

Abstract
Monogenic diseases that result in early pregnancy loss or neonatal death are genetically and phenotypically highly variable. This often poses significant challenges in arriving at a molecular diagnosis for reproductive planning. Molecular autopsy by proxy (MABP) refers to the genetic testing of relatives of deceased individuals to deduce the cause of death. Here, we specifically tested couples who lost one or more children/pregnancies with no available DNA. We developed our testing strategy using whole exome sequencing data from 83 consanguineous Saudi couples. We detected the shared carrier state of 50 pathogenic variants/likely pathogenic variants in 43 families and of 28 variants of uncertain significance in 24 families. Negative results were seen in 16 couples after variant reclassification. In 10 families, the risk of more than one genetic disease was documented. Secondary findings were seen in 10 families: either genetic variants with potential clinical consequences for the tested individual or a female carrier for X‐linked conditions. This couple‐based approach has enabled molecularly informed genetic counseling for 52% (43/83 families). Given the predominance of autosomal recessive causes of pregnancy and child death in consanguineous populations, MABP can be a helpful approach to consanguineous couples who seek counseling but lack molecular data on their deceased offspring.
Keywords: consanguinity, molecular autopsy by proxy, neonatal deaths, recurrent pregnancy loss, whole exome sequencing

1. INTRODUCTION
Advances in genomic data utilization have encouraged their adoption in increasingly diverse clinical settings. Reproductive medicine is one of the areas where genomic tools have proven to be useful. Genetic tests in reproductive medicine are typically pursued for three main purposes: to identify infertility causes, determine genetic diseases transmissible to offspring, and optimize assisted reproductive technology (ART). 1
Many monogenic disorders have a lethal phenotype either early in utero or later in life. Genetic and chromosomal disorders contribute significantly to neonatal and infant mortality and morbidity with congenital malformations and metabolic crisis are leading causes of death. 2 , 3 , 4 Indeed, 28% of deaths in neonatal intensive care unit (NICU) are caused by confirmed genetic diagnoses, a third of which are only diagnosed post‐mortem, which tend to lead to more time spent in the NICU in Boston Children's Hospital. 4 Furthermore, the incidence of inborn errors of metabolism (IEM) in the population of patients admitted to the PICU was 2.2%–3% in different populations, a figure quite similar to the reported incidence for patients with septic shock. 5 Wojcik et al. demonstrates the mortality burden of genetic diseases in infancy using NGS technology in prenatal, postnatal and post‐mortem samples. This study reveals a higher prevalence of genetic disorders up to 22% in 573 deceased infants; 54% had chromosomal disorders and 47% had monogenic disorders with one infant had both chromosomal disorder and monogenic disorder. The proportion of genetic diagnoses made by NGS technologies increased over the years. For counseling purpose, a confirmed molecular diagnosis is required to provide a family with the reproductive options. 6
Additionally, early pregnancy loss can represent the severe end of phenotypic spectrum of several monogenic disorders although this remains poorly addressed in the literature. 7 , 8 , 9 , 10 The incidence of pregnancy loss from implantation to clinically recognized spontaneous abortion (SAB) has been reported to be approximately 30%. Pregnancy loss includes SAB or fetal death prior to 20 weeks (miscarriage) and fetal death at 20 weeks of gestation or greater (stillbirth or intrauterine fetal demise [IUFD]). Approximately 50% of the spontaneous pregnancy loss results from chromosomal abnormalities such as aneuploidy. 11 It has been suggested that 86% of these abnormalities are numerical chromosomal abnormalities, 6% are structural abnormalities, and 8% are due to other genetic mechanisms, such as chromosomal mosaicism and molar pregnancies. 9 Due to the significant psychological consequences of recurrent pregnancy loss on families, determining the underlying genetic etiologies helps in providing the family with informed reproductive options for normal future pregnancies as well as minimizing the guilt felt among those losing pregnancies. 12 , 13
Consanguinity is known to be a possible risk factor for birth defects, as it results in the expression of rare and deleterious genes causing autosomal recessive disorders. Saudi Arabia is considered to be among the countries with the highest rates of consanguineous marriages, leading to high rates of birth defects and even neonatal deaths. 14 Many families in Saudi Arabia prefer consanguineous marriages in hopes of having already been acquainted with the spouse and as a way to keep the property within the family and the tradition alive. 15 Warsy et al. studied the consanguinity prevalence in well‐educated Saudi females in two generations. The study concludes that even though there is an awareness that certain genetic disorders occur at a higher frequency in cousin marriages, there is no decrease in the prevalence of consanguinity over a generation. 16 Although the contribution of genetics to neonatal deaths and IUFD is not fully understood, NGS has made it possible to increase awareness of monogenic diseases in embryonic stages, in addition to uncovering novel genes in embryonic lethality in humans. 8 , 10 , 12
Several strategies have been established to provide families, particularly high‐risk families, with accurate risk estimates of having a child with a genetic condition. Preconception exome‐based parental screening is the process of testing couples for their risk of having a child with a genetic disease, particularly autosomal recessive and X‐linked recessive conditions. 17 Sallevelt et al. proposed exome‐based preconception carrier screening (PCS) and a filtering strategy to rapidly identify the majority of relevant pathogenic mutations. 17 Additionally, several studies have investigated the utilization of WES in the prenatal setting in cases of structurally abnormal pregnancies revealed by prenatal scans as well as in cases of pregnancy loss and developmental disorders. These studies demonstrated the clinical applications of WES in pregnancy loss or IUFD and structurally abnormal pregnancy and have revealed the lethal Mendelian genes that might contribute to RPL. The few studies using WES in both parents looked for carrier status of genetic mutation and established a risk in each family. 8 , 18 , 19 , 20
MABP is a term we coined to describe genetic testing of couples or relatives with a deceased offspring (other relatives) which indicates a priori increased risk of having a child with a recessive genetic disease before they attempt to conceive. 8 In most families, there is no clear genetic etiology for the phenotype of early loss without genetic testing or incomplete genetic testing and therefore it is difficult to characterize the phenotype early in life or in utero. Furthermore, early neonatal death without a genetic diagnosis requires parental testing and family counseling. Therefore, MABP through PCS aims to reveal the genetic cause for an identified phenotype to provide families with informed reproductive options. 21
In this study, we describe a couple‐based approach using WES in 83 consanguineous Saudi couples with early pregnancy loss, IUFD, neonatal death or family history of an unidentified genetic condition without established genetic diagnosis to determine familial monogenic diseases. Our goal is to provide high‐risk couples with variable reproductive options, including prenatal diagnosis, preimplantation genetic diagnosis, acceptance of the genetic risk and preparation for the possibility of having a child with a certain disease, and avoidance of further conception.
2. METHODS
2.1. Human subjects
We counseled 83 Saudi consanguineous couples who sought medical genetics services with a previous history of neonatal and infantile death, IUFD, and/or pregnancy loss without an established genetic diagnosis to determine the familial monogenic diseases that could have been due to a recessive disorder. Detailed clinical information was gathered, including family history of monogenic disease, consanguinity, past medical history, and death reports. As applicable, couples and their children were recruited by written informed consent forms approved by the Internal Review Board of King Saud University Medical Center and King Faisal Specialist Hospital and Research Centre.
2.2. Whole exome sequencing
Double‐stranded DNA capture baits against approximately 36.5 Mb of the human coding exome (targeting >98% of the coding RefSeq and Gencode v28 regions, which were obtained from the human genome build GRCh37/hg19 on May 2018) were used to enrich target regions from fragmented genomic DNA with the Twist Human Core Exome Plus kit (Twist Bioscience). The generated library was sequenced on an Illumina platform to obtain at least 20× coverage depth for >98% of the targeted bases. An in‐house bioinformatics pipeline, including read alignment to GRCh37/hg19 genome assembly, variant calling and annotation, and comprehensive variant filtering, was applied. All disease‐causing variants reported in HGMD® in ClinVar as well as all variants with minor allele frequencies (MAFs) below 1% in the genomAD database were considered.
The investigation for relevant variants was focused on coding exons and flanking ±20 intronic bases. All potential modes of inheritance patterns were considered. In addition, family histories and clinical information were used to evaluate identified variants with respect to their pathogenicity and causality; the variants were categorized as diagnostic, inconclusive and unremarkable. All variants related to the phenotype of the patient, except benign or likely benign variants, were reported.
Our lab has established stringent quality criteria and validation processes for variants detected by NGS. Low‐quality single nucleotide variants and all relevant deletion/insertion variants were confirmed by Sanger sequencing. Consequently, we warrant a specificity of >99.9% for all reported variants.
2.3. Interpretation strategy
Causative pathogenic variants that were detected in both partners in the same autosomal recessive gene are classified as diagnostic. This indicates an increased risk for their progeny to be affected by autosomal recessive disease if both are heterozygous.
Variants of uncertain clinical significance (VUS) detected in both partners in the same autosomal recessive gene are classified as inconclusive. According to the American College of Medical Genetics (ACMG) recommendation, VUS were studied further by family segregation and/or clinical consistency (Figure 3). In selected cases, we have performed deletion and duplication analysis in the other partner in the case of a pathogenic variant detected in one partner consistent with the phenotype for further clarification.
The variants in the latter category were carefully chosen to qualify as much as possible as pathogenic, likely pathogenic if the genes were established in the online Mendelian inheritance in man database. Regarding the genetic variation to in candidate research genes, we have considered only genes with compelling biological candidacy (special emphasis was made on animal models, but other lines of evidence were also pursued).
Variants with no clinical relevance to the described phenotype and/or VUS excluded by family segregation or by lack of clinical consistency were classified as unremarkable.
2.4. Multiplex ligation‐dependent probe amplification
Multiplex ligation‐dependent probe amplification (MLPA) is a technique used to identify variations in the copy number of genes and if there are deletions or duplications in specific genes. We have used specific MLPA probes to recognize adjacent target‐specific sequences, and only in the presence of a perfect match without a single gap, after hybridization, the probes ligated and amplified after which PCR amplification is performed using only one PCR primers pair, which is fluorescently labeled followed by separation by size by capillary electrophoresis. 22 In one of the family, we have found a pathogenic variant in PEX12 gene in the father and negative maternal exome. So, we performed quantitative PCR assay (qPCR) by using six gene‐specific amplicons encompassing the coding exons 1, 2, 3 (or part of it) of the PEX12: NM_000286.2 genes.
3. RESULTS
3.1. Human subjects
All 83 counseled families were consanguineous couples. Clinical information was completed for 77% of the families, and 61% of the families had recurrent neonatal deaths with a reported phenotype.
As demonstrated in Figure 1, couples with nephews or nieces who died with of undetermined genetic diseases represented 7% (N = 6 couples) of the cases; RPL including IUFD and miscarriages represented 24% (N = 20 couples) of the cases; neonatal deaths in the offspring represented 60% (N = 50 couples) of cases; both neonatal death and RPL represented about 9% of our cohort (N = 7 couples).
FIGURE 1.
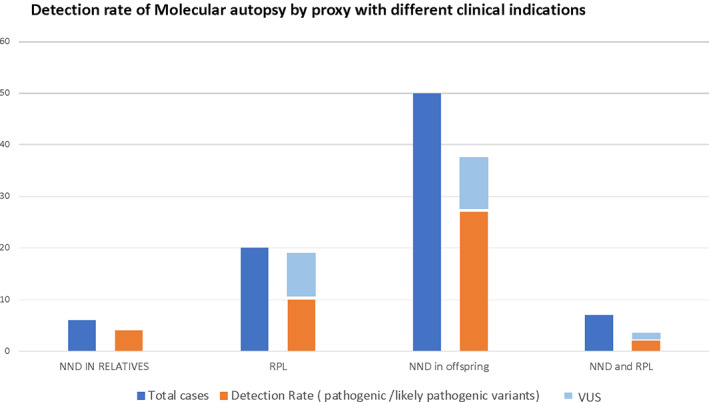
Clinical summary for indication of parental exome testing and the detection rate for pathogenic variants and VUS [Colour figure can be viewed at wileyonlinelibrary.com]
3.2. Whole exome sequencing and interpretation of the results
Duo WES was performed in 83 Saudi consanguineous parents and showed high‐diagnostic yield (65% total). Eighty‐one variants were found in 67 families, including pathogenic, likely pathogenic variants and VUS.
3.2.1. Diagnostic
Pathogenic or likely pathogenic variants that segregated in the family and showed clinical consistency were found in 52% of the families (N = 43 couples) (Figure 2). Diagnostic result with the variants in both parents were identified in the following genes: POLR3A, ECHS1, CTU2, ACAD9, GUSB, MMUT, IBA57, BCS1L, STXBP2, HSPG2, CANT1, MMAB, MALT1, EVC2, FRAS1, CEP290, TCTN2, NPHS1, ISPD, CRIPT, MKS1, CC2D2A, EML1, PEX26, LGI4, KLHL7, TMEM231, GAA, DNAH5, CDT1, LAMB2, MRAP, ACADVL TRIP11, PAX1, AK2, and SLC26A3 KIAA0586, LZTR1, MPDZ, NPHP3 (Table 1).
FIGURE 2.
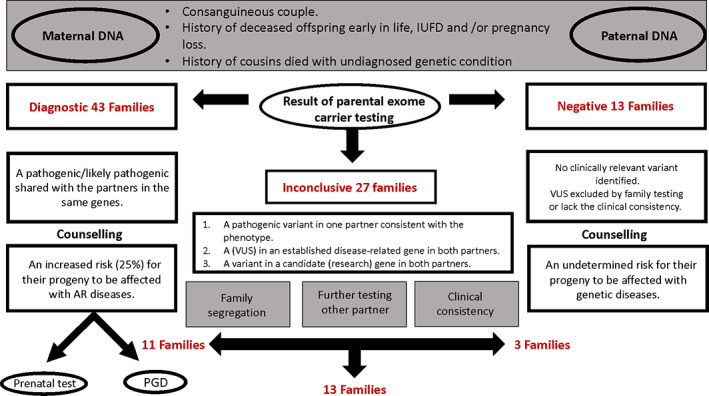
Workflow for all cases with different indications, including the result and the analysis of the result [Colour figure can be viewed at wileyonlinelibrary.com]
TABLE 1.
Pathogenic/likely pathogenic variants with the reported phenotype
| Phenotype | Indications | Gene | Variants | Classification | Serial no. | |
|---|---|---|---|---|---|---|
| 1 | Hx of second‐ and third‐degree relative died with GDD, hypotonia, and cardiac condition. | ND | POLR3A | c.1909 + 22G > A | Pathogenic | 1 |
| 2 | Parent of two children deceased with lactic acidosis and severe hypotonia. | ND | ECHS1 | c.88þ5G | Pathogenic | 2 |
| NEB | c.17358T > A:p.N5786K | Likely pathogenic | 3 | |||
| 3 | VSD, kidney malformation abnormal limb posture, cerebellar hypoplasia, hypertelorism and severe IUGR. | IUFD | CTU2 | c.1086 + 5G > A | Pathogenic | 4 |
| 4 | Persistent lactic acidosis, hypertrophic cardiomyopathy, high‐liver enzyme level. | ND | ACAD9 | c.1240C > T (p.Arg414Cys) | Pathogenic | 5 |
| CYP21A2 | c.92C > T | Pathogenic | 6 | |||
| 5 | Fetal hydrops fetalis, pleural effusion, pericardial effusion, ascites ended by fetal demise. | ND | GUSB | c.1429C > T (p.Arg477Trp) | Pathogenic | 7 |
| 6 | Second‐ or third‐degree nephews/nieces who died early in life (2nd or 3rd day of life). | ND | MMUT | c.329A > G (p.Tyr110Cys) | Pathogenic | 8 |
| 7 | Recurrent neonatal deaths. | ND | IBA57 | c.316A > G (p.Thr106Ala) | Pathogenic | 9 |
| 8 | Recurrent neonatal deaths. | ND | BCS1L | c.385G > A (p.Gly129Arg) | Pathogenic | 10 |
| 9 | Recurrent neonatal deaths | ND | STXBP2 | c.1485 + 1G > A | Pathogenic | 11 |
| 10 | Recurrent neonatal deaths. | ND | HSPG2 | c.790C > T (p.Arg264*) | Likely pathogenic | 12 |
| 11 | Recurrent neonatal deaths. | ND | CANT1 | c.902_906dup (p.Ser303Alafs*21 | Pathogenic | 13 |
| 12 | Recurrent neonatal deaths. | ND | MMAB | c.197‐1G > T | Pathogenic | 14 |
| 13 | Recurrent neonatal deaths. | ND | MALT1 | c.1240G > A (p.Gly414Arg) | Likely pathogenic | 15 |
| 14 | Dysmorphic, short neck, narrow restricted chest, faint heart sound, short four limbs with polydactyly of the upper limb. | ND | EVC2 | c.2017_2021del (p.Thr673Glufs*14) | Likely pathogenic | 16 |
| 15 | Early neonatal death with bilateral renal agenesis. | ND | FRAS1 | c.1226dup (p.Gln411Thrfs*26) | Likely pathogenic | 17 |
| 16 | Early death, microcephaly, Potter‐like facies, sloping head, microphthalmia, low‐set ears, short webbed neck, occipital encephalocele, polycystic kidneys, ambiguous genitalia, polydactyly (suspected diagnosis ‐ Meckel‐Gruber syndrome). | ND | CEP290 | c.613C > T (p.Arg205*) | Pathogenic | 18 |
| 17 | Microcephaly, Potter‐like facies, sloping head, microphthalmia, low‐set ears, hypertelorism, occipital encephalocele, polycystic kidneys, oligohydramnios, talipes, polydactyly. | ND | TCTN2 | c.1506‐2A > G | Pathogenic | 19 |
| 18 | Recurrent neonatal death due to polycystic kidney disease. | ND | NPHS1 | c.2540_2543delCTAA (p.Thr847ArgfsTer57) | Likely pathogenic | 20 |
| 19 | Severe hydrocephalus, encephalocele and lissencephaly in two pregnancies; mother is healthy apart from hypothyroidism with no other medical issues. | ND | ISPD | c.1186G > T (p.Glu396*) | Pathogenic | 21 |
| 20 | Characteristic facies, hypoplastic terminal phalanges, osteopenia, albinoid fundus, markedly impaired retinal function, recurrent infections, persistent anemia with anisopoikilocytosis. | ND | CRIPT | c.141delT (p.Phe47Leufs*84) | Pathogenic | 22 |
| 21 | Anencephaly, polydactyly, cystic kidney (suspected diagnosis Meckel syndrome). | ND | PEX26 | c.228C > T (p.Gly76Alafs*5) | Pathogenic | 23 |
| 22 | Microcephaly, agenesis of corpus callosum, occipital encephalocele, multicystic kidney, hypotonia, IUFR (suspected Meckel syndrome). | ND | MKS1 | c.261 + 2T > A | Pathogenic | 24 |
| 23 | Meckel syndrome‐affected fetuses. | IUFD | CC2D2A | c.3084delG (p.Lys1029Argfs*3) | Pathogenic | 25 |
| 24 | Four neonatal deaths with congenital hydrocephalus. | ND | CC2D2A | c.3084delG (p.Lys1029Argfs*3) | Pathogenic | 26 |
| 25 | Bilateral genu recurvatum, narrow chest with mild to moderate inspiratory stridor, no organomegaly, generalized hypotonia, hyporeflexia, mild microcephaly, respiratory distress. | ND | PEX26 | c.228C > T (p.Gly76Alafs*5) | Pathogenic | 27 |
| 26 | Hypotonia, dysmorphic feature, low‐set ears, micrognathia, high‐arched palate, bilateral talipes, equinovarus, poor feeding. | ND | EML1 | c.2233G > A (p.Val745Ile) | Pathogenic | 28 |
| 27 | Recurrent miscarriage. | RPL | LGI4 | c.834del (p.Ser279Alafs*191) | Likely pathogenic | 29 |
| 28 | Recurrent spontaneous abortion. Abnormal facial shape; CHD, atrial septal defect; CNS abnormality; cerebellar hypoplasia; congenital onset; depressed nasal bridge; dilation of lateral ventricles; enlarged cisterna magna; hernia of the abdominal wall; high, narrow palate; hypertonia; low‐set ears; microcephaly; optic atrophy; optic disc hypoplasia; overlapping fingers; ventriculomegaly. | RPL+ IUFD | KLHL7 | c.807C > A | Likely pathogenic | 30 |
| 29 |
Abnormal vertebral morphology, renal abnormality, aplasia/hypoplasia of the cerebellar vermis, clinodactyly, hydrocephalus, low‐set ears, micrognathia, multiple renal cysts, oligohydramnios, polydactyly, rocker bottom foot and ventriculomegaly. Asymptomatic parents are consanguineous, and they lost two children at the ages 33 and 32 G.W. |
IUFD | TMEM231 | c. 930‐5_930‐2delinsTGTC | Likely pathogenic | 31 |
| 30 | Recurrent pregnancy loss. | RPL | CHRNG | c.1019C > T | Likely pathogenic | 32 |
| GAA | c.1430delT | Likely pathogenic | 33 | |||
| 31 | One child died with unilateral renal agenesis, recurrent spontaneous abortion. Hx of previous hydatidiform mole. | RPL | DNAH5 | c.5503C > T(p.Gln1835Ter) | Likely pathogenic | 34 |
| AMHR2 | c.994C > T | Pathogenic | 35 | |||
| PTPRQ | c.4155 + 1G > A | Pathogenic | 36 | |||
| PCDH15 | c.4604_4608dup | Pathogenic | 37 | |||
| 32 | Recurrent miscarriages and IUFD 3X skeletal phenotype in the aborted fetus. | RPL | TRIP11 | c.3082C > T (p.Arg1028*) | Pathogenic | 38 |
| 33 | Multiple malformations. | IUFD | CDT1 | c.1393dupT (p.V464fs) | Likely pathogenic | 39 |
| 34 | Recurrent neonatal deaths with renal phenotype. | ND | LAMB2 | c.4276dupG (p.Ala1426Glyfs*6) | Likely pathogenic | 40 |
| 35 | Second‐ or third‐degree cousin died with phenotype consistent with liver disease, and hypoglycemia | ND | MRAP | c.105_106 + DEL P.(His36Phefs*84) | Pathogenic | 41 |
| 36 | Recurrent failed implantation after IVF and 2 miscarriages, one died with recurrent infection | RPL | SLC26A3 | c.79G > C p.(Gly27Arg) | Pathogenic | 42 |
| AK2 | c.559G > T p.(Gly187*) | Likely pathogenic | 43 | |||
| 37 | Abnormal stomach, congenital arthrogryposis multiplex, talipes equinovarus. | ND | LGI4 | c.639G > A P.(Trp213*) | Likely pathogenic | 44 |
| 38 | Son 1: Intracranial cystic lesion, Neonatal death, Premature birth; Son 2: Abnormal heart morphology, Abnormality of the kidney, Neonatal death, Premature birth. | ND | NPHP3 | c.2694‐2_2694‐1del | Pathogenic | 45 |
| 39 | Two previous IUFD at 24 weeks and 27 weeks with scan showing skeletal changes | ND | TRIP11 | c.763C > T p.R255X | Pathogenic | 46 |
| 40 |
Two offspring passed away with 1‐ Coarctation of aorta; Hydrocephalus and brain anomalies 2‐ Atrial septal defect, Hydrocephalus; Severe, other anomalies + recurrent pregnancy loss. |
ND IUFD |
MPDZ | c.628C > T. p.(Gln210*) | Pathogenic | 47 |
| 41 | Hx of cousins with early death with cardiomyopathy. | ND | LZTR1 | c.639del P.(Cys214Alafs*38) | Likely pathogenic | 48 |
| 42 | Recurrent neonatal deaths. | ND | KIAA0586 | c.78dup (p.Lys27fs*) | Likely pathogenic | 49 |
| 43 | Neonatal death at 3 DOL with positive NBS VLCAD | ND | ACADVL | c.65C > A;p.Ser22X | Pathogenic | 50 |
Abbreviations: RPL: recurrent pregnancy loss including miscarriage and IUFD; ND: neonatal death; PCS: preconception screening in the presence of cousins with genetic condition.
3.2.2. Inconclusive
VUS were found in 32% of the families (N = 27 couples) (Figure 2). Segregation, clinical consistency and/or duplication, and deletion analyses were performed to confirm the pathogenicity of these variants and resulted in the reclassification of these variants as follows: in 41% of the families with inconclusive variants (N = 11 couples), the variants (DOCK7, CHAT, KIAA0586, NDUFAF3, GBA, PEX1, NDUFAF5, HTRA2, CCDC88C, CPS1, POLR3A) were reclassified as diagnostic; in 11% of the families (N = 3 couple), the variants were reclassified as benign; c.1768G > A:p.(Val590Met) at KIAA0556 gene, c.4552C > G p.(Arg1518Gly) at DCHS1 gene, and c.685C > G:p.(Pro229Ala) at TMEM231 gene; and in 48% of the families (N = 13 couples), the variants remained uncertain (KIAA0556, WDR34, CC2D2A, AGRN, GORAB, QARS, KLHL40, FH, ACACA, PAX1, FKRP, DOCK6, FKTN, UNC80) (Figure 2 and Table 2). Therefore, the diagnostic yield increased to 65% (N = 54) (Figure 3).
TABLE 2.
Variants of unknown clinical significance likely related to the phenotype and shared by both partners
| Case ID | Phenotype | Indication | Gene | Variants | OMIM disease | Serial no. |
|---|---|---|---|---|---|---|
| 1 | Recurrent neonatal deaths | ND | WDR34 | c.544C > T p.R182W | Short‐rib thoracic dysplasia 11 with or without polydactyly | 1 |
| 2 | Oligohydramnios, dysmorphic facial features, midline cleft lip, agenesis of corpus callosum, occipital encephalocele. | ND | CC2D2A | c.4531T > C (p.Trp1511Arg) | Joubert syndrome‐9 | 2 |
| 3 | IUGR, subcutaneous edema, abnormal position of upper and lower limbs with no movement. | RPL | AGRN | c.5948C > T (p.Thr1983Met) | Congenital myasthenic syndrome‐8 | 3 |
| 4 | Intrauterine fetal death, IUGR. | RPL | ZMIZ2/KIAA1886 | c.1270C > T (p.Gln424*) | 4 | |
| 5 | Recurrent neonatal deaths. | ND | GORAB | c.306dup (p.Pro103Thrfs*20) | Gerodermia osteodysplastica | 5 |
| 6 | Recurrent neonatal deaths. | ND | QARS1 | c.316G > A (p.Asp106Asn) | Progressive microcephaly with seizures and cerebral and cerebellar atrophy (MSCCA) | 6 |
| ACACA | c.81A > G (p.Ile27Met) | Acetyl‐CoA carboxylase deficiency | 7 | |||
| 7 | Central hypotonia, scoliosis, cerebellar hypoplasia, 2–3 toe syndactyly, atrial septal defect, PDA, abnormal ear morphology, high palate. | ND | PAX1 | c.95C > T p.(A32V) | Otofaciocervical syndrome 2 | 8 |
| MSRB3 | c.2T > G/likely pathogenic | Autosomal recessive deafness‐74 | 9 | |||
| MOCOS | c.894CA (p.Y298*) likely pathogenic | Molybdenum cofactor sulfurase | 10 | |||
| 8 | 2X Hydrops fetalis, polyhydramnios, multiple congenital anomalies. | RPL | KLHL40 | c.35G > T p.(Arg12Leu) | AR nemaline myopathy type 8 | 11 |
| 9 |
1: Dandy‐Walker malformation, polyhydramnios, stillbirth; 2: IUFD with CNS anomalies. 3: Absent septum pellucidum; agenesis of corpus callosum; aplasia/hypoplasia of the corpus callosum; cerebellar vermis hypoplasia; Dandy‐Walker malformation; dilated third ventricle; echogenic fetal bowel; enlarged cisterna magna; ventriculomegaly. |
RPL | FH | c.1043G > A p.(Gly348Asp) | Fumarate hydratase | 12 |
| FKRP | c.1061G > A p.(Gly354Glu) | Congenital muscular dystrophy‐dystroglycanopathy with or without impaired intellectual development | 13 | |||
| 10 | Transposition of great arteries, abnormal heart valves, agenesis of corpus callosum, holoprosencephaly, hydrocephalus, IUGR. | IUFD | FKTN | c.44T > G P.(Leu15Arg) | Muscular dystrophy‐dystroglycanopathy (congenital with brain and eye anomalies), type A, 4 | 14 |
| DOCK6 | c.356A > G P.(Asp119Gly) | Adams‐Oliver syndrome‐2 | 15 | |||
| 11 | IUFD with increased NT, ventriculomegaly, multicyclic dysplastic kidney and short long bone. | IUFD | DHX34 | c. 1399G > A P. D467N | 16 | |
| 12 | Brain anomalies, FTT, dilated lateral ventricles, partial agenesis of the corpus callsum, EEG abnormalities, IUGR, bilateral hearing loss, optic atrophy. | IUFD | DOCK7 | c.1591G > A | Developmental and Epileptic Encephalopathy‐23 (DEE23) | 17 |
| 13 | 5X; IUFD at 7 months, boy with short limbs and hydrocephalus; IUFD at 6 months with short limbs; IUFD at 24 weeks with massive fetal edema, abnormal skeletal system and bilateral dilated renal pelvis; IUFD at 27 weeks, fetal edema, ascites and short limbs. | IUFD | CHAT | c.1300G > A (p.Gly434Ser) | Myasthenic syndrome, congenital, 6, presynaptic | 18 |
| 14 | Recurrent spontaneous abortions. Couple had 2 children affected with seizures, hypotonia, global developmental delay, recurrent aspiration pneumonia, constipation, both children deceased. | ND RPL | KIAA0556 | c.1777T > C p.(Tyr593His) | Joubert syndrome‐26 | 19 |
| 15 | Recurrent neonatal deaths. | ND | NDUFAF3 | c.481C > G (p.Arg161Gly) | Mitochondrial respiratory chain complex I | 20 |
| 16 | Recurrent neonatal deaths. | ND | GBA | c.520T > A (p.Tyr174Asn) | Gaucher disease | 21 |
| 17 | Abnormal VLCFA (? diagnosis‐Zellweger syndrome). | ND | PEX1 | c.1240_1359del (p.Ile414_Leu453del) | Peroxisomal biogenesis disorders 1A | 22 |
| 18 | Agenesis of corpus callosum associated with white matter and brainstem abnormal signal. | ND | NDUFAF5 | c.737T > A (p.Leu246Gln) | Mitochondrial respiratory chain complex I deficiency nuclear type 16 | 23 |
| 19 | Microcephaly, hypotonia, encephalopathy, increased CSF lactate level and 3‐methylglutaconic aciduria. | ND | HTRA2 | c.818_820del (p.Leu273del) | 3‐Methylglutaconic aciduria type VIII (MGCA8 | 24 |
| 20 | Neonatal death with hyperammonemia. | ND | CPS1 | c.211T > C p.(Ser71Pro) | Carbamoyl phosphate synthetase I | 25 |
| 21 | Recurrent miscarriage. | RPL | CCDC88C | c.5059‐2A > G | congenital hydrocephalus‐1 | 26 |
|
22 23 |
2X early neonatal death with hypotonia and recurrent respiratory infections and respiratory distress. | ND | POLR3A | c.1895G > T (p.Cys632Phe) | Leukodystrophy, hypomyelinating, 7 (HLD7) | 27 |
| 24 |
Abnormal facial features; Failure to thrive; feeding difficulty,Laryngomalacia; Motor delay; passed away with Respiratory compromise. 1: Stillbirth; Brother 2: Stillbirth; Brother 3: Stillbirth; Mother: Spontaneous abortion; Sister 1: Stillbirth; Sister 2: Stillbirth; Sister 3: Stillbirth Siblings affected. |
ND | UNC80 | c.5254C > T p.(Leu1752Phe) | Infantile hypotonia with psychomotor retardation and characteristic facies‐2 | 28 |
| 15 | Recurrent neonatal deaths. | ND | TMEM231 | c.685C > G:p.(Pro229Ala) |
Ciliopathic diseases Meckel syndrome 11 |
29 |
| 25 | Previous 3X IUFD Multiple congenital anomalies and neonatal death with brain atrophy, esophageal atresia, heterotaxy; hypoplastic right heart IUFD | ND | DCHS1 | c.4552C > G p.(Arg1518Gly) | Multiple congenital anomalies | 30 |
| 26 | Multiple congenital anomalies and early death. | ND | KIAA0556 | NM_015202.3:c.1768G > A:p.(Val590Met) | AR Joubert syndrome type 26 | 31 |
Abbreviations: RPL: recurrent pregnancy loss including miscarriage and IUFD; ND: neonatal death; OMIM, online Mendelian inheritance in man; PCS: preconception screening in the presence of cousins with a genetic condition.
FIGURE 3.
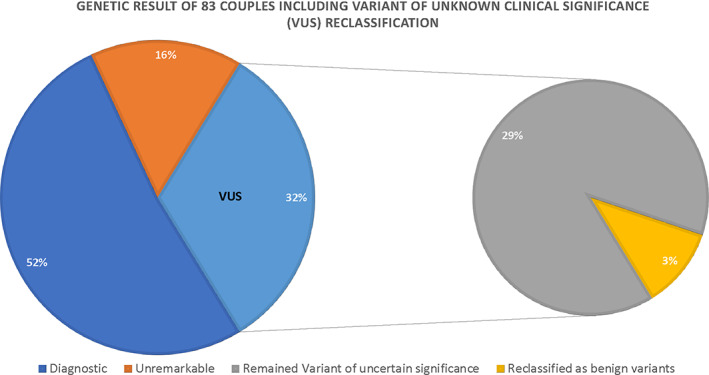
Parental exome sequencing results and VUS reclassification (pie chart) [Colour figure can be viewed at wileyonlinelibrary.com]
Further analysis revealed two variants in candidate genes, ZMIZ2 and DHX34 (novel genes), that might cause RPL.
3.2.3. Negative
Fifteen percentage of the families had negative results (N = 13 couples) (Figure 2, Table 1), which accounted for the lack of variants relevant to the phenotype and/or that only one of the partners was a carrier (no family segregation). Table 1 summarizes the WES findings for the 83 families, the phenotypes, whether the exome findings matched the phenotype or not, and the novel variants discovered. Including the three families with benign variants makes the undiagnosed percentage 19% (N = 16 couples).
3.3. Secondary findings
Few families showed secondary findings, even though these cases were solved, and, therefore, need counseling in the future. We detected 10 couples who shared the carrier status of several autosomal recessive disease‐associated genes, which indicates a risk of offspring having more than one genetic disease of 12%. In family 4, a well‐known pathogenic variant for congenital adrenal hyperplasia, CYP21A2: c.92C > T, was identified, and even though it segregated in the family, it was not consistent with the phenotype, as the child's death was due to persistent lactic acidosis, hypertrophic cardiomyopathy, and high‐liver enzyme, which is associated with the ACAD9 gene. Female carriers for an X‐linked condition were seen in three families (1, 43, and 65), and there was a common pathogenic variant found in both families, G6PD: c.233T > C p. (Ile78Thr); where only the female was a carrier, we performed further counseling regarding the 50% male offspring risk of G6PD deficiency. In family 71, ES of both parents revealed that the female partner carried an X‐linked condition AR gene variant p.(Gln58Leu), but no detected variants could explain recurrent acrania and IUFD. Furthermore, autosomal‐dominant mutations with potential clinical consequences for the tested individuals were found in five couples (family 36, 52, 65, 66) which changed our approach to presymptomatic testing for the affected individuals (Table 3).
TABLE 3.
Autosomal‐dominant mutations with potential clinical consequences for the tested individual
| Gene | Partner | Variants | Classification | OMIM phenotype |
|---|---|---|---|---|
| SLC5A2 | Male partner | c.1035_1062del.p.(Val346Alafs*1) | Pathogenic | Renal glucosuria |
| FLT4 | Female partner | c.2740G > C p.(Gly 914Arg) | Pathogenic | Pedal edema |
| TTN | Male partner | c.60451delp.(Ile20151Serfs*12) | Likely pathogenic | Cardiomyopathy, dilated |
| ASCC1 | Female partner | c.495del p.(Ala166Profs*14) | Likely pathogenic | Barrett esophagus/esophageal adenocarcinoma 23 |
| HTRA2 | Both partners | c.818_820del (p.Leu273del) | Likely pathogenic | Susceptibility to the development of autosomal dominant Parkinson disease‐13 |
| GBA | Both partners | c.520T > A (p.Tyr174Asn) | Likely pathogenic | Susceptibility of Parkinson disease |
Abbreviation: OMIM, online Mendelian inheritance in man.
Furthermore, MABP identify a carrier status of a pathogenic genetic variant in one partner that might explain a phenotype in several families, but the other partner was not a carrier; for example, MCPH1 c.2595‐1G > C/likely pathogenic p.(Arg497*) was found in a female partner and DDX11 NM_001257144.1:c.1489C > T likely pathogenic in a male partner, but the variants were not found in the other partner. Also, WES revealed a heterozygous variant in the PEX12 gene, c.616C > T p.(Gln206*) in the male partner only. Consequently, we performed MLPA to exclude deletions or duplications in the other partner that were negative in the couple (Table 3).
3.4. Diseases‐causing genes observed
The most observed genetic etiology of RPL was complex dysmorphology disorders; eight genes—including CTU2, KLHL7, DCHS1, CTD1, and DOCK6—were identified in five families. Additionally, variants in the TMEM231, DNAH5, and CC2D2A genes associated with ciliopathy, which is a specific group of multisystem disorders, were identified in two cases each. Diagnostic results with the detection of variants in the LGI4 gene causing neurogenic arthrogryposis were identified in two couples, and AGRN and CHAD, which are associated with congenital myasthenic syndrome, were identified in two couples each. Additionally, two couples each was carrier of DOCK7 or CCDC88C. Congenital muscular dystrophy genes FKRP and FKTN in other two families, each is carrier for one variant. Variants in three genes—GAA, FH, and AK2—associated with enzyme and metabolic diseases were noted in three cases. Variants were also identified in other disease categories, including myopathy (KLHL40), skeletal disorders (TRIP11), and gastrointestinal disorders (SLC26A3), in four cases.
Additionally, candidate genes that are not consistent with a given phenotype were identified that might be of clinical significance, including ZMIZ2 and DHX34.
On the other hand, our analyses showed that inborn errors in metabolism are the most common causes of neonatal deaths, including mitochondrial disorders associated with CPS1, HTRA2, NDUFAF5, PEX1, GBA, NDUFAF3, PEX26, MMAB, BCSIL, IBA57, MMUT, GUSB, ACAD9, ECHSI, ACADVL, and AK2. Ciliopathy, associated with EVC2, CEP290, TCTN2, MKS1, CC2D2A, TMEM231, DNAH5, KIAA0586, and TCTN2, was the second most common category. The third category involved neurologic phenotypes (POLR3A, DOCK7, CCDC88C, EML1, MACF1, MPDZ). Several variants were identified in different disease categories, including neuromuscular diseases (NEB, ISPD, CHRNG, KLHL40), complex dysmorphology syndrome (CRIPT, LZTR1, FRAS1), and renal (NPHS1, NPHP3, LAMB2), skeletal (CANT1, HSPG2, TRIP11), hematological (STXBP2), and immunological (MALT1) diseases (Table 1).
4. DISCUSSION
WES was first introduced for clinical diagnostic purposes in 2009 and has since been applied in different clinical settings as a highly valuable diagnostic approach mainly in postnatal and prenatal genetic diagnosis of Mendelian disorders. WES has provided an opportunity to affordably screen a patient's exome to establish the genetic basis of diseases. 24 , 25 , 26 The reported diagnostic yield of WES generally ranges between 25% and 35%, with a maximum yield of 40% in trio analysis 27 , 28 , 29 and a high‐diagnostic yield of 43%–49% in large consanguineous cohorts from Saudi Arabia. 30 , 31 Another study also reported a diagnostic yield of 60% in Middle Eastern patients from Qatar. 32
Our results provide 51% of the families with a genetic diagnosis, with an additional 13% of the families if we consider VUS with potential clinical usefulness. VUS in both parents were found in 31% of our cohort, and after further analysis of the reports describing the phenotype and segregation of the family, we could exclude three (11%) and consider 12 variants (46%) as potentially disease causing. A negative result was seen in 16 families (19%). We have counseled our families who consented to pretesting for the potential for identifying and reporting incidental (or secondary) findings, which are results that are not related to the indication for ordering the sequencing but that may nonetheless be of medical value or utility to the physician and the patient. In nine families, the results identified a risk of more than one genetic condition in the family; in three families, the female partner was determined to carry an X‐linked genetic disease; and in six families, we discovered that one of the partners carried a heterozygous mutation with potential clinical consequences for the tested individual.
The first report of the yield of WES of a couple (Duo WES) of 44 families with at least one death or lethal fetal malformation at any stage of in utero development and this strategy identified pathogenic/likely pathogenic variants that was shared by both of the couple and resulted in cause embryonic or perinatal lethality. 8 Further utilization of WES for trio analysis using cultured amniocytes or product of conception from the affected fetuses determined a genetic cause in four of seven cases of IUFD, 13 and compound heterozygous variants in DYNC2H1 and ALOX15 were identified in miscarriages from two of four families with RPL. 33 More recent trio‐WES studies in fetuses with ultrasound anomalies that resulted in IUFD or pregnancy termination identified positive variants in 20%, possible variants in 45%, and candidate variants in 9% of 84 fetal deaths with ultrasound anomalies. 34 In a similar work, WES in 15 of 19 POC cases with missed abortion revealed novel variants potentially associated with early embryonic lethality. 35 A study using different filtering strategies proved the applicability of parental WES in eight consanguineous and 25 nonconsanguineous couples for identifying the genetic variants shared by the couples. 17 These results supported the clinical utility of ES in reproductive medicine to assess in couples planning a pregnancy the risk of those couples having children affected with a genetic disease as well as to detect the monogenic etiology of pregnancy loss. The identification of disease‐associated variants provided information for follow‐up genetic counseling regarding recurrence risk and management of subsequent pregnancies. The discovery of novel variants could provide insight into the underlying molecular mechanisms of fetal death.
Several disease categories were noted for the 21 genes carrying variants of diagnostic value. The most prevalent disease category with recurrent pregnancy loss was multisystem disorders; eight genes—including CTU2, KLHL7, DCHS1, CTD1, and DOCK6—were identified in five families. Additionally, variants in the TMEM231, DNAH5 and CC2D2A genes of ciliopathy, which is a specific group of multisystem disorders, were identified in two cases. The second most common category was neurological disorders, including neurogenic arthrogryposis in two couples with the LGI4 gene, congenital myasthenic syndrome in two families with AGRN and CHAD, other neurological disorders associated with genes including DOCK7 and CCDC88C in two couples, and muscular dystrophy associated with FKRP and FKTN in two couples. In contrast, in another study, the second most common cause of RPL after multisystem disorders was cardiac anomalies. 36 In another study, a molecular panel of 70 genes associated with cardiac channelopathies and cardiomyopathies in stillbirth cases was applied to identify pathogenic variants in 12% of 290 cases of IUFD, which indicates that cardiac anomalies are one of the known causes of IUFD. 37 Variants in three genes—GAA, FH, and AK2—associated with enzyme and metabolic diseases were noted in three cases. Variants were also identified in other disease categories, including myopathy (KLHL40), skeletal disorders (TRIP11), and gastrointestinal disorders (SLC26A3), in three cases.
Two novel genes that might have clinical implications and embryonic lethality are ZMIZ2: c.1270C > T; p. (Gln424*), which causes embryonic lethality in mouse (https://www.mousephenotype.org/data/genes/MGI:106374#phenotypesTab), and DHX34: c.1399G > A P.(D467N), which is predicted to be deleterious in silico. Dhx34 protein deficiency in zebrafish has been shown to result in severe neurodevelopmental defects and embryonic lethality. 38
Among the solved cases, the most observed causes of neonatal death were inborn errors in metabolism, mainly mitochondrial disorders due to enzyme deficiencies, followed by ciliopathy and congenital anomaly disorders.
Other similar efforts with different strategies to reduce the neonatal morbidity and mortality as well as the pregnancy loss were conducted in different populations. Variable premarital carrier screening programs were established in different populations based on carrier frequency and disease frequency in that population either as obligatory or voluntary screening. 39 In Saudi Arabia, the premarital screening program that was instituted in 2002, includes sickle cell anemia and thalassemia as a mandatory screening, as well as in Iran and Tunisia the premarital test is a mandatory program. 39 Preconception carrier screening program was established in the Jews population since 2013. This program includes several fatal diseases, with a carrier frequency of at least 1:60 and/or a disease frequency of 1:15 000 live births. 40 The program resulted in a significant reduction of these diseases as shown in the Singer study 2020. 41 Furthermore, Preconception Carrier Screening Programs were also started in the Netherlands since 2016 for the couples wishing to start a family, to know their genetic carrier status for 50 severe genetic diseases. 40
Some ethical issues that were considered during the counseling of these couples included stigmatization and discrimination, knowing that the individual is a carrier for the autosomal recessive genetic condition and at risk of having affected offspring might cause a negative view to and about people with those traits. As well as, potential findings of a dominant trait in the tested individual that harbored a reduced penetrance or variable expressivity, however, it is not yet expressed clinically. Additionally, as expected this strategy influences how the couples perceive the planning of a pregnancy and making a family in a positive light but some undesirable consequences might impact the family like divorce or remarriage of another wife. Another concern is that if this test is routine or are they obligated to do it? Are they guilty if they opt not to?. Some of these ethical dilemmas were also considered in preconception carrier screening in different populations. 42
We acknowledge several limitations of MABP. First, this approach only works to reveal the carrier status of familial variants. Although our work focused on single gene cases, we note that MABP can also reveal the carrier status of balanced chromosomal rearrangements, which were found in a recent very large study to account for a substantial fraction of recurrent pregnancy loss. 43 Second, MABP may only identify the carrier status for autosomal recessive diseases in one of the couples, although this is less of a problem in consanguineous couples who tend to share the same variants. Third, as with other diagnostic applications of ES, VUS remain a formidable challenge. However, it is hoped that data sharing efforts will contribute to the successful reclassification of these variants and it is hoped that this study will contribute toward this goal.
In conclusion, we show that MABP is a highly effective testing strategy in consanguineous populations where autosomal recessive variants tend to be a more common cause of premature death among offspring of couples seeking preconception counseling. Our work highlights the additional benefit of uncovering additional pathogenic variants that can empower couples to make reproductive choices for diseases beyond the ones they are seeking preconception counseling for.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/cge.14049.
ACKNOWLEDGMENTS
This Project was funded by the National Plan for Science, Technology and Innovation (MAARIFAH), King Abdulaziz City for Science and Technology, Kingdom of Saudi Arabia, Award Number (5‐18‐03‐001‐0010).
Ali Alghamdi M, Alrasheedi A, Alghamdi E, et al. Molecular autopsy by proxy in preconception counseling. Clinical Genetics. 2021;100(6):678-691. doi: 10.1111/cge.14049
[Correction added on 12 October 2021, after first online publication: Funding Information section and Acknowledgement have been added.]
Funding information National Plan for Science, Technology and Innovation (MAARIFAH), King Abdulaziz City for Science and Technology, Grant/Award Number: 5‐18‐03‐001‐0010
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the article material and references of this article.
REFERENCES
- 1. Cariati F, D'Argenio V, Tomaiuolo R. The evolving role of genetic tests in reproductive medicine. J Transl Med. 2019;17:267. 10.1186/s12967-019-2019-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Meng M, Zhang Y‐P. Impact of inborn errors of metabolism on admission in a neonatal intensive care unit: a 4‐year report. J Pediatr Endocr Met. 2013;26(7–8):689‐693. 10.1515/jpem-2013-0021 [DOI] [PubMed] [Google Scholar]
- 3. Lin Y, Liu ZL, Zhang P, et al. Genetic aetiology of early infant deaths in a neonatal intensive care unit. J Med Genet. 2020;57:169‐177. [DOI] [PubMed] [Google Scholar]
- 4. Wojcik MH, Schwartz TS, Yamin I, et al. Genetic disorders and mortality in infancy and early childhood: delayed diagnoses and missed opportunities. Genet med. 2018;20(11):1396‐1404. 10.1038/gim.2018.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jouvet P, Touati G, Lesage F, et al. Impact of inborn errors of metabolism on admission and mortality in a pediatric intensive care unit. Eur J Pediatr. 2007;166:461‐465. 10.1007/s00431-006-0265-2 [DOI] [PubMed] [Google Scholar]
- 6. Wojcik MH, Schwartz TS, Thiele KE, et al. Infant mortality: the contribution of genetic disorders. J Perinatol. 2019;39(12):1611‐1619. 10.1038/s41372-019-0451-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shamseldin HE, Tulbah M, Kurdi W, et al. Identification of embryonic lethal genes in humans by autozygosity mapping and exome sequencing in consanguineous families. Genome Biol. 2015;16:116. 10.1186/s13059-015-0681-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shamseldin H, Kurdi W, Almusafri F, et al. Molecular autopsy in maternal–fetal medicine. Genet Med. 2018;20:420‐427. 10.1038/gim.2017.111 [DOI] [PubMed] [Google Scholar]
- 9. Goddijn M, Leschot N. Genetic aspects of miscarriage. Best Pract Res Clin Obstet Gynaecol. 2000;14(5):855‐865. 10.1053/beog.2000.0124 [DOI] [PubMed] [Google Scholar]
- 10. Alazami AM, Awad SM, Coskun S, et al. TLE6 mutation causes the earliest known human embryonic lethality. Genome Biol. 2015;16:240. 10.1186/s13059-015-0792-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hyde KJ, Schust DJ. Genetic considerations in recurrent pregnancy loss. Cold Spring Harb Perspect Med. 2015;5(3):a023119. 10.1101/cshperspect.a023119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maddirevula S, Awartani K, Coskun S, et al. A genomics approach to females with infertility and recurrent pregnancy loss. Hum Genet. 2020;139(5):605‐613. 10.1007/s00439-020-02143-5 [DOI] [PubMed] [Google Scholar]
- 13. Alamillo CL, Powis Z, Farwell K, et al. Exome sequencing positively identified relevant alterations in more than half of cases with an indication of prenatal ultrasound anomalies. Prenat Diagn. 2015;35:1073‐1078. [DOI] [PubMed] [Google Scholar]
- 14. Majeed‐Saidan MA, Ammari AN, AlHashem AM, et al. Effect of consanguinity on birth defects in Saudi women: results from a nested case‐control study. Birth Defects Res A Clin Mol Teratol. 2015;103(2):100‐104. [DOI] [PubMed] [Google Scholar]
- 15. el‐Hazmi MA, al‐Swailem AR, Warsy AS, al‐Swailem AM, Sulaimani R, al‐Meshari AA. Consanguinity among the Saudi Arabian population. J Med Genet. 1995;32(8):623‐626. 10.1136/jmg.32.8.623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Warsy AS, Al‐Jaser MH, Albdass A, Al‐Daihan S, Alanazi M. Is consanguinity prevalence decreasing in Saudis?: a study in two generations. Afr Health Sci. 2014;14(2):314‐321. 10.4314/ahs.v14i2.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sallevelt S, de Koning B, Szklarczyk R, et al. A comprehensive strategy for exome‐based preconception carrier screening. Genet Med. 2017;19:583‐592. 10.1038/gim.2016.153 [DOI] [PubMed] [Google Scholar]
- 18. Carss KJ, Hillman SC, Parthiban V, et al. Exome sequencing improves genetic 808 diagnosis of structural fetal abnormalities revealed by ultrasound. Hum Mol Genet. 2014;809(23):3269‐3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wright CF, Fitzgerald TW, Jones WD, et al. Genetic diagnosis of developmental disorders in the DDD study: 1022 a scalable analysis of genome‐wide research data. Lancet. 2015;385:1305‐1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Colley E, Hamilton S, Smith P, Morgan NV. Arri Coomarasamy, Stephanie Allen, potential genetic causes of miscarriage in euploid pregnancies: a systematic review. Hum Reprod Update. 2019;25(4):452‐472. 10.1093/humupd/dmz015 [DOI] [PubMed] [Google Scholar]
- 21. Godard B, ten Kate L, Evers‐Kiebooms G, Ayme S. Population genetic screening programmes: principles, techniques, practices, and policies. Eur J Hum Genet. 2003;11(Suppl 2):S49‐S87. [DOI] [PubMed] [Google Scholar]
- 22. Stuppia L, Antonucci I, Palka G, Gatta V. Use of the MLPA assay in the molecular diagnosis of gene copy number alterations in human genetic diseases. Int J Mol Sci. 2012;13(3):3245‐3276. 10.3390/ijms13033245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Orloff M, Peterson C, He X, et al. Germline mutations in MSR1, ASCC1, and CTHRC1 in patients with Barrett esophagus and esophageal adenocarcinoma. JAMA. 2011;306(4):410‐419. 10.1001/jama.2011.1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ng SB, Turner EH, Robertson PD, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461(7261):272‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bainbridge MN, Wiszniewski W, Murdock DR, et al. Whole‐genome sequencing for optimized patient management. Sci Transl Med. 2011;3(87):87re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gonzaga‐Jauregui C, Lupski JR, Gibbs RA. Human genome sequencing in health and disease. Annu Rev Med. 2012;63:35‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang Y, Muzny DM, Reid JG, et al. Clinical whole‐exome sequencing for the diagnosis of mendelian disor‐ ders. N Engl J Med. 2013;369(16):1502‐1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang DM, Muzny F, Xia Z, et al. Eng, molecular find‐ ings among patients referred for clinical whole‐exome sequencing. JAMA. 2014;312(18):1870‐1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee H, Deignan JL, Dorrani N, et al. Clinical exome sequencing for genetic identification of rare Mendelian dis‐ orders. JAMA. 2014;312(18):1880‐1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alfares A, Alfadhel M, Wani T, et al. A multicenter clinical exome study in unselected cohorts from a consanguineous population of Saudi Arabia demonstrated a high diagnostic yield. Mol Genet Metab. 2017;121(2):91‐95. 10.1016/j.ymgme.2017.04.002 [DOI] [PubMed] [Google Scholar]
- 31. Monies D, Abouelhoda M, Assoum M, et al. Lessons learned from large‐scale, first‐tier clinical exome sequencing in a highly consanguineous population. Am J Hum Genet. 2019;104(6):1182‐1201. 10.1016/j.ajhg.2019.04.011 Erratum in: Am J Hum Genet. 2019;105(4):879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yavarna T, Al‐Dewik N, Al‐Mureikhi M, et al. High diagnostic yield of clinical exome sequencing in middle eastern pa‐ tients with Mendelian disorders. Hum Genet. 2015;134(9):967‐980. [DOI] [PubMed] [Google Scholar]
- 33. Qiao Y, Wen J, Tang F, et al. Whole exome sequencing in recurrent early pregnancy loss. Mol Hum Reprod. 2016;22:364‐372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yates CL, Monaghan KG, Copenheaver D, et al. Whole‐exome sequencing on deceased fetuses with ultrasound anomalies: expanding our knowledge of genetic disease during fetal development. Genet Med. 2017;19:1171‐1178. [DOI] [PubMed] [Google Scholar]
- 35. Fu M, Mu S, Wen C, et al. Whole‐exome sequencing analysis of products of conception identifies novel mutations associated with missed abortion. Mol Med Rep. 2018;18:2027‐2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhao C, Chai H, Zhou Q, et al. Exome sequencing analysis on products of conception: a cohort study to evaluate clinical utility and genetic etiology for pregnancy loss. Genet Med. 2021;23:435‐442. 10.1038/s41436-020-01008-6 [DOI] [PubMed] [Google Scholar]
- 37. Sahlin E, Gréen A, Gustavsson P, et al. Identification of putative pathogenic single nucleotide variants (SNVs) in genes associated with heart disease in 290 cases of stillbirth. PLoS One. 2019;14:e0210017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Anastasaki C, Longman D, Capper A, Patton EE, Cáceres JF. Dhx34 and Nbas function in the NMD pathway and are required for embryonic development in zebrafish. Nucleic Acids Res. 2011;39(9):3686‐3694. 10.1093/nar/gkq1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zlotogora J. Population programs for the detection of couples at risk for severe monogenic genetic diseases. Hum Genet. 2009;126:247‐253. [DOI] [PubMed] [Google Scholar]
- 40. University of Groningen . Groningen: Testing for serious genetic diseases now possible before pregnancy (updated January 14, 2016). http://www.rug.nl/news-and-events/news/archief2016/nieuwsberichten/umcg-dragerschapstest?lang=en. (cited March 14, 2016) (Google Scholar).
- 41. Singer A, Sagi‐Dain L. Impact of a national genetic carrier‐screening program for reproductive purposes. Acta Obstet Gynecol Scan. 2020;99(6):802‐808. 10.1111/aogs.13858.Epub [DOI] [PubMed] [Google Scholar]
- 42. Kihlbom U. Ethical issues in preconception genetic carrier screening. Upsala J Med Sci. 2016;121(4):295‐298. 10.1080/03009734.2016.1189470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dong Z, Yan J, Xu F, et al. Genome sequencing explores complexity of chromosomal abnormalities in recurrent miscarriage. Am J Hum Genet. 2019;105(6):1102‐1111. 10.1016/j.ajhg.2019.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that supports the findings of this study are available in the article material and references of this article.