

Abstract

We describe the synthesis of triazole-containing carboline derivatives and their utility as bromodomain and extra-terminal (BET) inhibitors. A convergent synthetic route permitted the detailed investigation of deuteration and fluorination strategies to reduce clearance while maintaining a favorable in vitro profile. This work led to the identification of a potent BET inhibitor, 2-{8-fluoro-3-[4-(2H3)methyl-1-methyl-1H-1,2,3-triazol-5-yl]-5-[(S)-(oxan-4-yl)(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl}propan-2-ol (15), which demonstrated reduced clearance and an improved pharmacokinetic (PK) profile across preclinical species. Importantly, no major metabolite was observed when 15 was incubated with human hepatocytes (hHEP) for 2 h. This study culminated with the evaluation of 15 in a mouse triple-negative breast cancer (TNBC) tumor model where it demonstrated robust efficacy at low doses.
Keywords: Bromodomain, BET inhibitor, gene transcription, cancer, carboline
MYC is an oncogene that encodes c-Myc and is implicated in most human cancers.1 In cancer cells, c-Myc expression is independent of typical regulatory mechanisms and leads to amplified intracellular and extracellular transcription. Despite significant effort, the pharmaceutical industry has failed to identify a promising clinical candidate that inhibits c-Myc directly.1 Studies have shown that MYC transcription is under superenhancer control in some cancers.2 Superenhancers are genomic regions that cooperatively bind transcription factors and coactivators. The resulting synergy is highly sensitive to even small perturbations in factor concentration. Consequently, disruption of these enhancer–activator interactions represents a promising and indirect approach to inhibit c-Myc.
BET proteins have emerged as promising targets for cancer therapy.3−6 The BET family consists of four proteins (BRD2, BRD3, BRD4, and BRDT) that each contains two bromodomain binding domains (BD1 and BD2) capable of recognizing acetylated lysine residues found on histone tails.7−9 Once bound to a histone, BET proteins modulate chromatin architecture and recruit transcriptional regulators, which lead to gene expression. Importantly, when MYC is under superenhancer control, BET inhibitors selectively inhibit its transcription.2,3
Early BET inhibitors, JQ1 and GSK525762 (I-BET762), inspired work on similar scaffolds (e.g., MK8628 and CPI-0610) as well as structurally differentiated series (e.g., ABBV-075; Figure 1). BET inhibitors have shown broad utility, demonstrating preclinical efficacy in a variety of cancers, HIV, inflammation, acute heart failure, and other diseases.10−12 In oncology, the most advanced compounds are currently being investigated for clinical efficacy in both hematologic malignancies and solid tumors. Our clinical candidate, BMS-986158, is under evaluation in clinical trials for the treatment of myelofibrosis, lymphoma, and solid tumors.13 This compound was identified as part of an effort to find structurally distinct, pan-BET leads that were more potent than MK8628 and GSK525762. Preclinical studies on BMS-986158 revealed broad-spectrum targeted cytotoxicity in multiple cancers, tolerability at efficacious doses, and projected human pharmacokinetics (PK) suitable for oral dosing.
Figure 1.

Representative BET inhibitors.
Following the advancement of BMS-986158, we initiated a backup effort with a goal of improving the overall PK profile by improving absorption and reducing clearance for the existing carboline-based chemotype. A three-pronged approach was adopted to (1) increase liver microsomal (LM) oxidative stability across species, (2) reduce liver microsomal glucuronidation across species, and (3) reduce active transport as measured using Caco-2 cells (efflux ratio of ≤2). It was anticipated that advances across these parameters would translate into lower orally administered minimum efficacious doses (MEDs) in rodent efficacy models and ultimately in the treatment of human cancers.
Early attempts to improve LM oxidative stability centered around the triazole (Tz) moiety on pyridine X (Figure 2A). The 1,4-dimethyltriazole mimics an acetylated lysine and makes key anchoring interactions with Asn140 and a conserved water molecule within the binding pocket.13 Reaction phenotyping indicated that CYP3A4- and CYP3A5-mediated oxidation of the C4Tz methyl (Me) was the primary route of metabolism in human.14 In an effort to mitigate this oxidative pathway, both Tz isosteres and Me alternatives were investigated. Unfortunately, those strategies failed to provide highly potent analogs with improved LM stability.15
Figure 2.

(A) LM oxidation and glucuronidation of lead series. (B) Deuteration strategy to block LM oxidation.
Given these results, we shifted our approach and replaced the C4Tz Me with Me-d3 to harness the kinetic isotope effect (KIE). The C–H/C–D bond breaking event must be rate limiting to achieve a KIE, which is the case for LM oxidation of alkyl groups.16 Gratifyingly, this simple switch from BMS-986158 to analog 1 provided a 64% improvement in human LM T1/2 (from 36 to 59 min, Table 1).17 However, BMS-986158 contains a second major metabolic soft spot (the N5 phenyl/tetrahydropyran group) in mouse, an important species for evaluation of PK and efficacy. Therefore, 1 did not offer an improvement in mouse LM T1/2 (24 and 25 min, respectively). Methyl sulfone 2 and its Me-d3 analog (compound 3) were subsequently identified as a matched pair to study the correlation between oxidative LM T1/2 and exposure in a 7 h coarse mouse PK study. These results were expected to translate across species. Gratifyingly, the 120% increase in mouse LM T1/2 of 3 relative to 2 (22 vs 10 min) provided a 125% increase in exposure at 10 mg/kg, PO (AUC = 22 vs 9.6 μM·h).17
Table 1. BRD4 and c-Myc Inhibition, MM Proliferation, and Metabolic and Caco-2 Data for Various Substituted Carbolines.
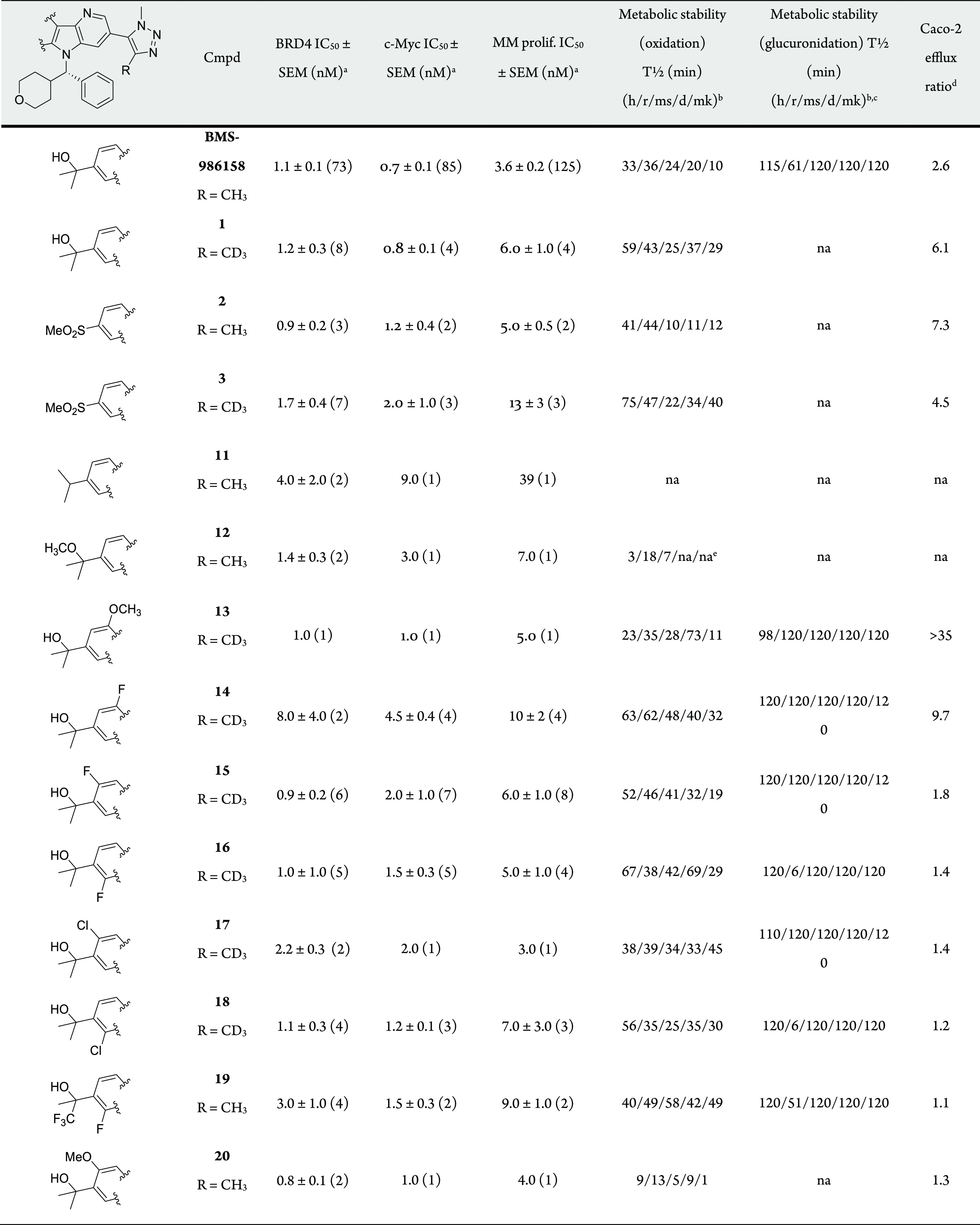
Number of determinations in parentheses.
Human = h, rat = r, mouse = ms, dog = d, monkey = mk.
Measurement limit = 120 min.
Efflux ratio = (A–B)/(B–A).
Percent remaining after 10 min incubation.17
The 1-methyl-4-(trideuteriomethyl)triazole moiety was installed on the carboline core (intermediate 4) using a Stille coupling (Scheme 1A). Organostannane 5 was readily synthesized in four steps (Scheme 1B).17 This protocol involved alkylation of trimethylsilylacetylene with iodomethane-d3 (≥99.5 atom % D) followed by a Huisgen 1,3-dipolar cycloaddition with trimethylsilylmethyl azide.18 Importantly, the Huisgen reaction afforded a single regioisomer (Tz 9). Deprotection with tetrabutylammonium fluoride and stannylation furnished intermediate 5.
Scheme 1. (A) Installation of the Tz Moiety via Stille Coupling13 and (B) Synthetic Route to Organostannane 5(17).

Subsequent efforts focused on carboline core (benzene Z) modification to limit glucuronidation of the carbinol, a structural element required for optimal inhibitor potency (Table 1, analog 11). When BMS-986158 was subjected to human hepatocytes (hHEP) for 1 h, glucuronidation on the carbinol by UGT1A4 and UGT2B17 accounted for approximately 20% of total metabolism (Figure 2A). However, glucuronides were minimally detectable in our higher throughput LM assay. To limit the potential for patient metabolic variability, we hoped to eliminate this pathway.19 We focused our attention on glucuronidation in rat because of the increased turnover in that species and its downstream importance in PK and toxicology studies.
Glucuronidation was precluded by O-alkylation, but analogs suffered from considerably diminished oxidative metabolic stability across species (e.g., methyl ether 12). Therefore, we focused on inductive modulation of carbinol nucleophilicity, a strategy that demonstrated dramatic influence on glucuronidation. As expected, electron rich 9-methoxy 13 did not provide improved human glucuronidation T1/2 (98 min), but a 9-fluoro substituent (14) prevented glucuronidation in all species (T1/2 ≥ 120 min). Unfortunately, C9-substitution (13, 14) was accompanied by a significant increase in Caco-2 efflux (ratios of >35 and 9.7, respectively).20 8-Fluoro 15 provided a similar glucuronidation benefit (T1/2 ≥ 120 min; all species) but without the increase in efflux (ratio = 1.8). 6-Fluoro 16 had a comparable efflux profile (ratio = 1.4) but was highly glucuronidated in rat (T1/2 = 6 min). Replacement of the fluorine with chlorine at C8 or C9 provided no additional benefit (compounds 17 and 18).21 Even a trifluoromethyl, installed directly adjacent to the hydroxyl, did not completely override the influence of 6-fluoro substitution on glucuronidation (19).
Notably, Caco-2 efflux was most influenced by substituent placement on benzene Z (Table 1). This was best illustrated by comparing C9-substituted analogs 13 and 14 (ratio > 35 and ratio = 9.7, respectively) with both C8 halo- and methoxybenzenes 15, 17, and 20 (ratio = 1.8, 1.4, and 1.3, respectively) as well as C6 halobenzenes 16 and 18 (ratio = 1.4 and 1.2, respectively). To a lesser extent, substituent identity affected efflux at C9 (e.g., 9-methoxy 13 vs 9-fluoro 14). No difference was observed when C6 and C8 were varied (e.g., 8-fluoro 15 vs 8-chloro 17 vs 8-methoxy 20). We found efflux ratios for C6- or C8-substituted analogs generally achieved our goal of efflux ≤2.
These structural modifications had little impact on inhibition in BRD4 binding and c-Myc functional assays (generally <3.0 nM, Table 1).17 An X-ray crystal structure of 15 bound to BRD4-BD1 showed that it recapitulated the interactions observed with BMS-986158, with the 8-fluoro projecting into solution. Specifically, deuterating the methyl attached to the triazole had no impact on the hydrogen bond network that includes Asn 140 ND2 and a water network involving interactions with among other atoms Gln 85 O and Tyr 97 OH. The carboline nitrogen forms a hydrogen bond through a water molecule to Gln 85 NE2. The tetrahydropyran ring interacts with the WPF shelf and the phenyl ring interacts with residues of the ZA-loop, including Leu 92 and Leu 94 side chains (Figure 3). However, modest differences were observed in cell proliferation as measured using a Velcade-resistant multiple myeloma (MM) cell line, JJN3R.22 Importantly, carbinol-containing compounds typically provided ≤10 nM inhibition in MM proliferation.
Figure 3.
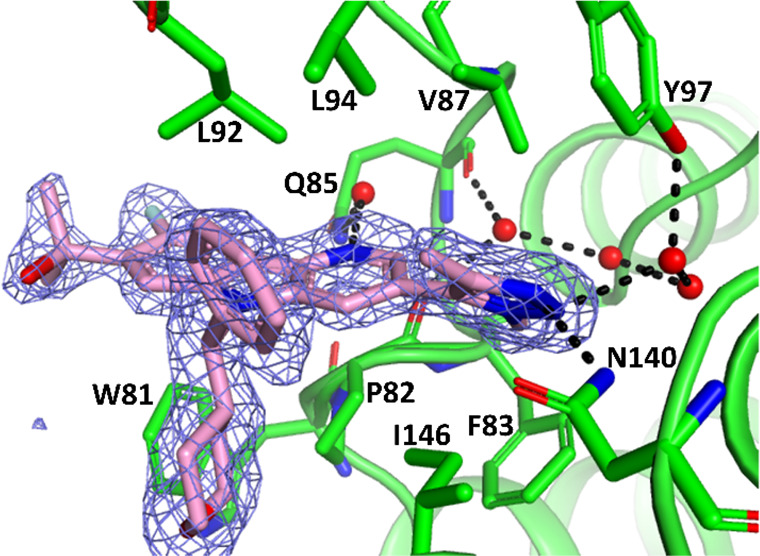
X-ray structure of compound 15 with BRD4 (BD1). PDB code 7UZN. Close-up of the acetylated lysine binding site with omit Fo – Fc electron density contoured at 3 RMSD (blue mesh).
2-{8-Fluoro-3-[4-(2H3)methyl-1-methyl-1H-1,2,3-triazol-5-yl]-5-[(S)-(oxan-4-yl)(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl}propan-2-ol (15) emerged as a promising lead, demonstrating increased stability toward oxidation and glucuronidation, reduced Caco-2 efflux compared to BMS-986158, and potent binding to all BET family members in FRET binding assays (IC50 BRD2/3/4/T = 0.2/1.6/0.9/0.1 nM). To validate the metabolic findings, 15 (10 μM) was incubated with hHEP (Figure 4).17 To our delight, 99% of parent drug was observed after a 2 h incubation. Oxidative metabolism occurred primarily on the N5 phenyl/tetrahydropyran group; 1% of this metabolite was present. Importantly, installation of the C4Tz Me-d3 almost completely eliminated oxidation at that site (<1%), and glucuronides were not detected. For comparison, two major BMS-986158 metabolites were formed following a 1 h incubation of parent compound (5 μM) with hHEP.
Figure 4.

hHEP metabolic profiles of BMS-986158 (1 h incubation) and 15 (2 h incubation).
Encouraged by these data, 15 was progressed into preclinical PK studies (Table 2). Across species, plasma clearances (CLp) of 15 were reduced compared to BMS-986158, with the largest improvement in cynomolgus monkey (CLp = 32 → 4.3 (mL/min)/kg). Half-life was also increased in all species except rat, again with the largest increase in monkey (2.0 → 4.0 h). Both BMS-986158 and 15 demonstrated 100% oral bioavailability in mouse. However, 15 showed significant increases in rat (47% → 74%), dog (59% → 73%), and monkey (16% → 85%). Taken together, the results in monkey were especially exciting since total exposure (po) increased from 85 nM·h for BMS-986158 to 1900 nM·h for 15. Significant increases in exposures were also observed in mouse (AUC = 18 000 and 25 000 nM·h, respectively) and rat (AUC = 1900 and 6600 nM·h, respectively). A small increase in total exposure was also noted in dog (470 and 550 nM·h, respectively).
Table 2. Comparison of BMS-986158 and 15 Preclinical PK Parameters17.
| mouse |
rat |
dog |
monkey |
|||||
|---|---|---|---|---|---|---|---|---|
| BMS-986158 | 15 | BMS-986158 | 15 | BMS-986158 | 15 | BMS-986158 | 15 | |
| dose (iv/po, mg/kg) | 1.75/5.0 | 1.0/3.0 | 1.0/5.0 | 1.0/5.0 | 0.1/0.3 | 0.1/0.3 | 0.1/0.3 | 0.1/0.3 |
| CLp ((mL/min)/kg) | 12 | 4.2 | 41 | 18 | 15 | 13 | 32 | 4.3 |
| Vd (L/kg) | 1.9 | 1.4 | 4.3 | 2.9 | 5.3 | 5.8 | 4.2 | 1.3 |
| half-life (iv, h) | 1.9 | 3.4 | 4.2 | 4.1 | 5.6 | 7.6 | 2.0 | 4.0 |
| F (%) | 100 | 100 | 47 | 74 | 59 | 73 | 16 | 85 |
| AUC (po, nM·h) | 18000 | 25000 | 2000 | 6600 | 410 | 550 | 85 | 1900 |
To explore in vivo BET activity, 15 was advanced into a TNBC (cell line BR1077F) patient-derived xenograft (PDX) tumor model that recapitulates human tumor physiology.23 In this experiment, mice were dosed (po, QD) beginning 26 days following tumor-cell implantation and followed a 5-day-on/2-day-off regimen for 2 weeks. Compound 15 treatment impaired the growth of established PDX tumors (TGI = 88%) at MED = 2 mg/kg (AUC = 4800 nM·h) compared to MED = 3 mg/kg (TGI = 99%, AUC = 2600 nM·h) for BMS-986158.
In summary, deuteration of the primary site of metabolism (C4Tz Me) led to significantly diminished oxidative metabolism of the triazole moiety and increased LM T1/2 across species. Importantly, in vitro–in vivo correlation between increased LM stability and increased exposure was established in a mouse PK study using a molecular matched pair (C4Tz Me vs C4Tz Me-d3). Modification of carboline substituents in proximity to the carbinol was found to impact human and rat glucuronidation as well as Caco-2 efflux. 8-Fluoro carboline 15 emerged from this work as a potent analog of BMS-986158 with improved stability toward oxidative metabolism and glucuronidation and with an improved permeability profile. Following incubation of 15 with hHEP, no major metabolites were observed. The data package for this compound triggered a full preclinical PK evaluation where compound 15 demonstrated enhanced exposure across species relative to BMS-986158. The improvements in PK profile led to robust activity at low doses in a TNBC PDX tumor model. Additional characterization of compound 15 will be reported in due course.
Acknowledgments
We thank Chiradeep Panja, Premsai Neithnadka, Manivel Pitchai, Subhabrata Chaudhury, and Ananta Karmakar for large-scale preparation of chemical intermediates.
Glossary
Abbreviations
- AUC
area under the curve
- BD
binding domain
- BET
bromodomain and extra-terminal
- BRD
bromodomain
- CLp
plasma clearance
- hHEP
human hepatocyte
- iv
intravenous administration
- LM
liver microsome
- Me
methyl
- MED
minimum efficacious dose
- MM
multiple myeloma
- PDX
patient-derived xenograft
- PK
pharmacokinetic
- po
oral administration
- QD
administered once daily
- TNBC
triple-negative breast cancer
- Tz
triazole
- Vd
volume of distribution
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00219.
Details for the synthesis and characterization of 1–20; experimental details for in vitro and in vivo assays; protein science and X-ray crystallography with 15 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Whitfield J. R.; Beaulieu M.-E.; Soucek L. Strategies to inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 2017, 5, 1–13. 10.3389/fcell.2017.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén J.; Hoke H. A.; Lin C. Y.; Lau A.; Orlando D. A.; Vakoc C. R.; Bradner J. E.; Lee T. I.; Young R. A. Selective Inhibition of Tumor Oncogenes by Disruption of Superenhancers. Cell 2013, 153, 320–334. 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore J. E.; Issa G. C.; Lemieux M. E.; Rahl P. B.; Shi J.; Jacobs H. M.; Kastritis E.; Gilpatrick T.; Paranal R. M.; Qi J.; Chesi M.; Schinzel A. C.; McKeown M. R.; Heffernan T. P.; Vakoc C. R.; Bergsagel P. L.; Ghobrial I. M.; Richardson P. G.; Young R. A.; Hahn W. C.; Anderson K. C.; Kung A. L.; Bradner J. E.; Mitsiades C. S. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell 2011, 146, 904–917. 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinjha R. K.; Witherington J.; Lee K. Place your BETs: the therapeutic potential of bromodomains. Trends Pharmacol. Sci. 2012, 33, 146–153. 10.1016/j.tips.2011.12.002. [DOI] [PubMed] [Google Scholar]
- Chung C.-w. Small Molecule Bromodomain Inhibitors: Extending the Druggable Genome. Prog. Med. Chem. 2012, 51, 1–55. 10.1016/B978-0-12-396493-9.00001-7. [DOI] [PubMed] [Google Scholar]
- Fu L.-L.; Tian M.; Li X.; Li J.-J.; Huang J.; Ouyang L.; Zhang Y.; Liu B. Inhibition of BET Bromodomains as a Therapeutic Strategy for Cancer Drug Discovery. Oncotarget 2015, 6, 5501–5516. 10.18632/oncotarget.3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C.; Kumar C.; Gnad F.; Nielsen M. L.; Rehman M.; Walther T. C.; Olsen J. V.; Mann M. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 2009, 325, 834–840. 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- High levels of lysine acetylation are linked to MYC transcription:; Patel J. H.; Du Y.; Ard P. G.; Phillips C.; Carella B.; Chen C.-J.; Rakowski C.; Chatterjee C.; Lieberman P. M.; Lane W. S.; Blobel G. A.; McMahon S. B. The c-MYC Oncoprotein Is a Substrate of the Acetyltransferases hGCN5/PCAF and TIP60. Mol. Cell. Biol. 2004, 24, 10826–10834. 10.1128/MCB.24.24.10826-10834.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Martile M. D.; Del Bufalo D. D.; Trisciuoglio D. The multifaceted role of lysine acetylation in cancer: prognostic biomarker and therapeutic target. Oncotarget 2016, 7, 55789–55810. 10.18632/oncotarget.10048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri E.; Petosa C.; McKenna C. E. Bromodomains: Structure, function and pharmacology of inhibition. Biochem. Pharmacol. 2016, 106, 1–18. 10.1016/j.bcp.2015.12.005. [DOI] [PubMed] [Google Scholar]
- Romero F. A.; Taylor A. M.; Crawford T. D.; Tsui V.; Côté A.; Magnuson S. Disrupting Acetyl-Lysine Recognition: Progress in the Development of Bromodomain Inhibitors. J. Med. Chem. 2016, 59, 1271–1298. 10.1021/acs.jmedchem.5b01514. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Wang P.; Chen H.; Wold E. A.; Tian B.; Brasier A. R.; Zhou J. Drug Discovery Targeting Bromodomain-Containing Protein 4. J. Med. Chem. 2017, 60, 4533–4558. 10.1021/acs.jmedchem.6b01761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavai A. V.; Norris D.; Delucca G.; Tortolani G.; Tokarski J. S.; Dodd D.; O’Malley D.; Zhao Y.; Quesnelle C.; Gill P.; Vaccaro W.; Huynh T.; Ahuja V.; Han W.-C.; Mussari C.; Harikrishnan L.; Kamau M.; Poss M.; Sheriff S.; Yan C.; Marsilio F.; Menard K.; Wen M. L.; Rampulla R.; Wu D.-R.; Li J.; Zhang H.; Li P.; Sun D.; Yip H.; Traeger S. C.; Zhang Y.; Mathur A.; Zhang H.; Huang C.; Yang Z.; Ranasinghe A.; Everlof G.; Raghavan N.; Tye C. K.; Wee S.; Hunt J. T.; Vite G.; Westhouse R.; Lee F. Y. Discovery and Preclinical Pharmacology of an Oral Bromodomain and Extra-Terminal (BET) Inhibitor Using Scaffold-Hopping and Structure-Guided Drug Design. J. Med. Chem. 2021, 64, 14247–14265. 10.1021/acs.jmedchem.1c00625. [DOI] [PubMed] [Google Scholar]
- Metabolic soft spots varied between species.
- Acetylated lysine mimic SAR will be communicated in due course.
- Gant T. G. Using Deuterium in Drug Discovery: Leaving the Label in the Drug. J. Med. Chem. 2014, 57, 3595–3611. 10.1021/jm4007998. [DOI] [PubMed] [Google Scholar]
- See Supporting Information for experimental details.
- Nulwala H. B.; Tang C. N.; Kail B. W.; Damodaran K.; Kaur P.; Wickramanayake S.; Shi W.; Luebke D. R. Probing the structure-property relationship of regioisomeric ionic liquids with click chemistry. Green Chem. 2011, 13, 3345–3349. 10.1039/c1gc16067b. [DOI] [Google Scholar]
- Dorne J. L. C. M.; Walton K.; Renwick A. G. Human variability in glucuronidation in relation to uncertainty factors for risk assessment. Food Chem. Toxicol. 2001, 39, 1153–1173. 10.1016/S0278-6915(01)00087-4. [DOI] [PubMed] [Google Scholar]
- Sun H.; Pang K. S. Permeability, Transport, and Metabolism of Solutes in Caco-2 Cell Monolayers: A Theoretical Study. Drug Metab. Dispos. 2008, 36, 102–123. 10.1124/dmd.107.015321. [DOI] [PubMed] [Google Scholar]
- Gillis E. P.; Eastman K. J.; Hill M. D.; Donnelly D. J.; Meanwell N. A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- Kuhne M. R.; Mulvey T.; Belanger B.; Chen S.; Pan C.; Chong C.; Cao F.; Niekro W.; Kempe T.; Henning K. A.; Cohen L. J.; Korman A. J.; Cardarelli P. M. BMS-936564/MDX-1338: A Fully Human Anti-CXCR4 Antibody Induces Apoptosis In Vitro and Shows Antitumor Activity In Vivo in Hematologic Malignancies. Clin. Cancer Res. 2013, 19, 357–366. 10.1158/1078-0432.CCR-12-2333. [DOI] [PubMed] [Google Scholar]
- Hidalgo M.; Amant F.; Biankin A. V.; Budinská E.; Byrne A. T.; Caldas C.; Clarke R. B.; de Jong S.; Jonkers J.; Mælandsmo G. M.; Roman-Roman S.; Seoane J.; Trusolino L.; Villanueva A. Patient Derived Xenograft Models: An Emerging Platform for Translational Cancer Research. Cancer Discovery 2014, 4 (9), 998–1013. 10.1158/2159-8290.CD-14-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.