6.71 – −3.19 |
| recommended range | –8.00 – −1.00 | –2.00–6.50 | –1.50–1.50 | 0.00–0.05 | –6.50–0.50 |
QPlogKp: Predicted skin permeability, log Kp; QPlogPo/w: Predicted octanol/water partition coefficient. QPlogKhsa: Prediction of binding to human serum albumin. Cohesive index: Index of cohesive interaction in solids, (number of hydrogen bond acceptors × number of hydrogen bond donors × 0.5/surface area);44 CIQPlogS: Conformation-independent predicted aqueous solubility, log S with S in mol dm–3 being the concentration of the solute in a saturated solution that is in equilibrium with the crystalline solid. R (Pearson correlation) and R2 were calculated using the percentage of inhibition of the T. brucei brucei Lister 427 bloodstream form at a 10 μM compound concentration as defined in the SI. Only descriptors with at least a Pearson correlation/anticorrelation of 0.40/–0.40 and two-tailed P-values lower than the chosen significance level α of 0.05 are reported. The covered range lists property values obtained for the studied compounds, while the recommended range lists values the properties take for typical drug-like molecules. The resampling recovery rate indicates in how many cases (expressed as percentage) the same property was identified when leaving a single compound out of the data set. The optimization direction indicates whether higher or lower values would putatively lead to improved anti-parasitic effects.
Combined Modifications Yield Pteridines with Both Improved Target Inhibition and Improved Antiparasitic Activity
To explore further derivatives of the studied pteridines, we next designed a merged compound library as follows. The pteridine core scaffold was retained, and the studied compounds were decomposed into fragments of their N10, PABA, and Tail regions and recombined in silico in all possible combinations to yield 2014 derivatives (see SI for details). These derivatives were evaluated in docking studies against targets and off-targets and additionally prioritized by the physicochemical marker properties that showed correlations with the anti-T. brucei effect (Figure S9). Of the remaining 600 candidates, 6 were selected by expert opinion as representative compounds for synthesis and experimental evaluation (5a–5f, Figure 11).
Figure 11.

Inhibitory activities, selectivities, and structures of the merged series of six pteridine derivatives. (A) Activity heatmap in the top panel shows IC50 values for the targets TbPTR1, LmPTR1, TbDHFR, and LmDHFR and the off-target hDHFR. All values, as well as data for hTS, are reported in Table S1. $ indicates that a precise activity value could not be determined as the tight binding limit was approached. In the bottom panel, selectivity indices are reported. (B) Structures of the selected and synthesized pteridines in the merged series.
Two compounds, 5a and 5b, were chosen for their favorable interaction patterns and scores predicted by docking simulations. 5a combines the N10 hydroxypropyl fragment of 2e, benzyl in place of the PABA phenyl of 3a, the tail amide of reference compound 1g, and, for 5b, in addition, the tail pyrrolidine of ref 1h, which replaces the tail piperidine. The activities and predicted interactions in all parasite targets are most similar to 2e, suggesting the key importance of the hydroxy-propyl substituent to N10 for the target inhibition. Notably, while 5a is poorly selective for TbDHFR (2-fold) and modestly selective for LmDHFR (31-fold), 5b is inactive against hDHFR, resulting in SI values of 169 and 113 for TbDHFR and LmDHFR, respectively. Moreover, 5b has SI values over hDHFR of about 25 000 for TbPTR1 and 588 for LmPTR1. However, in contrast to most compounds, 5b displays a weak inhibition of hTS (IC50 29 μM, Table S1).
Four additional compounds (5c–5f, Figure 11) were prioritized based on the physicochemical marker properties. Compound 5c combines fragments of ethyl modification to N10 of 2a and the tail ethyl ester of 4a. Due mainly to the tail ester, this modification improves the inhibition for both TbPTR1 (IC50 1 nM) and LmPTR1 (IC50 0.1 μM). The activity against TbDHFR is similar to that of the N10-modified parent 2a, whereas LmDHFR and hDHFR inhibition are again influenced by the tail modification (IC50LmDHFR 5c: 0.2 μM, 4a: 0.1 μM; hDHFR 5c: 13 μM, 4a: 12 μM). Compound 5d merges the ethyl N10 fragment of 2a with the unsubstituted benzene of 4d. In TbPTR1, this boosts the nanomolar IC50 of 4d to the subnanomolar range, while the activity toward LmPTR1 remains similar to 4d. This profile can be related to the N10 ethyl, which seems disfavored in LmPTR1 as judged by the modest inhibition of the parent 2a (IC50 13.3 μM).
Compounds 5e and 5f combine the ethylphenyl(4-methoxyphenyl) ether scaffold of 4i with the benzyl and methyl N10 modifications from 2c or 1b, respectively. Both compounds are nanomolar inhibitors of both PTR1 variants. The parent compounds, 2c and 1b, inhibit the parasite DHFR variants at micromolar to submicromolar levels, while 4i is inactive against all variants of DHFR. The combination with a favorable N10 substitution is able to restore medium micromolar anti-DHFR activity for the altered scaffold of parent 4i in the parasite enzymes in 5e and 5f. Thus, combined N10 and tail modifications allowed for the species-specific optimization of the target inhibition profile.
Compounds 5d–5f show an improved percentage of T. brucei inhibition at 10 μM, in line with their selection for synthesis being motivated by altered marker properties (Figure 10B). For these compounds, EC50 values were determined; see Table 2. Indeed, the more lipophilic compounds were found to have low micromolar EC50s against T. brucei brucei, with 5d being the best (EC50 0.66 ± 0.48 μM), and they have SIs of 3–38 based on their cytotoxicity on THP-1-derived macrophages.
Table 2. Properties with a Significant Correlation with the Observed Inhibitory Effect on T. brucei for Compounds in the Merged Series Calculated with QikProp43a.
| compound | QPlogKp | QPlogPo/w | QPlogKhsa | cohesive index | CIQPlogS | %inhibition of T. brucei at 10 μM ± SD | EC50T. brucei [μM] ± SD | CC50 [μM] | selectivity index |
|---|---|---|---|---|---|---|---|---|---|
| 5a | –6.74 | –1.16 | 0.05 | 0.04 | –4.53 | 30 ± 8 | N.D. | N.D. | N.D. |
| 5b | –6.48 | –1.32 | 0.43 | 0.04 | –6.35 | 23 ± 4 | N.D. | N.D. | N.D. |
| 5c | –5.18 | 2.02 | 0.04 | 0.03 | –5.32 | 57 ± 10 | N.D. | N.D. | N.D. |
| 5d | -3.91 | 2.19 | 0.07 | 0.02 | –5.43 | 78 ± 3 | 0.66 ± 0.48 | 25 < CC50 < 50 | 38 |
| 5e | –4.60 | 3.36 | –1.23 | 0.02 | –3.20 | 100 ± 0 | 4.53 ± 0.42 | 12.5 < CC50 < 25 | 3 |
| 5f | –5.16 | 2.09 | –1.14 | 0.02 | –3.44 | 100 ± 0 | 1.30 ± 0.05 | 12.5 < CC50 < 25 | 10 |
| pentamidine | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | 0.0019 ± 0.0005 | 10 | 5263 |
The properties are defined as in Table 1. Values shown in bold face are within 90% of the previously determined top value or exceeded the previously obtained range for the reference compounds and compounds in the N10-, PABA-, and Tail-modified series; see Table 1. The activity against the T. brucei brucei Lister 427 bloodstream form at a 10 μM compound concentration (%inhibition) is given. For the most promising compounds, 5d–5f, in addition, measured EC50 values, CC50 interval estimations, and selectivity indices are reported and compared to pentamidine, a reference compound with activity against T. brucei. EC50 represents the arithmetic average of at least two independent measurements done in triplicate. CC50 estimation was done by at least three independent cytotoxicity assessments on THP-1-derived macrophages by a colorimetric MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay, as previously reported.45 The selectivity index is determined as the CC50 or lower CC50 interval estimation divided by EC50. N.D.: Not determined.
Bulky Compounds with Hydrophobic Substituents Often Display Liabilities
Potential liabilities were assessed by determining the inhibition of the hERG potassium channel as well as five isoforms of CYP450 (1A2, 2C9, 2C19, 2D6, and 3A4), cytotoxicity against A459 cells (human lung adenocarcinoma epithelial cell line), and mitochondrial toxiticity against 786-O cells (renal carcinoma cell line) for all compounds at a concentration of 10 μM. The results are shown in Figure 12. Further, the compounds were assessed for and passed a check for being pan-assay interference compounds (PAINS).
Figure 12.

Heatmap representation of the liability assessment results for all the compounds studied. Inhibition of hERG as well as five CYP isoforms (1A2, 2C9, 2C19, 2D6, and 3A4), mitochondrial toxicity (MITO), and growth inhibition of A549 cells were determined at 10 μM. The data are represented as percentages on a color scale from white (desired) to orange (undesired) with values reported in the map. For the inhibitory activities against hERG, CYP isoforms, and mitochondrial toxicity, white = 0% and orange = 100% inhibition/toxicity, while for A549 cell growth inhibition, white = 100% and orange = 0% growth. The values are reported in Tables S8 and S9.
The reference compounds and the N10 series mostly exhibit a safe profile. In contrast, aromatic modifications to the compound tail region, for instance in 3d, 4j, and 5f (PABA, Tail, and Merged series, respectively) were associated with notable hERG liabilities. Increasing the hydrophobicity of the compounds further led to liabilities against some CYP isoforms, in particular, 2C9 and 2C19. The shortened tails of 3c and 3d resulted in a strong effect on CYP isoform 2D6. Finally, several of the bulky, more hydrophobic compounds resulted in a cytostatic or cytotoxic effect on A549 cells. Overall, the liability assessment suggests that increasing hydrophobicity is associated with greater compound liabilities.
In line with these observations, two of the best inhibitors of T. brucei bloodstream forms, 5e and 5f, show 54 and 81% hERG inhibition, respectively. 5e, and in many cases 5f, affects various CYP isoforms. Finally, 5f is cytostatic with A549 cell growth reduced to 15%, and 5d shows cytotoxicity, effectively completely inhibiting cell growth. Thus, the most active inhibitors of T. brucei bloodstream forms were found to suffer from liabilities associated with their greater hydrophobicity and would require careful optimization of their cellular specificity.
Conclusions
We applied a multitarget-based approach to the development of novel therapies for HAT and leishmaniasis, in which we focused on pteridine-based inhibitors of L. major and T. brucei PTR1 and DHFR, and successfully designed the first known apparent picomolar inhibitors of TbPTR1. While LmPTR1/LmDHFR inhibition was previously explored for this compound class,26 we here demonstrated the potential of pteridine-based inhibitors against TbPTR1 and TbDHFR. We solved a crystal structure of the reference compound 1b bound to TbPTR1 to confirm the overlap in observed binding modes between LmPTR1 and TbPTR1 and the preference of the methotrexate inhibitor-like bound orientation in TbPTR1. Guided by our detailed comparative study of on- and off-targets in the parasitic and human folate pathway,31 crystal structures of reference compounds, and enzymatic evaluation of published reference pteridines,26,30 we designed 26 new pteridine derivatives that mostly have improved activity and selectivity. For their synthesis, we made use of an advanced MW-assisted protocol to improve the reaction yield of the pteroid step with reduced reaction time compared to previous synthetic procedures.33 Further determination of the crystal structures of complexes and computational docking enabled us to obtain a complete characterization of the binding modes of the pteridines to their molecular targets and supported the derivation of a SAR. The compounds were also tested against the human off-targets hDHFR and hTS. While they were sometimes only modestly selective for the parasitic DHFR variant, many showed 1000-fold and higher selectivities for PTR1 over the off-targets and thus, the novel PTR1 inhibitors can overall be considered selective for the parasite proteins.
While many compounds exhibited excellent inhibitory activity at the target level, they were often only modest inhibitors of T. brucei brucei bloodstream forms and inactive toward L. infantum intracellular amastigotes in vitro. We found that increased lipophilicity correlated with improved inhibitory effects on T. brucei. We were able to prioritize compounds for synthesis from a designed combinatorial in silico library by using predicted ADMET-related properties, which suggested a likely improvement of the trypanocidal effect. In this way, we identified three improved compounds, 5d, 5e and 5f, with low micromolar inhibition of T. brucei brucei (EC50 0.66–4.53 μM).
The modulated on-target/off-target activities and selectivities of the above compounds showed that specific combinations of the N10 and tail modifications allow a fine-tuning of the target inhibition profile for enzymes of specific parasite species. Furthermore, the strategy employed here of combining property prediction correlation with multitarget-based compound design was found to be a useful approach to discovering antiparasitic agents, even when the antiparasitic data are available only as a percentage of inhibition determined at a single compound concentration. We also note that the presence of high folate concentrations in the HMI-9 medium used in the parasite assays may have resulted in underestimated antiparasitic activity due to competition between folic acid and folate-analogue inhibitors of DHFR. However, our main aim in this work was to identify potent PTR1 inhibitors with antiparasitic activity that are capable of targeting multiple enzymes. In future work, these compounds can be progressed to more in-depth mechanistic studies in various parasite and mice models. Further, integration of transport-related considerations in the design,31 or using, for instance, structurally related scaffolds reported in the literature, which show inhibition of the Leishmania parasite, and a similar property-based correlation concept to that presented here, may help to overcome the current limitations of the pteridine-based compounds as inhibitors of intracellular parasites. Our data show that, overall, optimization for increased lipophilicity leads to more potent pteridine-based T. brucei inhibitors. However, increased lipophilicity can also introduce compound liabilities, e.g. for hERG and CYPs. Strategies to avoid these, for instance by making use of a similar property-based optimization strategy, should thus be incorporated in future design efforts.
Experimental Procedures
General Synthesis Information
Reagent grade chemicals and solvents were purchased from commercial suppliers and used without further purification. Reactions were monitored by TLC on silica gel plates (Kieselgel 60, F254, Merck) and visualized using UV light, cerium ammonium sulfate, or alkaline KMnO4 aqueous solution. Solvents are abbreviated as follows: tetrahydrofuran (THF), ethyl ether (Et2O), dimethyl sulfoxide (DMSO), dimethylacetamide (DMA), ethyl acetate (EtOAc), dichloromethane (DCM), dimethylformamide (DMF), methanol (MeOH), and acetonitrile (ACN). The structures of the isolated compounds were confirmed by NMR spectroscopy and mass spectrometry. NMR spectra were recorded on Bruker 400 and 600 spectrometers with 1H at 400.134–600 MHz and 13C at 100.62–151 MHz and are given in the Supporting Information. The purity of all synthesized compounds was determined by elemental analyses performed on a PerkinElmer 240C instrument and liquid chromatography–mass spectrometry (LC–MS) on UHPLC–HRMS (Agilent 6500 QTOF mass spectrometer) under electrospray ionization mode, with 4800 V of ion voltage at Centro Interdipartimentale Grandi Strumenti, CIGS UniMoRe). The purity of the reported compounds is >95%. Exact monoisotopic masses are reported in the Supporting information along with the melting point intervals of all compounds, which were measured on a Stuart SMP3 instrument.
General Synthetic Procedure A: Reductive Alkylation of Amines Using Nitriles (32, 33, 51, 74)
After two vacuum/H2 cycles to remove air from the reaction tube, the stirred mixture of the amine (1.0 equiv), Pd\C catalyst (10 wt % of the amine), the respective RCN (5.0 equiv), and NH4OAc (1.0 equiv) in MeOH (5.0 mL) was hydrogenated under ambient pressure (balloon) at room temperature (rt) for the appropriate time (24–36 h). The reaction mixture was filtrated using Celite cake before the filtrate was concentrated under reduced pressure. The residue was partitioned between Et2O (10 mL) and water (10 mL). The aqueous phase was extracted thrice with Et2O (10 mL), and the combined organic phases were washed with brine (10 mL), dried with anhydrous Na2SO4, filtered, and concentrated under reduced pressure to yield the amines without further purification.
General Synthetic Procedure B: Amide Coupling Reaction for the Synthesis of 27, 28, 40–49, 52–54, 56, 57, 71, 72, 75
Carboxylic acid compounds (1 equiv), EDC·HCl (1.1 equiv), and HOBt (0.1 equiv) were added to a dried round-bottomed flask and dissolved in DMF dry under N2. The reaction mixture was cooled down to 0 °C and stirred for 30 min before it was added to the respective amine (1 equiv) with/without TEA (2–3 equiv). After mixing overnight at rt, the mixture was washed 1× with saturated NaHCO3, 1× with H2O, and 1× with brine. The washed organic mixture was then dried with Na2SO4, concentrated in vacuo, and purified using column chromatography (SiO2, eluent: Cy/EtOAc or DCM/MeOH or DCM/EtOAc/MeOH) to give the desired amide.
General Synthetic Procedure C: MW Alkylation 1b,c, 2a–e, 3a–c, 4a–j, 5a–f
To a suspension of amine intermediates (1 equiv) in DMA (3 mL) in a microwave Biotage vial, 29 (1.2 equiv), K2CO3 (3 equiv), and KI (0.1 equiv) were added. The vial was sealed and heated by microwave irradiation in a Biotage Initiator+ microwave at 60 °C for 20 min (30′ for compounds 3c, 4d, and 5d), before cooling to rt and diluting with water (20 mL). The precipitate was then collected by filtration and dried before the final compound was purified by fractional crystallization from methanol, DCM, and Et2O.
General Synthetic Procedure D: SNAr for the Preparation of 4-Substituted Benzaldehyde (63–65)
A mixture of substituted phenol 58-60 (1 equiv), 4-fluorobenzaldehyde (1 equiv), and K2CO3 (3 equiv) in DMF (10 mL) was refluxed for 16–18 h under nitrogen. After cooling, the solution was concentrated in vacuo to give a crude residue, which was purified by crystallization in 1 M NaHCO3. The obtained crystal was washed with H2O to obtain the desired benzaldehyde derivatives.
General Synthetic Procedure E: Preparation of Primary Amines from 4-Substituted Benzaldehyde (66–68)
A solution of carbonyl (aldehyde) compounds 66–68 (1 equiv) and hydroxylammonium chloride (1.2 equiv) in ethanol (30 mL) was stirred for 1 h at rt. Subsequently, 12 M hydrochloric acid (4 equiv) and zinc dust (2.5 equiv) were slowly added to the solution and left to stir at rt for 15 min. To the resulting slurry, a solution of ammonia (30%, 14 mL) and sodium hydroxide (6 M, 30 mL) was added dropwise, and the mixture was stirred at rt for another 15 min. Then, the resulting solution was extracted with DCM, dried over anhydrous Na2SO4, and filtered. The solvent was removed under vacuum to give the amines without further purification.
Protein Expression and Purification
Recombinant TbPTR1, LmPTR1, TbDHFR-TS, LmDHFR-TS, hDHFR, and hTS were expressed and purified according to previously reported procedures.26,45,46
Crystallization of TbPTR1 and LmPTR1
Well-ordered monoclinic crystals of histidine-tagged TbPTR1 were obtained by the vapor diffusion hanging drop technique at rt.47 Drops were prepared by mixing equal volumes of protein and precipitant solution (2–2.5 M sodium acetate and 0.1 M sodium citrate pH 5) according to a previously described procedure.45 The TbPTR1–cofactor–inhibitor ternary complexes were obtained by the soaking technique. The compounds, solubilized in DMSO, were diluted in the cryoprotectant solution (30% vol/vol glycerol added to the precipitant solution) to a final concentration of 2–4 mM (keeping the DMSO concentration below 10% vol/vol). Crystals were then transferred in the resulting soaking/cryoprotectant solution and flash frozen in liquid nitrogen after 8–24 h of exposure.
Crystals of LmPTR1 were prepared as described elsewhere.38 The LmPTR1–cofactor–2e ternary complex was obtained by the soaking technique via the addition of 2 mM compound (solubilized in DMSO, without exceeding the 10% drop volume) directly into the crystallization drop. After 5 h, crystals were transferred to the cryoprotectant solution and flash frozen in liquid nitrogen.
Data Collection, Structure Solution, and Refinement
X-ray crystallographic data were collected using synchrotron radiation at the Diamond Light Source (DLS, Didcot, United Kingdom) beamlines I04-1 and I03 equipped with a Dectris Pilatus 6M-F and a Pilatus3 6 M detector, respectively. Reflections were integrated using MOSFLM and scaled with Scala (CCP4 suite).48−52 Data collection and processing statistics are reported in Table S2. The crystals of TbPTR1 and LmPTR1 belonged to the primitive monoclinic space group P21 and the primitive orthorhombic space group P212121, respectively. Both had a functional enzyme tetramer in the asymmetric unit. The structures were solved by molecular replacement using MOLREP and either a TbPTR1 (PDB-ID 5jdc) or a LmPTR1 tetramer (PDB-ID 5l4n) as the searching model (all nonprotein atoms were excluded).38,45,53 Models were refined using REFMAC5 (CCP4 suite).54 Visual inspection and manual rebuilding of missing atoms was performed using Coot.55,56 Water molecules were added with the automated standard procedures implemented in the software ARP/wARP and checked with Coot.57 In the higher-resolution complexes of TbPTR1 with compounds 2a and 2e, all atoms were refined anisotropically in the final refinement cycles, and hydrogen atoms were added in the calculated positions. The occupancies of exogenous ligands were individually adjusted to values resulting in atomic displacement parameters comparable to those of surrounding protein atoms in fully occupied sites. The final models were checked with Coot and Procheck.58 Statistics for data refinement are reported in Table S3. Figures were generated using CCP4mg.59 Coordinates and structure factors were deposited in the Protein Data Bank under the PDB-IDs 6rx5 (TbPTR1-NADPH/NADP+-1b), 6rx0 (TbPTR1-NADPH/NADP+-2a), 6rx6 (TbPTR1-NADPH/NADP+-2e), and 6rxc (LmPTR1-NADPH/NADP+-2e).
TbPTR1, TbDHFR, LmPTR1, LmDHFR, hDHFR, and hTS Target/Off-Target Enzyme Assays
In vitro assays for TbPTR1 and LmPTR1 were based on the coupled assay reported by Shanks et al.60 The assay nonenzymatically links the reduction of cytochrome c (Cc) with the reduction of dihydrobiopterin to tetrahydrobiopterin, catalyzed by PTR1. The formation of reduced Cc (Fe2+) results in a signal increase in the photometric readout at 550 nm wavelength. TbPTR1 and LmPTR1 assays were performed in a buffer containing 20 nM sodium citrate (pH 6.0) in a well-plate-based format as previously reported.45LmDHFR, TbDHFR, hDHFR, and hTS activities were assessed spectrophotometrically according to published procedures.61,62 Each inhibitory compound was assayed at 11 different concentrations in triplicate, and IC50 values were calculated as described in the SI.
Computational Preparation of Pteridine Compounds and Protein Receptors and SiteMap Calculation of DHFR Pocket Volumes
The 3D structures of the reference and designed compounds were generated from SMILES strings and optimized with the OPLS_2005 force field using LigPrep of Maestro (Schrödinger, LLC) as described previously, except that tautomers were created for the pH range 5.0–8.0, and both N1-deprotonated and N1-protonated tautomers were considered for every compound.45,63−66 In addition, all different substituents to the N10 position, PABA modifications, and compound tail alterations present in compounds 1b–4j were combined in all possible permutations in silico in a “merged” series and prepared similarly.
All receptors were prepared in the presence of MTX (from the following PDB-IDs for TbPTR1: 2c7v, LmPTR1: 1e7w, TbDHFR and LmDHFR: 3cl9 and hDHFR: 1u72) to improve the interactions of binding site residues and the conserved water molecules with the pteridine core. Receptor preparation was following published procedures with minor modifications.45,63,64,67−69 For the LmPTR1 (PDB-ID 1e92) and TbPTR1 (PDB-ID 2x9g) receptors, an energy minimization with a harmonic restraint of 25 kcal mol–1 Å–2 on heavy atoms and no restraint on hydrogens was performed until the heavy atom RMSD relative to the previous minimization step was less than 0.30 Å.70 For the TbDHFR receptor, PDB-ID 3rg9 was used; for LmDHFR, our previously published homology model based on a TcDHFR-TS template (PDB-ID 3inv) was chosen.31 For off-target docking, we used the hDHFR structure 1u72. For PTR1, we also considered the previously described set of conserved water molecules identified by a WatCH clustering approach.45,67 Further, using WatCH, we identified conserved water sites in hDHFR as described in the SI.67 Except for the parasite DHFR variants, where the identification of a conserved water set was not possible, all receptors were prepared both with the identified set of conserved structural waters and without explicit water molecules. Grid preparation was done as described before for LmPTR1 and TbPTR145 with the following grid centers and rotatable groups: (i) LmPTR1: center Phe113, rotatable OH in Ser111, Thr184, Tyr191, Tyr194, Thr195, Tyr283, and NADP+ ribose; (ii) TbPTR1: center Phe97, rotatable OH/SH in Ser95, Cys168, Tyr174, and NADP+ ribose; (iii) LmDHFR: center Phe31, rotatable OH/SH in Thr35, Thr36, Ser61, Cys130, Tyr137, Thr155, and NADP+ ribose; (iv) TbDHFR: center Phe58, rotatable OH in Thr46, Thr62, Thr86, Ser89, Ser98, Tyr166, Thr184, and NADP+ ribose; and (v) hDHFR: center Phe34, rotatable OH in Thr38, Thr39, Ser59, Tyr121, and NADP+ ribose.
The volumes of the binding pockets of TbDHFR (PDB-ID 3rg9), hDHFR (1u72), and the LmDHFR homology model were computed with Schrödinger SiteMap63,71,72 as described in the SI.
Computational Docking Studies
Docking studies were performed using a rigid receptor in Glide standard precision (SP) and extra precision (XP) modes and employing the Induced Fit (IF) protocol to allow for refinement of binding site residues.63,73−79 For rigid receptor docking, van der Waals radii scaling of ligand atoms and settings for sampling, addition of Epik state penalties to the docking score, rewarding of intramolecular hydrogen bonds, and enhancement of the planarity of conjugated π-groups were chosen as described previously,45 but a total of 50 poses per ligand were subjected to postdocking energy minimization. For the in silico library, we used SP docking with a constraint on all heavy atoms of the pteridine core to match the orientation of MTX in the corresponding protein receptor with a tolerance of 1 Å.
In addition, since some compounds showed major variation in substituent size when compared to the starting scaffold and explicit water molecules are treated as frozen in the standard SP/XP docking, additional studies allowing protein side chain and water reorganization in response to ligand binding were performed using the standard protocol for the IF workflow implemented in Maestro. The planarity of conjugated π-groups was enhanced, and a Prime refinement was performed for residue side chains within 5 Å of ligand atoms. XP redocking was done as previously described, yielding up to 20 receptor–ligand complexes per compound.45
The validation of the docking protocol is presented in the SI.
Computational Property Prediction, Pan-assay Interference Compounds (PAINS), and Correlation Analysis with Antiparasitic Data
Physicochemical descriptors and parameters related to ADMET were computed for all prepared compounds using QikProp (Schrödinger).43 Pearson correlations (R), R2 values, and two-tailed P-values for each property with the measured percentage of inhibition of T. brucei at the 10 μM compound concentration were computed using SciPy and Python scripts written for the purpose. Only properties with a P-value equal to or below the statistical significance level α = 0.05 were considered further. To ensure robustness, a resampling analysis was performed by leaving every compound out once before recomputing the correlations. Properties with R > 0.40 or < −0.40 and a P-value ≤ α in >50% of the resampling correlation analyses were considered to be the most robust markers for the optimization for antiparasitic effect. These properties were employed to prioritize compounds for synthesis as part of the Merged series; for details, see the SI and Figure S9.
In addition, a multivariate correlation coefficient between parasite target protein inhibition and antiparasitic activity was computed; for details, see the SI.
Finally, all synthesized compounds were checked for PAINS filters A, B, and C, undesirable substructure moieties, covalent inhibition, and compliance with the rule-of-five with the FAF-Drugs4 Web server (https://fafdrugs4.rpbs.univ-paris-diderot.fr) by inputing SMILES strings for the compounds.80
In Vitro Biological Evaluation against T. brucei and L. infantum Intramacrophage Amastigotes
The efficacy against T. brucei brucei Lister 427 bloodstream forms was evaluated in a modified resazurin-based assay as previously described.81 Cells were grown at 37 °C and 5% CO2 in a complete HMI-9 medium supplemented with 10% fetal calf serum (FCS) and 100 UI/mL of penicillin/streptomycin. The HMI-9 medium was selected due to its high folic acid content (9 μM), resulting in efficient parasite growth and enabling process standardization.82 The same medium was used in our previous experiments and allowed maintenance of similar relative folate metabolism levels between models studied in high- and low-throughput systems, namely, between Trypanosoma cells and mouse models, and between mouse models and human plasma.83,84 Cultures were then diluted to a cell density of 2 × 106 /mL. For the assay, compounds were prepared in 10 mM DMSO and diluted in HMI-9 to a 40 μM solution (0.4% DMSO). The assay solution was further used to perform serial dilutions (1:2) in a 96-well plate. Mid log bloodstream forms (100 μL) were added in complete HMI-9 medium at a final cell density of 1 × 104 /mL in a well volume of 200 μL after compound addition, leading to a maximum DMSO concentration of 0.2%. Following incubation for 72 h at 37 °C and 5% CO2, 20 μL of 0.5 mM resazurin solution was added, and plates were further incubated for 4 h under similar conditions. Fluorescence was then measured using a Synergy 2 multimode reader (BioTek) at 540 and 620 nm excitation and emission wavelength, respectively. The efficacy of compounds against L. infantum intracellular amastigotes was determined according to Sereno et al. with slight modifications described in detail in the SI.85
Liability Assays
The hERG cardiotoxicity assay was performed using the Invitrogen Predictor hERG fluorescence polarization (FP) assay. A membrane fraction containing hERG (Predictor hERG membrane) was used together with a red fluorescent high-affinity ligand of the hERG channel (Predictor hERG Tracer Red). Displacement of the latter from hERG by binding of the test compound can be determined in an FP-based format.45
Cytochrome P450 (CYP450) assays against isoforms 1A2, 2C9, 2C19, 2D6, and 3A4 were performed using the Promega P450-Glo assay platform. Microsomal preparations of cytochrome P450s from baculovirus-infected insect cells were used. In this assay, light is generated when a CYP450 enzyme acts on its substrate and a decrease thereof was indicative of inhibitory effects of the tested compound on the respective isoform.45
For monitoring mitochondrial toxicity caused by the test compounds in the 786-O cell line, uptake of MitoTracker Red (chloromethyl-X-rosamine) combined with high content imaging was used. Cells were maintained in Roswell Park Memorial Institute (RPMI)-1640 medium containing 2 mM glutamine, FCS (10% v/v), streptomycin (100 μg/mL), and penicillin G (100 U/mL).45
The cytotoxicity assay against A549 cells was performed using the CellTiter-Glo assay from Promega. The number of viable cells present is directly proportional to the cellular ATP content, which is detected. The A549 cells were obtained from DSMZ (German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany) and grown in Dulbecco’s modified Eagle medium (DMEM) with FCS (10% v/v), streptomycin (100 μg/mL), and penicillin G (100 U/mL).45
Acknowledgments
This work has received funding from the European Union’s Seventh Framework Programme for research, technological development, and demonstration under grant agreement no. 603240 (NMTrypI, New Medicines for Trypanosomatidic Infections, https://fp7-nmtrypi.eu/). We thank Prof. Antonio Carta, University of Sassari, for providing the reference compounds 1d–1h. I.P., J.P.-H., and R.C.W. gratefully acknowledge the support of the Klaus Tschira Foundation. J.P.-H. acknowledges support from the Polish National Science Centre (grant no. 2016/21/D/NZ1/02806), the BIOMS program at the Interdisciplinary Center for Scientific Computing (IWR), Heidelberg University, and the Interdisciplinary Centre for Mathematical and Computational Modelling (ICM), University of Warsaw (grant no. G70-13, GB70-11, GA73-25).
Glossary
Abbreviations Used
- DHFR
dihydrofolate reductase
- HAT
human African trypanosomiasis
- MTX
methotrexate
- NTDs
neglected tropical diseases
- PABA
para-amino benzoic acid
- PAINS
pan-assay interference compounds
- PTR1
pteridine reductase 1
- SI
selectivity index
- TS
thymidylate synthase
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c00232.
Accession Codes
Crystal structures described in this paper are available in the Protein Data Bank with identifiers: 6rx5 (TbPTR1-NADPH/NADP+-1b), 6rx0 (TbPTR1-NADPH/NADP+-2a), 6rx6 (TbPTR1-NADPH/NADP+-2e), 6rxc (LmPTR1-NADPH/NADP+-2e).
Author Present Address
⊥⊥ School of Pharmacy, University of Eastern Finland, 70211 Kuopio, Finland. (I.P.)
Author Contributions
§§ I.P. and A.Q. are joint first authors.
Author Contributions
Conceptualization, M.P.C., A.V., R.C.W.; computational methodology and investigation, I.P., J.P.-H.; chemical synthesis methodology and investigation, A.Q.; enzyme assay methodology and investigation, R.L., M.S., P.L.; crystallography methodology and investigation, G.L., F.D.P., L.D.I., C.P.; ADMET methodology and investigation, S.G., G.W., B.E., M.K.; parasite assay methodology and investigation, N.S.; writing – original draft, I.P., writing – review & editing, I.P., J.P.-H., M.P.C., R.C.W.; supervision, S.M., A.C.S., M.P.C., A.V., R.C.W.
The authors declare no competing financial interest.
Notes
Additional supplementary data are freely available at https://doi.org/10.15490/fairdomhub.1.investigation.417.1: QikProp prediction results for synthesized and in silico pteridines and corresponding SOP. PAINS filtering results, Python modules for correlating QikProp data with experimental activities and for computing a multiple correlation between target and parasite inhibition. Compound library construction data and SOP, prepared docking receptors (PDB) with SOP, all Glide XP rigid-body docking results as PDB files of the receptor–ligand complexes and SOP as well as selected discussed induced fit docking results and corresponding SOP.
Supplementary Material
References
- Neglected tropical diseases. https://www.who.int/neglected_diseases/en/ (accessed May 2020).
- The WHO Strategic and Technical Advisory Group for Neglected Tropical Diseases (WHO STAG). https://www.who.int/publications/i/item/9789240015098.
- Barrett M. P.; Burchmore R. J. S.; Stich A.; Lazzari J. O.; Frasch A. C.; Cazzulo J. J.; Krishna S. The trypanosomiases. Lancet. 2003, 362, 1469–1480. 10.1016/S0140-6736(03)14694-6. [DOI] [PubMed] [Google Scholar]
- Blum J.; Schmid C.; Burri C. Clinical aspects of 2541 patients with second stage human African trypanosomiasis. Acta Trop. 2006, 97, 55–64. 10.1016/j.actatropica.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Stuart K.; Brun R.; Croft S.; Fairlamb A.; Gürtler R. E.; McKerrow J.; Reed S.; Tarleton R. Kinetoplastids: related protozoan pathogens, different diseases. J. Clin. Invest. 2008, 118, 1301–1310. 10.1172/JCI33945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham A. C. Parasitic adaptive mechanisms in infection by Leishmania. Exp. Mol. Pathol. 2002, 72, 132–141. 10.1006/exmp.2002.2418. [DOI] [PubMed] [Google Scholar]
- Herwaldt B. L. Leishmaniasis. Lancet. 1999, 354, 1191–1199. 10.1016/S0140-6736(98)10178-2. [DOI] [PubMed] [Google Scholar]
- Castillo E.; Dea-Ayuela M. A.; Bolás-Fernández F.; Rangel M.; González-Rosende M. E. The kinetoplastid chemotherapy revisited: current drugs, recent advances and future perspectives. Curr. Med. Chem. 2010, 17, 4027–4051. 10.2174/092986710793205345. [DOI] [PubMed] [Google Scholar]
- Machado-Silva A.; Goulart Guimarães P. P.; Pereira Tavares C. A.; Sinisterra R. D. New perspectives for leishmaniasis chemotherapy over current anti-leishmanial drugs: a patent landscape. Expert Opin. Ther. Pat. 2015, 25, 247–260. 10.1517/13543776.2014.993969. [DOI] [PubMed] [Google Scholar]
- Gilbert I. H. Drug discovery for neglected diseases: molecular target-based and phenotypic approaches. J. Med. Chem. 2013, 56, 7719–7726. 10.1021/jm400362b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller J.; Hemphill A. Drug target identification in protozoan parasites. Expert Opin. Drug Discovery 2016, 11, 815–824. 10.1080/17460441.2016.1195945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsari C.; Quotadamo A.; Ferrari S.; Venturelli A.; Cordeiro-da-Silva A.; Santarem N.; Costi M. P. Chapter Two - Scaffolds and biological targets avenue to fight against drug resistance in leishmaniasis. Annu. Rep. Med. Chem. 2018, 51, 39–95. [Google Scholar]
- Shuvalov O.; Petukhov A.; Daks A.; Fedorova O.; Vasileva E.; Barlev N. A. One-carbon metabolism and nucleotide biosynthesis as attractive targets for anticancer therapy. Oncotarget 2017, 8, 23955–23977. 10.18632/oncotarget.15053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson A. C.; Wright D. L. Antifolate agents: a patent review (2010 - 2013). Expert Opin. Ther. Pat. 2014, 24, 687–697. 10.1517/13543776.2014.898062. [DOI] [PubMed] [Google Scholar]
- Hawser S.; Lociuro S.; Islam K. Dihydrofolate reductase inhibitors as antibacterial agents. Biochem. Pharmacol. 2006, 71, 941–948. 10.1016/j.bcp.2005.10.052. [DOI] [PubMed] [Google Scholar]
- Yuthavong Y.; Yuvaniyama J.; Chitnumsub P.; Vanichtanankul J.; Chusacultanachai S.; Tarnchompoo B.; Vilaivan T.; Kamchonwongpaisan S. Malarial (Plasmodium falciparum) dihydrofolate reductase-thymidylate synthase: structural basis for antifolate resistance and development of effective inhibitors. Parasitology 2005, 130, 249–259. 10.1017/S003118200400664X. [DOI] [PubMed] [Google Scholar]
- Christensen K. E.; MacKenzie R. E. Mitochondrial one-carbon metabolism is adapted to the specific needs of yeast, plants and mammals. Bioessays 2006, 28, 595–605. 10.1002/bies.20420. [DOI] [PubMed] [Google Scholar]
- Cullia G.; Tamborini L.; Conti P.; De Micheli C.; Pinto A. Folates in Trypanosoma brucei: Achievements and opportunities. ChemMedChem. 2018, 13, 2150–2158. 10.1002/cmdc.201800500. [DOI] [PubMed] [Google Scholar]
- Bello A. R.; Nare B.; Freedman D.; Hardy L.; Beverley S. M. PTR1: A reductase mediating salvage of oxidized pteridines and methotrexate resistance in the protozoan parasite Leishmania major. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 11442–11446. 10.1073/pnas.91.24.11442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson A.; Gibellini F.; Sienkiewicz N.; Tulloch L. B.; Fyfe P. K.; McLuskey K.; Fairlamb A. H.; Hunter W. N. Structure and reactivity of Trypanosoma brucei pteridine reductase: inhibition by the archetypal antifolate methotrexate. Mol. Microbiol. 2006, 61, 1457–1468. 10.1111/j.1365-2958.2006.05332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers T. J.; Beverley S. M. Folate metabolic pathways in Leishmania. Essays Biochem. 2011, 51, 63–80. 10.1042/bse0510063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong H. B.; Sienkiewicz N.; Wyllie S.; Fairlamb A. H. Dissecting the metabolic roles of pteridine reductase 1 in Trypanosoma brucei and Leishmania major. J. Biol. Chem. 2011, 286, 10429–10438. 10.1074/jbc.M110.209593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sienkiewicz N.; Ong H. B.; Fairlamb A. H. Trypanosoma brucei pteridine reductase 1 is essential for survival in vitro and for virulence in mice. Mol. Microbiol. 2010, 77, 658–671. 10.1111/j.1365-2958.2010.07236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mpamhanga C. P.; Spinks D.; Tulloch L. B.; Shanks E. J.; Robinson D. A.; Collie I. T.; Fairlamb A. H.; Wyatt P. G.; Frearson J. A.; Hunter W. N.; Gilbert I. H.; Brenk R. One scaffold, three binding modes: Novel and selective pteridine reductase 1 inhibitors derived from fragment hits discovered by virtual screening. J. Med. Chem. 2009, 52, 4454–4465. 10.1021/jm900414x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinks D.; Ong H. B.; Mpamhanga C. P.; Shanks E. J.; Robinson D. A.; Collie I. T.; Read K. D.; Frearson J. A.; Wyatt P. G.; Brenk R.; Fairlamb A. H.; Gilbert I. H. Design, synthesis and biological evaluation of novel inhibitors of Trypanosoma brucei pteridine reductase 1. ChemMedChem. 2011, 6, 302–308. 10.1002/cmdc.201000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavazzuti A.; Paglietti G.; Hunter W. N.; Gamarro F.; Piras S.; Loriga M.; Allecca S.; Corona P.; McLuskey K.; Tulloch L.; Gibellini F.; Ferrari S.; Costi M. P. Discovery of potent pteridine reductase inhibitors to guide antiparasite drug development. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 1448–1453. 10.1073/pnas.0704384105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanetich K. M.; Santi D. V. Bifunctional thymidylate synthase-dihydrofolate reductase in protozoa. FASEB J. 1990, 4, 1591–1597. 10.1096/fasebj.4.6.2180768. [DOI] [PubMed] [Google Scholar]
- Schormann N.; Senkovich O.; Walker K.; Wright D. L.; Anderson A. C.; Rosowsky A.; Ananthan S.; Shinkre B.; Velu S.; Chattopadhyay D. Structure-based approach to pharmacophore identification, in silico screening, and three-dimensional quantitative structure-activity relationship studies for inhibitors of Trypanosoma cruzi dihydrofolate reductase function. Proteins 2008, 73, 889–901. 10.1002/prot.22115. [DOI] [PubMed] [Google Scholar]
- Schormann N.; Velu S. E.; Murugesan S.; Senkovich O.; Walker K.; Chenna B. C.; Shinkre B.; Desai A.; Chattopadhyay D. Synthesis and characterization of potent inhibitors of Trypanosoma cruzi dihydrofolate reductase. Bioorg. Med. Chem. 2010, 18, 4056–4066. 10.1016/j.bmc.2010.04.020. [DOI] [PubMed] [Google Scholar]
- Corona P.; Gibellini F.; Cavalli A.; Saxena P.; Carta A.; Loriga M.; Luciani R.; Paglietti G.; Guerrieri D.; Nerini E.; Gupta S.; Hannaert V.; Michels P. A. M.; Ferrari S.; Costi P. M. Structure-based selectivity optimization of piperidine-pteridine derivatives as potent Leishmania pteridine reductase inhibitors. J. Med. Chem. 2012, 55, 8318–8329. 10.1021/jm300563f. [DOI] [PubMed] [Google Scholar]
- Panecka-Hofman J.; Pöhner I.; Spyrakis F.; Zeppelin T.; Di Pisa F.; Dello Iacono L.; Bonucci A.; Quotadamo A.; Venturelli A.; Mangani S.; Costi M. P.; Wade R. C. Comparative mapping of on-targets and off-targets for the discovery of anti-trypanosomatid folate pathway inhibitors. Biochim. Biophys. Acta, Gen. Subj. 2017, 1861, 3215–3230. 10.1016/j.bbagen.2017.09.012. [DOI] [PubMed] [Google Scholar]
- Tulloch L. B.; Martini V. P.; Iulek J.; Huggan J. K.; Lee J. H.; Gibson C. L.; Smith T. K.; Suckling C. J.; Hunter W. N. Structure-based design of pteridine reductase inhibitors targeting African sleeping sickness and the leishmaniases. J. Med. Chem. 2010, 53, 221–229. 10.1021/jm901059x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quotadamo A.; Linciano P.; Costi M. P.; Venturelli A. Optimization of N-alkylation in the synthesis of methotrexate and pteridine-based derivatives under microwave-irradiation. ChemistrySelect 2019, 4, 4429–4433. 10.1002/slct.201900721. [DOI] [Google Scholar]
- Sajiki H.; Ikawa T.; Hirota K. Reductive and catalytic monoalkylation of primary amines using nitriles as an alkylating reagent. Org. Lett. 2004, 6, 4977–4980. 10.1021/ol047871o. [DOI] [PubMed] [Google Scholar]
- Ikawa T.; Fujita Y.; Mizusaki T.; Betsuin S.; Takamatsu H.; Maegawa T.; Monguchi Y.; Sajiki H. Selective N-alkylation of amines using nitriles under hydrogenation conditions: facile synthesis of secondary and tertiary amines. Org. Biomol. Chem. 2012, 10, 293–304. 10.1039/C1OB06303K. [DOI] [PubMed] [Google Scholar]
- Ayedi M. A.; Le Bigot Y.; Ammar H.; Abid S.; El Gharbi R.; Delmas M. Synthesis of primary amines by one-pot reductive amination of aldehydes. Synth. Commun. 2013, 43, 2127–2133. 10.1080/00397911.2012.714830. [DOI] [Google Scholar]
- Gourley D. G.; Schüttelkopf A. W.; Leonard G. A.; Luba J.; Hardy L. W.; Beverley S. M.; Hunter W. N. Pteridine reductase mechanism correlates pterin metabolism with drug resistance in trypanosomatid parasites. Nat. Struct. Biol. 2001, 8, 521–525. 10.1038/88584. [DOI] [PubMed] [Google Scholar]
- Di Pisa F.; Landi G.; Dello Iacono L.; Pozzi C.; Borsari C.; Ferrari S.; Santucci M.; Santarem N.; Cordeiro-da-Silva A.; Moraes C. B.; Alcantara L. M.; Fontana V.; Freitas-Junior L. H.; Gul S.; Kuzikov M.; Behrens B.; Pöhner I.; Wade R. C.; Costi M. P.; Mangani S. Chroman-4-one derivatives targeting pteridine reductase 1 and showing anti-parasitic activity. Molecules 2017, 22, 426. 10.3390/molecules22030426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd M. D. High-Throughput Screening for the Discovery of Enzyme Inhibitors. J. Med. Chem. 2020, 63, 10742–10772. 10.1021/acs.jmedchem.0c00523. [DOI] [PubMed] [Google Scholar]
- Holdgate G.; Meek T.; Grimley R. Mechanistic enzymology in drug discovery: a fresh perspective. Nat. Rev. Drug Discovery 2018, 17, 115–132. 10.1038/nrd.2017.219. [DOI] [PubMed] [Google Scholar]
- Synold T. W.; Willits E. M.; Barredo J. C. Role of folylpolyglutamate synthetase (FPGS) in antifolate chemotherapy; a biochemical and clinical update. Leuk. Lymphoma 1996, 21, 9–15. 10.3109/10428199609067573. [DOI] [PubMed] [Google Scholar]
- Gibson M. W.; Dewar S.; Ong H. B.; Sienkiewicz N.; Fairlamb A. H. Trypanosoma brucei DHFR-TS revisited: characterisation of a bifunctional and highly unstable recombinant dihydrofolate reductase-thymidylate synthase. PLoS Negl. Trop. Dis. 2016, 10, e0004714. 10.1371/journal.pntd.0004714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger Release 2015-4: QikProp v4.6, Schrödinger, LLC: New York, NY, 2015.
- Jorgensen W. L.; Duffy E. M. Prediction of drug solubility from Monte Carlo simulations. Bioorg. Med. Chem. Lett. 2000, 10, 1155–1158. 10.1016/S0960-894X(00)00172-4. [DOI] [PubMed] [Google Scholar]
- Borsari C.; Luciani R.; Pozzi C.; Poehner I.; Henrich S.; Trande M.; Cordeiro-da-Silva A.; Santarem N.; Baptista C.; Tait A.; Di Pisa F.; Dello Iacono L.; Landi G.; Gul S.; Wolf M.; Kuzikov M.; Ellinger B.; Reinshagen J.; Witt G.; Gribbon P.; Kohler M.; Keminer O.; Behrens B.; Costantino L.; Tejera Nevado P.; Bifeld E.; Eick J.; Clos J.; Torrado J.; Jiménez-Antón M. D.; Corral M. J.; Alunda J. M.; Pellati F.; Wade R. C.; Ferrari S.; Mangani S.; Costi M. P. Profiling of flavonol derivatives for the development of antitrypanosomatidic drugs. J. Med. Chem. 2016, 59, 7598–7616. 10.1021/acs.jmedchem.6b00698. [DOI] [PubMed] [Google Scholar]
- Cardinale D.; Guaitoli G.; Tondi D.; Luciani R.; Henrich S.; Salo-Ahen O. M.; Ferrari S.; Marverti G.; Guerrieri D.; Ligabue A.; Frassineti C.; Pozzi C.; Mangani S.; Fessas D.; Guerrini R.; Ponterini G.; Wade R. C.; Costi M. P. Protein-protein interface-binding peptides inhibit the cancer therapy target human thymidylate synthase. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, E542–E549. 10.1073/pnas.1104829108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benvenuti M.; Mangani S. Crystallization of soluble proteins in vapor diffusion for X-ray crystallography. Nat. Protoc. 2007, 2, 1633–1651. 10.1038/nprot.2007.198. [DOI] [PubMed] [Google Scholar]
- Battye T. G. G.; Kontogiannis L.; Johnson O.; Powell H. R.; Leslie A. G. W. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol Crystallogr 2011, 67, 271–281. 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell H. R.; Johnson O.; Leslie A. G. W. Autoindexing diffraction images with iMosflm. Acta Crystallogr. D Biol Crystallogr 2013, 69, 1195–1203. 10.1107/S0907444912048524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Crystallogr. D Biol Crystallogr 2006, 62, 72–82. 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Evans P. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol Crystallogr. 2011, 67, 282–292. 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol Crystallogr. 1994, 50, 760–763. 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol Crystallogr. 2010, 66, 22–25. 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol Crystallogr 2011, 67, 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol Crystallogr 2004, 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol Crystallogr 2010, 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer G. G.; Cohen S. X.; Lamzin V. S.; Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008, 3, 1171–1179. 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W.; Thornton J. M. Validation of protein models derived from experiment. Curr. Opin. Struct. Biol. 1998, 8, 631–639. 10.1016/S0959-440X(98)80156-5. [DOI] [PubMed] [Google Scholar]
- Potterton L.; McNicholas S.; Krissinel E.; Gruber J.; Cowtan K.; Emsley P.; Murshudov G. N.; Cohen S.; Perrakis A.; Noble M. Developments in the CCP4 molecular-graphics project. Acta Crystallogr. D Biol Crystallogr 2004, 60, 2288–2294. 10.1107/S0907444904023716. [DOI] [PubMed] [Google Scholar]
- Shanks E. J.; Ong H. B.; Robinson D. A.; Thompson S.; Sienkiewicz N.; Fairlamb A. H.; Frearson J. A. Development and validation of a cytochrome c-coupled assay for pteridine reductase 1 and dihydrofolate reductase. Anal. Biochem. 2010, 396, 194–203. 10.1016/j.ab.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari S.; Morandi F.; Motiejunas D.; Nerini E.; Henrich S.; Luciani R.; Venturelli A.; Lazzari S.; Calò S.; Gupta S.; Hannaert V.; Michels P. A. M.; Wade R. C.; Costi M. P. Virtual screening identification of nonfolate compounds, including a CNS drug, as antiparasitic agents inhibiting pteridine reductase. J. Med. Chem. 2011, 54, 211–221. 10.1021/jm1010572. [DOI] [PubMed] [Google Scholar]
- Tan X.; Huang S.; Ratnam M.; Thompson P. D.; Freisheim J. H. The importance of loop region residues 40–46 in human dihydrofolate reductase as revealed by site-directed mutagenesis. J. Biol. Chem. 1990, 265, 8027–8032. 10.1016/S0021-9258(19)39034-9. [DOI] [PubMed] [Google Scholar]
- Schrödinger Release 2015-4: LigPrep v3.6; Epik v3.4; Protein Preparation Wizard (PrepWizard); SiteMap v3.7; Glide v6.9; Induced Fit Docking Protocol; Prime v4.2, Schrödinger, LLC: New York, NY, 2015.
- Linciano P.; Dawson A.; Pöhner I.; Costa D. M.; Sá M. S.; Cordeiro-da-Silva A.; Luciani R.; Gul S.; Witt G.; Ellinger B.; Kuzikov M.; Gribbon P.; Reinshagen J.; Wolf M.; Behrens B.; Hannaert V.; Michels P. A. M.; Nerini E.; Pozzi C.; Di Pisa F.; Landi G.; Santarem N.; Ferrari S.; Saxena P.; Lazzari S.; Cannazza G.; Freitas-Junior L. H.; Moraes C. B.; Pascoalino B. S.; Alcântara L. M.; Bertolacini C. P.; Fontana V.; Wittig U.; Müller W.; Wade R. C.; Hunter W. N.; Mangani S.; Costantino L.; Costi M. P. Exploiting the 2-Amino-1,3,4-thiadiazole scaffold to inhibit Trypanosoma brucei pteridine reductase in support of early-stage drug discovery. ACS Omega 2017, 2, 5666–5683. 10.1021/acsomega.7b00473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelley J. C.; Cholleti A.; Frye L. L.; Greenwood J. R.; Timlin M. R.; Uchimaya M. Epik: a software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- Greenwood J. R.; Calkins D.; Sullivan A. P.; Shelley J. C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. 10.1007/s10822-010-9349-1. [DOI] [PubMed] [Google Scholar]
- Sanschagrin P. C.; Kuhn L. A. Cluster analysis of consensus water sites in thrombin and trypsin shows conservation between serine proteases and contributions to ligand specificity. Protein Sci. 1998, 7, 2054–2064. 10.1002/pro.5560071002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavi Sastry G.; Adzhigirey M.; Day T.; Annabhimoju R.; Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Li H.; Robertson A. D.; Jensen J. H. Very fast empirical prediction and rationalization of protein pKa values. Proteins 2005, 61, 704–721. 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- Banks J. L.; Beard H. S.; Cao Y.; Cho A. E.; Damm W.; Farid R.; Felts A. K.; Halgren T. A.; Mainz D. T.; Maple J. R.; Murphy R.; Philipp D. M.; Repasky M. P.; Zhang L. Y.; Berne B. J.; Friesner R. A.; Gallicchio E.; Levy R. M. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. 10.1002/jcc.20292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. 10.1111/j.1747-0285.2007.00483.x. [DOI] [PubMed] [Google Scholar]
- Halgren T. A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. 10.1021/ci800324m. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. Extra Precision Glide: Docking and scoring incorporating a model of hydrophobic wnclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Sherman W.; Beard H. S.; Farid R. Use of an induced fit receptor structure in virtual screening. Chem. Biol. Drug Des. 2006, 67, 83–84. 10.1111/j.1747-0285.2005.00327.x. [DOI] [PubMed] [Google Scholar]
- Sherman W.; Day T.; Jacobson M. P.; Friesner R. A.; Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- Jacobson M. P.; Pincus D. L.; Rapp C. S.; Day T. J. F.; Honig B.; Shaw D. E.; Friesner R. A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- Jacobson M. P.; Friesner R. A.; Xiang Z.; Honig B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 2002, 320, 597–608. 10.1016/S0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- Lagorce D.; Sperandio O.; Baell J. B.; Miteva M. A.; Villoutreix B. O. FAF-Drugs3: a web server for compound property calculation and chemical library design. Nucleic Acids Res. 2015, 43, W200–W207. 10.1093/nar/gkv353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowling T.; Mercer L.; Don R.; Jacobs R.; Nare B. Application of a resazurin-based high-throughput screening assay for the identification and progression of new treatments for human African trypanosomiasis. Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 262–270. 10.1016/j.ijpddr.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirumi H.; Hirumi K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 1989, 75, 985–989. 10.2307/3282883. [DOI] [PubMed] [Google Scholar]
- Linciano P.; Cullia G.; Borsari C.; Santucci M.; Ferrari S.; Witt G.; Gul S.; Kuzikov M.; Ellinger B.; Santarém N.; Cordeiro da Silva A.; Conti P.; Bolognesi M. L.; Roberti M.; Prati F.; Bartoccini F.; Retini M.; Piersanti G.; Cavalli A.; Goldoni L.; Bertozzi S. M.; Bertozzi F.; Brambilla E.; Rizzo V.; Piomelli D.; Pinto A.; Bandiera T.; Costi M. P. Identification of a 2,4-diaminopyrimidine scaffold targeting Trypanosoma brucei pteridine reductase 1 from the LIBRA compound library screening campaign. Eur. J. Med. Chem. 2020, 189, 112047. 10.1016/j.ejmech.2020.112047. [DOI] [PubMed] [Google Scholar]
- Leamon C. P.; Reddy J. A.; Dorton R.; Bloomfield A.; Emsweller K.; Parker N.; Westrick E. Impact of high and low folate diets on tissue folate receptor levels and antitumor responses toward folate-drug conjugates. J. Pharmacol. Exp. Ther. 2008, 327, 918–925. 10.1124/jpet.108.143206. [DOI] [PubMed] [Google Scholar]
- Sereno D.; Cavaleyra M.; Zemzoumi K.; Maquaire S.; Ouaissi A.; Lemesre J. L. Axenically grown amastigotes of Leishmania infantum used as an in vitro model to investigate the pentavalent antimony mode of action. Antimicrob. Agents Chemother. 1998, 42, 3097–3102. 10.1128/AAC.42.12.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.