

Abstract
Altered metabolic activity contributes to the pathogenesis of a number of diseases, including diabetes, heart failure, cancer, fibrosis and neurodegeneration. These diseases, and organismal metabolism more generally, are only partially recapitulated by cell culture models. Accordingly, it is important to measure metabolism in vivo. Over the past century, researchers studying glucose homeostasis have developed strategies for the measurement of tissue-specific and whole-body metabolic activity (pathway fluxes). The power of these strategies has been augmented by recent advances in metabolomics technologies. Here, we review techniques for measuring metabolic fluxes in intact mammals and discuss how to analyse and interpret the results. In tandem, we describe important findings from these techniques, and suggest promising avenues for their future application. Given the broad importance of metabolism to health and disease, more widespread application of these methods holds the potential to accelerate biomedical progress.
The mammalian body breaks down nutrients from food and converts them into energy and biomolecules using metabolic enzymes (Fig. 1). One of the greatest achievements of biochemistry has been the identification of nearly all mammalian metabolic pathways. However, knowledge of how these pathways are used and regulated in vivo, including the quantitative determinants of pathway fluxes, remains incomplete.
Fig. 1 |. Mammalian metabolic fluxes.
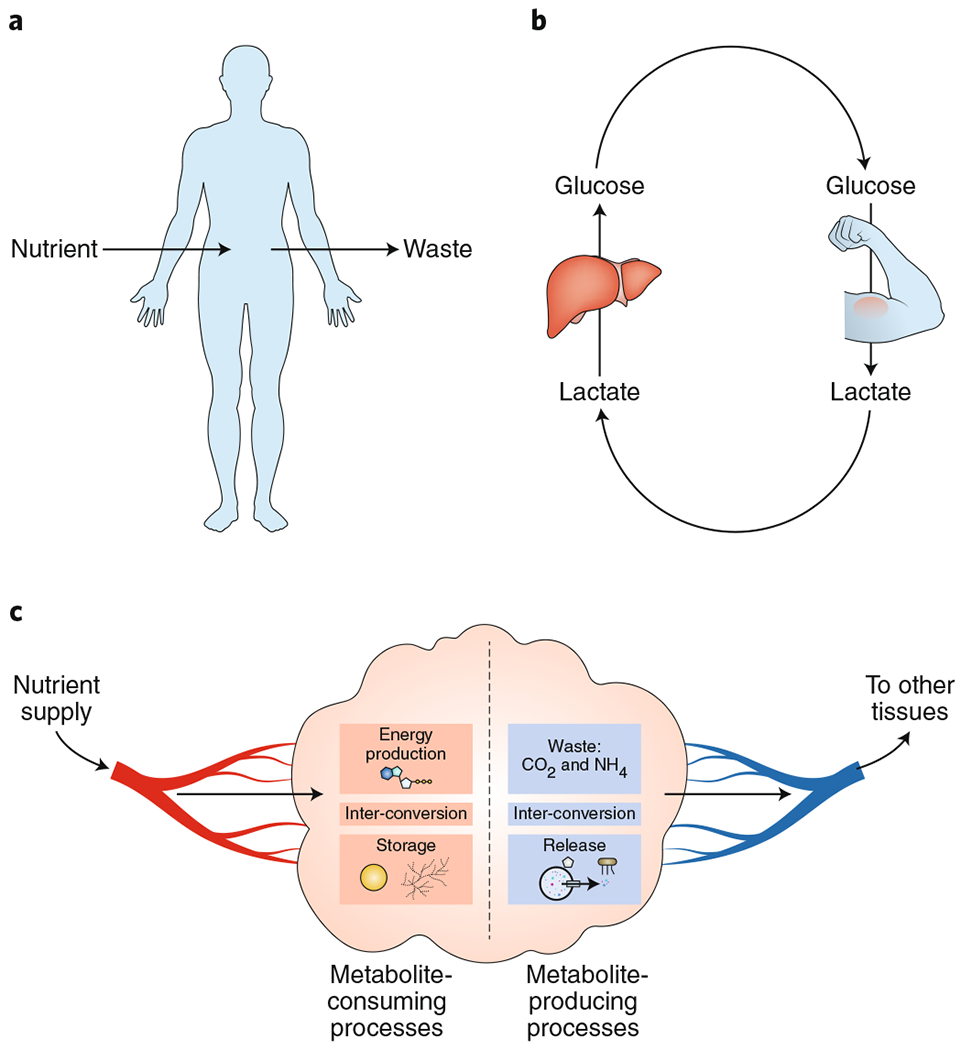
a, Organismal-level metabolism. Mammals convert oxygen and dietary nutrients into CO2, urea and other waste products. In healthy adults, these processes precisely balance at the whole-body level. b, Metabolic tasks are divided among tissues. For example, in the Cori cycle, the liver uses circulating lactate, made by muscle, to produce circulating glucose. c, At the tissue level, metabolic inputs and outputs are also subject to mass balance constraints, with net chemical transformations within the tissue measurable by sampling incoming and outgoing blood.
Beginning in the early 1900s, investigators developed approaches to measure metabolic fluxes in mammals, and their studies have helped connect metabolism to physiology. However, despite the long history of the field, measurements have remained limited to certain aspects of metabolism and disease. Motivated by diabetes, glucose has been a focal point in many studies.
Recently, there has been rising interest in more broadly and quantitatively probing metabolic flux in vivo. This interest has been catalysed by analytical advances in metabolite measurement, especially mass spectrometry1,2. It builds on a wave of research identifying metabolism as a biological outcome driver in many tissues and disease contexts.
This Perspective presents the most widely used approaches for in vivo metabolic flux measurement. We explain the underlying concepts and experimental design challenges and highlight key discoveries made using these methods (Table 1). Specifically, we describe three experimental approaches: (1) arteriovenous gradients (comparing venous with arterial metabolite levels to measure net metabolite production and consumption across a tissue); (2) circulatory fluxes (determining the circulating enrichment of an infused metabolite tracer to reveal its gross whole-body production and consumption fluxes); and (3) tissue metabolite sources (quantifying downstream metabolite labelling from infused tracers to determine the metabolite’s sources). More extensive discussion is available in Wolfe’s excellent Tracers in Metabolic Research: Radioisotope and Stable Isotope/Mass Spectometry Methods3 and several helpful reviews4,5. We close with our ideas for how these approaches can be extended in the future to push towards flux measurements for the full scope of metabolic reactions at single-cell resolution.
Table 1 |.
Summary of experimental and analytical considerations
| Section | Interpretation | Experimental design questions |
|---|---|---|
| 1. Measuring net tissue fluxes using arteriovenous sampling | This measurement yields a net flux: the imbalance between tissue production and consumption of a metabolite | Which vein should be sampled? How should internal veins be accessed with minimal metabolic perturbation (for example, using anaesthesia or catheterization)? Which tissues/cell types are drained by the vein? What is the blood flow rate to the tissue drained by the vein? How can the (often small) arteriovenous concentration differences be measured precisely enough to draw conclusions (many replicates may help)? |
| 2. Measuring circulatory turnover flux using tracer infusion | This measurement yields a gross flux: total production or total consumption of a metabolite, rather than the difference between production and consumption | Which nutrient should be infused? What form of tracer should be used (for example, which element is labelled, which position is labelled, radioactive or stable isotope used)? Should the measurement be taken at labelling pseudo-steady state? Is the individual at metabolic pseudo-steady state? What is the infusion rate (should be minimally perturbative)? Where should the blood be sampled (artery or vein)? |
| 3. Measuring sources of tissue metabolites using tracer infusion | This measurement identifies the contribution of a circulating nutrient to a tissue metabolite (not a flux) | Same choices regarding tracer, infusion and steady states as for circulatory fluxes. Which tissues should be sampled (in addition to blood, the sampling of which is required to normalize the labelling of the tissue metabolite to the infused nutrient)? Is the observed labelling directly derived from the tracer metabolite or indirectly derived after an intermediary metabolic transformation (see the section ‘Data interpretation’ under ‘Measuring sources of tissue metabolites using tracer infusion’)? |
Measuring net tissue fluxes using arteriovenous sampling
Introduction.
Quantifying organ metabolite production and consumption is fundamental to understanding metabolism. For example, the liver and kidney produce glucose, which is then used as fuel by other tissues such as skeletal muscle. How are such fluxes altered in different disease states? More generally, can we systematically measure which organs produce or consume different metabolites? In cultured cells, comparing spent media with fresh media identifies metabolites produced or consumed by the cells6,7. In the same way, comparing the venous blood exiting an organ with the arterial blood feeding it identifies metabolites that are net produced or consumed (Fig. 2). If a metabolite is being produced by an organ, it will be present at a higher concentration in the draining vein than in arterial blood, whereas if a metabolite is being consumed, the opposite will be true.
Fig. 2 |. Measuring net tissue fluxes using arteriovenous sampling.
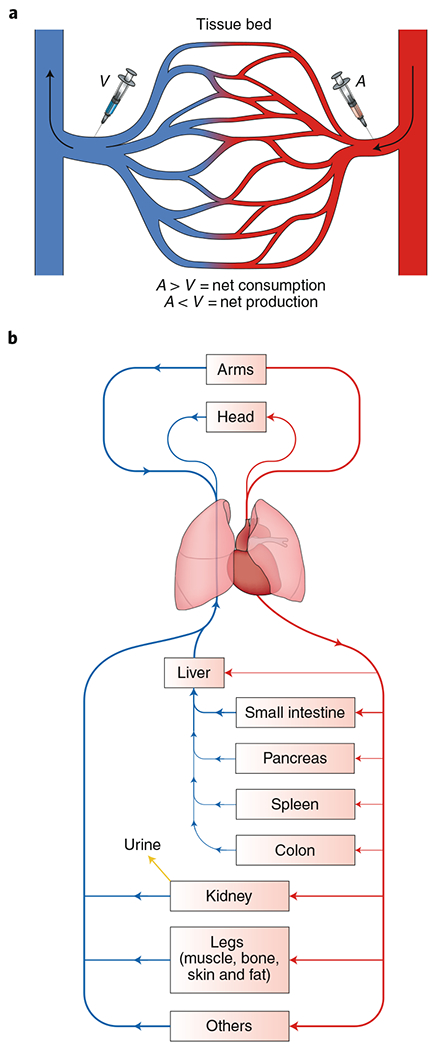
a, The net production or consumption of metabolites by an organ can be measured by sampling arterial (A) and venous (V) blood metabolite concentrations. If the metabolite concentration in the vein is higher than in the artery, the metabolite is net produced in the tissue bed. If the concentration is lower in the vein than the artery, the metabolite is net consumed. b, To quantify fluxes from arteriovenous sampling, the blood supply and flow rate to each vascular bed must be known. For example, blood supplied to the liver comes from both the hepatic artery and portal vein. Several major vascular beds involve multiple organs. For example, the portal vein drains the intestine, colon, spleen and pancreas. The femoral vein drains the muscle, bone, skin and fat in the leg.
Historical results.
Arteriovenous sampling has yielded two major types of information: how tissues exchange metabolites and how different physiological states (such as starvation, exercise or injury) alter nutrient use by specific tissues. One major nutrient exchange, identified from arteriovenous measurements of 20 amino acids, is the Cahill (glucose–alanine) cycle, in which muscle releases alanine and liver consumes it to make glucose8. Siuzdak and colleagues used modern metabolomic approaches to extend the arteriovenous measurement technique to hundreds of metabolites and found that the human arm releases the tricarboxylic acid (TCA) cycle metabolites succinate and malate9. Our group recently applied a similar metabolomic approach to a wider set of tissue-draining veins in pigs10. The study identified organ-specific production or consumption of over 250 different circulating metabolites, including multiple new inter-organ metabolic cycles. For example, most body sites, including the head and leg, produce citrate, which is in turn consumed by the kidney.
Other studies have focused on how physiological state alters tissue nutrient use. Using jugular vein blood draws, Cahill identified that the brain decreases its use of glucose and instead consumes ketones in starving human volunteers11. Studies in patients with leg burns showed that the affected limb increases its conversion of glucose to lactate, while the liver increases glucose production to balance out this fermentation12,13. During exercise, muscle increases its alanine and glutamine release, and these amino acids are then used by the liver to regenerate glucose14. More recently, measurement of arteriovenous differences across the human heart revealed that failing hearts consume ketones15. Collectively, arteriovenous studies suggest that major stresses (for example, severe burns or starvation), as well as minor metabolic changes (for example, meal consumption, mild exercise or taking common medications), can alter tissue substrate usage (for example, broadening nutrient use by inducing glucose or ketone usage)14,16–20.
Technical approach.
To measure net metabolite production or consumption using arteriovenous sampling, the investigator draws blood from one or more veins of interest and from any artery (as arterial blood is uniform). The net flux (J) of a given nutrient across the capillary bed drained by the sampled vein is given by:
| (1) |
where Q is the blood flow rate through an organ, and A and V are the arterial and venous concentrations, respectively, of the nutrient of interest. Using this equation, a positive flux J (which has units of metabolite amount per time) indicates that the organ is net consuming the metabolite, while a negative flux indicates net metabolite production (Fig. 2a). The blood flow to each organ can be measured directly (for example, by injecting beads into the artery and measuring the fraction that deposits in each tissue21,22, or by contrast ultrasound23,24) or values can be taken from the literature25. Note that blood flow, especially to organs like the heart and muscle, can change dramatically depending on physiological status (for example, exercise) or disease22,26–28.
Experimental considerations and pitfalls.
Measuring arteriovenous differences requires precise measurement of metabolite levels: for example, even a small decrease in glucose concentration in a vein compared with an artery implies biologically meaningful glucose metabolism since the total circulating glucose concentration is high. To achieve adequate confidence in such small changes, both high-quality analytical methods and many experimental replicates may be required.
Arteriovenous samples can be collected from the full range of mammals, including mice and humans. Human participants are generally cooperative, facilitating studies of behaviours such as exercise. Moreover, they have relatively large veins. In awake humans, several important veins can be accessed by catheters (for example, the brachial vein (draining the arm), femoral vein (draining the leg) and jugular vein (draining the head)). Other important veins can only be sampled in anaesthetized patients undergoing surgeries or catheterization for therapeutic purposes (for example, the coronary sinus (draining the heart), renal vein (draining the kidney), portal vein (draining the intestine, pancreas and spleen) and hepatic vein (draining the liver)).
In smaller animals, vein accessibility is limited by the skill of the investigator, since the smaller the animal, the smaller its veins. Both stress and anaesthesia can alter metabolism, so animal handling and the choice and dose of anaesthetic should be considered in the experimental design. For example, anaesthesia reduces blood flow to muscles, reduces brain glucose use and can induce global hyperglycaemia, whereas stress causes hyperglycaemia and increases TCA flux and thus whole-body oxygen consumption19,29–31.
Data interpretation.
Arteriovenous blood measurement identifies net metabolic fluxes rather than gross fluxes: if a body region both uses and produces a given metabolite in equal amounts, such metabolic activity is not detected with this approach. This is an important point given that many veins drain a mixture of tissues. For example, the femoral vein drains leg muscle, fat, bone and skin, so measured arteriovenous differences reflect the integrated activity of these diverse tissue types. Moreover, even single tissues contain diverse cell types. For example, kidney cortex produces glucose by gluconeogenesis while kidney medulla may use glucose. As these processes offset, both could be active even if the net glucose flux measured by sampling the renal vein is low. For this reason, isotope tracer measurements that measure gross flux are a useful complement to arteriovenous net flux measurements.
Measuring circulatory turnover flux using tracer infusion
Introduction.
Isotope-labelled tracers, which allow a nutrient’s atoms to be followed as they undergo enzymatic transformations, are key tools for metabolism research. Tracer infusion was first used in the 1930s at Columbia University after Harold Urey, a professor there, discovered the deuterium isotope of hydrogen32. (As an aside, for those who feel the world is currently changing quickly, it was less than 20 years between the discovery of deuterium and its weaponization in fusion bombs.) Tracers are metabolite molecules composed of atoms with excess neutrons and can be either stable or radioactive. For example, 99% of naturally occurring carbon atoms are 12C, with six neutrons to accompany their six protons. Carbon atoms with one additional neutron (13C) are stable but approximately 1 dalton heavier and thus readily distinguished by mass spectrometry; 13C’s magnetic spin also allows it to be detected by nuclear magnetic resonance (see Box 1 for a comparison of the different measurement approaches). Carbon with two additional neutrons (14C) is radioactive; radioactive tracers boost the signal-to-noise ratio but come with safety concerns and other technical considerations. To probe metabolic activity, investigators intravenously infuse such heavy-isotope tracers, typically using a venous catheter3. We will describe two major applications of tracer infusion: measuring whole-body production and consumption of the infused nutrient (this section) or measuring the contribution of the infused nutrient to downstream tissue metabolites (see the section ‘Measuring sources of tissue metabolites using tracer infusion’).
Box 1 |. Measuring metabolites and their enrichment from isotope-labelled tracers.
A variety of analytical methods can be used to measure blood and tissue metabolites and their labelling71,88.
Mass spectrometry detection of labelling.
Mass spectrometry directly detects gas-phase metabolite ions, usually after chromatographic separation, as described below. Metabolites with heavy-isotope-labelled atoms appear at higher mass-to-charge ratios (m/z), with each extra neutron in a molecule increasing its mass by approximately 1 dalton. The exact mass difference depends on the nuclear binding energy (based on E = mc2, where E is energy, m is mass and c is the speed of light), with subtle mass differences between different isotopes (for example, 15N – 14N versus 13C – 12C) sometimes feasible to distinguish using high-resolution mass spectrometry instruments such as the Orbitrap. By detecting signals at the mass of the unlabelled metabolite and every potential labelled form, mass spectrometry can reveal the fraction of the metabolite with different numbers of labelled atoms (commonly notated M + 0, M + 1, M + 2 and so on), but does not reveal the labelled atoms’ positions within the molecule. Raw isotopic distributions should be corrected for natural isotope abundance, with subsequent calculations based on the corrected labelling pattern. Software packages to perform natural isotope correction are freely available89.
Liquid chromatography–mass spectrometry (LC–MS).
LC–MS is our main method for measuring stable isotope labelling of metabolites. Blood or tissue samples extracted with organic solvent are injected onto a column that is either hydrophobic (reversed phase) or hydrophilic (hydrophilic interaction liquid chromatography (HILIC)). Metabolites are eluted by a solvent gradient and then ionized, commonly using electrospray ionization—a soft ionization technique that mostly leaves metabolite structures intact. The resulting ions’ m/z values are then measured, often by time-of-flight or Orbitrap mass spectrometry, which both broadly quantitate incoming ions with high mass resolution (that is, they have the ability to distinguish between different ions based on tiny mass differences). By separating and identifying compounds by both column retention time and m/z, LC–MS enables unambiguous identification of hundreds of metabolites. Additional information can be achieved by tandem mass spectrometry, which fragments a targeted m/z and measures the resulting fragment ions9,33,60,90.
Gas chromatography–mass spectrometry.
Metabolites are typically first derivatized to increase volatility, then vapourized and separated based on partitioning between heated gas and the gas chromatography column. The column is then eluted by ramping the temperature. Compounds exiting the column are ionized, typically by electron ionization—a hard ionization technique that tends to break up metabolites into fragment ions. The m/z ratios of the resulting ions are then measured by mass spectrometry. The strengths of this method are measuring intrinsically volatile analytes such as ethanol and enabling position-specific labelling measurement based on metabolite fragment ions45,65,71,91,92.
Isotope ratio mass spectrometry.
This approach measures the ratio of labelled to unlabelled elements (for example, 13C/12C) very precisely. The measurement requires combustion to a gas species for measurement (such as H2, N2 or CO2); thus, metabolite identity is typically lost, limiting its utility for tracing downstream metabolic transformations3,93,94.
Radioactive tracer and scintillation counter.
This approach uses radioactive rather than stable isotopes. The commonly used radioactive isotopes for metabolite tracing studies, 3H and 14C, have a shelf life of years, so storage stability is not a concern. Due to the high specific activity of tritium, the detection of 3H radioactive isotopes is much (100- to 1,000-fold) more sensitive than the detection of stable isotopes, while 14C detection is comparable to that of stable isotopes. This sensitivity enables the use of minuscule amounts of 3H tracer, minimizing metabolic perturbations. A scintillation counter measures the total abundance of labelled atoms, including fully and partially labelled forms. Like isotope ratio mass spectrometry, radioactive approaches lack the intrinsic metabolite specificity of mass spectrometry and NMR. For this reason, radioactive tracers are less useful for tracking downstream metabolic transformations, although they remain useful for Fcirc measurements and studies measuring terminal catabolism into CO2 or H2O48,56,95.
NMR.
NMR detects nuclei with a non-zero magnetic spin, including 13C, 15N and 1H. It is non-destructive and can be performed on intact animals in the form of magnetic resonance imaging. While intrinsically less sensitive than mass spectrometry, NMR can provide positional 13C-labelling information, with the abundant TCA-related amino acid glutamate often used as a primary readout in tracing studies88,96–101.
Hyperpolarized NMR.
The sensitivity of NMR can be dramatically increased using hyperpolarized 13C tracer, in which nuclear spins are aligned by dynamic nuclear polarization. Hyperpolarization decays rapidly at ambient temperatures: t1/2 = ~1 min for [1-13C] pyruvate in blood and is even shorter for other metabolites. Accordingly, most studies involve bolus injection of hyperpolarized [1-13C]pyruvate and dynamic monitoring of lactate and alanine production in vivo by magnetic resonance imaging102–104.
Positron emission tomography.
An alternative approach to measuring metabolic flux directly in vivo involves injecting a radioactive tracer that decays primarily through positron emission, with 18F and 11C being the main options. While both of these isotopes decay relatively rapidly (109 and 20 min, respectively), the greater stability of 18F has enabled widespread clinical utilization. Fluorodeoxyglucose positron emission tomography imaging tracks glucose uptake and trapping as glucose-6-phosphate within cells. It is among the most sensitive ways to detect and monitor tumours105,106.
The steady-state labelling of an infused tracer metabolite in the blood reveals its rate of appearance (Ra; the rate at which the metabolite is released from diet or tissues into the bloodstream) and rate of disappearance (Rd; the rate at which it is consumed from the bloodstream by tissues) (see Table 2 for definitions of all of the key terms used in this Perspective). In the absence of exogenous metabolite infusion, rates of appearance and disappearance of each metabolite are identical at metabolic steady state (that is, when metabolite pool sizes are not changing). This steady-state flux of metabolite between tissues and the bloodstream, as measured using minimally perturbative tracer infusion, defines the metabolite’s circulatory turnover flux (Fcirc) (Fig. 3)3,33.
Table 2 |.
Terminology
| Related literature terms | Meaning | Distinction between the terms |
|---|---|---|
| [U-13C] metabolite; [1-13C] metabolite; M + 1 metabolite |
Notations to convey the labelled form of a metabolite | [U-13C]: a metabolite uniformly labelled with 13C in all carbon positions; [1-13C]: has 13C in the 1 position, as determined by International Union of Pure and Applied Chemistry nomenclature: M + 1: has a single heavy-isotope-labelled atom in any one position |
| Circulatory turnover flux (Fcirc); Endogenous production rate; Rate of appearance (Ra); Rate of disappearance (Rd); |
The rate at which a circulating metabolite is produced or consumed by the whole body, as measured by dilution of infused isotope tracer |
Ra = turnover = whole-body production of circulating metabolite; Endogenous production rate = Ra; Rd = Ra + infusion rate; Fcirc = Ra for a minimally perturbative infusion |
| , | Circulatory turnover flux, measured by either intact tracer or average labelling remaining in the tracer |
: rate of any transformations that alter the labelling pattern of the infused metabolite; : discounts fluxes that recycle labelled tracer atoms (radioactive tracer infusions measure ) |
| Enrichment Labelling Lintact or Lavg); Atom percent excess; Specific activity |
Fractional labelling of a metabolite in the blood or tissue resulting from providing isotope-labelled tracer | Atom percent excess = Lavg: the fraction of a metabolite made up of heavy-isotope atoms from the tracer; Lintact: the fraction of a metabolite with the same number of heavy atoms as the original infused tracer; Enrichment and labelling are more general terms; Specific activity refers to radioactivity |
| Tracee; Unlabelled metabolite; Endogenous metabolite |
Metabolite endogenously present in the blood and tissues, including naturally occurring isotope forms | Tracee: unlabelled circulating form of the metabolite that is infused as the tracer; Unlabelled and endogenous metabolite are more general terms |
| Normalized labelling; Fractional contribution |
Enrichment of the downstream metabolite divided by enrichment of the precursor (for example, the tissue labelling of a metabolite divided by the circulating labelling of infused tracer nutrient) | Normalized labelling: metabolite enrichment divided by infused tracer circulating enrichment; Fractional contribution: fraction of a metabolite produced from an upstream source |
| Secondary tracer; Direct contribution; Indirect contribution |
Infused tracers can label downstream metabolites either directly or via other circulating metabolites | Secondary tracer: other circulating metabolites labelled by the tracer; Direct contribution: contribution of the infused tracer to downstream metabolite without passing through other circulating metabolites; Indirect contribution: contribution of the tracer to the downstream metabolite via a secondary tracer |
Fig. 3 |. Measuring circulatory turnover flux using tracer infusion.
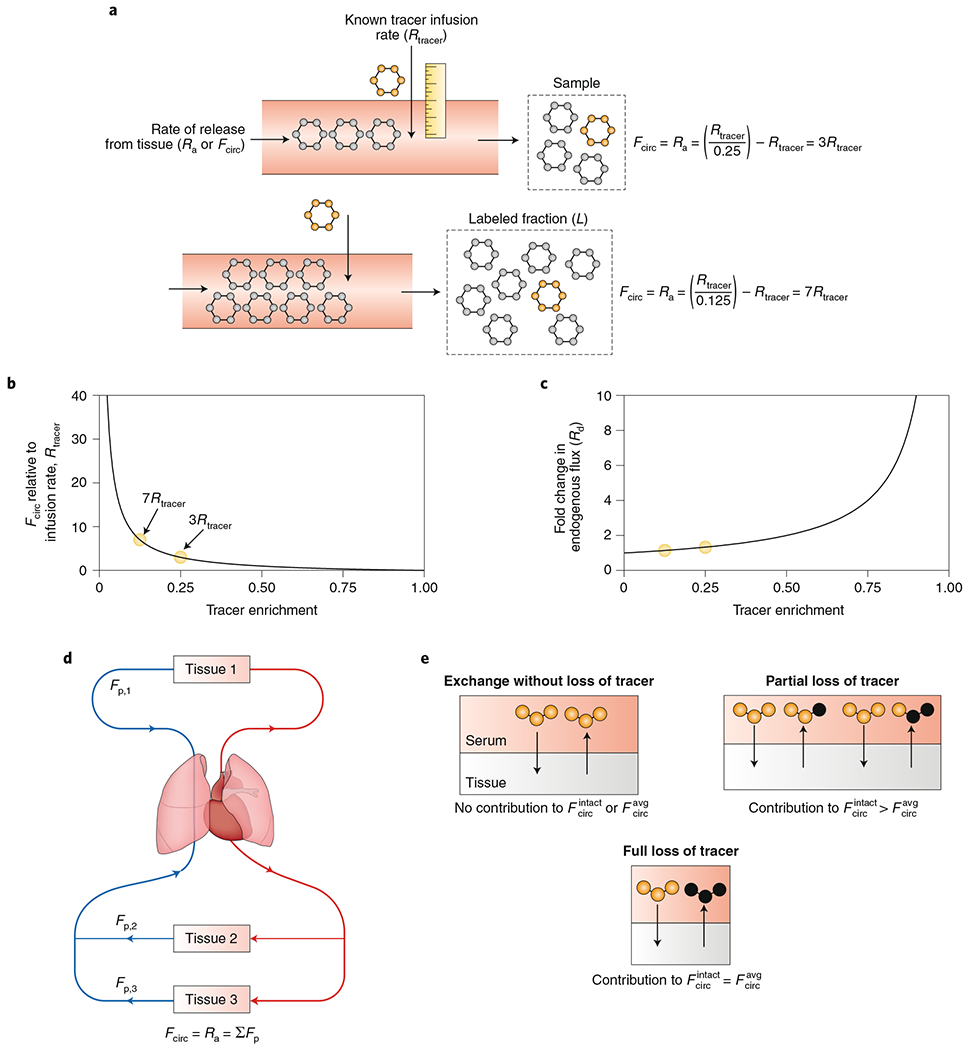
a, The circulatory turnover flux (Fcirc) of a metabolite, also known as its rate of appearance (Ra), is the whole-body rate of release of that metabolite from tissues into the circulation. To calculate Fcirc, an investigator infuses an isotope-labelled metabolite and measures the labelled fraction of that metabolite in the blood. b, Quantitative relationship between Fcirc and observed labelling for a fixed labelled-metabolite infusion rate. c, Quantitative relationship between observed labelling and perturbation of the endogenous metabolic flux. This graph assumes that the body compensates for the labelled-metabolite infusion by increasing Rd. Irrespective of the form of compensation, infusions that produce high circulating metabolite labelling are always perturbative, while those that produce low labelling are not. The degree of metabolic perturbation is modest for labelling of <25% and increases rapidly for labelling of >50%. d, Fcirc is the sum of the gross production fluxes of the metabolite in each tissue. e, Mass spectrometry measurements of labelled forms define two variants of Fcirc. measures all reactions that reduce the number of atoms labelled in the infused nutrient, while discounts reaction pathways that recycle portions of certain tracer atoms.
Historical results.
The faster a metabolite is taken up and released by tissues, the higher its Fcirc. Therefore, metabolites with high carbon Fcirc are the major players in carbon transport in the circulation and thus energy metabolism. Recently, our group measured the Fcirc of 31 different carbon-labelled metabolites to identify which had the highest flux33. These metabolites were selected based on their abundance in the blood, since the Fcirc for a metabolite cannot exceed the amount pumped by the heart (calculated by the product of cardiac output and the metabolite’s blood concentration). Due to this limitation, low-abundance metabolites cannot be major carbon carriers.
Glucose is the main dietary carbohydrate source for mammals. It is the most abundant nutrient in the blood (5 mM or 30 mM of carbon atoms as glucose contains six carbons). Consistent with its central metabolic role, glucose exhibits a high turnover flux. Blood glucose concentration is tightly regulated by insulin and dysregulated in diabetes, and its turnover has been studied extensively. Early studies showed that depancreatized dogs have increased glucose turnover, probably due to the lack of insulin causing uncontrolled high glucose in their bloodstream3,34,35. To study the effects of insulin without the confounding factor of altered glucose levels, investigators developed the hyperinsulinaemic–euglycaemic clamp technique (Box 2)4,19,36,37. This differs from typical Fcirc measurement protocols in intentionally infusing perturbative quantities of both insulin and glucose, with the insulin infusion rate fixed and the glucose infusion rate varied to ‘clamp’ (that is, maintain) glucose at its normal level. Clamp studies have shown that insulin increases the disposal of glucose, while decreasing its release from tissues via glycogenolysis and gluconeogenesis.
Box 2 |. Measuring whole-body glucose metabolism using a glucose clamp.
Glucose is the major component of dietary carbohydrates and of the carbohydrate storage polymer glycogen. Its level in the blood is tightly regulated: a relatively modest elevation in blood glucose after a meal stimulates insulin secretion107. Insulin both increases tissue glucose uptake via glucose transporter type 4 (GLUT4) translocation and reduces tissue glucose release via suppression of glycogenolysis and gluconeogenesis.
Starting in the 1960s, investigators developed the glucose clamp model, which enables measurement of the rates of circulating glucose production and consumption while controlling for the confounding effects of time-varying glucose levels4,19,36,37,108. The most common type of glucose clamp—the hyperinsulinaemic-euglycaemic clamp—measures glucose metabolism in response to insulin. Here, we provide a concise overview of this approach, which has been reviewed more extensively elsewhere4,19,36.
The hyperinsulinaemic–euglycaemic clamp reveals two pieces of information: how fast tissues are disposing glucose in response to insulin and how much residual glucose production (gluconeogenesis and/or glycogenolysis) occurs in the presence of insulin. A typical glucose clamp experiment consists of a priming bolus followed by a steady infusion of labelled glucose (often [3-3H] radioactive glucose), as well as a steady infusion of insulin, with an additional, varying-rate infusion of unlabelled glucose to maintain target blood glucose. Blood is sampled from an arterial catheter every 5–10 min. The glucose level is tested quickly with a hand-held glucose monitor and the rate of unlabelled glucose infusion is modified if the glucose level is lower or higher than the target level. When conducted in mice, heparinized blood from another mouse is also infused to replace the blood volume removed for the frequent glucose testing109.
To evaluate glucose metabolism in response to hyperinsulinaemia using a clamp, a key piece of data is the rate of unlabelled glucose infusion required during the experiment to maintain euglycaemia. As the pool size of glucose in the blood is held steady during the clamp:
| (11) |
where Rd = Rtracer/Ltracer, Ra (also in some cases referred to as endogenous Ra, endogenous glucose production or hepatic glucose production) is the glucose production by tissues via gluconeogenesis or glycogenolysis, and Rtracer and Runlabelled glucose are labelled and unlabelled glucose infused by the investigator. Because of the substantial unlabelled glucose infusion, Rd >> Ra during a hyperinsulinaemic–euglycaemic clamp. Some investigators omit tracer infusion and classify an individual as insulin resistant based solely on the glucose infusion rate required to maintain euglycaemia. Tracing can separate whether insulin resistance is due to lower Rd (slower glucose disposal by tissues) or higher Ra (failure to suppress glucose production).
A common addition to the glucose clamp is a bolus of radioactive 2-deoxyglucose towards the end of the clamp, with the goal of probing the rate of glucose uptake and phosphorylation in tissues of interest110. The compound 2-deoxyglucose is an analogue of glucose that is taken up by glucose transporters, phosphorylated by hexokinase and trapped in tissues in the phosphorylated state (except for tissues with glucose-6-phosphatase activity). The glucose uptake rate is typically calculated as the tissue 2-deoxyglucose-phosphate concentration 30 min after 2-deoxyglucose bolus delivery divided by the area under the curve of blood 2-deoxyglucose during this time. This method assumes that the glucose analogue is metabolized equivalently to glucose.
The clamp technique has been powerfully applied to quantify how insulin and other stimuli regulate glucose metabolism. For example, Shulman and colleagues showed that infusing high rates of free fatty acids decreased Rd during hyperinsulinaemic clamp in humans, while exercise training increased it92,98. Hyperinsulinaemic clamp has also been applied in humans to validate the widely used homeostasis model assessment (HOMA) calculation that predicts insulin resistance from fasting glucose and insulin levels111, as well as to show the efficacy of thiazolidinediones in increasing insulin sensitivity112.
In genetically modified mice, investigators have applied the clamp approach to investigate how different proteins in specific tissues influence insulin sensitivity. For example, mice lacking the insulin receptor in the liver or those that are heterozygous for glucokinase in the liver displayed low Rd in response to hyperinsulinaemic clamp, as well as hyperglycaemia in the fed state, similarly to humans with diabetes, suggesting a major role for hepatic glycogen storage in glucose disposition113–115.
Lactate is also one of the most abundant circulating metabolites, but its blood concentration is markedly lower than that of glucose (1 mM or 3 mM carbon atoms). Nevertheless, lactate displays high circulatory flux33,38–40. Indeed, even after correcting for the twofold greater number of carbon atoms in glucose, the carbon turnover flux of lactate is yet higher than that of glucose in fasting rodents33,41. In fasting humans, on a per-carbon basis, the turnover of glucose is slightly greater than that of lactate, and heavy exercise increases the per-carbon turnover of lactate so that it is yet greater than that of glucose41–43. The biological meaning of this high lactate flux has been debated extensively. A fundamental conclusion is that the carbohydrate oxidation pathway, from glucose to CO2, passes through circulating lactate: cells convert glucose into circulating lactate, which is then used as a TCA cycle fuel33. However, the lactate turnover value is too high to solely represent lactate oxidation in the TCA cycle, as in some contexts it exceeds the total rate of whole-body carbohydrate burning44. Therefore, the high lactate flux also encompasses metabolism of circulating lactate by pyruvate cycling and gluconeogenesis. Lactate turnover flux does not necessarily represent net transfer of lactate between tissues; rather, it represents gross tissue uptake and production. Some of the flux reflects cells that simultaneously produce and consume circulating lactate. In addition, lactate may be exchanged between tissues within the same vascular bed; for example, between fat, fast twitch muscle and slow twitch muscle within the leg41.
Three fatty acids are abundant in the circulation: palmitate, oleate and linoleate. These are next in line behind glucose and lactate in terms of carbon atom flux. Non-esterified fatty acids in the blood chiefly come from adipose tissue lipolysis45. Around two-thirds of fatty acid flux is consumed by re-esterification into triglyceride, while the remaining one-third is burned by tissues46. Fatty acids have higher flux in the fasted state and lower flux in the fed state. This inverse pattern to glucose flux reflects insulin’s dual role as a master promoter of tissue uptake of glucose and a suppressor of lipolysis47,48.
The other essential category of circulating nutrients are amino acids, which carry both carbon and nitrogen and serve as the building blocks of proteins. The carbon Fcirc of most amino acids correlates to their abundance in protein, because they are chiefly produced by protein degradation49,50. Glutamine, alanine, glycine and serine display higher Fcirc values, reflecting their extensive production from de novo synthesis in addition to protein degradation, as well as their consumption in nucleotide synthesis, gluconeogenesis and nitrogen shuttling in addition to protein synthesis33. Whole-body fluxes of essential amino acids are increased in the fed state compared with the fasted state and are also increased by sepsis or severe burns50–52.
Collectively, Fcirc measurements suggest that much of the circulating metabolic flux is futile, supporting cycles of glycolysis and gluconeogenesis, triglyceride hydrolysis and fat re-esterification, and protein degradation and synthesis. A benefit of these apparently futile fluxes is maintaining a robust supply of circulating nutrients53.
Technical approach.
To measure circulatory fluxes, investigators infuse a heavy-isotope-labelled nutrient and measure its resulting pseudo-steady-state circulatory labelling by either mass spectrometry or radioactivity measurement (Box 1). Fcirc, Ra and Rd are then determined by the following equations:
| (2) |
| (3) |
For minimally perturbative infusions (Ltracer ≪ 1), Fcirc = Ra where Rtracer is the rate of tracer infusion and Ltracer is the fraction of the infused metabolite in the blood retaining labelling. Assuming that Ltracer is small, Fcirc, Ra and Rd are all about the same (Fig. 3b,c).
What is the logic underlying equations (5) and (3)? Consider the example of glucose infusion in a fasting mammal. Blood glucose comes from two sources: unlabelled glucose produced by the tissues; and labelled glucose from the infusion (Fig. 3a). For a fixed infusion rate, if endogenous glucose production is fast, the infused labelled glucose will be extensively diluted and, as a result, labelling in the bloodstream will be low. In contrast, if endogenous production is slower, dilution will be less and labelling will be greater. In essence, the infusion serves as a flux measuring stick: since the rate of labelled infusion is known, using the ratio of labelled to unlabelled metabolite in the blood, the investigator can calculate the rate of unlabelled-metabolite production by the tissues (Ra and Fcirc).
Experimental considerations and pitfalls.
To determine Fcirc, circulating enrichment Ltracer should be measured at the metabolic and isotopic pseudo-steady state and ideally in arterial blood. To avoid the tracer infusion itself changing metabolite levels or fluxes, the infusion rate should be set low enough to limit perturbation of metabolism (Fig. 3b,c). Low circulating enrichments (<25%) are associated with minimal metabolic perturbation and high enrichments (>50%) are associated with marked perturbation. Yet, the tracer infusion rate has to be high enough to enable robust detection of blood labelling (and, if desired, downstream tissue metabolite labelling; see the section ‘Measuring sources of tissue metabolites using tracer infusion’). For 13C and 15N stable isotope tracers, we have found that a circulating enrichment of 5–25% typically balances these demands.
How long should tracer be infused before tracer enrichment is measured? As mentioned above, equations (5) and (3) assume an isotopic steady state, meaning that tracer enrichment in the circulation is not changing. The duration of tracer infusion required to reach the isotopic steady state depends on Fcirc (greater flux results in a faster approach to the steady state), as well as the metabolite’s overall abundance in the body (greater abundance results in a slower approach to the steady state)3,33. To determine Fcirc, an effective approach is to sample the blood serially until blood tracer enrichment plateaus. Strictly speaking, initial plateau labelling typically reflects the isotopic pseudo-steady state rather than a true steady state. Over longer infusions (for example, >4 h), labelling will rise further as endogenous precursors (such as protein for amino acids or glycogen for glucose) become labelled, giving rise to apparent decreases in Fcirc51,54. Normally, however, experimenters are interested in the total nutrient production rate, including rates of production from long-term storage depots, which is captured by the initial plateau attained within a few hours of infusion.
Arterial blood sampling is the best approach to measuring Fcirc, as the infused tracer and endogenous metabolites converge and mix thoroughly in the heart and then pass into the arteries. When an organ produces the metabolite of interest, the produced metabolite will dilute labelling in the draining vein, leading to overestimation of Fcirc33,38,55. In practice, substantial label dilution between the arterial and venous circulation occurs only for a few metabolites that have fast turnover relative to their circulating concentration, including lactate, acetate and alanine, and for these metabolites arterial sampling is preferred33,38,55,56.
The precision and accuracy of Fcirc measurements depend both on animal handling or patient stress and tracer measurement in the resulting blood samples. Especially for glucose and lactate, stress can augment metabolic fluxes, leading to elevated Fcirc. Because blood sampling causes stress (in rodents29 and also many patients), blood collection with minimal handling and discomfort (that is, through an arterial catheter) is ideal. In our experience with mice, a familiar experimenter who calmly enters the room and waits silently for the mice to be relaxed before drawing from a pre-implanted catheter tends to measure glucose and lactate Fcirc values up to twofold below those of a laboratory member who touches the mice to collect blood. On the analytical chemistry side, Fcirc values depend solely on isotope ratios, not absolute signals, and this makes mass spectrometry measurements robust and precise, with Fcirc changes of around 1.5-fold typically reproducible and biologically meaningful.
A final note on Fcirc experimental design: the above discussion assumes infusion through a catheter. Catheterized animals can be purchased from major animal vendors, or animal catheters can be surgically placed in house. Alternative strategies, such as intravenous or intraperitoneal injection, while tempting, generate non-steady-state data confounded by tracer distribution, and thus do not reliably determine Fcirc3. Catheter placement is worth the effort!
Data interpretation.
Tracers can be labelled by radioactive or stable isotopes, in one or multiple atom positions. The advent of modern mass spectrometry has led to expanded use of stable isotopes. Beyond avoiding concerns with radioactivity, mass spectrometry measurement of stable isotope labelling enables detection of the quantitative extent of labelling in specific metabolites—a major advantage compared with measuring overall radioactivity (Box 1). This comes with the downside of less sensitive tracer detection and thus a requirement for a higher tracer infusion rate.
Interpretation depends on the specific labelled atoms in the tracer, with carbon labelling most common. Special care must be taken when labelling hydrogen because: (1) heavy forms of hydrogen (deuterium or tritium) can slow down metabolic reactions due the kinetic isotope effect, which is minimal for other elements57; and (2) deuterium or tritium can be lost through exchange with water spontaneously or in an enzyme-catalysed manner58.
Importantly, tracers labelled in different atom positions probe different metabolic reactions. For example, reversible flux through the lactate dehydrogenase reaction does not alter the labelling of 13C-lactate but will remove 2H from lactate’s central position. Accordingly, [2-2H] lactate shows a higher Fcirc than [U-13C]lactate39,40. Thus, although it is convenient to discuss Fcirc as a property of a metabolite (implicitly referring to the uniformly 13C-labelled form), Fcirc is more precisely a property of a particular metabolite atom or atoms.
Relatedly, circulatory flux can be defined either in terms of any reactions that modify the exact infused labelled form (, calculated based on Lintact; that is, the fraction of circulating tracer in the infused form) or in terms of the infused tracer’s average circulatory labelling (, calculated based on Lavg; that is, the average labelling of the circulating tracer) (Fig. 3e)33. For tracers containing a single labelled atom, these are identical, but for [U-13C] tracers, they differ. For example, infusion of [U-13C]glucose (with all six carbons labelled) can generate circulating M + 3 glucose by being broken down into lactate, which then recombines with an unlabelled triose during gluconeogenesis. Radioactive experiments measure , which can be calculated in stable isotope tracer experiments, for a tracer with n atoms labelled:
| (4) |
| (5) |
Here, M + i labelling conveys the number of labelled atoms in a molecule of the nutrient; for example, M + 2 is a nutrient with two labeled atoms. discounts flux that recycles atoms from the infused tracer back into the traced nutrient; thus, it is less than or equal to (Fig. 3e).
Fcirc measures gross metabolite flux: when a tissue takes in the tracer and excretes the same metabolite in unlabelled or partially labelled form, this generates circulatory turnover flux (Fig. 3). Tracer entry into tissues followed by subsequent release without chemical modification does not generate circulatory turnover flux. Therefore, Fcirc does not measure mere transport of a metabolite into and out of tissues, but instead gross biochemical production and consumption of the metabolite by the whole body (Fig. 3d). For some metabolites, gross flux far exceeds net flux. For example, lactate is both produced and consumed by many organs, so lactate Fcirc is greater than the summed net lactate production across organs10,38.
Measuring sources of tissue metabolites using tracer infusion
Introduction.
Metabolism consists of enzymatic transformations: nutrients we eat pass from the intestine into the bloodstream and are then ultimately burned (transformed into CO2) or used to construct biomolecules (for example, protein, fat or glycogen). Along the way, nutrients may be transformed into a number of intermediate metabolites.
We can determine the inputs to tissue metabolism by infusing isotope-labelled forms of putative source nutrients and monitoring downstream tissue metabolite labelling. It is important to note that such measurements, when conducted at pseudo-steady state, do not reveal metabolic fluxes; rather, they measure the fraction of the labelled metabolite derived from the infused circulating nutrient. In contrast, pre-steady-state measurements can reveal fluxes (Box 3). The very first studies with labelled tracers were conducted to track metabolic transformations. For example, investigators fed mice deuterium-labelled stearate (C18:0 fatty acid) and found that the body transformed it to palmitate (C16:0)59. Recently, the study of contributions of circulating metabolites to tissue products has been rapidly expanding, due in large part to advances in the measurement of stable-isotope-labelled products by mass spectrometry60–62.
Box 3 |. Quantifying tissue production fluxes by measuring pre-steady-state labelling of metabolic products.
When a tracer is infused, labelling initially increases linearly and is followed by saturation (Fig. 4). Measuring pre-steady-state labelling of a metabolite produced from a circulating tracer enables calculation of the production flux of that metabolite, in contrast with measuring pseudo-steady-state labelling, which does not measure flux but only the fractional sources of the metabolite (see the section ‘Measuring sources of tissue metabolites using tracer infusion’). Flux calculation from pre-steady-state labelling requires that tracer enrichment in the blood approaches the steady state more rapidly than target analyte enrichment. Such a flux measurement has been achieved for TCA intermediates99,100, nucleotides, fats45,116,117, cholesterol118, glycogen119 and protein18,49,120. Labelling data can be fitted either with a line (focusing solely on pre-steady-state measurements) or with a rising exponential (including data as the labelling approaches the steady state)121.
Historical results.
A number of important studies have examined the contribution of circulating nutrients to tissue metabolites3,5. We will focus on two selected examples: inputs to tissue TCA cycle metabolism and inputs to liver triglycerides. Since most ATP is made by the TCA cycle, knowledge of which nutrients are the main TCA fuels is fundamental. Studies infusing nutrient tracers (including glucose, lactate, amino acids and fatty acids) revealed that, in a fasted individual, most tissues fill their TCA cycle with carbon from circulating fatty acids and lactate33,53. The brain is an exception to this pattern, relying chiefly on circulating glucose. Among the non-brain tissues, there are striking differences in amino acid consumption. For example, glutamine contributes 40% of TCA cycle carbon in the pancreas but almost none in the heart33,53.
Starting from Otto Warburg’s work on aerobic glycolysis, there has been avid interest in understanding how tumour metabolism differs from that of normal tissues. In vitro, glutamine is the dominant TCA fuel in cancer cells63. However, in vivo, tumours use similar nutrients to fuel their TCA cycle as their tissues of origin do33,64–66. Moreover, despite their glycolytic nature, some tumours also use circulating lactate as a TCA input33,65,67.
Like cancer, liver triglyceride excess is a major health problem in developed countries. Non-alcoholic fatty liver disease, which may afflict up to half of American adults68, occurs when triglyceride accumulates in the liver, and can progress to fibrosis and loss of liver function. The fatty acids in liver triglycerides can come from three sources: dietary fat, adipose lipolysis or de novo lipogenesis. To determine which source contributes the most to liver fat, human volunteers received infusions of [1,2,3,4-13C]palmitate and [1-13C] acetate for 4 d, then consumed deuterated triglycerides in a meal45. Their livers were then biopsied. In this strategy, deuterated liver fat comes from the meal, M + 1 13C-labelled liver fat comes from de novo lipogenesis and M + 4 13C-labelled liver fat comes from adipose lipolysis. This study revealed that roughly two-thirds of fatty acids in liver triglycerides come from adipose tissue lipolysis45,69. The contribution of de novo lipogenesis to liver fat, however, more than doubles (to about 20%) in individuals with fatty liver disease45,70.
Technical approach.
To measure how much of a tissue metabolite y derives from a circulating nutrient x, an investigator infuses x until y has reached the labelling pseudo-steady state (Fig. 4). This typically occurs within minutes to a few hours if the tissue metabolite of interest is a central carbon metabolite (for example, in glycolysis or the TCA cycle)3,33, but may take much longer for storage biomolecules such as triglycerides45. Tissues are then harvested and metabolites extracted. To prevent continued metabolic activity after tissue harvesting, which can alter metabolite levels and labelling, we cool tissues immediately with a liquid nitrogen-cooled metal clamp. Samples are then kept at −70°C or lower during storage and grinding with a liquid nitrogen-cooled cryomill, until they are extracted with organic solvent with formic acid to denature enzymes71.
Fig. 4 |. Pre-steady-state versus steady-state measurements of tissue metabolite labelling.
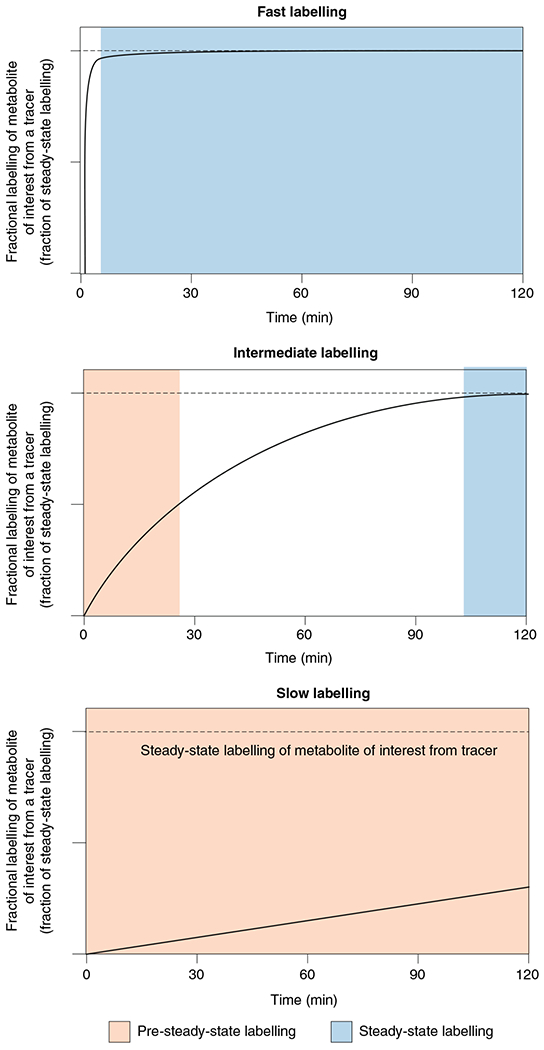
Pre-steady-state (slow) labelling of a metabolite from tracer infusion (bottom) can directly measure the production flux of the metabolite (for example, in protein synthesis or de novo lipogenesis). Steady-state (fast) metabolite labelling (top) does not measure production flux but rather the fraction of metabolite contributed by the circulating tracer nutrient (for example, in glycolysis). Intermediate labelling (middle) can measure both, depending on the time point at which tissues are sampled (for example, in the TCA cycle).
The circulating tracer nutrient typically consists of a mix of unlabelled, fully labelled and partially labelled forms due to scrambling by tissue metabolism. The fractional carbon contribution of nutrient x to metabolite y (Ly←x) can be calculated based on their average carbon atom labellings using equation (5):
| (6) |
For example, if we infuse [U-13C]glutamine and find that 10% of carbons in circulating glutamine are labelled while 2% of carbons in small intestinal malate are labelled, we divide 0.02 by 0.1 to find that 20% of small intestinal malate is derived from circulating glutamine (Fig. 5a). The remaining 80% is derived from other nutrients, such as circulating lactate and free fatty acids. By performing infusion experiments with different 13C tracers, and using the calculations described below to dissect the contributions of blood metabolites that interchange with each other (see the section ‘Data interpretation’ under ‘Measuring sources of tissue metabolites using tracer infusion’), one can also calculate the direct carbon sources of tissue metabolites33,53.
Fig. 5 |. Measuring sources of tissue metabolites using tracer infusion.
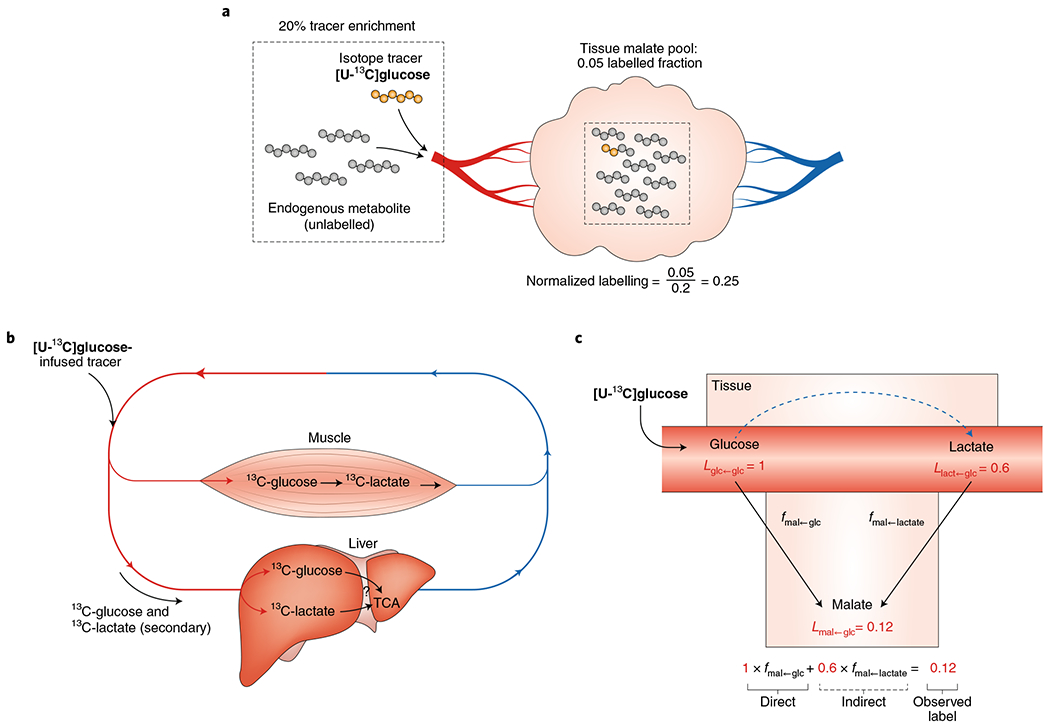
a, Normalized labelling of tissue metabolites is calculated by normalizing the labelled fraction of the tissue metabolite to the labelled fraction of the infused tracer in the blood. b, Infused metabolite tracers are often converted by tissues into other circulating metabolites, generating secondary tracers that indirectly label downstream metabolite pools. For example, when [U-13C] glucose is infused, tissues such as muscle will catabolize it and release 13C-labelled lactate. c, Both the infused tracer ([U-13C]glucose) and the secondary tracer (circulating 13C-lactate) may contribute labelled carbon to downstream tissue metabolite pools such as the TCA cycle. The observed labelled fraction of a TCA cycle metabolite such as malate equals the sum of the direct contribution from the infused tracer and the indirect contribution via the secondary tracer. To solve for the direct contribution of the infused tracer, the secondary tracer metabolite is infused in a separate experiment. An equation can be written for each infused tracer in which the direct fractional contribution of each considered metabolite (two in this example) serves as a variable. This forms a system of linear equations in which the direct contribution of each metabolite can be calculated.
Experimental considerations and pitfalls.
Measuring the nutrient sources of a tissue metabolite requires many of the same considerations as measuring circulatory fluxes. For quantifying metabolite sources, enrichment of the infused nutrient and of the downstream tissue metabolite of interest should be measured at the metabolic and isotopic pseudo-steady state. Infused nutrient enrichment should ideally be measured in arterial blood, just as when measuring Fcirc, and infusion rates should be chosen to allow detection of labelling of the downstream metabolite without excessively perturbing circulating nutrient levels. Preferably, this can be achieved by circulating enrichments in the range of 5–25%, although higher enrichments are sometimes required. If the Fcirc (Ra) of a given tracer is known, equation (5) can be rearranged and used to select the infusion rate Rtracer to yield the desired circulating enrichment Ltracer.
| (7) |
In some cases, downstream metabolites can take more than a few hours to reach steady-state labelling3. One cause is slow labelling of the circulating tracer itself, which can be accelerated by a priming dose of a tracer at the beginning of the infusion. The labelling steady state for the tracer in theory can be reached almost instantaneously if the priming dose is just right. This optimal priming dose can be calculated as the product of the whole-body pool of the tracer metabolite and the desired label enrichment.
Note that tissue metabolites may derive not just from circulating nutrients but also from internal nutrient reservoirs such as glycogen (which feeds into glycolysis), protein (whose degradation yields amino acids) or fat (with lipolysis producing glycerol and free fatty acids). These depots can in some cases be labelled by long-term infusions and their contributions can be quantified by pulse-chase infusion experiments72.
Similarly to measuring Fcirc from circulating metabolite labelling, assuming high-quality sample handling and LC–MS protocols, measurements of tissue metabolite labelling tend to be reproducible, with variation primarily from biological and not technical factors.
Data interpretation.
One challenge in interpreting which blood nutrient gives rise to a particular tissue metabolite is the secondary tracer problem73 (Fig. 5b). For example, infusion of uniformly labelled 13C-glucose results in over 60% of carbons in lactate being labelled, so in this context lactate becomes a secondary tracer. When tissue metabolites are labelled from glucose infusion, we would like to know whether the tissue generated them directly from circulating glucose or via circulating lactate (that is, what is the direct contribution of circulating glucose to a tissue metabolite). These possibilities can be mathematically dissected given data from infusing, one at a time, all interconverting nutrients of interest33,53 (Fig. 5c).
For example, let us calculate the direct contributions of glucose and lactate to tissue malate. Two experiments are required: (1) infuse 13C-lactate and measure the labelling of lactate and glucose in the blood, as well as of malate in the tissue of interest; and (2) infuse 13C-glucose and perform the same three measurements. Using these data, the direct contributions of glucose and lactate to malate can be calculated by solving the below system of two equations with two unknowns:
| (8) |
| (9) |
The six L values are calculated from the experimental data using equation (7). For example,
| (10) |
where the two f values are the unknowns, representing the fraction of tissue malate derived directly from circulating glucose (fmal←gluc) and the fraction of tissue malate derived directly from circulating lactate (fmal←lact).
This system of two equations can also be written as a matrix, for ease of solving using software such as MATLAB, Octave, R or Python. This matrix is scalable and generalizable to account for any number of circulating metabolites: with n circulating nutrients, there will be n equations with n unknowns. While this method is useful for accounting for indirect labelling via known secondary tracers, it is blind to unmeasured secondary tracers. Any direct contributions of such tracers will be assigned to their upstream inputs.
Using this approach to deconvolute the direct sources of tissue TCA intermediates, we found that, in fasted mice, glucose mainly contributes to the TCA cycle indirectly through circulating lactate (except in the brain, which directly uses glucose)33. This observation aligns with the high lactate Fcirc and squarely places circulating lactate as a key intermediate in carbohydrate catabolism. An open question is whether the same cells both produce and consume circulating lactate (perhaps using this exchange for redox buffering74), which would contribute to the high lactate Fcirc without any net carbon transfer between tissues. Otherwise, could there be new lactate shuttles carrying net carbon between cells to be discovered?
Conclusions and perspectives
We have described approaches to measure and interpret in vivo metabolic fluxes, as well as how flux measurements reveal biology and disease mechanisms. However, current methods reveal only the tip of the iceberg. They assume that tissues are homogeneous, while in reality they are composed of different cell types. The ability to measure metabolic fluxes at cellular resolution will probably reveal a new world of metabolic biology. For example, in vitro studies suggest that dividing cells tend to display Warburg metabolism, and accordingly that perhaps the small population of stem cells that renew tissues such as the intestine and skin might display high glycolytic flux not seen in the tissue as a whole75–77. Approaches such as imaging mass spectrometry78 or sorting cell populations after in vivo tracing60,61 are making progress in this direction.
The methods described here also fail to differentiate among subcellular compartments, despite the known compartmentation of metabolic processes into organelles such as mitochondria and lysosomes. A combination of experimental and informatic approaches may help to identify compartment-specific fluxes79–81.
Moreover, many fluxes still cannot be reliably quantified, even at the whole-tissue level. For example, glycolytic fluxes in tissues are too fast to measure using pre-steady-state approaches (Box 3). Systems-level modelling may help, especially as steady-state fluxes must balance82. This mass balance constraint has been applied effectively at the genome scale in microorganisms and in cultured mammalian cells83–85, but has not yet been extensively used in vivo86. Integrating arteriovenous metabolite measurements, circulatory turnover measurements and tissue labelling data holds great promise in this regard.
A further challenge for the field is developing a quantitative understanding of how metabolic fluxes are regulated. The role of insulin in controlling metabolic fluxes has been examined carefully, both at the organismal level (Box 2) and in terms of biochemical mechanisms. Fluxes also change in response to other hormones (for example, glucocorticoids, thyroid hormone and adrenaline) and physiological stimuli (for example, infection, injury, exercise and cancer), but the flux regulation in these contexts is less well understood. Applying CRISPR and other genetic engineering approaches to manipulate regulators (kinases and transcription factors) or enzymes themselves will help to reveal how metabolite availability and/or enzyme levels are controlled to lead to altered fluxes87.
This is an exciting time for in vivo metabolism, since combining tracer approaches developed over the past century with modern metabolomic technologies yields measurements of greater specificity and breadth. Applying these approaches will ultimately provide whole-body metabolic flux maps and identify the metabolic alterations driving human disease, and thereby pave the way to new diagnostic and therapeutic strategies.
Acknowledgements
We thank Yihui Shen, Wenyun Lu, Michael Neinast, Asael Roichman and other members of the Rabinowitz laboratory for helpful discussion. This work was supported by NIH Pioneer award 5DP1DK113643, Diabetes Research Center grant P30 DK019525 and Paul G. Allen Family Foundation grant 0034665 to J.D.R., NIH fellowship F32DK118856 to T.T. and Damon Runyon Cancer Research Foundation fellowship DRG-2373-19 to C.R.B.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Fenn JB, Mann M, Meng CK, Wong SF & Whitehouse CM Electrospray ionization–principles and practice. Mass Spectrom. Rev 9, 37–70 (1990). [Google Scholar]
- 2.Tanaka K et al. Protein and polymer analyses up to m/z 100 000 by laser ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom 2, 151–153 (1988). [Google Scholar]
- 3.Wolfe RR Tracers in Metabolic Research: Radioisotope and Stable Isotope/Mass Spectometry Methods (A.R. Liss, 1984). [PubMed] [Google Scholar]
- 4.McCabe BJ & Previs SF Using isotope tracers to study metabolism: application in mouse models. Metab. Eng 6, 25–35 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Fernández-García J, Altea-Manzano P, Pranzini E & Fendt S-M Stable isotopes for tracing mammalian-cell metabolism in vivo. Trends Biochem. Sci 45, 185–201 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Jain M et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 336, 1040–1044 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frauwirth KA et al. The CD28 signaling pathway regulates glucose metabolism. Immunity 16, 769–777 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Felig P, Pozefsk T, Marlis E & Cahill GF Alanine: key role in gluconeogenesis. Science 167, 1003–1004 (1970). [DOI] [PubMed] [Google Scholar]
- 9.Ivanisevic J et al. Arteriovenous blood metabolomics: a readout of intra-tissue metabostasis. Sci. Rep 5, 12757 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jang C et al. Metabolite exchange between mammalian organs quantified in pigs. Cell Metab. 30, 594–606.e3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Owen OE et al. Brain metabolism during fasting. J. Clin. Invest 46, 1589–1595 (1967). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilmore DW, Aulick LH, Mason AD & Pruitt BA Influence of the burn wound on local and systemic responses to injury. Ann. Surg 186, 444–456 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilmore DW et al. Effect of injury and infection on visceral metabolism and circulation. Ann. Surg 192, 491–504 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wahren J, Felig P, Ahlborg G & Jorfeldt L Glucose metabolism during leg exercise in man. J. Clin. Invest 50, 2715–2725 (1971). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murashige D et al. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science 370, 364–368 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rennie MJ et al. Effect of exercise on protein turnover in man. Clin. Sci 61, 627–639 (1981). [DOI] [PubMed] [Google Scholar]
- 17.Taylor R et al. Direct assessment of liver glycogen storage by 13C nuclear magnetic resonance spectroscopy and regulation of glucose homeostasis after a mixed meal in normal subjects. J. Clin. Invest 97, 126–132 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wagenmakers AJM Tracers to investigate protein and amino acid metabolism in human subjects. Proc. Nutr. Soc 58, 987–1000 (1999). [DOI] [PubMed] [Google Scholar]
- 19.Ayala JE, Bracy DP, McGuinness OP & Wasserman DH Considerations in the design of hyperinsulinemic–euglycemic clamps in the conscious mouse. Diabetes 55, 390–397 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Hundal RS et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes 49, 2063–2069 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee-Young RS et al. Skeletal muscle AMP-activated protein kinase is essential for the metabolic response to exercise in vivo. J. Biol. Chem 284, 23925–23934 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laughlin MH & Armstrong RB Muscular blood flow distribution patterns as a function of running speed in rats. Am. J. Physiol. Heart Circ. Physiol 243, H296–H306 (1982). [DOI] [PubMed] [Google Scholar]
- 23.Sjøberg KA, Rattigan S, Hiscock N, Richter EA & Kiens B A new method to study changes in microvascular blood volume in muscle and adipose tissue: real-time imaging in humans and rat. Am. J. Physiol. Heart Circ. Physiol 301, H450–H458 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Wei K et al. Quantification of myocardial blood flow with ultrasound-induced destruction of microbubbles administered as a constant venous infusion. Circulation 97, 473–483 (1998). [DOI] [PubMed] [Google Scholar]
- 25.Brown RP, Delp MD, Lindstedt SL, Rhomberg LR & Beliles RP Physiological parameter values for physiologically based pharmacokinetic models. Toxicol. Ind. Health 13, 407–484 (1997). [DOI] [PubMed] [Google Scholar]
- 26.Berne RM Regulation of coronary blood flow. Physiol. Rev 44, 1–29 (1964). [DOI] [PubMed] [Google Scholar]
- 27.Høst U et al. Haemodynamic effects of eating: the role of meal composition. Clin. Sci 90, 269–276 (1996). [DOI] [PubMed] [Google Scholar]
- 28.Lang CH, Bagby GJ, Ferguson JL & Spitzer JJ Cardiac output and redistribution of organ blood flow in hypermetabolic sepsis. Am. J. Physiol. Regul. Integr. Comp. Physiol 246, R331–R337 (1984). [DOI] [PubMed] [Google Scholar]
- 29.Tabata H, Kitamura T & Nagamatsu N Comparison of effects of restraint, cage transportation, anaesthesia and repeated bleeding on plasma glucose levels between mice and rats. Lab. Anim 32, 143–148 (1998). [DOI] [PubMed] [Google Scholar]
- 30.Ensinger H, Weichel T, Lindner KH, Grünert A & Ahnefeld FW Effects of norepinephrine, epinephrine, and dopamine infusions on oxygen consumption in volunteers. Crit. Care Med 21, 1502–1508 (1993). [DOI] [PubMed] [Google Scholar]
- 31.Ghosal S et al. Mouse handling limits the impact of stress on metabolic endpoints. Physiol. Behav 150, 31–37 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schoenheimer R & Rittenberg D Deuterium as an indicator in the study of intermediary metabolism. 3. The role of the fat tissues. J. Biol. Chem 111, 175–181 (1935). [DOI] [PubMed] [Google Scholar]
- 33.Hui S et al. Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Searle GL, Strisower EH & Chaikoff IL Glucose pool and glucose space in the normal and diabetic dog. Am. J. Physiol 176, 190–194 (1954). [DOI] [PubMed] [Google Scholar]
- 35.Searle GL, Strisower EH & Chaikoff IL Determination of rates of glucose oxidation in normal and diabetic dogs by a technique involving continuous Injection of C14-glucose. Am. J. Physiol 185, 589–594 (1956). [DOI] [PubMed] [Google Scholar]
- 36.Ayala JE et al. Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis. Models Mech 3, 525–534 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sherwin RS et al. A model of the kinetics of insulin in man. J. Clin. Invest 53, 1481–1492 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stanley WC et al. Lactate extraction during net lactate release in legs of humans during exercise. J. Appl. Physiol 60, 1116–1120 (1986). [DOI] [PubMed] [Google Scholar]
- 39.Okajima F, Chenoweth M, Rognstad R, Dunn A & Katz J Metabolism of 3H- and 14C-labelled lactate in starved rats. Biochem. J 194, 525–540 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Donovan CM & Brooks GA Endurance training affects lactate clearance, not lactate production. Am. J. Physiol. Endocrinol. Metab 244, E83–E92 (1983). [DOI] [PubMed] [Google Scholar]
- 41.Brooks GA The science and translation of lactate shuttle theory. Cell Metab. 27, 757–785 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Bergman BC et al. Muscle net glucose uptake and glucose kinetics after endurance training in men. Am. J. Physiol. Endocrinol. Metab 277, E81–E92 (1999). [DOI] [PubMed] [Google Scholar]
- 43.Bergman BC et al. Active muscle and whole body lactate kinetics after endurance training in men. J. Appl. Physiol 87, 1684–1696 (1999). [DOI] [PubMed] [Google Scholar]
- 44.Sahlin K Lactate production cannot be measured with tracer techniques. Am. J. Physiol. Endocrinol. Metab 252, E439–E440 (1987). [DOI] [PubMed] [Google Scholar]
- 45.Donnelly KL et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest 115, 1343–1351 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Landau BR et al. Glycerol production and utilization in humans: sites and quantitation. Am. J. Physiol. Endocrinol. Metab 271, E1110–E1117 (1996). [DOI] [PubMed] [Google Scholar]
- 47.Klein S, Young VR, Blackburn GL, Bistrian BR & Wolfe RR Palmitate and glycerol kinetics during brief starvation in normal weight young adult and elderly subjects. J. Clin. Invest 78, 928–933 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perry RJ et al. Leptin mediates a glucose–fatty acid cycle to maintain glucose homeostasis in starvation. Cell 172, 234–248.e17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neinast MD et al. Quantitative analysis of the whole-body metabolic fate of branched-chain amino acids. Cell Metab. 29, 417–429.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matthews DE et al. Regulation of leucine metabolism in man: a stable isotope study. Science 214, 1129–1131 (1981). [DOI] [PubMed] [Google Scholar]
- 51.Waterlow JC Whole-body protein turnover in humans—past, present, and future. Annu. Rev. Nutr 15, 57–92 (1995). [DOI] [PubMed] [Google Scholar]
- 52.Wolfe RR, Goodenough RD, Burke JF & Wolfe MH Response of protein and urea kinetics in burn patients to different levels of protein intake. Ann. Surg 197, 163–171 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hui S et al. Quantitative fluxomics of circulating metabolites. Cell Metab. 32, 676–688.e4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sprinson DB & Rittenberg D The rate of interaction of the ammo acids of the diet with the tissue proteins. J. Biol. Chem 180, 715–726 (1949). [PubMed] [Google Scholar]
- 55.Golden S, Chenoweth M, Dunn A, Okajima F & Katz J Metabolism of tritium- and 14C-labeled alanine in rats. Am. J. Physiol. Endocrinol. Metab 241, E121–E128 (1981). [DOI] [PubMed] [Google Scholar]
- 56.Katz J, Okajima F, Chenoweth M & Dunn A The determination of lactate turnover in vivo with 3H- and 14C-labelled lactate. The significance of sites of tracer administration and sampling. Biochem. J 194, 513–524 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rendina AR, Hermes JD & Cleland WW Use of multiple isotope effects to study the mechanism of 6-phosphogluconate dehydrogenase. Biochemistry 23, 6257–6262 (1984). [DOI] [PubMed] [Google Scholar]
- 58.Zhang Z, Chen L, Liu L, Su X & Rabinowitz JD Chemical basis for deuterium labeling of fat and NADPH. J. Am. Chem. Soc 139, 14368–14371 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schoenheimer R & Rittenberg D Deuterium as an indicator in the study of intermediary metabolism. 9. The conversion of stearic acid into palmitic acid in the organism. J. Biol. Chem 120, 155–165 (1937). [Google Scholar]
- 60.Lau AN et al. Dissecting cell-type-specific metabolism in pancreatic ductal adenocarcinoma. eLife 9, e56782 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ma EH et al. Metabolic profiling using stable isotope tracing reveals distinct patterns of glucose utilization by physiologically activated CD8+ T cells. Immunity 51, 856–870.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Zhang L et al. Spectral tracing of deuterium for imaging glucose metabolism. Nat. Biomed. Eng 3, 402–413 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.DeBerardinis RJ et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl Acad. Sci. USA 104, 19345–19350 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Davidson SM et al. Environment impacts the metabolic dependencies of Ras-driven non-small cell lung cancer. Cell Metab. 23, 517–528 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hensley CT et al. Metabolic heterogeneity in human lung tumors. Cell 164, 681–694 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mayers JR et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 353, 1161–1165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Faubert B et al. Lactate metabolism in human lung tumors. Cell 171, 358–371.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brunt EM et al. Nonalcoholic fatty liver disease. Nat. Rev. Dis. Prim 1, 15080 (2015). [DOI] [PubMed] [Google Scholar]
- 69.Hudgins LC et al. Relationship between carbohydrate-induced hypertriglyceridemia and fatty acid synthesis in lean and obese subjects. J. Lipid Res 41, 595–604 (2000). [PubMed] [Google Scholar]
- 70.Lambert JE, Ramos–Roman MA, Browning JD & Parks EJ Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 146, 726–735 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lu W et al. Metabolite measurement: pitfalls to avoid and practices to follow. Annu. Rev. Biochem 86, 277–304 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.TeSlaa T et al. The source of glycolytic intermediates in mammalian tissues. Cell Metab. 33, 367–378 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Previs SF & Kelley DE Tracer-based assessments of hepatic anaplerotic and TCA cycle flux: practicality, stoichiometry, and hidden assumptions. Am. J. Physiol. Endocrinol. Metab 309, E727–E735 (2015). [DOI] [PubMed] [Google Scholar]
- 74.Patgiri A et al. An engineered enzyme that targets circulating lactate to alleviate intracellular NADH:NAD+ imbalance. Nat. Biotechnol 38, 309–313 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cham CM & Gajewski TF Glucose availability regulates IFN-γ production and p70S6 kinase activation in CD8+ effector T cells. J. Immunol 174, 4670–4677 (2005). [DOI] [PubMed] [Google Scholar]
- 76.Flores A et al. Lactate dehydrogenase activity drives hair follicle stem cell activation. Nat. Cell Biol 19, 1017–1026 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schell JC et al. Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat. Cell Biol 19, 1027–1036 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ryan DJ, Spraggins JM & Caprioli RM Protein identification strategies in MALDI imaging mass spectrometry: a brief review. Curr. Opin. Chem. Biol 48, 64–72 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Abu-Remaileh M et al. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 358, 807–813 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen WW, Freinkman E, Wang T, Birsoy K & Sabatini DM Absolute quantification of matrix metabolites reveals the dynamics of mitochondrial metabolism. Cell 166, 1324–1337.e11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee WD, Mukha D, Aizenshtein E & Shlomi T Spatial-fluxomics provides a subcellular-compartmentalized view of reductive glutamine metabolism in cancer cells. Nat. Commun 10, 1351 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Orth JD, Thiele I & Palsson BØ What is flux balance analysis? Nat. Biotechnol 28, 245–248 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Metallo CM et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lu H et al. A consensus S. cerevisiae metabolic model Yeast8 and its ecosystem for comprehensively probing cellular metabolism. Nat. Commun 10, 3586 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Blank LM, Kuepfer L & Sauer U Large-scale 13C-flux analysis reveals mechanistic principles of metabolic network robustness to null mutations in yeast. Genome Biol. 6, R49 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shlomi T, Cabili MN, Herrgård MJ, Palsson BØ & Ruppin E Network-based prediction of human tissue-specific metabolism. Nat. Biotechnol 26, 1003–1010 (2008). [DOI] [PubMed] [Google Scholar]
- 87.Kacser H, Burns JA, Kacser H & Fell DA The control of flux. Biochem. Soc. Trans 23, 341–366 (1995). [DOI] [PubMed] [Google Scholar]
- 88.Emwas A-HM The strengths and weaknesses of NMR spectroscopy and mass spectrometry with particular focus on metabolomics research. In Metabonomics: Methods and Protocols Vol. 1277 (ed. Bjerrum JT) 161–193 (Humana, 2015). [DOI] [PubMed] [Google Scholar]
- 89.Su X, Lu W & Rabinowitz JD Metabolite spectral accuracy on orbitraps. Anal. Chem 89, 5940–5948 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bajad SU et al. Separation and quantitation of water soluble cellular metabolites by hydrophilic interaction chromatography-tandem mass spectrometry. J. Chromatogr. A 1125, 76–88 (2006). [DOI] [PubMed] [Google Scholar]
- 91.Zhang Y et al. Comparing stable isotope enrichment by gas chromatography with time-of-flight, quadrupole time-of-flight, and quadrupole mass spectrometry. Anal. Chem 93, 2174–2182 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Perseghin G et al. Increased glucose transport—phosphorylation and muscle glycogen synthesis after exercise training in insulin-resistant subjects. N. Engl. J. Med 335, 1357–1362 (1996). [DOI] [PubMed] [Google Scholar]
- 93.Zabielski P et al. Comparison of different mass spectrometry techniques in the measurement of L-[ring-13C6]phenylalanine incorporation into mixed muscle proteins. J. Mass Spectrom 48, 269–275 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wolfe RR Measurement of urea kinetics in vivo by means of a constant tracer infusion of di-15N-urea. Am. J. Physiol. Endocrinol. Metab 240, E428–E434 (1981). [DOI] [PubMed] [Google Scholar]
- 95.Institute of Medicine (US) Committee on Military Nutrition Research. Emerging Technologies for Nutrition Research: Potential for Assessing Military Performance Capability (US National Academies Press, 1997). [PubMed] [Google Scholar]
- 96.Emwas A-H et al. NMR spectroscopy for metabolomics research. Metabolites 9, 123 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lin P, Lane AN & Fan TW-M Stable isotope-resolved metabolomics by NMR. Methods Mol. Biol 2037, 151–168 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Roden M et al. Mechanism of free fatty acid-induced insulin resistance in humans. J. Clin. Invest 97, 2859–2865 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Befroy DE et al. Direct assessment of hepatic mitochondrial oxidative and anaplerotic fluxes in humans using dynamic 13C magnetic resonance spectroscopy. Nat. Med 20, 98–102 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mason GF et al. Simultaneous determination of the rates of the TCA cycle, glucose utilization, α-ketoglutarate/glutamate exchange, and glutamine synthesis in human brain by NMR. J. Cereb. Blood Flow. Metab 15, 12–25 (1995). [DOI] [PubMed] [Google Scholar]
- 101.Landau BR et al. 14C-labeled propionate metabolism in vivo and estimates of hepatic gluconeogenesis relative to Krebs cycle flux. Am. J. Physiol 265, E636–E647 (1993). [DOI] [PubMed] [Google Scholar]
- 102.Brindle KM Imaging metabolism with hyperpolarized 13C-labeled cell substrates. J. Am. Chem. Soc 137, 6418–6427 (2015). [DOI] [PubMed] [Google Scholar]
- 103.Witney TH & Brindle KM Imaging tumour cell metabolism using hyperpolarized 13C magnetic resonance spectroscopy. Biochem. Soc. Trans 38, 1220–1224 (2010). [DOI] [PubMed] [Google Scholar]
- 104.Deh K et al. Dynamic volumetric hyperpolarized 13C imaging with multi-echo EPI. Magn. Reson. Med 85, 978–986 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kernstine KH et al. Does tumor FDG-PET avidity represent enhanced glycolytic metabolism in non-small cell lung cancer? Ann. Thorac. Surg 109, 1019–1025 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen DL et al. Increased T cell glucose uptake reflects acute rejection in lung grafts. Am. J. Transplant 13, 2540–2549 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Frayn KN, Coppack SW, Humphreys SM, Clark ML & Evans RD Periprandial regulation of lipid metabolism in insulin-treated diabetes mellitus. Metabolism 42, 504–510 (1993). [DOI] [PubMed] [Google Scholar]
- 108.DeFronzo RA, Tobin JD & Andres R Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am. J. Physiol. Endocrinol. Metab 237, E214–E223 (1979). [DOI] [PubMed] [Google Scholar]
- 109.Elahi D In praise of the hyperglycemic clamp. A method for assessment of β-cell sensitivity and insulin resistance. Diabetes Care 19, 278–286 (1996). [DOI] [PubMed] [Google Scholar]
- 110.Kraegen EW, James DE, Jenkins AB & Chisholm DJ Dose–response curves for in vivo insulin sensitivity in individual tissues in rats. Am. J. Physiol. Endocrinol. Metab 248, E353–E362 (1985). [DOI] [PubMed] [Google Scholar]
- 111.Bonora E et al. Homeostasis model assessment closely mirrors the glucose clamp technique in the assessment of insulin sensitivity: studies in subjects with various degrees of glucose tolerance and insulin sensitivity. Diabetes Care 23, 57–63 (2000). [DOI] [PubMed] [Google Scholar]
- 112.Miyazaki Y et al. Effect of pioglitazone on circulating adipocytokine levels and insulin sensitivity in type 2 diabetic patients. J. Clin. Endocrinol. Metab 89, 4312–4319 (2004). [DOI] [PubMed] [Google Scholar]
- 113.Michael MD et al. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol. Cell 6, 87–97 (2000). [PubMed] [Google Scholar]
- 114.Bali D et al. Animal model for maturity-onset diabetes of the young generated by disruption of the mouse glucokinase gene. J. Biol. Chem 270, 21464–21467 (1995). [DOI] [PubMed] [Google Scholar]
- 115.Petersen MC, Vatner DF & Shulman GI Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol 13, 572–587 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hellerstein MK & Neese RA Mass isotopomer distribution analysis: a technique for measuring biosynthesis and turnover of polymers. Am. J. Physiol. Endocrinol. Metab 263, E988–E1001 (1992). [DOI] [PubMed] [Google Scholar]
- 117.Stanhope KL et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Invest 119, 1322–1334 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bloch K & Rittenberg D On the utilization of acetic acid for cholesterol formation. J. Biol. Chem 145, 625–636 (1942). [Google Scholar]
- 119.Shulman GI et al. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N. Engl. J. Med 322, 223–228 (1990). [DOI] [PubMed] [Google Scholar]
- 120.Daurio NA et al. Spatial and temporal studies of metabolic activity: contrasting biochemical kinetics in tissues and pathways during fasted and fed states. Am. J. Physiol. Endocrinol. Metab 316, E1105–E1117 (2019). [DOI] [PubMed] [Google Scholar]
- 121.Yuan J, Bennett BD & Rabinowitz JD Kinetic flux profiling for quantitation of cellular metabolic fluxes. Nat. Protoc 3, 1328–1340 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]