

Abstract
Despite continued interest in the development of nonsteroidal estrogens and antiestrogens, there are only a few chemotypes of estrogen receptor ligands. Using targeted screening in a ligand sensing assay, we identified a phenolic thieno[2,3-d]pyrimidine with affinity for estrogen receptor α. An efficient three-step synthesis of the heterocyclic core and structure-guided optimization of the substituents resulted in a series of potent nonsteroidal estrogens. The chemical tractability of the thieno[2,3-d]pyrimidine chemotype will support the design of new estrogen receptor ligands as therapeutic hormones and antihormones.
Keywords: Estrogen receptor, Hormone, Nonsteroidal, Heterocycle, Structure
Estrogens are important endocrine sex hormones that control many aspects of female physiology and have important actions in males.1 Estrogen pharmacology is primarily mediated through the estrogen receptor α (ERα), a ligand-activated transcription factor and member of the nuclear receptor superfamily.2,3 A second estrogen receptor β (ERβ), which shares high sequence identity with ERα, has also been characterized. However, unlike ERα, the contribution of ERβ to the pharmacology of estrogens remains contentious, despite extensive study over a 20-year period.4 ERα functions as a homodimer to bind to palindromic DNA sequences, known as estrogen responsive elements (EREs), in the enhancer region of its target genes. The binding of an estrogen agonist (hormone) to the ligand binding domain (LBD) of the ERα homodimer facilitates the recruitment of co-activators to the receptor–DNA complex and initiation of gene transcription. ERβ can form heterodimers with ERα, and one of its functions may be to modulate the breadth of target gene activation in tissues where the receptors are co-expressed.5,6
The main circulating estrogens in women are estrone (E1) and estradiol (E2), although additional metabolites with lower activity have been detected.7 All of the naturally occurring steroidal estrogens have an aromatic A-ring with a phenol at C-3 (Figure 1). Chemical modification of the steroid core at C-7 with a long lipophilic chain results in compounds with antagonist (antihormone) activity, such as the breast cancer drug fulvestrant (FULV), which blocks the action of endogenous estrogens and has the additional effect of promoting receptor turnover in cells.8 Diethylstilbestrol (DES) is a synthetic nonsteroidal estrogen that was first reported in 1938.9 DES contains two phenols to mimic the steroid A- and D-rings, and its affinity for ERα is similar to that of E2. Many nonsteroidal antiestrogens have been developed from the DES core, including 4-hydroxytamoxifen (4OHT), where addition of a third aromatic substituent was used to modulate receptor activation.10 Several cyclic analogs of 4OHT that block estrogen action or induce ERα degradation remain in clinical development as breast cancer drugs.11 A wide range of phenol-containing natural products have also been shown to possess estrogenic activity,12 including the phytoestrogen ferutinin and the mycotoxin β-zearalenol. In addition, several phenolic plasticizers and detergents, such as butylated hydroxyanisole (BHA) and bisphenol A (BPA), have weak estrogenic activity and are labeled as potential xenoestrogens due to their occurrence in the environment.13 It is notable that most synthetic estrogens and antiestrogens contain a phenol as a common pharmacophore (Figure 1).10,14 Remarkably, despite decades of pharmaceutical research on synthetic ERα ligands, there is a dearth of chemically tractable chemotypes. Most nonsteroidal ligands are still based on the DES/4OHT core, with only limited chemical diversity in their structures.10,14 Given the continued interest in development of nonsteroidal ERα ligands for hormone replacement or breast cancer therapy, we sought to identify new chemotypes of synthetic estrogens.
Figure 1.
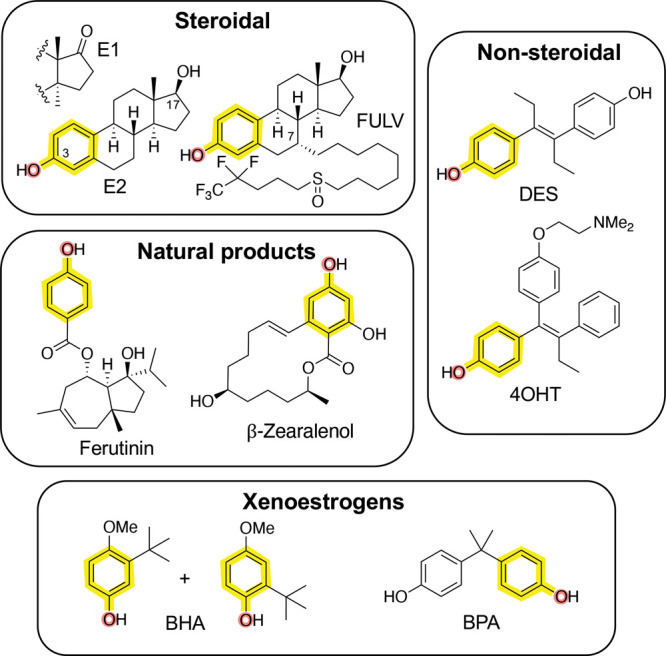
Chemotypes of estrogen receptor ligands: E1, estrone; E2, 17β-estradiol; FULV, fulvestrant; DES, diethylstilbestrol; 4OHT, 4-hydroxytamoxifen; BHA, butylated hydroxyanisole; BPA, bisphenol A. The phenol pharmacophore is highlighted in yellow and red in each compound.
To identify new cell-active ERα ligands, we developed a sensitive, high-throughput screening assay in 384-well format. We had previously identified an 11 amino acid peptide (αII, SSLTSRDFGSWYASR) that was recruited to ERα when bound by small-molecule agonists, partial agonists, and antagonists.15,16 To configure the assay in a two-hybrid format, HepG2 cells were transiently transfected with expression vectors for ERα-VP16 and αII-Gal4 fusions as well as a luciferase reporter construct under the control of five copies of a Gal4 upstream enhancer element. Addition of a wide range of standard hormones and antihormones resulted in an increase in luciferase (Figure 2A), demonstrating that the assay was sensitive to ER ligands independent of their agonist or antagonist functional activity. The ERα ligand sensing assay (ERα LiSA) demonstrated a robust Z′ = 0.7 in a 384-well format that was suitable for high-throughput screening.
Figure 2.

(A) ERα LiSA in HepG2 cells transfected with Gal4-αII, ERα-VP16, and 5x-gal-tata-Luc. After 24 h, cells were lysed, and luciferase assays were performed. (B) ERα LiSA screen. Representative data from plate 1/28. Compounds with luciferase >4× (gray shading) over background (green shading) were selected as primary hits. (C) Dose-response of the thieno[2,3-d]pyrimidine hits 1 and analog 2a in the ERα LiSA. (D) Whole-cell ERα affinity determined by competition binding with 3H-E2. (E) Agonist activity in HepG2 cells transfected with ERα, (ERE)7-tk-luciferase, and Renilla luciferase. All data: error bars represent ± SD of triplicate points. BZE, bazedoxifene; AZD, AZD-9496; RAD, RAD-1901; LASO, lasofoxifene.
We opted to perform a targeted diversity screen, based on the principle that natural and synthetic ER ligands often contain a phenol in their core structure (Figure 1). A library of 8400 phenols arrayed across 28 384-well plates was obtained from Enamine (Monmouth Jct., NJ), representing a subset of their >2.9 million compound screening collection. All compounds were screened at 10 μM final concentration in the ERα LiSA, and the results are presented as a scatter plot (Figure 2B). Screening the full library of 8400 compounds yielded 324 primary hits that were confirmed in follow-up four-point dose–response studies (Table S1). Blinded positive control compounds raloxifene (RAL, ERα antagonist) and diethylstilbestrol (DES, ERα agonist) scored as the top hits during the screening process, validating our general approach (Table S1). The primary hits were triaged by potency to yield 34 compounds with EC50 < 1.0 μM in the ERα LiSA. A thieno[2,3-d]pyrimidine (1) (Figure 2C) (EC50 = 180 nM) was selected for further optimization based on the novelty of its core structure and the commercial availability of 26 analogs from Chemspace (Monmouth Jct., NJ). While these additional analogs did not provide a systematic evaluation of the thieno[2,3-d]pyrimidine core, some initial structure–activity trends were observed (Table S2): switching the isobutyl group on C-5 on the thieno[2,3-d]pyrimidine core to a phenyl group resulted in a 2-fold increase in potency; the 4-hydroxybenzylamine at C-4 on the thienopyrimidine appeared to be essential for activity, since analogs where the 4′-OH was replaced with 3′-OH, 4′-NH2, or 4′-CO2H were >10-fold less active; and at C-2, chlorine was the only replacement for methyl that had improved activity. Thieno[2,3-d]pyrimidine (2a) was the most potent of these analogs, with EC50 = 14 nM in the ERα LiSA (Figure 2C). To confirm that the activity of 2a in the ERα LiSA was consistent with direct interaction on the receptor, a whole-cell competition binding assay was performed using 3H-E2 as a radioligand. Thienopyrimidine 2a competed for 3H-E2 binding to ERα with IC50 = 65 nM (Figure 2D). For comparison, unlabeled E2 competed with IC50 = 0.2 nM.
The functional activity of 2a was determined in an estrogen-responsive reporter gene assay using HepG2 cells that were transiently transfected with expression plasmids for ERα and an (ERE)7-tk-luciferase reporter. Thieno[2,3-d]pyrimidine (2a) induced a dose-responsive increase in luciferase activity to 60–75% of the maximal efficacy of the natural hormone E2 (Figure 2E). The nanomolar agonist activity of thienopyrimidine 2a, EC50 = 14 nM, was consistent with its affinity as an ERα ligand. The binding and functional data characterized thieno[2,3-d]pyrimidine (2a) as a new chemotype of nonsteroidal estrogens.
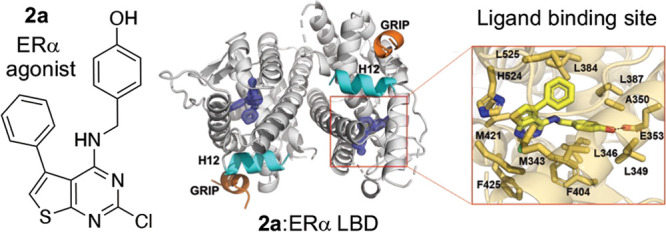
To understand the molecular basis of its potent estrogenic activity, the X-ray co-crystal structure of 2a in complex with the ERα LBD was determined. A Y537S mutant of the ERα LBD and a short peptide from the GRIP co-activator, which together favor the agonist conformation of the receptor,17 were used to facilitate crystallization. The structure was solved to 1.55 Å using molecular replacement (Table S3) and showed a canonical ERα LBD dimer with its C-terminal helix 12 (H12) in the agonist conformation over the ligand binding pocket and with the co-activator GRIP peptide in the cleft that forms the activating function 2 (AF-2) (Figure 3A). Within the ligand binding pocket, thieno[2,3-d]pyrimidine (2a) was clearly identified in the electron density map (Figure S1). The ligand adopted a binding pose perpendicular to H12 and formed a single hydrogen bond between its phenolic oxygen and E353, but with no additional interaction to R394 or the water molecule that are utilized by E2 (Figure 3B). The 5-phenyl group of 2a sat adjacent to helix 3 near L384, L387, A350, and L525 and adopted a similar vector to the quaternary methyl group of E2 that is pointed toward H12. The 2-chloro group of 2a sat in a hydrophobic binding pocket formed by M343, M421, and F425. Both M343 and F425 were displaced away from their conformation in the E2 structure by the presence of the 2-chloro group of 2a. Although the thieno[2,3-d]pyrimidine 2a induced a canonical agonist conformation where H12 is folded over the ligand binding pocket, the thiophene ring perturbed the H11–H12 connecting loop and slightly reduced the contact surface of H12 with the ligand binding pocket compared to the E2 structure. The reduced burial of the H12 may explain why 2a was marginally less efficacious than E2 in the transcriptional activation assay (Figure 2E). Unlike the 2-, 4-, and 5-substituents of the thieno[2,3-d]pyrimidine that formed direct contacts with amino acids lining the ERα ligand binding pocket, the polar 1- and 3-pyrimidine nitrogens and the secondary amino group of the 4-substituent did not form any H-bond interactions with the receptor. In comparison to other ERα co-crystal structures, the 2a complex most closely resembled the phytoestrogen ferutinin (PDB 4MG7), which occupied a similar region of the ligand binding pocket while also forming only a single polar interaction with E353 (Figure 3D).
Figure 3.

Structural basis of ERα agonist activity of thieno[2,3-d]pyrimidines 2a and 6b. (A) Overview of the co-crystal structure with ERα LBD with 2mFo – DFc difference maps of 2a in the ligand binding pocket contoured to 2σ. (B) Thieno[2,3-d]pyrimidine binding pocket. Amino acid side chains within 4 Å of 2a are shown in bold. (C) Superposition with E2 (green) and 2a (yellow), highlighting the differences between ligand binding poses. (D) Expanded view of the differences in phenolic hydrogen-bonding patterns between 2a and E2. (E) Superposition of ferutinin (gray) and 2a (yellow), showing the similar binding poses. (F) Superposition of E2 (green) and 6b (purple), highlighting the H-bond interactions with E353 and H524. All highlighted protein features are color-coded to their respective ligands. Dashed lines represent hydrogen bonds. The PDB codes are 6CBZ and 4MG7 for the E2 and ferutinin structures, respectively.
To perform a systematic evaluation of the structure–activity relationship of 2a as a new chemotype of nonsteroidal estrogens, we developed an efficient synthesis of the 2-chlorothieno[2,3-d]pyrimidine core that allowed for sequential modification of the 4- and 5-substituents. The first step involved a three-component Gewald reaction to generate trisubstituted thiophene 3a from acetophenone, malononitrile, and sulfur.18,19 Using the one-pot base catalysis conditions reported by Zeng et al.,20,21 the desired thiophene 3a was isolated, but only in 10% yield (Table 1, entry 1). We performed a systematic optimization of the reaction by varying the base catalyst, solvent, and reaction time. Increasing the reaction temperature to 80 °C and the reaction time to 24 h led to 14% isolated yield of 3a (entry 2). Increasing the amount of base to 0.25 equiv of imidazole at the original 60 °C temperature also gave an increase in the yield of 3a to 18% (entry 3). Switching the solvent to 2-Me-THF (entry 4) resulted in a near doubling of the yield to 32%. Under these conditions, successive increase in the equivalents of imidazole to 0.5 and 1.0 (entries 5 and 6) gave 36% and 40% yields, respectively, of 3a. Increasing the temperature to 80 °C (entry 7) did not give any further increase in yield. Switching the solvent from 2-Me-THF to dioxane (entry 8) resulted in a small decrease in the yield of 3a to 35%. Some authors have reported the use of the bases morpholine and piperidine for catalysis of the Gewald reaction.18,19 However, in our hands, neither of these bases yielded more than traces of 3a with either 2-Me-THF or DMF as solvent (entries 9–12). The use of proline as the base in DMF21 at 0.1 and 0.25 equiv gave 26% of 3a (entries 13 and 14) but was still inferior to the use of imidazole. Finally, after consideration of the reaction mechanism for the three-component base-catalyzed Gewald reaction (Scheme S1), which involves an initial Knoevenagel condensation of malononitrile with acetophenone, we opted to delay addition of the sulfur until formation of the Knoevenagel product was observed by TLC.18 Our goal was to minimize the formation of sulfur-containing acyclic intermediates that were isolated as various byproducts under the lower yielding reaction conditions (data not shown). Accordingly, when the sulfur addition was delayed for 1.5 h after the mixing of acetophenone and malononitrile with 1.0 equiv of imidazole in 2-Me-THF at 60 °C, the trisubstituted thiophene 3a was isolated in 57% yield (entry 15). Having optimized the synthesis of thiophene 3a, we employed an acetonitrile-assisted diphosgene reaction, as reported by Chi22 and by Roecker,23 to form the 2,4-dichlorothieno[2,3-d]pyrimidine 4a in high yield (Scheme 1). Finally, 4a was subjected to SNAr reaction with 4-hydroxybenzylamine to obtain 2a. Under the optimized conditions, the efficient three-step synthesis of thieno[2,3-d]pyrimidine 2a from acetophenone was achieved in 28% overall yield on a gram scale.
Table 1. Optimization of the One-Pot Gewald Reaction.

| entry | base (equiv) | solvent | temp (°C) | time (h) | yield of 3a (%) |
|---|---|---|---|---|---|
| 1 | imidazole (0.1) | DMF | 60 | 18 | 10 |
| 2 | imidazole (0.1) | DMF | 80 | 24 | 14 |
| 3 | imidazole (0.25) | DMF | 60 | 24 | 18 |
| 4 | imidazole (0.25) | 2-Me-THF | 60 | 24 | 32 |
| 5 | imidazole (0.5) | 2-Me-THF | 60 | 24 | 36 |
| 6 | imidazole (1.0) | 2-Me-THF | 60 | 24 | 40 |
| 7 | imidazole (1.0) | 2-Me-THF | 80 | 24 | 40 |
| 8 | imidazole (1.0) | dioxane | 60 | 24 | 35 |
| 9 | morpholine (1.0) | 2-Me-THF | 60 | 24 | <1 |
| 10 | piperidine (1.0) | 2-Me-THF | 60 | 24 | <1 |
| 11 | morpholine (1.0) | DMF | 60 | 24 | <1 |
| 12 | piperidine (1.0) | DMF | 60 | 24 | <1 |
| 13 | proline (0.1) | DMF | 60 | 24 | 26 |
| 14 | proline (0.25) | DMF | 60 | 24 | 26 |
| 15 | imidazole (1.0) | 2-Me-THF | 60 | 24 | 57a |
Addition of sulfur 1.5 h after the reaction of acetophenone with malononitrile.
Scheme 1. Optimized Three-Step Synthesis of Thienopyrimidine 2a.

Reagents and conditions: (a) (i) malononitrile (1.1 equiv), imidazole (1.0 equiv), 2-Me-THF, 60 °C, 1.5 h, then (ii) S8 (1.25 equiv), 60 °C, 24 h, yield 57%; (b) diphosgene (1.5 equiv), MeCN, sealed tube, 110 °C, 24 h, yield 77%; (c) 4-hydroxybenzylamine, Et3N, CHCl3, 75 °C, yield 66%.
Guided by the co-crystal structure of 2a in complex with the ERα LBD (Figure 3), we explored the roles of the phenolic group at the 4-position and the hydrophobic substituent at the 5-position of the thieno[2,3-d]pyrimidine core. The commercially available analogs (Table S1) indicated that the 2-chloro substituent was already optimal, so we opted to leave this position constant. Six new analogs (2b–g) were obtained using the three-step 2-chlorothieno[2,3-d]pyrimidine synthesis from the corresponding methyl ketones (Scheme 2). The analogs were tested for their ERα binding affinity using the two-hybrid LiSA and for their agonist functional activity in the ERE-luciferase reporter assay in HepG2 cells. A resynthesized sample of 2a gave an ERα binding affinity of 11 nM and an EC50 = 48 nM in the reporter gene assay (Table 2). The 5-methyl analog 2b and 5-isopropyl analog 2c were both >100-fold less active in the receptor binding and reporter gene functional assays. In contrast, the 5-isobutyl analog 2d was only 3-fold less active than 2a in the binding assay and 5-fold less active in the reporter gene assay. This result mirrors the structure–activity relationship observed in the 2-methylthieno[2,3-d]pyrimidines (Table S1), where the corresponding 5-isobutyl and 5-phenyl analogs had similar activity on ERα. The 5-tert-butyl-2-chlorothieno[2,3-d]pyrimidine 2e was less active than 2a. However, the 5-cyclohexyl analog 2f showed a relatively small decrease in potency, which was notable, given the poor activity of the 5-isopropyl analog 2c. Finally, the analog 2g, with an ortho-chloro substituent on the 5-phenyl group, had relatively potent ERα binding and activation, which was consistent with the rotation of the 5-phenyl group out of the plane of the 2-chlorothieno[2,3-d]pyrimidine core in the co-crystal structure (Figure 3B). Overall, while the phenyl group remained the optimal 5-substituent, the receptor was able to accommodate branched alkyl substituents and ortho-substituted phenyl groups, which indicated the potential for further optimization of the receptor binding and functional activity.
Scheme 2. Synthesis of Thienopyrimidine Analogs 2b–g and 5a–g.

Reagents and conditions: (a) (i) malononitrile (1.1 equiv), imidazole (1.0 equiv), 2-Me-THF, 60 °C, 1.5 h, then (ii) S8 (1.25 equiv), 60 °C, 24 h; (b) diphosgene (1.5 equiv), MeCN, sealed tube, 110 °C, 18–24 h; (c) amine, Et3N, CHCl3*, 75 °C; *for 5bt-BuOH/DIPEA at 130 °C in microwave, for 5et-BuOH.
Table 2. ERα Activity of Compounds 2a–g.
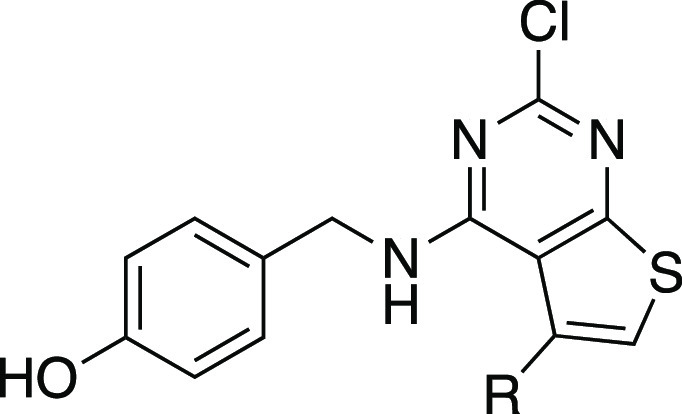
| EC50 (nM) |
|||
|---|---|---|---|
| compd | R | ERα bindinga | ERα agonismb |
| 2a | Ph | 11 | 48 |
| 2b | Me | 1700 | 6600 |
| 2c | i-Pr | 6200 | >10 000 |
| 2d | i-Bu | 31 | 240 |
| 2e | t-Bu | 890 | 2000 |
| 2f | c-C6H11 | 95 | 350 |
| 2g | 2-Cl-Ph | 43 | 170 |
ERα LiSA assay.
(ERE)7-TK-Luc reporter assay in HepG2 cells; all data in triplicate ±15%.
A second series of analogs (5a–g) explored the role of the 4′-phenol in binding to ERα (Table 3). The replacement of the 4′-hydroxyl of 2a with 4′-amine (5a) led to a decrease in activity, while the 4′-carboxylic acid (5b), and 4′-methyl ester (3b) groups gave analogs that were inactive in the binding and functional assays at 10 μM. Switching from the 4′-phenol of 2a to the 3′-phenol of 5d led to a 1000-fold loss in activity. This result was, on one hand, surprising, given the comparison of 2a with E2 in the X-ray co-crystal structures (Figure 3C), yet was consistent with the observations in the 5-methylthieno[2,3-d]pyrimidine analogs (Table S1). The detrimental effect of the 3′-phenol was confirmed in the 3′,4′-catechol analog (5e) that was inactive in the binding and functional assays. The unsubstituted phenyl analog (5f), while retaining sub-μM binding and functional activity, provided quantitative evidence that addition of the 4′-hydroxyl onto the 2-chlorothieno[2,3-d]pyrimidine core in 2a contributed a 20- to 40-fold increase in potency on ERα. Homologation of the linker between the amine and phenol by an extra methylene unit in 5g resulted in a large decrease in ERα activity.
Table 3. ERα Activity of Compounds 5a–g.
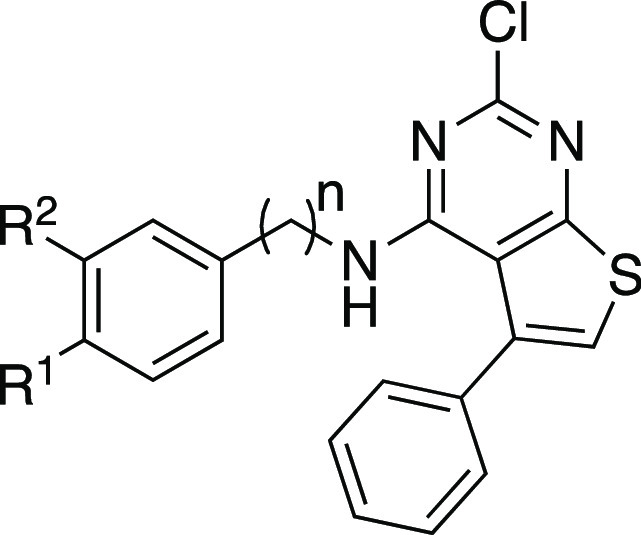
| EC50 (nM) |
|||||
|---|---|---|---|---|---|
| compd | R1 | R2 | n | ERα bindinga | ERα agonismb |
| 5a | NH2 | H | 1 | 238 | 1430 |
| 5b | CO2H | H | 1 | i.a. | i.a. |
| 5c | CO2Me | H | 1 | i.a. | i.a. |
| 5d | H | OH | 1 | 7790 | >3000 |
| 5e | OH | OH | 1 | >10 000 | >10 000 |
| 5f | H | H | 1 | 490 | 900 |
| 5g | OH | H | 2 | 2600 | 9500 |
ERα LiSA assay.
(ERE)7-TK-Luc reporter assay in HepG2 cells. i.a. = inactive at 10 μM. All data in triplicate ±15%.
To more closely mimic the binding of E2 to ERα, where the 17β-hydroxyl group forms an H-bond with the H524 residue (Figure 3F),10 we sought to add polar functionality at C-6 of the thieno[2,3-d]pyrimidine core. The C-6-substituted analogs were synthesized from 2-methoxyacetophenone using the imidazole-catalyzed one-pot Gewald reaction and acetonitrile-assisted diphosgene reactions to give the corresponding 6-methoxy-2,4-dichlorothieno[2,3-d]pyrimidine 4h (Scheme 3). SNAr reaction with 4-hydroxybenzylamine yielded the 6-methoxy analog 6a. Demethylation with BBr3 produced a mixture of 6-hydroxythieno[2,3-d]pyrimidine 6b and its keto tautomer 6b′. The ratio of 6b′:6b favored the keto form by a 9:1 ratio in d6-DMSO, as determined by the 1H NMR assignment (Figure S2), and it is likely that they readily interconvert in protic solvents. When tested in the ERα LiSA (Table 4), the 6-methoxy analog (6a) was 10-fold less potent than the unsubstituted thieno[2,3-d]pyrimidine (2a). The 6-hydroxy/keto analog (6b/6b′) showed improved potency compared to 6a, with EC50 = 40 nM, although it was 3-fold less potent than 2a and also showed slightly lower binding affinity (Figure 2D). Notably, 6b/6b′ had potency equivalent to that of 2a in the ERE reporter assay. X-ray crystallography was used to determine the preferred tautomer of 6b/6b′ for binding to ERα (Figure 3E and Table S3). The electron density map (Figure S1) clearly showed the enol form 6b in the binding pocket, with an sp2 center at C-5 on the thieno[2,3-d]pyrimidine and the 6-hydroxyl group forming an H-bond with H524. The crystal structure showed that thieno[2,3-d]pyrimidine (6b) was able to engage both of the polar residues (E353 and H524) that are utilized by the natural hormone E2 for molecular recognition of ERα. The small decrease in potency of 6b/6b′ compared to that of unsubstituted 2a in the ERα LiSA and binding assay may be due to the ratio of enol:keto tautomers in the assay media. It is notable that estrone, the 17-keto analog of E2 (Figure 1), is 10–100-fold less potent as a hormone,24 consistent with a preference for an H-bond donor over an acceptor in the interaction with H524.
Scheme 3. Synthesis of Thienopyrimidine Analogs 6a and 6b.

Reagents and conditions: (a) (i) malononitrile (1.1 equiv), imidazole (1.0 equiv), 2-Me-THF, 60 °C, 1.5 h, then (ii) S8 (1.25 equiv), 60 °C, 24 h, yield 75%; (b) diphosgene (1.5 equiv), MeCN, sealed tube, 110 °C, 24 h, yield 70%; (c) 4-hydroxybenzylamine, Et3N, CHCl3, 75 °C, 24 h, yield 63%; (d) BBr3, CH2Cl2, 0 °C, 4 h, yield 51%.
Table 4. ERα Activity of Compounds 6a and 6b.

ERα LiSA assay.
(ERE)7-TK-Luc reporter assay in HepG2 cells.
The bioactive tautomer is assumed to be the enol form (6b) based on the X-ray co-crystal structure. All data in triplicate ±15%.
In conclusion, 2-chloro-4-((4-hydroxybenzyl)amino)-5-phenylthieno[2,3-d]pyrimidin-6-ol (6b) exemplifies a new chemotype of potent nonsteroidal estrogens. While there are no prior reports of thieno[2,3-d]pyrimidines with activity as steroid receptor ligands, these and related heterocycles have been identified as ATP-competitive kinase inhibitors.25,26 However, when screened at a concentration of 20 μM, across a panel of 100 human protein kinases, 2a showed little or no effect on protein thermal stability27 (Table S4), indicating that it is unlikely to have significant kinase inhibitory activity. Importantly, thieno[2,3-d]pyrimidines are synthetically accessible through a modular three-step synthesis (Schemes 1–3) that supports chemical modification at each position on the heterocyclic core. Furthermore, X-ray crystallography revealed the detailed interactions of 2a and 6b within the ERα ligand binding pocket and identified the molecular interactions contributing to their potency and efficacy as estrogens. These molecular insights, combined with the chemical tractability of the thieno[2,3-d]pyrimidine chemotype, will support the design and synthesis of new nonsteroidal estrogen agonists, partial agonists, and antagonists as potential therapeutic hormones and antihormones.
Acknowledgments
We thank L. Elson, A. Kraemer, and S. Knapp at the SGC-Frankfurt for the kinase selectivity data.
Glossary
Abbreviations
- ER
estrogen receptor
- ERE
estrogen response element
- LBD
ligand binding domain
- E1
estrone
- E2
17β-estradiol
- FULV
fulvestrant
- DES
diethylstilbestrol
- 4OHT
4-hydroxytamoxifen
- BHA
butylated hydroxyanisole
- BPA
bisphenol A
- LiSA
ligand sensing assay
- BZE
bazedoxifene
- AZD
AZD-9496
- RAD
RAD-1901
- LASO
lasofoxifene
- AF-2
activating function 2
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00180.
List of primary hits from ERα LiSA screen, structure–activity of commercial thienopyrimidine analogs, X-ray refinement data, kinase selectivity screen of 2a, electron density maps of 2a and 6b, determination of the 6b/6b′ keto/enol ratio by 1H NMR, Gewald reaction mechanism, experimental procedures for compounds and assays, and NMR spectra of final compounds 2a–g, 5a–g, and 6a,b, including Tables S1–S4, Figures S1 and S2, and Scheme S1 (PDF)
Accession Codes
Atomic coordinates for the X-ray structure of ERα LBD Y537S in complex with 2a (PDB: 7RKE) and 6b (PDB: 7T2X) are available from the RCSB Protein Data Bank (www.rscb.org).
Author Contributions
T.M.W., V.R.S., J.D.N., D.P.M., and S.W.F. conceived of the study. V.R.S. designed compounds and performed the synthesis. J.D.N. led the biological studies. J.D.N., S.A., C.D.T., and J.B. performed the biological assays. C.J. and S.W.F. performed the X-ray crystallography studies. T.M.W., V.R.S., and S.W.F. wrote the manuscript with edits from J.D.N. and D.P.M. All authors read and approved the manuscript.
The Structural Genomics Consortium is a registered charity (no. 1097737) that receives funds from Bayer AG, Boehringer Ingelheim, Bristol Myers Squibb, Genentech, Genome Canada through Ontario Genomics Institute [OGI-196], EU/EFPIA/OICR/McGill/KTH/Diamond Innovative Medicines Initiative 2 Joint Undertaking [EUbOPEN grant 875510], Janssen, Merck KGaA (aka EMD in Canada and US), Pfizer. and Takeda. This work was supported by the U.S. Department of Defense through the FY 17 BRCP Innovator Award under Award No. BC170954. Research reported in this publication was supported in part by the NC Biotech Center Institutional Support Grant 2018-IDG-1030 and funding from Loyola University Chicago Startup Funds and Susan G. Komen CCR 19608597. Results shown in this report are derived from work performed at Argonne National Laboratory (ANL), Structural Biology Center (SBC) at the Advanced Photon Source (APS), under U.S. Department of Energy, Office of Biological and Environmental Research, contract DE-AC02-06CH11257.
Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense.
The authors declare no competing financial interest.
Supplementary Material
References
- Hamilton K. J.; Hewitt S. C.; Arao Y.; Korach K. S. Estrogen Hormone Biology. Curr. Top. Dev. Biol. 2017, 125, 109–146. 10.1016/bs.ctdb.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldring N.; Pike A.; Andersson S.; Matthews J.; Cheng G.; Hartman J.; Tujague M.; Strom A.; Treuter E.; Warner M.; Gustafsson J. A. Estrogen receptors: how do they signal and what are their targets. Physiol. Rev. 2007, 87 (3), 905–31. 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- Deroo B. J.; Korach K. S. Estrogen receptors and human disease. J. Clin. Invest. 2006, 116 (3), 561–70. 10.1172/JCI27987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deroo B. J.; Buensuceso A. V. Minireview: Estrogen receptor-β: mechanistic insights from recent studies. Mol. Endocrinol. 2010, 24 (9), 1703–14. 10.1210/me.2009-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang E. C.; Charn T. H.; Park S. H.; Helferich W. G.; Komm B.; Katzenellenbogen J. A.; Katzenellenbogen B. S. Estrogen Receptors α and β as determinants of gene expression: influence of ligand, dose, and chromatin binding. Mol. Endocrinol. 2008, 22 (5), 1032–43. 10.1210/me.2007-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg M. K.; Moverare S.; Skrtic S.; Gao H.; Dahlman-Wright K.; Gustafsson J. A.; Ohlsson C. Estrogen receptor (ER)-β reduces ERα-regulated gene transcription, supporting a ″ying yang″ relationship between ERα and ERβ in mice. Mol. Endocrinol. 2003, 17 (2), 203–8. 10.1210/me.2002-0206. [DOI] [PubMed] [Google Scholar]
- Simpson E. R. Sources of estrogen and their importance. J. Steroid Biochem. Mol. Biol. 2003, 86 (3–5), 225–230. 10.1016/S0960-0760(03)00360-1. [DOI] [PubMed] [Google Scholar]
- Carlson R. W. The history and mechanism of action of fulvestrant. Clin. Breast Cancer 2005, 6 (Suppl 1), S5–S8. 10.3816/CBC.2005.s.008. [DOI] [PubMed] [Google Scholar]
- Veurink M.; Koster M.; Berg L. T. The history of DES, lessons to be learned. Pharm. World Sci. 2005, 27 (3), 139–43. 10.1007/s11096-005-3663-z. [DOI] [PubMed] [Google Scholar]
- Jordan V. C. Antiestrogens and selective estrogen receptor modulators as multifunctional medicines. 1. Receptor interactions. J. Med. Chem. 2003, 46 (6), 883–908. 10.1021/jm020449y. [DOI] [PubMed] [Google Scholar]
- McDonnell D. P.; Wardell S. E.; Norris J. D. Oral Selective Estrogen Receptor Downregulators (SERDs), a Breakthrough Endocrine Therapy for Breast Cancer. J. Med. Chem. 2015, 58 (12), 4883–7. 10.1021/acs.jmedchem.5b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branham W. S.; Dial S. L.; Moland C. L.; Hass B. S.; Blair R. M.; Fang H.; Shi L.; Tong W.; Perkins R. G.; Sheehan D. M. Phytoestrogens and mycoestrogens bind to the rat uterine estrogen receptor. J. Nutr. 2002, 132 (4), 658–64. 10.1093/jn/132.4.658. [DOI] [PubMed] [Google Scholar]
- Nilsson R. Endocrine modulators in the food chain and environment. Toxicol. Pathol. 2000, 28 (3), 420–31. 10.1177/019262330002800311. [DOI] [PubMed] [Google Scholar]
- Katzenellenbogen J. A. The 2010 Philip S. Portoghese Medicinal Chemistry Lectureship: addressing the ″core issue″ in the design of estrogen receptor ligands. J. Med. Chem. 2011, 54 (15), 5271–82. 10.1021/jm200801h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris J. D.; Paige L. A.; Christensen D. J.; Chang C. Y.; Huacani M. R.; Fan D.; Hamilton P. T.; Fowlkes D. M.; McDonnell D. P. Peptide antagonists of the human estrogen receptor. Science 1999, 285 (5428), 744–6. 10.1126/science.285.5428.744. [DOI] [PubMed] [Google Scholar]
- Paige L. A.; Christensen D. J.; Gron H.; Norris J. D.; Gottlin E. B.; Padilla K. M.; Chang C. Y.; Ballas L. M.; Hamilton P. T.; McDonnell D. P.; Fowlkes D. M. Estrogen receptor (ER) modulators each induce distinct conformational changes in ERα and ERβ. Proc. Natl. Acad. Sci. U.S.A. 1999, 96 (7), 3999–4004. 10.1073/pnas.96.7.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanning S. W.; Jeselsohn R.; Dharmarajan V.; Mayne C. G.; Karimi M.; Buchwalter G.; Houtman R.; Toy W.; Fowler C. E.; Han R.; Lainé M.; Carlson K. E.; Martin T. A.; Nowak J.; Nwachukwu J. C.; Hosfield D. J.; Chandarlapaty S.; Tajkhorshid E.; Nettles K. W.; Griffin P. R.; Shen Y.; Katzenellenbogen J. A.; Brown M.; Greene G. L. The SERM/SERD bazedoxifene disrupts ESR1 helix 12 to overcome acquired hormone resistance in breast cancer cells. eLife 2018, 7, e37161 10.7554/eLife.37161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Domling A. The Gewald multicomponent reaction. Mol. Divers 2011, 15 (1), 3–33. 10.1007/s11030-010-9229-6. [DOI] [PubMed] [Google Scholar]
- Sabnis R. W. The Gewald Synthesis. Sulfur Reports 1994, 16 (1), 1–17. 10.1080/01961779408048964. [DOI] [Google Scholar]
- Huang X.-G.; Liu J.; Ren J.; Wang T.; Chen W.; Zeng B.-B. A facile and practical one-pot synthesis of multisubstituted 2-aminothiophenes via imidazole-catalyzed Gewald reaction. Tetrahedron 2011, 67 (34), 6202–6205. 10.1016/j.tet.2011.06.061. [DOI] [Google Scholar]
- Wang T.; Huang X.-G.; Liu J.; Li B.; Wu J.-J.; Chen K.-X.; Zhu W.-L.; Xu X.-Y.; Zeng B.-B. An Efficient One-Pot Synthesis of Substituted 2-Aminothiophenes via Three-Component Gewald Reaction Catalyzed by L-Proline. Synlett 2010, 1351–1354. 10.1055/s-0029-1219917. [DOI] [Google Scholar]
- Lee J. H.; Lee B. S.; Shin H.; Nam D. H.; Chi D. Y. Acetonitrile-mediated synthesis of 2,4-dichloroquinoline from 2-ethynylaniline and 2,4-dichloroquinazoline from anthranilonitrile. Synlett 2006, 1, 65–68. 10.1055/s-2005-922790. [DOI] [Google Scholar]
- Roecker A. J.; Mercer S. P.; Harrell C. M.; Garson S. L.; Fox S. V.; Gotter A. L.; Prueksaritanont T.; Cabalu T. D.; Cui D.; Lemaire W.; Winrow C. J.; Renger J. J.; Coleman P. J. Discovery of dual orexin receptor antagonists with rat sleep efficacy enabled by expansion of the acetonitrile-assisted/diphosgene-mediated 2,4-dichloropyrimidine synthesis. Bioorg. Med. Chem. Lett. 2014, 24 (9), 2079–85. 10.1016/j.bmcl.2014.03.052. [DOI] [PubMed] [Google Scholar]
- Escande A.; Pillon A.; Servant N.; Cravedi J. P.; Larrea F.; Muhn P.; Nicolas J. C.; Cavailles V.; Balaguer P. Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor α or β. Biochem. Pharmacol. 2006, 71 (10), 1459–69. 10.1016/j.bcp.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Elrazaz E. Z.; Serya R. A. T.; Ismail N. S. M.; Albohy A.; Abou El Ella D. A.; Abouzid K. A. M. Discovery of potent thieno[2,3-d]pyrimidine VEGFR-2 inhibitors: Design, synthesis and enzyme inhibitory evaluation supported by molecular dynamics simulations. Bioorg. Chem. 2021, 113, 105019. 10.1016/j.bioorg.2021.105019. [DOI] [PubMed] [Google Scholar]
- Ghith A.; Ismail N. S. M.; Youssef K.; Abouzid K. A. M. Medicinal Attributes of Thienopyrimidine Based Scaffold Targeting Tyrosine Kinases and Their Potential Anticancer Activities. Arch. Pharm. (Weinheim) 2017, 350 (11), 1700242. 10.1002/ardp.201700242. [DOI] [PubMed] [Google Scholar]
- Fedorov O.; Niesen F. H.; Knapp S. Kinase inhibitor selectivity profiling using differential scanning fluorimetry. Methods Mol. Biol. 2012, 795, 109–18. 10.1007/978-1-61779-337-0_7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.