

Abstract
Non‐alcoholic fatty liver disease (NAFLD) is the hepatic manifestation of metabolic syndrome, being a common comorbidity of type 2 diabetes and with important links to inflammation and insulin resistance. NAFLD represents a spectrum of liver conditions ranging from steatosis in the form of ectopic lipid storage, to inflammation and fibrosis in nonalcoholic steatohepatitis (NASH). Macrophages that populate the liver play important roles in maintaining liver homeostasis under normal physiology and in promoting inflammation and mediating fibrosis in the progression of NAFLD toward to NASH. Liver macrophages are a heterogenous group of innate immune cells, originating from the yolk sac or from circulating monocytes, that are required to maintain immune tolerance while being exposed portal and pancreatic blood flow rich in nutrients and hormones. Yet, liver macrophages retain a limited capacity to raise the alarm in response to danger signals. We now know that macrophages in the liver play both inflammatory and noninflammatory roles throughout the progression of NAFLD. Macrophage responses are mediated first at the level of cell surface receptors that integrate environmental stimuli, signals are transduced through multiple levels of regulation in the cell, and specific transcriptional programmes dictate effector functions. These effector functions play paramount roles in determining the course of disease in NAFLD and even more so in the progression towards NASH. The current review covers recent reports in the physiological and pathophysiological roles of liver macrophages in NAFLD. We emphasise the responses of liver macrophages to insulin resistance and the transcriptional machinery that dictates liver macrophage function.
Keywords: inflammation, liver, macrophages, NAFLD, NASH
Non‐alcoholic fatty liver disease (NAFLD) is the hepatic manifestation of metabolic syndrome. Insulin resistance and inflammation are major drivers of NAFLD progression. Inflammation in the liver is driven by macrophages, a heterogenous population of cells that undergo polarisation controlled by intricate molecular mechanisms. Some of these molecular mechanisms controlling inflammation are promising therapeutic targets. Here, we discuss the state‐of‐the‐art in the regulation of inflammation and liver macrophage polarisation in NAFLD.
Abbreviations
- AP‐1
activator protein 1
- BMDM
bone marrow‐derived macrophages
- CCL
chemokine ligand 2
- CCR
C‐C motif chemokine receptor
- CD
cluster of differentiation
- CD‐HFD
choline‐deficient high‐fat diet
- CETP
cholesteryl ester transfer protein
- CLEC4F
C‐type lectin domain family 4 member F
- CXCL
C‐X‐C motif ligand
- CXCR
C‐X‐C chemokine receptor
- DAMP
damage‐associated molecular patterns
- DR5
death receptor 5
- ER
endoplasmic reticulum
- GLUT4
glucose transporter type 4
- GM‐CSF
granulocyte‐macrophage colony‐stimulating factor
- HCC
hepatocellular carcinoma
- HFD
high‐fat diet
- HIF
hypoxia‐inducible factor
- HRG
histidine‐rich glycoprotein
- HSC
hepatic stellate cell
- IFN
interferon
- IGFBP7
insulin‐like growth factor‐binding protein 7
- IL
interleukin
- iNOS
inducible nitric oxide synthase
- Insr−/−
insulin receptor knockout
- IRF
interferon regulatory factor
- IκB
inhibitor of kappa‐B
- JNK
c‐Jun N‐terminal kinase
- KC
Kupffer cell
- LDLR
low density lipoprotein receptor
- LPS
lipopolysaccharides
- LSEC
liver sinusoidal epithelial cells
- LXR
liver X receptor
- Ly6
lymphocyte antigen
- MAPK
mitogen‐activated protein kinase
- MCD
methionine‐choline deficient
- MCP‐1
monocyte chemoattractant protein 1
- M‐CSF
macrophage colony‐stimulating factor
- Mo‐
monocyte‐derived
- MP
macrophage
- MUP‐uPA
methionine adenosyl transferase 1A knockout mouse with high transient expression of urokinase plasminogen activator in hepatocytes
- MyD88
myeloid differentiation primary response 88
- NAFLD
non‐alcoholic fatty liver disease
- NAM
Nash‐associated macrophage
- NASH
nonalcoholic steatohepatitis
- NF‐κB
nuclear factor kappa‐B
- PD‐L1
programmed death ligand 1
- PI3K
phosphoinositide 3‐kinase
- PPAR
peroxisome proliferator‐activated receptor
- PTPROt
protein tyrosine phosphatase receptor type O truncated isoform
- RHM
recruited hepatic macrophages
- scRNA‐seq
single‐cell RNA sequencing
- SREBP1c
sterol regulatory element‐binding protein‐1c
- STAT
signal transducer and activator of transcription
- STING
stimulator of interferon genes
- T2D
type 2 diabetes
- TGF
transforming growth factor
- TH
helper T‐cell
- TH/TR
thyroid hormone/thyroid hormone receptor
- TIM4
T‐cell immunoglobulin and mucin domain containing 4
- TLR
Toll‐like receptor
- TNF
tumour necrosis factor
- Treg
regulatory T cell
- TREM2
triggering receptor expressed on myeloid cells
- UPR
unfolded protein response
- WD
Western diet
- XBP1
X‐box binding protein 1
Introduction: Inflammation and metabolic decline in non‐alcoholic fatty liver disease
Non‐alcoholic fatty liver disease (NAFLD) is the most common form of chronic liver disease with an estimated worldwide prevalence of 25% [1, 2]. NAFLD is the hepatic manifestation of metabolic syndrome and common comorbidity of type 2 diabetes (T2D), obesity and hypertension. Indeed, around 55% of patients with T2D also have NAFLD [3]. Metabolic and inflammatory disturbances are important parts of the aetiology of NAFLD and of its comorbidities [4, 5].
Non‐alcoholic fatty liver disease represents a spectrum of conditions ranging from fatty liver, relatively benign steatosis in the form ectopic lipid storage, to nonalcoholic steatohepatitis (NASH) where inflammation and tissue remodelling can impair tissue function and whole‐body metabolism. NASH represents the last reversible step of NAFLD, before progression to hepatocellular carcinoma (HCC) [6, 7] (Fig. 1).
Fig. 1.
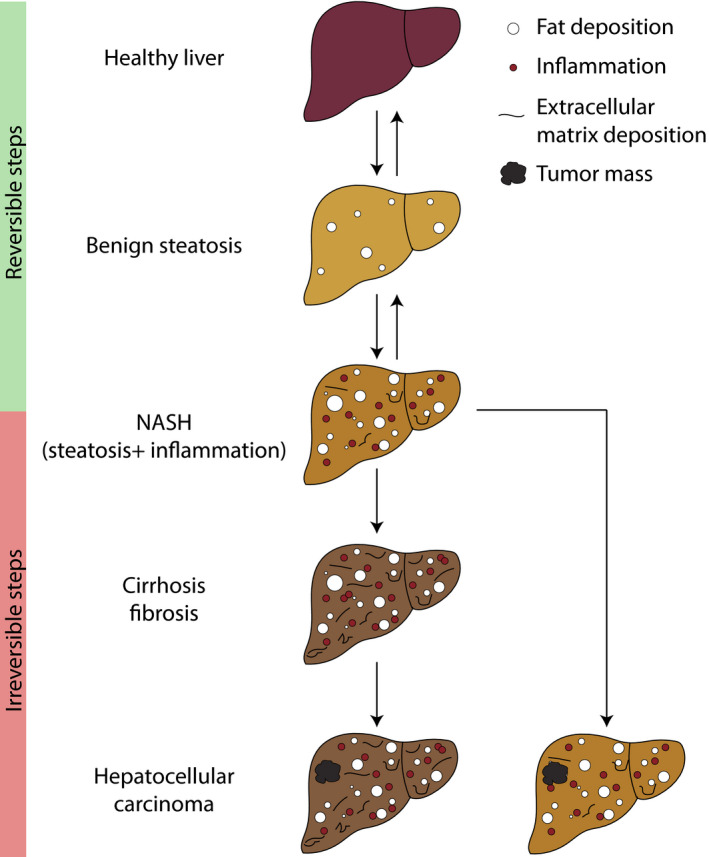
NAFLD progression. Benign steatosis (fat accumulation in hepatocytes) can trigger inflammation in the liver (starting point for NASH). As inflammation worsens, hepatic stellate cell activation leads to extracellular matrix deposition and fibrosis. Eventually, this process facilitates tumorigenesis and development of hepatocellular carcinoma. A tumour mass can also arise directly from NASH without need for progressive fibrosis. Fibrosis in NASH is the last reversible step of NAFLD.
Over recent decades, considerable progress has been made in understanding the mechanisms of NAFLD development and progression [5]. An important milestone was published by Day and James in 1998 when they put forward their ‘two‐hit’ hypothesis. In this hypothesis, steatosis was considered the first hit and inflammation the second, causing progression through the spectrum of NAFLD towards NASH [8].
Given that T2D and NAFLD are frequent comorbidities, a relationship with insulin sensitivity or secretion was sought in the earliest studies [9]. Initial clinical work found associations between insulin resistance and NAFLD, even in the absence of frank T2D (compromised insulin secretion). Glucose disposal and insulin sensitivity were also found to be progressively impaired going from healthy subjects, to patients with steatotic livers and then in patients with NASH [10, 11]. At the cellular level, insulin resistance also contributes to steatosis through two main mechanisms: increased hepatic de novo lipogenesis [12] and ectopic lipid storage in response to systemic dyslipidaemia [13]. Dyslipidaemia arises early in disease course from increased lipolysis in adipose tissue [13].
When hepatocytes reach their lipid storage threshold, lipotoxicity and hepatocellular stress lead to apoptosis [14, 15]. Lipid overload and insulin resistance are associated with endoplasmic reticulum (ER) stress and the unfolded protein response (UPR) [15]. Physiologically, X‐box binding protein (XBP)‐1 mRNA splicing responds to ER stress and promotes cell survival by increasing ER protein folding capacity [16]. However, XBP‐1‐mediated cell survival fails in NAFLD, resulting in hepatocellular stress, inflammation, further loss of insulin sensitivity and apoptosis [17, 18].
The above step is key in initiating inflammation and the transition from benign steatosis to NASH. The initial inflammatory response is largely mediated by tissue‐resident macrophages [19]. Upon inflammatory signalling, tissue‐resident macrophages recruit other immune cells from circulation, including monocytes that differentiate into macrophages in situ and amplify inflammatory signalling [20]. When this cycle is sustained under chronic hepatocellular stress, a macrophage pro‐resolution response is also initiated. The resolution of inflammation is beneficial in response to acute inflammation; however, in response to chronic inflammation in the liver the resolution phase is associated with excessive deposition of collagen in extracellular matrix [21]. Fibrosis in later stages of NASH, from excessive collagen deposition, is the result of an exuberant scarring response, which over time significantly remodels the tissue and impedes liver function [21, 22].
Macrophages are central to the progression of NAFLD, and their proliferation, differentiation and polarisation are tightly controlled and dependent on extracellular stimuli as well as intracellular signalling cascades [23]. While initially acting as sentinel cells, macrophages are also very important effectors cells that secrete cytokines and chemokines, influencing cells in the microenvironment. This review covers the mechanisms of how liver macrophages undergo activation and contribute to the development and progression of NAFLD. Given the importance of insulin resistance in the pathogenesis of disease, we also address the role of insulin signalling, and insulin action on liver macrophages.
Insulin signalling and NAFLD
Insulin is an anabolic hormone secreted by pancreatic beta cells and is widely recognised for its role in regulating glucose homeostasis, lipid metabolism and cell growth. The effects of insulin are mediated through the insulin receptor [24, 25] and the insulin‐like growth factor 1 receptor [26]. When insulin binds to its receptor it activates two major downstream pathways: the phosphoinositide 3‐kinase (PI3K) pathway and the mitogen‐activated protein kinase (MAPK) pathway [26, 27]. The PI3K pathway mediates insulin's metabolic effects including the translocation of glucose transporter (GLUT)‐4 in metabolic tissues such as muscle, liver and adipose [26], while the MAPK pathway regulates mitogenesis and growth [27]. Recently, the insulin receptor has also been shown to directly interact with transcriptional machinery, an additional mechanism for effects in normal physiology and disease [28].
Insulin resistance is the term given to the lack of an appropriate response to physiological levels of insulin, typically determined through systemic metabolic measures such as blood glucose. Insulin resistance is a precursor syndrome to T2D and its comorbidities [29]. In humans, patients presenting with NAFLD are often insulin resistant; however, it is unclear whether insulin resistance is compensatory rather than causal – the challenges to addressing this important question have been recently reviewed [30]. Various murine models of NAFLD have been proposed (detailed below), and in order to reproduce human disease, the model applied will ideally display obesity, insulin resistance and NAFLD concurrently [31]. One of the early mouse models investigating insulin's function, through global targeted disruption of the insulin receptor (Insr−/− mouse), reported liver steatosis and hepatic insulin resistance. The model initially exhibits dramatic metabolic insulin resistance, which is followed by age‐dependent morphological and functional changes in the liver [32, 33]. This early model suggested that changes in insulin sensitivity are sufficient to initiate NAFLD [32, 33].
Insulin and macrophages
While macrophages are less associated with the roles of insulin compared with the majority cells of the metabolic tissues, macrophages do express insulin receptors and downstream intracellular signalling pathways [34, 35]. In vitro studies investigating the direct effects of insulin on macrophages have shown insulin to have a profound effect on macrophage activation including inflammatory or M1‐like polarisation (Table 1) or anti‐inflammatory or M2‐like polarisation (Table 2). Discrepancies in reports may be due to a lack of consistency in the model of macrophage investigated (different species/tissue type/cell line), concentration or duration of insulin used. The effects of insulin on macrophages are seemingly wide‐ranging and thus may reflect macrophage plasticity and ability to respond to the fluctuating nature of blood insulin levels.
Table 1.
Evidence supporting pro‐inflammatory role of insulin in macrophages. Where insulin resistance is anti‐inflammatory.
| Model | Summary | Ref |
|---|---|---|
| Cell lines ML‐1, THP‐1, PL‐21 | Insulin enhances LPS‐stimulated IL‐1β | [333] |
| Cell line THP‐1 | Insulin upregulates TNFα | [334] |
| Mouse myeloid/ macrophage insulin resistance | Protects against atherosclerosis | [37] |
| Mouse myeloid/ macrophage insulin resistance | Protects against obesity‐induced inflammation | [335] |
| Human macrophages | Insulin promotes foam cell formation | [336] |
| Mouse insulin‐resistant macrophages | Attenuation of atherosclerosis, promotion of M2‐type phenotype when stimulated with pro‐inflammatory cytokines | [337] |
| Mouse macrophages | Insulin and IL‐1β synergistically promote inflammation | [338] |
| Diabetic mouse bone marrow‐derived macrophages | Insulin increases TNFα and IL‐6 secretion in LPS‐stimulated macrophages | [339] |
| Mouse macrophages | Insulin resistance promotes M2‐like phenotype and reduced LPS responses | [340] |
Table 2.
Evidence supporting anti‐inflammatory role of insulin in macrophages. Where insulin resistance is pro‐inflammatory.
| Model | Summary | Ref |
|---|---|---|
| Rat peritoneal macrophages | Insulin enhances phagocytosis capacity and production of H2O2 | [341] |
| Obese human mononuclear cells | Insulin inhibits NFκB and stimulates IκB | [342] |
| Cell line THP‐1 | Insulin inhibits apoptosis | [343] |
| Cell line THP‐1 | Insulin inhibits apoptosis and reduces TNF and IL‐1β | [344] |
| Rat macrophages | Insulin suppresses LPS‐induced iNOS and COX‐2 expression and NK‐κB activation | [345] |
| Mouse myeloid/ macrophage insulin resistance | Increased macrophage apoptosis and atherosclerotic plaque necrotic core formation | [36] |
| Mouse insulin‐resistant macrophages | Increased macrophage apoptosis | [346] |
| Cell line THP‐1 | Insulin pretreatment delays endotoxin mediated macrophage activation | [347] |
| Mouse insulin‐resistant macrophages | Increased LPS IL‐1β production | [348] |
| Mouse insulin‐resistant macrophages | Enhanced monocyte adhesion | [349] |
| Mouse insulin‐resistant macrophages | Enhanced vascular wall adhesion and pro‐inflammatory mediator adhesion | [350] |
| Mouse insulin‐resistant macrophages | Increased apoptosis | [351] |
| Cell line RAW264.7 and high fat fed mice +insulin | Reduced foam cell formation, down‐regulation of pro‐inflammatory cytokines, decreased serum pro‐inflammatory mediators and macrophage infiltration | [352] |
| Mouse insulin‐resistant macrophages | Increased atherosclerosis through IFNγ‐regulated macrophage network | [353] |
| Mouse macrophages | Insulin promotes IL‐10 expression and attenuates LPS‐induced Tnf‐α, Il‐1β and iNOS expression | [354] |
| Rat macrophages | Insulin advances infiltration and resolution of macrophages | [355] |
| Diabetic mouse alveolar and peritoneal macrophages | Insulin reduces TNFα, IL‐6 and IL‐1β secretion in LPS‐stimulated macrophages | [339] |
| Mouse macrophages | Insulin resistance impairs M2a activation | [356] |
| Rat macrophages and cell line THP‐1 | Insulin polarises macrophages to M2 phenotype under high glucose conditions | [357] |
| Rat macrophages | Insulin restores abnormal macrophage infiltration, promotes efferocytosis and induces M1 to M2 transition | [358] |
Insulin and liver macrophages
Surprisingly, while the impact of macrophages on NAFLD development is appreciated, and the significance of insulin resistance on macrophages in cardiometabolic diseases such as atherosclerosis are recognised, studies investigating the specific role of insulin‐resistant macrophages on NAFLD have yet to be reported [36, 37, 38]. In obesity‐induced insulin resistance in mice, distinct subpopulations of hepatic macrophages, with Kupffer cells (KC), secrete high levels of chemokine ligand (CCL)‐2/monocyte chemoattractant protein (MCP)‐1. CCL2/MCP‐1 acts to recruit ‘recruited hepatic macrophages’ (RHMs). RHMs in turn enhance the severity of obesity‐induced inflammation and hepatic insulin resistance [39]. Recently, Morgantini et al. [40] have shown that in obesity‐induced insulin resistance in flies, humans and mice; liver macrophages produce noninflammatory factors including insulin‐like growth factor‐binding protein (IGFBP)‐7 that can bind to the insulin receptor, directly regulating liver metabolism independently of inflammation.
Macrophages in liver physiology
There are two major types of hepatic macrophages: monocyte‐derived and tissue‐resident macrophages. KCs, bona fide liver‐resident macrophages, are by far the most abundant in the healthy liver. In mice, KCs are identified by their expression of the pan‐macrophage marker F4/80, low expression of CD11b, as well as by expression of specific markers such as the C‐Type Lectin Domain Family 4 Member F (CLEC4F) or T‐Cell Immunoglobulin and Mucin Domain Containing 4 (TIM4) [41, 42, 43]. Their development occurs during embryogenesis, from yolk‐sac precursors that populate the foetal liver [44, 45, 46, 47]. Like other tissue‐resident macrophages, KCs are thought to persist in adult mice by self‐renewal [48]. From surveillance, to recycling iron and promoting immune tolerance, KCs play important homeostatic roles in normal liver physiology (Fig. 2A).
Fig. 2.
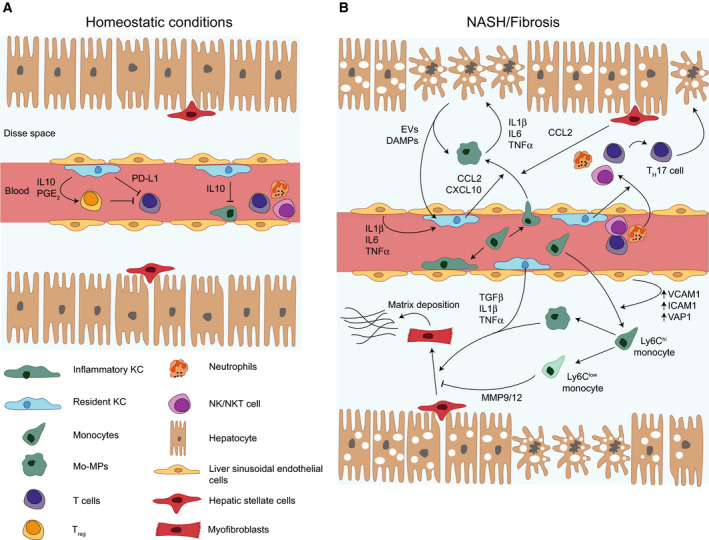
Cellular crosstalk during NASH. (A) In homeostatic conditions, Kupffer cells (KCs) inhibit monocyte and macrophage recruitment through interleukin 10 (IL‐10) secretion. They also promote immune tolerance from T cells by inducing regulatory T cells (Treg) and expressing programmed death ligand 1 (PD‐L1). (B) During NASH, apoptotic hepatocytes release danger‐associated molecular patterns (DAMPs) that activate KCs. Activated KCs secrete chemokines recruiting monocytes to the liver. Monocytes differentiate into macrophages in situ and produce pro‐inflammatory cytokines which drives hepatocyte death and reinforces their pro‐inflammatory phenotype. Liver macrophages are also fuelling inflammation by promoting recruitment of other immune cells and TH17 polarisation. Additionally, KCs and recruited Ly6Chi monocytes can trigger hepatic stellate cells (HSCs) activation through cytokine signalling. HSCs differentiation into myofibroblasts leads to production of extracellular matrix and fibrosis. On the contrary, Ly6Clow monocytes are able to inhibit this process. EV, extracellular vesicle; ICAM1, intercellular adhesion molecule 1; MMP, metalloproteinase; Mo‐MPs, monocyte‐derived macrophages; NK, natural killer; NKT cells, natural killer T cells; PGE2, prostaglandin E2; TGFβ, transforming growth factor β; TNF‐α, tumour necrosis factor α; VCAM1, vascular cell adhesion molecule 1; VAP1, vascular adhesion protein 1.
KCs are located in liver sinusoids, and they continuously survey blood for metabolites and microbial products [41]. Mice lacking KCs show impaired survival following Listeria monocytogenes infection, emphasising their importance for the depletion of blood‐borne bacteria [49, 50]. Similarly, KCs remove damaged or apoptotic cells [51, 52].
KCs also have important roles in iron and cholesterol metabolism. They are able to detect and phagocytose damaged erythrocytes and erythrocyte‐derived vesicles containing haemoglobin [53, 54]. KCs also influence iron reabsorption by regulating hepatocyte hepcidin expression [55]. With regard to cholesterol, all macrophages metabolise lipids, as required by their canonical function of phagocytosing cellular debris and processing lipid‐rich elements such as membranes. However, relative to other tissue‐resident macrophages, the KC transcriptome is enriched with genes that uptake, process and export cholesterol to extracellular high‐density lipoprotein acceptors [42]. Indeed, KCs highly express cholesteryl ester transfer protein (CETP) amongst other genes in lipid processing, which are controlled by well‐known transcription factors that regulate cellular lipids (e.g. PPARs, LXR) [42, 56]. Physiologically, KCs may require this high lipid processing capacity to cope with dynamic cholesterol synthesis in the liver or to cope with exposure to systemic lipids packaged into lipoproteins in the liver. While KCs are clear drivers of inflammation in NAFLD, [57] their activation spectrum remains to be defined (in the context of M1‐/M2‐like polarisation), similarly questions remain unanswered with regards to their capacity to accumulate lipids, such as adipose or vascular foams cells, and with regard to their persistence in later stages of NAFLD [58, 59, 60, 61].
Immunologically, KCs promote immune tolerance by diverse mechanisms, and their capacity to present antigens and activate T cells is very limited [62]. In mice, as well as in humans, KCs secrete anti‐inflammatory cytokines such as interleukin (IL)‐10 [63, 64]. They also express co‐inhibitory molecule Programmed Death Ligand (PD‐L)‐1, a potent inhibitor of T‐cell activity [64]. They can also induce regulatory T‐cell (TRegs) differentiation through secretion of prostaglandins [62, 64]. Monocyte‐derived macrophages (Mo‐MPs) or RHMs can also populate the liver and differentiate from (C‐C motif chemokine receptor (CCR)‐2+ C‐X‐C 3 chemokine receptor (CX3CR)‐1+ lymphocyte antigen (Ly)‐6C+ monocytes. Mo‐MPs account for a minority of macrophages in the healthy liver [65] but can be rapidly recruited upon liver injury [66] and can persist in chronic diseases such as NAFLD. Like KCs, they are important for erythrocyte clearance and iron recycling during homeostasis [67]. More recently, a third type of macrophage was reported in the liver capsule and these capsular macrophages are derived from bone marrow and play a role in peritoneal‐derived pathogen clearance [68].
Modelling NAFLD physiopathology
To study NAFLD physiopathology and allow the isolation of different cell fractions, including macrophages, murine models are indispensable. Modelling NAFLD in mice comes with its challenges and opportunities. Resistance of mice to spontaneously develop NAFLD upon high‐fat feeding had initially led scientists to develop various models that recapitulate isolated events in the disease. In this light, a high‐fat diet (HFD) can recapitulate simple steatosis and insulin resistance, while carbon tetrachloride (CCl4) induces inflammation and fibrosis without steatosis and a methionine‐and‐choline–deficient (MCD) diet results in fibrosis, inflammation and steatosis without insulin resistance [31, 69, 70]. Similarly, surgical ligation of the bile‐duct induces cholestatic injury, inflammation and fibrosis in mice, without insulin resistance [71]. Genetic models such as the widely adopted ob/ob or db/db mice can recapitulate obesity, insulin resistance and to a slight degree liver inflammation, but do not progress beyond steatosis [69]. These above models may be considered extreme and can only be interpreted as models due to their lacking holistic systemic representation of NAFLD and its comorbidities.
More holistic models exist today that recapitulate a larger part of the NAFLD spectrum, such as the choline‐deficient HFD (CD‐HFD), high fructose‐HFD (HF‐HFD) or genetic‐based models including the sterol regulatory element‐binding protein‐1c (SREBP1c) transgenic mouse, methionine adenosyl transferase 1A knockout mouse with high transient expression of urokinase plasminogen activator in hepatocytes (MUP‐uPA Tg) and the DIAMOND model [69]. These models, and others, have been recently reviewed in‐depth by Febbraio et al. [69]. Briefly, through different mechanisms, these models have been shown to recapitulate obesity, insulin resistance, steatosis, inflammation, ER stress and fibrosis in NASH, including a transition towards HCC [69]. Of these models, CD‐HFD is gaining popularity, where the lack of choline prevents cholesterol export from hepatocytes, resulting in lipotoxicity and progression of NAFLD. Mice on CD‐HFD develop obesity, insulin resistance, glucose intolerance and NAFLD. However, whether the status of other tissues is modified by the lack of choline has not been investigated. The MUP‐uPA model recapitulates obesity, insulin resistance and glucose intolerance on HFD, where the key mechanism of hepatocyte ER stress (due to uPA overexpression) leads to mice consistently developing NASH, and up to 85% spontaneously progressing to HCC [69, 72].
Choice of model and understanding the mechanisms by which NAFLD and NASH develop are critical to correct interpretation of results. In the case of macrophage responses, most models recapitulate the inflammatory hit, to varying degrees, and thus most are applicable. However, results must always be interpreted within the constraints and contexts of the given model, especially in the case of toxic models of fibrosis (CCl4) or in models of global knockout or knockin (e.g. ob/ob, db/db or SREBP1c Tg).
Macrophages in NAFLD physiopathology
Macrophages are drivers of NAFLD, and human studies show positive correlations between macrophage numbers in the liver and NAFLD severity [73, 74]. In mouse models, early depletion of KCs prevents progression of the disease, as well as insulin resistance [75, 76, 77]. In macrophage‐depleted mice, IL‐1β production was decreased while levels of the protective factor peroxisome proliferator‐activated protein (PPAR)‐α was increased in hepatocytes [78, 79]. Additionally, preventing monocyte entry into the liver through CCR2 blockade improves NASH [74, 80] and it is now widely accepted that monocyte recruitment and in situ differentiation into macrophages fuels NAFLD progression. In NASH, monocytes replace a fraction of the KC pool by differentiating into monocyte‐derived KCs (Mo‐KCs) [42, 43] which express KC markers, but are functionally different. Mo‐KCs express more inflammatory genes potentially contributing to disease progression [43].
Macrophages are at the heart of intense cellular crosstalk in NAFLD, interacting with many liver cell types (Fig. 2B). Macrophages recognise hepatocyte‐derived Danger‐Associated Molecular Patterns (DAMPs), they secrete cytokines that may alter hepatocyte physiology and promote NAFLD progression [79]. Macrophage‐derived cytokines also target hepatic stellate cells (HSCs). Tumour necrosis factor (TNF)‐α, IL‐1β and transforming growth factor (TGF)‐β can all induce HSC activation [81, 82]. In turn, HSCs up‐regulate several ligands able to attract macrophages and regulate their activity (like CCL2) in NASH [83]. Liver sinusoidal endothelial cells (LSECs) drive anti‐inflammatory polarisation of macrophages and down‐regulate cytokine and chemokine secretion through nitric oxide production [84]. However, LSECs can also promote monocyte infiltration and contribute to liver inflammation [85, 86]. Finally, macrophages can interact with other immune cells. Cytokine secretion by activated T cells can then reinforce macrophage pro‐inflammatory phenotype in a feed‐forward loop [87]. Additionally, chemokine secretion by activated macrophages leads to recruitment of several immune cell types in the liver [88].
Macrophage subtypes in the liver
The optimisation of single‐cell RNA sequencing (scRNA‐seq) in recent years has allowed the more precise identification of macrophage subpopulations. Several macrophage subpopulations have been defined in NAFLD. One study identified KCs and three different populations of Mo‐MPs. While it is surprising to see the presence of such Mo‐MPs already under a normal diet, these cells were enriched in mice fed a western diet (WD). They express less calprotectin, a marker of inflammation, in WD‐fed mice, suggesting that these subsets may be protective [65]. Another study in amylin diet‐induced NASH showed both KCs and Mo‐MPs displayed a pro‐inflammatory phenotype compared with controls. Two KC subsets were identified and segregated based on triggering receptor expressed on myeloid cells (TREM)‐2 expression. TREM2‐low KCs were predominant in mice fed a normal diet. TREM2‐high KCs were almost exclusively found in NASH and were therefore called ‘NASH‐associated macrophages’ (NAMs) [83]. These two reports independently identified different functionally and phenotypically diverse subsets of macrophages in NAFLD. Interestingly however, subsequent reanalysis demonstrated that similar macrophage subsets were found in sequencing data from both studies. This highlights the divergent views in the field with regard to the identity of resident macrophages [89]. Specifically addressing fibrosis, two populations of Mo‐MPs have been described based on Ly6C expression. Ly6Chi cells are pro‐fibrogenic and express cytokines that are able to activate HSCs such as IL‐1β and TGFβ whereas Ly6Clow cells derived from Ly6hi monocytes promote resolution of fibrosis and express matrix‐degrading factors such as metalloproteinases [90].
Macrophage polarisation in NAFLD
Depending on microenvironmental cues, macrophages display different functional phenotypes. Typically, macrophages have been divided into classically activated M1‐like or alternatively activated M2‐like macrophages. Macrophages adopt a M1 phenotype in response to Toll‐like receptor (TLR) stimulation for example with lipopolysaccharide (LPS), and in response to type 1 cytokines such as interferon (IFN)‐γ. These macrophages produce pro‐inflammatory cytokines such as IL‐1β or TNF‐α and are potent antigen presenting cells that are able to induce TH1/TH17 cell responses. As a consequence, they are involved in inflammatory responses and are responsible for pathogen killing. On the contrary, M2 macrophages secrete anti‐inflammatory cytokines such as IL‐10 or TGF‐β and elicit TH2/TReg responses. They respond to extracellular pathogens, are more tolerogenic and are associated with resolution of inflammation and tissue repair [91].
In a number of tissues, including the liver, M1 macrophages are generally characterised by the expression of CD11c, while CD206 is the common marker of M2 macrophages [92]. With increasing mechanistic studies and novel technologies, this framework has now developed to offer a more comprehensive representation of macrophage heterogeneity and functional diversity. For example, functionally diverse M2 macrophage subtypes have now been identified: M2a, M2b, M2c and M2d [93]. The emergence of transcriptomics and scRNA‐seq technologies has further refined that description by emphasising the diversity of tissue macrophages and the highly spectral nature of macrophage polarisation [94, 95].
Macrophage polarisation is an important parameter influencing NAFLD progression. Reports with regard to polarisation of liver macrophages in metabolic diseases have been conflicting. While high‐fat diet (HFD) has been predominantly associated with M1 polarisation of liver macrophages [58, 96], other groups have recently demonstrated that liver macrophages regulate systemic metabolism upon HFD through predominantly noninflammatory signalling [40]. Impaired M2 polarisation was associated with impaired hepatic lipid metabolism and steatosis [97]. In addition, M2 macrophages were shown to induce M1 macrophage apoptosis [96]. Overall, studies suggest that pro‐inflammatory macrophages are mostly detrimental in NAFLD while anti‐inflammatory macrophages are protective, this remains highly dependent on the stage of NAFLD progression. Indeed, studies have pointed out detrimental roles for M2 macrophages, for example during long‐term HFD [60, 98]. Polarisation state has different effects depending on disease stage, for example M2‐like macrophages may mitigate inflammation in the early stages of NAFLD but promote matrix deposition and fibrosis at later stages.
Molecular mechanisms of macrophage polarisation in NAFLD
Molecular drivers of macrophage inflammatory status
Macrophage polarisation, and more generally the role of macrophages in liver inflammation during NAFLD, is controlled by multiple pathways and transcription factors. The activity of these transcription factors is triggered by molecular cues coming from the liver but also from other organs of the body. While diverse in nature, in the context of NAFLD, these cues can be divided into two main categories: DAMPs and cytokines, both of which act through cell surface receptors and lead to transcriptional reprogramming in macrophages (Fig. 3).
Fig. 3.
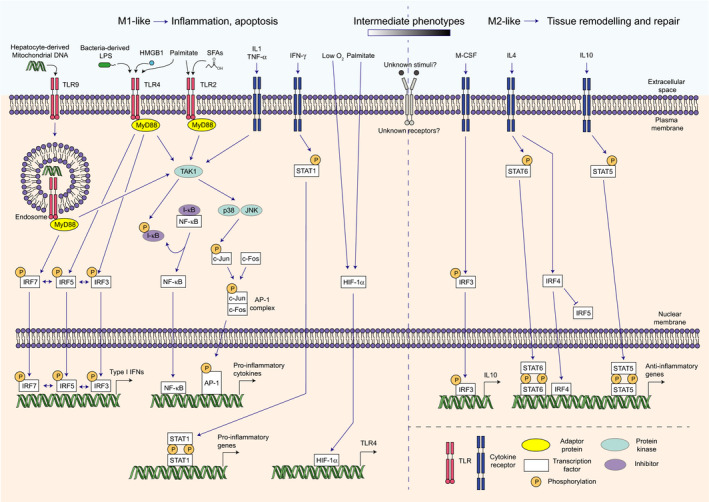
Transcriptional control of macrophage polarisation. Toll‐like receptor (TLR) stimulation by different ligands leads to activation of different intracellular pathways. Mitogen‐activated protein kinase kinase kinase 7 (TAK1) activation ultimately leads to phosphorylation of I‐κB and c‐Jun. Phosphorylation of I‐κB enables release of NF‐κB and its translocation to the nucleus. c‐Jun phosphorylation triggers formation of the activator protein 1 (AP‐1) complex through association with c‐Fos. NF‐κB and AP‐1 can then launch transcription of pro‐inflammatory genes and cytokines. TLR4 and TLR9 ligation additionally triggers activation of interferon regulatory factor (IRF) 3, 5 and/or 7 and subsequent type I interferon (IFN) production. Cytokine signalling also drives M1‐like polarisation, notably through activation of TAK1 and Signal Transducer and Activator of Transcription (STAT) 1. M2‐like phenotype is mainly driven by cytokine signalling leading to activation of STAT5/6 and IRF3/4. In vivo, a spectrum of intermediate phenotypes exists and it is likely still unknown signalling pathways are involved in this differentiation process. HIF‐1α, hypoxia‐inducible factor 1α; HMGB1, high‐mobility group box 1; JNK, c‐Jun N‐terminal kinase; M‐CSF, macrophage colony‐stimulating factor; MyD88, myeloid differentiation primary response 88; SFAs, saturated fatty acids.
TLRs and TLR ligands in liver macrophage polarisation
Toll‐like receptors are transmembrane proteins expressed by cells of the innate immune system, notably macrophages, and that are activated in response to DAMPs, thirteen TLRs have been identified in mice [99]. Several TLRs play important roles in macrophages during NAFLD progression. TLR4, which responds to bacterial LPS, is pivotal in KCs activation. Steatosis promotes lipid accumulation in macrophages, which become more sensitive to LPS‐mediated TLR4 stimulation, promoting inflammation [100]. This is of particular relevance since microbial dysbiosis and microbial products coming from the gut are increasingly shown to potentiate NAFLD [101]. Other TLRs and their ligands play a part in macrophage polarisation during NAFLD. Dying hepatocytes, for example, release DAMPs that are recognised by TLRs. Histidine‐rich glycoprotein (HRG), a protein that is abundantly produced by hepatocytes, induces pro‐inflammatory cytokines in macrophages, even though the receptor and transduction pathway implicated are yet to be discovered. As a result, HRG‐deficient mice were partially protected against steatohepatitis induced by a MCD diet [102]. Hepatocyte‐derived mitochondrial DNA and extracellular vesicles also trigger a pro‐inflammatory phenotype through respective ligation of TLR9 and Death receptor 5 (DR5) and therefore also drive NAFLD progression [103, 104]. Finally, free fatty acids, especially saturated fatty acids, have been associated with macrophage‐driven inflammation. Saturated fatty acids induce IL‐1 and inducible nitric oxide synthase (iNOS) expression in macrophages in vitro through TLR4‐dependent nuclear factor (NF)‐κB activity while unsaturated fatty acids inhibit this process [105]. In another report, palmitate was shown to induce IL‐1 expression after TLR2 stimulation [106]. TLR4‐palmitate interaction in infiltrating macrophages has also been shown to drive NAFLD, indicating that different macrophage subsets may have specificities in recognising different sources of free fatty acids [107].
Cytokine signalling and liver macrophage polarisation
During NAFLD, activated KCs become pro‐inflammatory and secrete cytokines such as IL‐1β, IL‐6 or TNF‐α which promote inflammation and monocyte recruitment to the liver [108]. Several cytokines play key roles in NAFLD. Early TNF‐α secretion by KCs drives hepatic steatosis and inflammation [108, 109]. Similarly, IL‐1β is a potent driver of NAFLD and produced by KCs in the early phases of the disease [108, 110]. It has an important role in promoting monocyte recruitment, inflammation and steatosis [79, 110]. In addition, both TNF‐α and IL‐1β, as well as TGFβ, promote liver fibrosis by activating HSCs and promoting their survival [81, 82]. IL‐6 has a more complex role in the course of NAFLD. It seems to have a protective effect against liver injury and hepatocyte death but may promote NASH progression and fibrosis at high levels [111, 112].
Other cytokines favouring M1 polarisation, while less studied, also contribute to NAFLD progression [113]. Interestingly, IFNγ deficiency has been associated with decreased production of pro‐inflammatory cytokines, decreased inflammation and fibrosis in a mouse model of NASH [114]. Granulocyte–macrophage colony‐stimulating factor (GM‐CSF) promotes M1 polarisation and subsequent fibrosis in a model of virus‐related fibrosis, suggesting that it could also play a similar role during NAFLD [115]. However, M2‐polarising cytokines such as IL‐10 also have a role in NAFLD. IL‐10 production was reported in livers from mice fed a HFD alongside pro‐inflammatory cytokines. Its blockade is associated with increased TNF‐α and IL‐1β levels and impaired insulin sensitivity [116].
Transcriptional control of liver macrophage polarisation
Transcriptional control downstream of TLR ligation
In response to their respective ligands, TLRs can trigger different intracellular pathways. TLR3 and TLR4 activate NF‐κB, Activator protein (AP)‐1 and Interferon Regulatory Factors (IRF)‐3 and ‐5 while TLRs 7, 8 and 9 activate IRF7 instead of IRF3 (Fig. 3) [99].
In quiescent macrophages, NF‐κB activity is hindered by inhibitor of κB (IκB). Upon TLR stimulation, phosphorylation of IκB releases NF‐κB, and NF‐κB then translocates to induce transcription of target genes [117]. NF‐κB is a key regulator of M1 polarisation [118, 119]. It is responsible for production of pro‐inflammatory cytokines such as IL‐1, IL‐6, IL‐12 or TNF‐α [118]. During NAFLD, NF‐κB seems to have an important role in triggering inflammation. Indeed, one upstream regulator of NF‐κB, glucocorticoid‐induced leucine zipper (Gilz), has been shown to be down‐regulated in macrophages during NAFLD. Its overexpression in macrophages results in decreased pro‐inflammatory cytokine secretion and decreased hepatic inflammation [120]. Decreased activity of NF‐κB due to loss of Protein tyrosine phosphatase receptor type O truncated isoform (PTPROt) activity in liver macrophages is also associated with decreased inflammation [121].
AP‐1 is a complex formed of 2 proteins, c‐Jun and c‐Fos. After TLR stimulation, AP‐1 is activated through c‐Jun phosphorylation by MAPK, specifically by p38 and c‐Jun N‐terminal kinase (JNK). AP‐1 and NF‐kB are closely linked and regulate similar transcriptional programmes [122]. JNK has been shown to promote M1 polarisation of adipose tissue macrophages, while pro‐inflammatory cytokine production in response to palmitate in vitro is JNK‐dependent [123, 124]. Studies in other macrophages populations indicate a strongly pro‐inflammatory role for AP‐1, yet its role and regulation in liver macrophages remains to be entirely elucidated. A recent study has reported that c‐Jun/AP‐1 plays different roles in hepatocytes and in nonparenchymal liver cells (NPLCs, a significant proportion of which is macrophages) [125]. Schulien et al report that while expression in hepatocytes correlated with transition from steatosis to NASH, c‐Jun expression in NPLCs specifically correlated with fibrosis. In hepatocytes, c‐Jun promotes survival, preventing the regenerative ductal reaction and fibrosis, whereas in NPLCs c‐Jun promotes ductal regeneration and fibrosis through regulating both osteopontin and CD44 expression [125]. Whether this mechanism is mediated wholly or in part by macrophages remains to be demonstrated. A recent study of macrophage‐specific p38 deficiency demonstrated that, as canonically described, p38 maintains its pro‐inflammatory actions in liver macrophages and this promotes the progression of NASH [126].
IRF3 has a more complex role in macrophage polarisation. BMDM differentiated with Macrophage Colony‐Stimulating Factor (M‐CSF) display are predominantly M2‐like and activate IRF3 [127]. Its overexpression in microglia blunts the production of IL‐1β and TNF‐α in response to IL‐1β and IFNγ, while boosting IL‐10 secretion [128]. However, IRF3 also triggers the expression of pro‐inflammatory factors CCL5 [129] and IFN‐β, as well as CXCL9 and CXCL10 [130]. Studies showed that the Stimulator of Interferon Genes (STING)‐IRF3 cascade is activated in livers of mice fed a HFD [131]. Upon inactivation of STING in myeloid cells, inflammation and steatosis are decreased, suggesting a potential role for this axis in regulating liver macrophage inflammatory status [132]. Likewise, IRFs 5 and 7 has been associated with M1 polarisation in response to LPS [133, 134]. Following TLR signalling, IRFs 3, 5 and 7 are responsible for type I IFNs production [99, 135]. Type I IFNs are increasingly regarded as important players in NAFLD. In particular, they have been shown to induce T‐cell recruitment, secretion of pro‐inflammatory cytokines and subsequent insulin resistance [92, 136].
Other transcription factors, such as hypoxia inducible factor (HIF)‐1α, can be indirectly involved in TLR‐mediated macrophage polarisation, HIF‐1α is stabilised and activated in response to hypoxia [137, 138]. It was shown to induce M1 polarisation in vitro and to have an important role in macrophage function, both under normoxic or hypoxic conditions [139, 140]. TLR stimulation can also induce HIF‐1α activity though transcriptional control by NF‐κB [141, 142]. Liver macrophages from mice fed a MCD diet have enhanced HIF‐1α expression. Mice overexpressing HIF‐1α in myeloid cells display increased levels of pro‐inflammatory cytokines and increased steatosis compared with controls, both under a chow diet or MCD [143, 144]. Moreover, palmitate was shown to induce HIF‐1α activity [143, 144]. These results suggest a macrophage‐specific role for TLR‐induced HIF‐1α in liver inflammation and pathology in NASH. A role for HIF‐1α in liver fibrosis has also been reported even if the precise mechanisms still remain to be elucidated [145]. Additionally, HIF‐1α is able to increase TLR4 in response to hypoxia, thereby sensitising macrophages to LPS stimulation [137]. Hypoxia has been reported to happen during NAFLD [146], which suggests that HIF‐1α may also increase hepatic inflammation indirectly through macrophage sensitisation to TLR ligands.
Transcriptional control through cytokine signalling
Cytokines such as IFN‐γ, IL‐1β, IL‐4 or IL‐10 can activate different transcription factors to orient macrophage polarisation, among which NF‐κB, AP‐1 or members of the IRF and Signal Transducer and Activator of Transcription (STAT) families.
Interferon regulatory factors
The IRF family comprises nine members of transcription factors [147]. IRFs 3, 5 and 7 have critical roles in M1 polarisation. GM‐CSF‐treated macrophages display an M1‐like phenotype and highly express IRF5 in particular. Macrophages transfected in vitro with a siRNA targeting IRF5 lose their ability to produce pro‐inflammatory cytokines like IL‐12 in response to LPS [148]. Alzaid et al. showed that IRF5 is also metabolically responsive and its expression in macrophages during NAFLD was responsible for M1 polarisation and secretion of pro‐inflammatory and pro‐apoptotic mediators. This translated into liver inflammation, Fas‐dependent hepatocyte death and fibrosis [92, 149].
Other members of the IRF family can induce M1 polarisation. IRF1 expression is induced in vitro in macrophages treated with IFN‐γ. IRF1 can then synergise with NF‐κB to trigger IL‐12, iNOS and IFN‐β expression [150]. IRF8 is activated through the Notch pathway during LPS stimulation and is crucial for transcription of typical M1‐related genes in this context [151]. It was shown to collaborate with other transcription factors, such as IRF1 or STAT1, to drive pro‐inflammatory genes transcription in response to IFN‐γ [152]. On the contrary, IRF4 has a clear role in M2 polarisation in response to IL‐4. IRF4‐deficient macrophages secrete more cytokines such as TNF‐α, IL‐6 but also IL‐10 [153]. Lysine demethylase 6B (KDM6B) was shown to enhance IRF4 production in IL‐4‐stimulated macrophages, an event directly promoting M2 polarisation [154]. Additionally, IRF4 can suppress IRF5 activity by competing for binding to the Myeloid differentiation primary response 88 (MyD88), a crucial adaptor protein in TLR signalling [155].
Signal transducers and activators of transcription
The STAT family is composed of seven members [156]. STAT1 is phosphorylated and activated in response to IFN‐γ, one of the canonical stimuli of M1 polarisation [157]. STAT1‐deficient macrophages lose the induction of IFN‐γ‐activated genes such as inducible nitric oxide synthase (iNOS) or Class II major histocompatibility complex transactivator (CIITA) [158]. STAT1 also plays a key role in type I IFNs ability to induce M1 macrophages through STAT1:STAT2 dimers and IFN‐stimulated gene factor 3 (ISGF3) [159, 160]. Likewise, STAT5 is classically regarded as inducing M1 polarisation in response to GM‐CSF [127]. However, broader assessment of GM‐CSF–responsive genes has revealed that STAT5 may in fact induce both M1‐ and M2‐related genes, resulting in an intermediate phenotype [161]. STAT3 and STAT6 promote M2‐like macrophage polarisation. STAT6‐deficient macrophages lose their ability to respond to IL‐4 [162] and the ability to induce a number of M2‐related genes [160]. IL‐6 and IL‐10 induce STAT3, and STAT3‐deficiency leads to greater accumulation of pro‐inflammatory macrophages and susceptibility to inflammatory conditions, namely enterocolitis [163, 164, 165].
Nuclear receptors in gene regulation and as therapeutic targets in NAFLD and NASH
The nuclear receptor (NR) superfamily of transcription factors control transcription in response to specific ligands [166]. Agonists for these receptors range from hormones to vitamins, to fatty acids and cholesterol [167] as well as synthetic and pharmaceutical ligands that currently represent about 16% of all approved drugs [168]. NRs play important functions in regulating hepatic lipid and glucose metabolism as well as multiple inflammatory pathways and immune responses. As such, they are prime candidates to modulate NAFLD development [169]. Indeed, many NRs have shown promising potential as targets for anti‐NAFLD therapeutics. To date, 48 NRs that share structural and functional characteristics have been described in humans [166]. Of these, 17 have been linked to NAFLD, either using synthetic ligands that target them in experimental models of disease or in models of global, hepatic‐ or, in few cases, myeloid‐specific NR deficiency showing changes in liver steatosis and/or the development of steatohepatitis. For a detailed description of how these receptors function and the roles they play, we refer the reader to a recent comprehensive review [169]. Here, we focus on receptors that are or have been drug targets in clinical trials for NAFLD and NASH.
Thyroid hormone receptor β
Thyroid hormone (TH) receptor β (TRβ) is the TR isoform thought to be responsible for the main beneficial effects of TH on liver [170]. TRβ regulates gene expression by binding to TH response elements (TREs) in regulatory regions within target genes, mostly as heterodimers with the retinoid X receptor or RXR [171]. Unliganded TR represses basal gene expression by recruiting a corepressor complex [172]. Ligand (TH) binding then leads to the dissociation of corepressors and favours the recruitment of coactivators promoting chromatin accessibility thereby increasing gene transcription [173]. In this manner, TR enhances the expression of genes involved in fatty acid metabolism [174]. TR also inhibits the expression of lipogenic genes promoting steatosis [175].
TH metabolism and TH status have been linked to various aspects of the immune response [176] and recent reports suggest that innate immune cells are important TH targets and that intracellular TH plays essential roles on several innate immune cell types, including monocytes and liver macrophages [176]. Functional studies have shown TH pro‐inflammatory actions in macrophages. A shift towards an M1 phenotype alongside an inhibition of M2 polarisation was reported in bone marrow‐derived macrophages [177]. Intriguingly, polarisation was associated with changes in TRα1 : TRβ1 ratio, suggesting the relative abundance of TR isoforms may be linked to macrophage phenotype [177]. However, this study contrasts with reports that found no effect on macrophage polarisation [178] and it has been speculated that this could be due to differences in the hormone concentrations used [179].
Some TH actions are mediated through signal transduction mechanisms, for instance, through cell surface integrins leading to PI3K activation followed by the iNOS upregulation, nitrite production and bacterial killing [180]. Other studies have concluded that higher levels of bioavailable TH increase macrophage phagocytic capacity [181]. The effects of intracellular TH are partly mediated via TRα [182] and unstimulated macrophages deficient in TRα show low‐grade inflammation suggesting a TRα‐mediated anti‐inflammatory response [182]. TH stimulation leads to KC hyperplasia and enhanced phagocytosis [183]. TH has shown pro‐inflammatory actions in KCs involving NFκB activation [184] and acute‐phase responses in liver involving increased STAT3 activation [185], in turn increasing hepatic iNOS activity, enhancing production of reactive oxygen species and hepatic oxidative stress [186]. However, another study showed conflicting results in models of endotoxemia [187]. Clearly, more research is needed to establish whether similar mechanisms occur in other inflammatory contexts including NAFLD and to establish better in vitro models to replicate not only the inflammatory, but also dyslipidaemic environment in this disease.
Overall, THs have been shown to be beneficial for liver metabolism through: (a) an increase in energy expenditure via ATP consumption, membrane permeability and effects on mitochondrial biogenesis and activation [188], and (b) lipid metabolism such as cholesterol clearance by LDLR, cholesterol biosynthesis and metabolism through regulation of CYP7A1, a key bile acid synthesis enzyme [189]. In addition, hypothyroidism has been considered a risk factor for NAFLD [190], while TH administration improves lipid profiles in experimental models of NAFLD [191]. Unfortunately, these beneficial effects are accompanied by thyrotoxicosis and harmful effects in the brain [192]. Work on animal models stresses the importance of TRs for the hepatic actions of TH [193]. Using individual TRα1 and TRβ, knockout mice treated with TH and dietary cholesterol showed that CYP7A1 regulation was lost only in TRβ knockout mice and that TH administration was not able to modulate cholesterol levels [194], suggesting a key role for TRβ. Consistently, TRβ mutant mice are unable to bind TH and develop liver steatosis [195].
As TRβ is the predominant isoform in liver, efforts have focussed on the development of TH analogs capable of uncoupling beneficial liver actions (triglyceride and cholesterol lowering) from deleterious effects [196]. TRβ agonists modulate lipid metabolism pathways and reduce hepatic steatosis and inflammation in animal models [197] as well as improve liver function in clinical trials in patients with NAFLD and NASH [191]. Unfortunately, Sobetirome (GC‐1) and Eprotirome (KB2115), early examples of TRβ‐selective thyromimetics showing encouraging effects against hypercholesterolemia and NASH, in the absence of adverse side effects, were stopped after Phase 1 and Phases 2–3 clinical trials, respectively [170]. Resmetirom (MGL‐3196), another liver‐directed and TRβ‐selective agonist, successfully reduced steatosis and was advanced to Phase 3 trials. Other compounds are being evaluated in Phase 2 trials [198] or have shown promising preclinical effects [199]. These studies suggest that the most recent classes of thyromimetics are promising alternatives to existing NASH therapies.
Peroxisome proliferator‐activated receptors
Peroxisome proliferator‐activated receptors (PPARs) are a nuclear receptor subfamily with key actions on glucose and lipid metabolism as well as on inflammatory and fibrotic processes. Thus, PPARs are considered interesting NAFLD therapeutic targets for improving liver function and showing beneficial liver, cardiovascular and diabetes‐related outcomes [200]. The role of PPARs in the development of NAFLD has been recently reviewed in detail [201].
PPARs were first described as ligand‐activated transcription factors that promote peroxisome proliferation [202], and subsequently, they have been shown to be involved in function of other organelles, mainly mitochondria, showing pleiotropic actions [203]. Three PPAR isotypes have been described – α, β/δ and γ, with two subtypes: γ1 and γ2, and with each isotype showing a specific pattern of tissue and cell‐type expression [203]. Additionally, substantial species‐specific differences, especially for PPARα, exist and must be considered when translating findings from experimental models [204]. Specifically, PPARα activity in human liver is lower compared with rodents with reported differences in PPARα expression, ligand activation and biological responses [205].
PPARα
PPARα, encoded by the NR1C1 gene, binds to several saturated and unsaturated fatty acids, whereas the other isotypes show affinity mostly restricted to polyunsaturated fatty acids [206]. PPARα is predominantly expressed in tissues with high fatty acid oxidation rates such as skeletal muscle, liver – mostly in hepatocytes – heart, kidney and brown adipose tissue [207]. Besides hepatocytes, PPARα is expressed in sinusoidal endothelial cells and in HSCs [208]. In the liver, this nuclear receptor acts as a nutrient sensor and its expression and activity are stimulated by fasting or a fat‐rich diet [209]. PPARα functions as a transcription factor mostly as a heterodimer with RXR and, upon ligand binding, activates genes associated with mitochondrial and peroxisomal fatty acid oxidation [210]. PPARα can also repress gene expression, by interfering with the glucocorticoid receptor [211] or by tethering to other transcription factors [212]. Regarding NAFLD development, it is worth noting that a fat‐rich diet elevates hepatic PPARα expression in a circadian rhythmic manner and that the lipid‐lowering effect of a PPARα agonist is more prominent when PPARα expression peaks [212, 213]. Additionally, PPARα dampens NASH fibrotic and inflammatory gene expression through protein–protein interactions with pro‐inflammatory transcription factors NF‐kB and AP‐1 [210, 214].
Multiple studies in preclinical models or PPARα‐deficient mice show PPARα is a critical regulator of target genes involved in fatty acid metabolism and ketogenesis. Specifically, it regulates fatty acid transport, peroxisomal and mitochondrial β‐oxidation and lipolysis, and influences the production of apolipoproteins [215]. This reduces triglyceride‐rich lipoproteins and triglyceride accumulation in the liver, whereas plasma HDL cholesterol is increased [215]. Consistently, preclinical studies show that deficiency in PPARα, either in global or liver‐specific‐deficient mice, leads to more severe NASH [216, 217], which can be improved or prevented by specific PPARα ligands [217, 218, 219]. Interestingly, expression of a PPARα mutant that only shows transrepressive activity in mice confers protection against NASH but not steatosis, whereas mice expressing wild‐type PPARα are protected from both NASH and steatosis [210] highlighting the importance of this activity in the overall effects of PPARα.
Considering that approximately 50% of PPARα target genes are conserved between mice and humans [220], it is relevant that this experimental evidence agrees with existing clinical findings (see below). Additionally, hepatic PPARα expression inversely correlates with severity in patients with NASH, visceral fat and insulin resistance, and improved liver histology positively correlates with increased PPARα expression [221]. Accordingly, PPARα was considered a promising therapeutic target for NAFLD, though the number of clinical studies evaluating single PPARα ligands is low [201, 218]. Drugs of the fibrate class that predominantly act as PPARα ligands such as Clofibrate and Fenofibrate have been used clinically to treat hypertriglyceridemia, without affecting insulin sensitivity or hepatic steatosis [222, 223, 224, 225]. Disappointingly, their effect on NASH was not proven [226, 227], which could be due in part to the species‐specific differences mentioned above. Exploiting the concept of selective PPAR modulators based on differences in receptor and coactivator binding, other fibrate compounds (Gemfibrozil, Pemafibrate) are currently being tested in clinical studies based on their promising clinical profiles [228, 229, 230]. In addition, targeting both PPARα and PPARβ/δ with Elafibranor has shown promising anti‐NASH properties in a clinical trial [231], reporting improved glycaemic control and lipid profile, reduction in hepatic and muscle insulin resistance and steatohepatitis [232]. Recruitment was recently terminated on the phase III RESOLVE‐IT clinical trial (NCT02704403) assessing Elafibranor for NASH resolution [233]. Results at termination of the study have not yet been published; however, interim analyses in May 2020 revealed a near significant (P = 0.066) resolution of NASH without worsening fibrosis in patients treated with Elafibranor compared with placebo [234]. Elafibranor was found to be safe and well tolerated, consistent with a previous study in biliary cholangitis that reported improvement in a number of disease markers [234, 235].
PPARβ/δ
PPARβ/δ which is encoded by NR1C2 and expressed in hepatocytes, sinusoidal endothelial cells, HSCs and KCs also has an important role in liver metabolism [236]. This receptor activates glucose utilisation, hepatic lipogenesis and lipoprotein metabolism, as confirmed by transcriptomic analyses in PPARβ/δ‐deficient mice [237]. In addition, PPARβ/δ increases the production of monounsaturated fatty acids and protects against lipotoxicity and saturated fatty acid cytotoxicity in vitro [238]. It appears PPARα is predominant in the fasting state whereas PPARβ/δ is equally involved in both fasting and fed states [239].
PPARβ/δ also regulates the expression of key genes in innate immunity and inflammation [237, 240]. In the absence of ligand, PPARβ/δ has pro‐inflammatory effects mainly in atherosclerotic models. Ligand binding exerts anti‐inflammatory effects, such as the suppression of pro‐inflammatory adhesion molecules on endothelial cells [241, 242]. PPARβ/δ ligands promote a more anti‐inflammatory phenotype in KCs resulting in improved metabolic and hepatic dysregulation [97]. In addition, PPARβ/δ may have a potential role in wound healing, as its activation in fibroblasts increases α‐smooth muscle actin production and myofibroblast differentiation [243, 244]. Importantly, synthetic PPARβ/δ ligands mimic the endogenous activation of PPARβ/δ, although different responses have been reported for different ligands [245]. Additionally, the selective PPARβ/δ agonist Seladelpar improves dyslipidaemic lipid profiles in overweight or obesity patients at risk of CVD [246] although these compounds have not been as broadly tested as the fibrate PPARα agonists.
PPARγ
Finally, PPARγ is encoded by NR1C3 and is expressed in the liver, yet less than adipose tissue where it is a master regulator of multiple metabolic pathways. As other PPARS, PPARγ forms a heterodimer with RXR to control gene expression. In addition, as shown in cistrome studies in peritoneal macrophages, macrophage lineage factors SPI1 (PU.1) and CEBPβ are present with PPARγ in regulatory sites [247, 248], enhancing permissive chromatin configurations. In liver, PPARγ is induced by obesity in mice [249] although this is not seen in patients with NASH [221]. Hepatocyte‐specific PPARγ deficiency protects mice from steatosis in diet‐induced or genetic obesity in mice by reducing expression of genes promoting lipogenesis and lipid transport [250, 251]. In contrast, PPARγ agonists, including antidiabetic thiazolidinedione drugs (TZDs), improve NAFLD partly by reshuffling fatty acids and triglycerides to privilege storage in adipose tissue [252].
PPARγ is also present in macrophages, KCs and HSCs. In liver and other tissues, PPARγ binds to the p65 subunit of the NF‐κB complex to dampen NF‐κB‐driven inflammatory gene expression [253]. A PPARγ sumoylation‐dependent pathway was described to mediate some of the anti‐inflammatory actions of this receptor [254]. In KCs, PPARγ agonists inhibit pro‐inflammatory gene expression leading to lower inflammation and hepatosteatosis [58]. Consistently, inhibition of PPARγ with a specific antagonist promotes the M2c anti‐inflammatory phenotype in human monocyte‐derived macrophages [255], although despite the concomitant induction of MerTK expression, cells do not show enhanced efferocytosis. In HSCs, PPARγ is predominantly expressed in the quiescent state and lowered in the activated state. Ligand activation in these cells or in experimental models of fibrosis reduces collagen levels, but the mechanism underlying this regulation still need to be refined [256, 257]. Finally, PPARγ also improves endothelial cell inflammation and function in patients with diabetes and atherosclerosis [258], controls vascular homeostasis and decreases blood pressure in patients with diabetes, leading to reduced CVD risk [259].
Hepatic PPARγ expression is elevated in patients with NAFLD and NASH [260], and PPARγ agonists are promising therapeutics. The TZD class of PPARγ agonist antidiabetics, including rosiglitazone and pioglitazone. TZDs ameliorate steatosis and inflammation, but have shown only minimal reduction in fibrosis [258, 259, 261, 262, 263]. A PPARα/γ dual agonist Saroglitazar improves cardiovascular risk profiles in diabetics [264, 265] and after promising results in animal models of NASH [266] is being tested in a randomised clinical trial [258].
This subfamily of nuclear receptors represents a great example of how simultaneous activation of multiple isotypes could be a more efficacious therapeutic approach by targeting multiple pathways that contribute to the development and progression of NASH. Early studies with Lanifibranor (IVA337), which activates all three PPAR subtypes and acts on multiple NASH‐affected pathways [267, 268], showed it was effective at preventing and even inducing regression of pre‐existing fibrotic lesions in different organs [269, 270]. This occurred in the absence of deleterious effects of TZDs while improving insulin sensitivity and lipid profiles in NASH [267]. Remarkably, Lanifibranor actions on inflammation, fibrosis and macrophage accumulation and activation seem stronger than single and dual PPAR agonists in several models of NASH [271]. Lanifibranor is now part of a phase IIb trial in patients with NASH without cirrhosis. To date, significant reductions in steatohepatitis, regression of fibrosis and improved glycaemic control and lipid profile have been reported [272], suggesting pan‐PPAR agonism could have a strong therapeutic potential and be a promising therapeutic strategy for NASH.
Farnesoid X receptor
Farnesoid X receptor (FXR), encoded by NR1H4 gene, whose expression is attenuated in NASH patients [273], was originally labelled as an orphan receptor and subsequently considered ‘adopted’ as free or conjugated bile acids were recognised to be endogenous ligands [274, 275, 276]. Its impact on the regulation of key aspects of metabolic, inflammatory and fibrotic pathways has been recently covered in detail [277]. FXR is highly expressed in liver [274] and acts, through FXR response elements, mainly as a heterodimer with RXR [278]. The liver receptor homolog‐1 (LRH‐1) is also present in a substantial number of FXR‐binding sites and induces gene transcription mostly in lipid metabolic pathways [279, 280]. Other studies have proposed direct transcriptional repression by FXR in the regulation of lipoprotein metabolism and as an important contributor to its anti‐inflammatory effects through a motif independent of the canonical one [281, 282, 283, 284].
FXR is a well‐established regulator of bile acid homeostasis showing tissue‐specific roles in the liver and intestine [285]. Upon activation, FXR reduces the levels of its ligands by suppressing bile acid synthesis through CYP7A1 [275], an example of a negative feedback regulatory loop. In addition, FXR is critical in regulating the enterohepatic circulation of bile acids by affecting the expression of several transporters [286, 287, 288, 289] and in regulating lipid and glucose homeostasis. FXR activation lowers blood lipid levels as it inhibits fatty acid synthesis [290, 291], decreases hepatic secretion of VLDL [292] and increases triglyceride hydrolysis and clearance as well as fatty acid oxidation [293, 294, 295, 296]. Conflicting evidence exists regarding FXR actions in glucose homeostasis [277] which could be due to species differences between humans and mice [297, 298]. Nevertheless, FXR likely plays an important role as FXR‐deficient mice develop steatosis, show elevated circulating FFAs and glucose levels, and are insulin resistant [299]. In addition, FXR activation may improve glucose dysregulation, as either FXR activation or hepatic overexpression significantly lowers blood glucose levels and FFA levels, and improves insulin sensitivity in both db/db diabetic and wild‐type mice [300].
FXR is also a homeostatic regulator that suppresses liver inflammation and fibrosis. Pretreatment of HepG2 cells and primary hepatocytes with FXR agonists suppresses NF‐κB‐mediated inflammation in an FXR‐dependent manner [301]. In NASH models, synthetic FXR agonists lower MCP‐1 chemokine expression leading to significantly reduced hepatic inflammatory cell infiltration [301, 302]. Moreover, FXR‐deficient mice display strong hepatic inflammation in response to LPS, concomitant liver necrosis and a significant increase in inflammatory molecules such as iNOS, COX‐2 and IFN‐γ [301]. A growing body of evidence suggests that bile acids modulate intestinal and liver immune cells [303, 304, 305] and the role played by bile acid receptors has been reviewed in detail [303]. Briefly, FXR is expressed by circulating monocytes and both intestinal and liver macrophages [306]. FXR activation in human and rodent macrophages shows effective anti‐inflammatory activities, and FXR is required for the TLR9‐dependent inhibition of pro‐inflammatory responses of intestinal macrophages [307]. Transrepression of inflammatory genes in macrophages by FXR ligands involves complex mechanisms that are both SHP‐dependent and SHP‐independent [306, 307, 308]. In addition, ligand‐induced sumoylation of FXR has also been implicated in the regulation of NF‐κB and AP1‐driven gene expression [309]. Beyond NF‐κB‐mediated mechanisms, FXR may exert anti‐inflammatory actions indirectly, for instance by reducing cholestasis and the levels of toxic bile acid production and accumulation in the liver [277].
Macrophage phenotypic shift has also been described for FXR. Treatment of an obese and diabetic mouse model of NAFLD mice with the semi‐synthetic bile acid and FXR agonist obeticholic acid (OCA) improves liver histology and increases expression of M2 markers and the proportion of intrahepatic anti‐inflammatory monocytes [310]. In addition, nonspecific ligands for FXR also acting on another bile acid receptor [311], reverse liver steatosis and fibrosis along with markers of inflammation, shifting macrophage polarisation towards an M2‐like phenotype [311, 312]. Whether these modulatory effects of the hepatic immune system add to the metabolic effects of FXR ligands in the clinic requires further investigations. Moreover, FXR activation suppresses the development of hepatic fibrosis both by reducing fibrosis and by inducing antifibrotic gene expression in HSCs. HSC inactivation is also achieved by ligand‐activated FXR inducing a transcriptional regulatory cascade involving other nuclear receptors, namely the small heterodimeric partner SHP and PPARγ [256, 313, 314].
Both steroidal and nonsteroidal FXR agonists have been developed for the treatment of liver diseases. Based on previous favourable results [315], OCA was investigated in the phase IIb Farnesoid X Receptor Ligand Obeticholic Acid in NASH Treatment (FLINT) multicentre trial in patients with noncirrhotic NASH [316]. OCA improved biochemical and histological features of NASH when compared with placebo without the worsening of fibrosis. Unfortunately, no difference was observed on the resolution of NASH and effects on ALP, lipids and blood glucose observed in the placebo group associated with weight loss were absent or even reversed in OCA‐treated patients [317]. In addition, unfavourable dyslipidaemia occurred in the OCA treatment group [316]. OCA, FDA approved for biliary cholangitis therapy, was further evaluated in a NASH phase III trial REGENERATE [318], with a disappointing outcome [319]. Nevertheless, FXR remains an attractive target for NAFLD. For instance, safety and efficacy of the nonsteroidal FXR agonist Cilofexor (GS‐9674) was evaluated in a phase II study for other liver conditions [320]. Cilofexor improved inflammatory biomarkers alongside significant reductions in serum markers of liver injury [320]. In a phase II trial in NASH noncirrhotic patients, Cilofexor significantly improved hepatic steatosis, liver biochemistry and bile acids without affecting serum lipids [321]. Moreover, Tropifexor significantly reduced oxidative stress, steatosis, inflammation and fibrosis in mouse models of NASH [322]. Time will tell whether this nonsteroidal FXR agonist also proves beneficial in NASH patients.
Closing remarks: Challenges and future perspectives
NAFLD is extremely complex, due to its multiple aetiologies and the large spectrum of liver states that ranges from steatosis to inflammation and fibrosis. The complexity of NAFLD is also present at the cellular and molecular levels, where we now know that macrophages play an important role in both maintaining normal physiology and in the pathophysiology of NAFLD. As reviewed, liver macrophages are central actors of NAFLD progression, they receive and respond to signals from systemic circulation, such as insulin or lipolysis products from adipose tissue, and they are exposed to nutrient‐rich blood from portal circulation as well as a multitude of signals from the liver microenvironment. As part of the innate immune system, macrophages must act primarily as sentinels, keeping the peace then raising the alarm when homeostasis is disturbed. Raising this alarm is a tightly regulated process where a number of molecular actors within the cell integrate afferent signals and coordinate efferent responses. The responses of these very important cells can dictate disease course in NAFLD. While recent decades have accumulated a wealth of knowledge, much is yet to be learned about how therapeutically target these cells in NAFLD.
In insulin dynamics, NAFLD and liver macrophages
Strong mechanistic associations have been drawn between insulin resistance and NAFLD, yet the physiological and physiopathological adaptations of liver macrophages remain to be fully understood. Insulin levels oscillate from low levels when fasting, to higher postprandial levels. Basal levels of insulin in the blood are also different in healthy people compared with people with prediabetes or diabetes (reviewed recently [323]). An added layer of complexity for investigating the physiological and pathophysiological levels of insulin on liver macrophage function include the location and action of the liver. The liver is immediately downstream of the pancreas and clears 40–80% of the insulin [324, 325]. The amplitude of insulin's oscillations are thought to be ~ 100‐fold higher in the portal vein than in the systemic circulation [326]. Therefore, even in health, liver macrophages are exposed to higher levels of insulin compared with other tissue‐resident macrophages which may have consequences, affecting trained immunity in macrophages for example, these effects of insulin have been recently reviewed [327].
Macrophage heterogeneity and challenges in therapeutic targeting
The plasticity of macrophage terminal differentiation is now widely recognised, similarly macrophage polarisation states and effector functions now exist on a sliding scale with the classical and alternative states being at the extremes. The technical advances of recent years, namely single‐cell sequencing and high‐density cytometric methods, have allowed this appreciation of macrophage heterogeneity. Novel functional classification of macrophage subsets is an area of active research, booming in a number of fields and specially in study of the liver and pathogenesis of NAFLD [94, 328]. Despite the currently wide application of these technologies, one technical hurdle that has been a long‐standing subject of discussion in the field is the in vivo modelling of NAFLD [31]. Given the range of models available that reproduce the different components of NAFLD, macrophage populations are also very likely to vary across these numerous in vivo models. Future studies are working towards consistency and specificity with regard to the different models available and in the way that data are reported, with the multiple models being increasingly applied as mechanistic representations of different stages of disease. This trend enables a more thorough understanding of the importance of different macrophage subsets in NAFLD. Accordingly, it is necessary to decipher macrophage heterogeneity across different models of NAFLD and to understand cellular and molecular drivers of this intra‐ and inter‐model heterogeneity. The respective role of embryonic KCs, inflammatory KCs and Mo‐MPs, and their different subsets, in liver disease can now be investigated with great precision. Understanding macrophage heterogeneity in kinetic studies will also be of value. Such an approach will allow understanding of how cellular diversity arises, leading to the therapeutic targeting of detrimental subsets at the appropriate time without influencing other potentially beneficial subsets.
Challenges in translatability and NAFLD clinical evaluation
One of the most important milestones of research in NAFLD was the proposal of the two‐hit hypothesis in 1998. Since then, clear experimental evidence has implicated insulin resistance, lipotoxicity and inflammation in the pathogenesis of NAFLD. Yet a number of well‐known barriers exist in the field with regard to the translatability of certain findings from basic research to clinical practice, which in itself has clear priorities to improve staging and diagnosis of NAFLD.
The main barrier to translatability is in the complex modelling of NAFLD. Today no single murine model recapitulates the whole spectrum of NAFLD, the models applied will allow at best the integration of one or two stages, taking into account one or two factors. For example, a high‐fat diet will robustly reproduce insulin resistance; however, the liver is not affected beyond simple steatosis. Diets deficient in certain amino acids (methionine/choline) may induce moderate steatohepatitis and when combined with high fat will also induce obesity and insulin resistance; however, the depletion of amino acids reduces the physiological relevance to human disease. Similarly, surgical (bile‐duct ligation) and toxic (CCl4) models that mimic steatohepatitis and fibrosis are far from physiological. Taking into account the above models and other genetic models, reviewed by [31, 69], the scientific community can still gain a lot of mechanistic insight with regard to discrete components of the disease that can be currently reproduced. While challenging to encapsulate the entire spectrum of NAFLD as well as its comorbidities, it is also of mechanistic value that these models allow the isolated study of the different components of NAFLD. The application of these models must in future be interpreted as such, until a holistic and physiologically relevant model is developed.
Two major clinical barriers that are areas of active work are the noninvasive staging and the detection of NAFLD. Currently, a very widely used and accepted staging method is the SAF score, which histologically grades steatosis, activity and fibrosis in NAFLD. However, establishing a SAF score requires an invasive biopsy and a recognised limit to this method is considerable heterogeneity in the staging of advanced fibrosis [329]. It is for this reason that a future priority in the field is the development of sensitive noninvasive virtual biopsies, notably through the application of imaging techniques (CT, MRI) in conjunction with serological and immunological parameters. A number of large‐scale trials are currently tackling this issue (LITMUS, NIMBLE, QUID‐NASH) [330, 331, 332].
Conflict of interest
The authors declare no conflict of interest.
Author contributions
RT, MCG, IP‐T, GC, NV and FA wrote the manuscript.
Acknowledgements
This work is supported by The French National Research Agency (Agence National de la Recherche) ANR‐JCJC grant for the MitoFLAME Project ANR‐19‐CE14‐0005, the French Society for Diabetes (Société Francophone du Diabète; SFD) Allocation Exceptionnelle and the European Foundation for the Study of Diabetes grant to FA.
References
- 1. Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM & Ahmed A (2015) Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 148, 547–555. [DOI] [PubMed] [Google Scholar]
- 2. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, George J & Bugianesi E (2018) Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 15, 11–20. [DOI] [PubMed] [Google Scholar]
- 3. Younossi ZM, Golabi P, de Avila L, Paik JM, Srishord M, Fukui N, Qiu Y, Burns L, Afendy A & Nader F (2019) The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: a systematic review and meta‐analysis. J Hepatol 71, 793–801. [DOI] [PubMed] [Google Scholar]
- 4. Gehrke N & Schattenberg JM (2020) Metabolic inflammation‐a role for hepatic inflammatory pathways as drivers of comorbidities in nonalcoholic fatty liver disease? Gastroenterology 158, 1929–1947.e6. [DOI] [PubMed] [Google Scholar]
- 5. Sheka AC, Adeyi O, Thompson J, Hameed B, Crawford PA & Ikramuddin S (2020) Nonalcoholic steatohepatitis: a review. JAMA 323, 1175–1183. [DOI] [PubMed] [Google Scholar]
- 6. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L & Wymer M (2016) Global epidemiology of nonalcoholic fatty liver disease‐meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84. [DOI] [PubMed] [Google Scholar]
- 7. Huang DQ, El‐Serag HB & Loomba R (2021) Global epidemiology of NAFLD‐related HCC: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 18, 223–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Day CP & James OF (1998) Hepatic steatosis: innocent bystander or guilty party? Hepatology 27, 1463–1466. [DOI] [PubMed] [Google Scholar]
- 9. Amiri Dash Atan N, Koushki M, Motedayen M, Dousti M, Sayehmiri F, Vafaee R, Norouzinia M & Gholami R (2017) Type 2 diabetes mellitus and non‐alcoholic fatty liver disease: a systematic review and meta‐analysis. Gastroenterol Hepatol Bed Bench 10, S1–S7. [PMC free article] [PubMed] [Google Scholar]
- 10. Marchesini G, Brizi M, Morselli‐Labate AM, Bianchi G, Bugianesi E, McCullough AJ, Forlani G & Melchionda N (1999) Association of nonalcoholic fatty liver disease with insulin resistance. Am J Med 107, 450–455. [DOI] [PubMed] [Google Scholar]
- 11. Sanyal AJ, Campbell‐Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML & Clore JN (2001) Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 120, 1183–1192. [DOI] [PubMed] [Google Scholar]
- 12. Smith GI, Shankaran M, Yoshino M, Schweitzer GG, Chondronikola M, Beals JW, Okunade AL, Patterson BW, Nyangau E, Field T et al. (2020) Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J Clin Invest 130, 1453–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cedo L, Santos D, Roglans N, Julve J, Pallares V, Rivas‐Urbina A, Llorente‐Cortes V, Laguna JC, Blanco‐Vaca F & Escola‐Gil JC (2017) Human hepatic lipase overexpression in mice induces hepatic steatosis and obesity through promoting hepatic lipogenesis and white adipose tissue lipolysis and fatty acid uptake. PLoS One 12, e0189834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mota M, Banini BA, Cazanave SC & Sanyal AJ (2016) Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 65, 1049–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim OK, Jun W & Lee J (2015) Mechanism of ER stress and inflammation for hepatic insulin resistance in obesity. Ann Nutr Metab 67, 218–227. [DOI] [PubMed] [Google Scholar]
- 16. Achard CS & Laybutt DR (2012) Lipid‐induced endoplasmic reticulum stress in liver cells results in two distinct outcomes: adaptation with enhanced insulin signaling or insulin resistance. Endocrinology 153, 2164–2177. [DOI] [PubMed] [Google Scholar]
- 17. Puri P, Mirshahi F, Cheung O, Natarajan R, Maher JW, Kellum JM & Sanyal AJ (2008) Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 134, 568–576. [DOI] [PubMed] [Google Scholar]
- 18. Brown M, Dainty S, Strudwick N, Mihai AD, Watson JN, Dendooven R, Paton AW, Paton JC & Schroder M (2020) Endoplasmic reticulum stress causes insulin resistance by inhibiting delivery of newly synthesized insulin receptors to the cell surface. Mol Biol Cell 31, 2597–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Davies LC & Taylor PR (2015) Tissue‐resident macrophages: then and now. Immunology 144, 541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wen Y, Lambrecht J, Ju C & Tacke F (2021) Hepatic macrophages in liver homeostasis and diseases‐diversity, plasticity and therapeutic opportunities. Cell Mol Immunol 18, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bataller R & Brenner DA (2005) Liver fibrosis. J Clin Invest 115, 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Weiskirchen R & Tacke F (2016) Liver fibrosis: from pathogenesis to novel therapies. Dig Dis 34, 410–422. [DOI] [PubMed] [Google Scholar]
- 23. Alisi A, Carpino G, Oliveira FL, Panera N, Nobili V & Gaudio E (2017) The role of tissue macrophage‐mediated inflammation on NAFLD pathogenesis and its clinical implications. Mediators Inflamm 2017, 8162421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ullrich A, Bell JR, Chen EY, Herrera R, Petruzzelli LM, Dull TJ, Gray A, Coussens L, Liao YC, Tsubokawa M et al. (1985) Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 313, 756–761. [DOI] [PubMed] [Google Scholar]
- 25. Ebina Y, Ellis L, Jarnagin K, Edery M, Graf L, Clauser E, Ou J‐H, Masiarz F, Kan YW, Goldfine ID et al. (1985) The human insulin receptor cDNA: the structural basis for hormone‐activated transmembrane signalling. Cell 40, 747–758. [DOI] [PubMed] [Google Scholar]
- 26. Ullrich A, Gray A, Tam AW, Yang‐Feng T, Tsubokawa M, Collins C, Henzel W, Le Bon T, Kathuria S, Chen E et al. (1986) Insulin‐like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J 5, 2503–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haystead CM, Gregory P, Shirazi A, Fadden P, Mosse C, Dent P & Haystead TA (1994) Insulin activates a novel adipocyte mitogen‐activated protein kinase kinase kinase that shows rapid phasic kinetics and is distinct from c‐Raf. J Biol Chem 269, 12804–12808. [PubMed] [Google Scholar]
- 28. Hancock ML, Meyer RC, Mistry M, Khetani RS, Wagschal A, Shin T, Ho Sui SJ, Naar AM & Flanagan JG (2019) Insulin receptor associates with promoters genome‐wide and regulates gene expression. Cell 177, 722–736.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boucher J, Kleinridders A & Kahn CR (2014) Insulin receptor signaling in normal and insulin‐resistant states. Cold Spring Harb Perspect Biol 6, a009191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Farese RV Jr, Zechner R, Newgard CB & Walther TC (2012) The problem of establishing relationships between hepatic steatosis and hepatic insulin resistance. Cell Metab 15, 570–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jahn D, Kircher S, Hermanns HM & Geier A (2019) Animal models of NAFLD from a hepatologist's point of view. Biochim Biophys Acta Mol Basis Dis 1865, 943–953. [DOI] [PubMed] [Google Scholar]
- 32. Joshi RL, Lamothe B, Cordonnier N, Mesbah K, Monthioux E, Jami J & Bucchini D (1996) Targeted disruption of the insulin receptor gene in the mouse results in neonatal lethality. EMBO J 15, 1542–1547. [PMC free article] [PubMed] [Google Scholar]
- 33. Michael MD, Kulkarni RN, Postic C, Previs SF, Shulman GI, Magnuson MA & Kahn CR (2000) Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell 6, 87–97. [PubMed] [Google Scholar]
- 34. Bar RS, Kahn CR & Koren HS (1977) Insulin inhibition of antibody‐dependent cytoxicity and insulin receptors in macrophages. Nature 265, 632–635. [DOI] [PubMed] [Google Scholar]
- 35. O'Rourke L, Yeaman SJ & Shepherd PR (2001) Insulin and leptin acutely regulate cholesterol ester metabolism in macrophages by novel signaling pathways. Diabetes 50, 955–961. [DOI] [PubMed] [Google Scholar]
- 36. Han S, Liang CP, DeVries‐Seimon T, Ranalletta M, Welch CL, Collins‐Fletcher K, Accili D, Tabas I & Tall AR (2006) Macrophage insulin receptor deficiency increases ER stress‐induced apoptosis and necrotic core formation in advanced atherosclerotic lesions. Cell Metab 3, 257–266. [DOI] [PubMed] [Google Scholar]
- 37. Baumgartl J, Baudler S, Scherner M, Babaev V, Makowski L, Suttles J, McDuffie M, Tobe K, Kadowaki T, Fazio S et al. (2006) Myeloid lineage cell‐restricted insulin resistance protects apolipoproteinE‐deficient mice against atherosclerosis. Cell Metab 3, 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kazankov K, Jorgensen SMD, Thomsen KL, Moller HJ, Vilstrup H, George J, Schuppan D & Gronbaek H (2019) The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol 16, 145–159. [DOI] [PubMed] [Google Scholar]
- 39. Morinaga H, Mayoral R, Heinrichsdorff J, Osborn O, Franck N, Hah N, Walenta E, Bandyopadhyay G, Pessentheiner AR, Chi TJ et al. (2015) Characterization of distinct subpopulations of hepatic macrophages in HFD/obese mice. Diabetes 64, 1120–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Morgantini C, Jager J, Li X, Levi L, Azzimato V, Sulen A, Barreby E, Xu C, Tencerova M, Naslund E et al. (2019) Liver macrophages regulate systemic metabolism through non‐inflammatory factors. Nat Metab 1, 445–459. [DOI] [PubMed] [Google Scholar]
- 41. Bleriot C & Ginhoux F (2019) Understanding the heterogeneity of resident liver macrophages. Front Immunol 10, 2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, Lippens S, Abels C, Schoonooghe S, Raes G et al. (2016) Bone marrow‐derived monocytes give rise to self‐renewing and fully differentiated Kupffer cells. Nat Commun 7, 10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tran S, Baba I, Poupel L, Dussaud S, Moreau M, Gelineau A, Marcelin G, Magreau‐Davy E, Ouhachi M, Lesnik P et al. (2020) Impaired Kupffer cell self‐renewal alters the liver response to lipid overload during non‐alcoholic steatohepatitis. Immunity 53, 627–640.e5. [DOI] [PubMed] [Google Scholar]
- 44. Schulz C, Gomez Perdiguero E, Chorro L, Szabo‐Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW et al. (2012) A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336, 86–90. [DOI] [PubMed] [Google Scholar]
- 45. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss‐Ayali D, Viukov S, Guilliams M, Misharin A et al. (2013) Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F et al. (2015) Tissue‐resident macrophages originate from yolk‐sac‐derived erythro‐myeloid progenitors. Nature 518, 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, Beaudin AE, Lum J, Low I, Forsberg EC et al. (2015) C‐Myb(+) erythro‐myeloid progenitor‐derived fetal monocytes give rise to adult tissue‐resident macrophages. Immunity 42, 665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D et al. (2013) Tissue‐resident macrophages self‐maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38, 792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ebe Y, Hasegawa G, Takatsuka H, Umezu H, Mitsuyama M, Arakawa M, Mukaida N & Naito M (1999) The role of Kupffer cells and regulation of neutrophil migration into the liver by macrophage inflammatory protein‐2 in primary listeriosis in mice. Pathol Int 49, 519–532. [DOI] [PubMed] [Google Scholar]
- 50. Gregory SH, Sagnimeni AJ & Wing EJ (1996) Bacteria in the bloodstream are trapped in the liver and killed by immigrating neutrophils. J Immunol 157, 2514–2520. [PubMed] [Google Scholar]
- 51. Shi J, Fujieda H, Kokubo Y & Wake K (1996) Apoptosis of neutrophils and their elimination by Kupffer cells in rat liver. Hepatology 24, 1256–1263. [DOI] [PubMed] [Google Scholar]
- 52. Shi J, Gilbert GE, Kokubo Y & Ohashi T (2001) Role of the liver in regulating numbers of circulating neutrophils. Blood 98, 1226–1230. [DOI] [PubMed] [Google Scholar]
- 53. Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK & Moestrup SK (2001) Identification of the haemoglobin scavenger receptor. Nature 409, 198–201. [DOI] [PubMed] [Google Scholar]
- 54. Willekens FL, Werre JM, Kruijt JK, Roerdinkholder‐Stoelwinder B, Groenen‐Dopp YA, van den Bos AG, Bosman GJ & van Berkel TJ (2005) Liver Kupffer cells rapidly remove red blood cell‐derived vesicles from the circulation by scavenger receptors. Blood 105, 2141–2145. [DOI] [PubMed] [Google Scholar]
- 55. Theurl M, Theurl I, Hochegger K, Obrist P, Subramaniam N, van Rooijen N, Schuemann K & Weiss G (2008) Kupffer cells modulate iron homeostasis in mice via regulation of hepcidin expression. J Mol Med (Berl) 86, 825–835. [DOI] [PubMed] [Google Scholar]
- 56. Wang Y, van der Tuin S, Tjeerdema N, van Dam AD, Rensen SS, Hendrikx T, Berbee JF, Atanasovska B, Fu J, Hoekstra M et al. (2015) Plasma cholesteryl ester transfer protein is predominantly derived from Kupffer cells. Hepatology 62, 1710–1722. [DOI] [PubMed] [Google Scholar]
- 57. Krenkel O & Tacke F (2017) Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol 17, 306–321. [DOI] [PubMed] [Google Scholar]
- 58. Luo W, Xu Q, Wang Q, Wu H & Hua J (2017) Effect of modulation of PPAR‐gamma activity on Kupffer cells M1/M2 polarization in the development of non‐alcoholic fatty liver disease. Sci Rep 7, 44612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Han YH, Kim HJ, Na H, Nam MW, Kim JY, Kim JS, Koo SH & Lee MO (2017) RORalpha induces KLF4‐mediated M2 polarization in the liver macrophages that protect against nonalcoholic steatohepatitis. Cell Rep 20, 124–135. [DOI] [PubMed] [Google Scholar]
- 60. Hart KM, Fabre T, Sciurba JC, Gieseck RL, Borthwick LA, Vannella KM, Acciani TH, de Queiroz Prado R, Thompson RW, White Sandra, et al. (2017) Type 2 immunity is protective in metabolic disease but exacerbates NAFLD collaboratively with TGF‐β. Sci Transl Med 9, eaal3694. doi: 10.1126/scitranslmed.aal3694 [DOI] [PubMed] [Google Scholar]
- 61. Devisscher L, Scott CL, Lefere S, Raevens S, Bogaerts E, Paridaens A, Verhelst X, Geerts A, Guilliams M & Van Vlierberghe H (2017) Non‐alcoholic steatohepatitis induces transient changes within the liver macrophage pool. Cell Immunol 322, 74–83. [DOI] [PubMed] [Google Scholar]
- 62. You Q, Cheng L, Kedl RM & Ju C (2008) Mechanism of T cell tolerance induction by murine hepatic Kupffer cells. Hepatology 48, 978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Knolle P, Schlaak J, Uhrig A, Kempf P, Meyer zum Buschenfelde KH & Gerken G (1995) Human Kupffer cells secrete IL‐10 in response to lipopolysaccharide (LPS) challenge. J Hepatol 22, 226–229. [DOI] [PubMed] [Google Scholar]
- 64. Heymann F, Peusquens J, Ludwig‐Portugall I, Kohlhepp M, Ergen C, Niemietz P, Martin C, van Rooijen N, Ochando JC, Randolph GJ et al. (2015) Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology 62, 279–291. [DOI] [PubMed] [Google Scholar]
- 65. Krenkel O, Hundertmark J, Abdallah AT, Kohlhepp M, Puengel T, Roth T, Branco DPP, Mossanen JC, Luedde T, Trautwein C et al. (2020) Myeloid cells in liver and bone marrow acquire a functionally distinct inflammatory phenotype during obesity‐related steatohepatitis. Gut 69, 551–563. [DOI] [PubMed] [Google Scholar]
- 66. Dal‐Secco D, Wang J, Zeng Z, Kolaczkowska E, Wong CH, Petri B, Ransohoff RM, Charo IF, Jenne CN & Kubes P (2015) A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J Exp Med 212, 447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Theurl I, Hilgendorf I, Nairz M, Tymoszuk P, Haschka D, Asshoff M, He S, Gerhardt LM, Holderried TA, Seifert M et al. (2016) On‐demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat Med 22, 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sierro F, Evrard M, Rizzetto S, Melino M, Mitchell AJ, Florido M, Beattie L, Walters SB, Tay SS, Lu B et al. (2017) A liver capsular network of monocyte‐derived macrophages restricts hepatic dissemination of intraperitoneal bacteria by neutrophil recruitment. Immunity 47, 374–388.e6. [DOI] [PubMed] [Google Scholar]
- 69. Febbraio MA, Reibe S, Shalapour S, Ooi GJ, Watt MJ & Karin M (2019) Preclinical models for studying NASH‐driven HCC: how useful are they? Cell Metab 29, 18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Oligschlaeger Y, Shiri‐Sverdlov R. (2020) NAFLD preclinical models: more than a handful, less of a concern?. Biomedicines 8, 28. doi: 10.3390/biomedicines8020028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tag CG, Sauer‐Lehnen S, Weiskirchen S, Borkham‐Kamphorst E, Tolba RH, Tacke F, Weiskirchen R (2015) Bile duct ligation in mice: induction of inflammatory liver injury and fibrosis by obstructive cholestasis. J Vis Exp. doi: 10.3791/52438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nakagawa H, Umemura A, Taniguchi K, Font‐Burgada J, Dhar D, Ogata H, Zhong Z, Valasek MA, Seki E, Hidalgo J et al. (2014) ER stress cooperates with hypernutrition to trigger TNF‐dependent spontaneous HCC development. Cancer Cell 26, 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Park JW, Jeong G, Kim SJ, Kim MK & Park SM (2007) Predictors reflecting the pathological severity of non‐alcoholic fatty liver disease: comprehensive study of clinical and immunohistochemical findings in younger Asian patients. J Gastroenterol Hepatol 22, 491–497. [DOI] [PubMed] [Google Scholar]
- 74. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, Huss S, Klussmann S, Eulberg D, Luedde T et al. (2012) Pharmacological inhibition of the chemokine CCL2 (MCP‐1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 61, 416–426. [DOI] [PubMed] [Google Scholar]
- 75. Huang W, Metlakunta A, Dedousis N, Zhang P, Sipula I, Dube JJ, Scott DK & O'Doherty RM (2010) Depletion of liver Kupffer cells prevents the development of diet‐induced hepatic steatosis and insulin resistance. Diabetes 59, 347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Neyrinck AM, Cani PD, Dewulf EM, De Backer F, Bindels LB & Delzenne NM (2009) Critical role of Kupffer cells in the management of diet‐induced diabetes and obesity. Biochem Biophys Res Commun 385, 351–356. [DOI] [PubMed] [Google Scholar]
- 77. Reid DT, Reyes JL, McDonald BA, Vo T, Reimer RA & Eksteen B (2016) Kupffer cells undergo fundamental changes during the development of experimental NASH and are critical in initiating liver damage and inflammation. PLoS One 11, e0159524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Stienstra R, Mandard S, Patsouris D, Maass C, Kersten S & Muller M (2007) Peroxisome proliferator‐activated receptor alpha protects against obesity‐induced hepatic inflammation. Endocrinology 148, 2753–2763. [DOI] [PubMed] [Google Scholar]
- 79. Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JE, van Rooijen N, Staels B, Kersten S & Muller M (2010) Kupffer cells promote hepatic steatosis via interleukin‐1beta‐dependent suppression of peroxisome proliferator‐activated receptor alpha activity. Hepatology 51, 511–522. [DOI] [PubMed] [Google Scholar]
- 80. Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M, Liepelt A, Lefebvre E, Luedde T, Hellerbrand C et al. (2018) Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 67, 1270–1283. [DOI] [PubMed] [Google Scholar]
- 81. Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, Jang MK, Guenther ND, Mederacke I, Friedman R et al. (2013) Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 58, 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tsuchida T & Friedman SL (2017) Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol 14, 397–411. [DOI] [PubMed] [Google Scholar]
- 83. Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, Zhao XY, Ji Y, Li C, Guo L et al. (2019) Landscape of intercellular crosstalk in healthy and NASH liver revealed by single‐cell secretome gene analysis. Mol Cell 75, 644–660.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tateya S, Rizzo NO, Handa P, Cheng AM, Morgan‐Stevenson V, Daum G, Clowes AW, Morton GJ, Schwartz MW & Kim F (2011) Endothelial NO/cGMP/VASP signaling attenuates Kupffer cell activation and hepatic insulin resistance induced by high‐fat feeding. Diabetes 60, 2792–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Weston CJ, Shepherd EL, Claridge LC, Rantakari P, Curbishley SM, Tomlinson JW, Hubscher SG, Reynolds GM, Aalto K, Anstee QM et al. (2015) Vascular adhesion protein‐1 promotes liver inflammation and drives hepatic fibrosis. J Clin Invest 125, 501–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hammoutene A & Rautou PE (2019) Role of liver sinusoidal endothelial cells in non‐alcoholic fatty liver disease. J Hepatol 70, 1278–1291. [DOI] [PubMed] [Google Scholar]
- 87. Sutti S & Albano E (2020) Adaptive immunity: an emerging player in the progression of NAFLD. Nat Rev Gastroenterol Hepatol 17, 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Heymann F & Tacke F (2016) Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol 13, 88–110. [DOI] [PubMed] [Google Scholar]
- 89. Remmerie A, Martens L & Scott CL (2020) Macrophage subsets in obesity, aligning the liver and adipose tissue. Front Endocrinol (Lausanne) 11, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, Hartland SN, Snowdon VK, Cappon A, Gordon‐Walker TT et al. (2012) Differential Ly‐6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci USA 109, E3186–E3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Mosser DM & Edwards JP (2008) Exploring the full spectrum of macrophage activation. Nat Rev Immunol 8, 958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Alzaid F, Lagadec F, Albuquerque M, Ballaire R, Orliaguet L, Hainault I, Blugeon C, Lemoine S, Lehuen A, Saliba DG et al. (2016) IRF5 governs liver macrophage activation that promotes hepatic fibrosis in mice and humans. JCI Insight 1, e88689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Shapouri‐Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, Seifi B, Mohammadi A, Afshari JT & Sahebkar A (2018) Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol 233, 6425–6440. [DOI] [PubMed] [Google Scholar]
- 94. MacParland SA, Liu JC, Ma XZ, Innes BT, Bartczak AM, Gage BK, Manuel J, Khuu N, Echeverri J, Linares I et al. (2018) Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun 9, 4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L et al. (2014) Transcriptome‐based network analysis reveals a spectrum model of human macrophage activation. Immunity 40, 274–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wan J, Benkdane M, Teixeira‐Clerc F, Bonnafous S, Louvet A, Lafdil F, Pecker F, Tran A, Gual P, Mallat A et al. (2014) M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 59, 130–142. [DOI] [PubMed] [Google Scholar]
- 97. Odegaard JI, Ricardo‐Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, Subramanian V, Mukundan L, Ferrante AW & Chawla A (2008) Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity‐induced insulin resistance. Cell Metab 7, 496–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Svendsen P, Graversen JH, Etzerodt A, Hager H, Roge R, Gronbaek H, Christensen EI, Moller HJ, Vilstrup H & Moestrup SK (2017) Antibody‐directed glucocorticoid targeting to CD163 in M2‐type macrophages attenuates fructose‐induced liver inflammatory changes. Mol Ther Methods Clin Dev 4, 50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kawai T & Akira S (2006) TLR signaling. Cell Death Differ 13, 816–825. [DOI] [PubMed] [Google Scholar]
- 100. Leroux A, Ferrere G, Godie V, Cailleux F, Renoud ML, Gaudin F, Naveau S, Prevot S, Makhzami S, Perlemuter G et al. (2012) Toxic lipids stored by Kupffer cells correlates with their pro‐inflammatory phenotype at an early stage of steatohepatitis. J Hepatol 57, 141–149. [DOI] [PubMed] [Google Scholar]
- 101. Kolodziejczyk AA, Zheng D, Shibolet O, Elinav E (2019) The role of the microbiome in NAFLD and NASH. EMBO Mol Med 11. doi: 10.15252/emmm.201809302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Bartneck M, Fech V, Ehling J, Govaere O, Warzecha KT, Hittatiya K, Vucur M, Gautheron J, Luedde T, Trautwein C et al. (2016) Histidine‐rich glycoprotein promotes macrophage activation and inflammation in chronic liver disease. Hepatology 63, 1310–1324. [DOI] [PubMed] [Google Scholar]
- 103. Garcia‐Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, Shlomchik MJ, Coffman RL, Candia A & Mehal WZ (2016) Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest 126, 859–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF, Werneburg NW, Charlton MR, Shah VH, Malhi H & Gores GJ (2016) Lipid‐induced signaling causes release of inflammatory extracellular vesicles from hepatocytes. Gastroenterology 150, 956–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lee JY, Sohn KH, Rhee SH & Hwang D (2001) Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase‐2 mediated through Toll‐like receptor 4. J Biol Chem 276, 16683–16689. [DOI] [PubMed] [Google Scholar]
- 106. Snodgrass RG, Huang S, Choi IW, Rutledge JC & Hwang DH (2013) Inflammasome‐mediated secretion of IL‐1beta in human monocytes through TLR2 activation; modulation by dietary fatty acids. J Immunol 191, 4337–4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kim SY, Jeong JM, Kim SJ, Seo W, Kim MH, Choi WM, Yoo W, Lee JH, Shim YR, Yi HS et al. (2017) Pro‐inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4‐MD2 complex. Nat Commun 8, 2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Tosello‐Trampont AC, Landes SG, Nguyen V, Novobrantseva TI & Hahn YS (2012) Kuppfer cells trigger nonalcoholic steatohepatitis development in diet‐induced mouse model through tumor necrosis factor‐alpha production. J Biol Chem 287, 40161–40172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. De Taeye BM, Novitskaya T, McGuinness OP, Gleaves L, Medda M, Covington JW & Vaughan DE (2007) Macrophage TNF‐alpha contributes to insulin resistance and hepatic steatosis in diet‐induced obesity. Am J Physiol Endocrinol Metab 293, E713–E725. [DOI] [PubMed] [Google Scholar]
- 110. Mirea AM, Tack CJ, Chavakis T, Joosten LAB & Toonen EJM (2018) IL‐1 family cytokine pathways underlying NAFLD: towards new treatment strategies. Trends Mol Med 24, 458–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Yamaguchi K, Itoh Y, Yokomizo C, Nishimura T, Niimi T, Fujii H, Okanoue T & Yoshikawa T (2010) Blockade of interleukin‐6 signaling enhances hepatic steatosis but improves liver injury in methionine choline‐deficient diet‐fed mice. Lab Invest 90, 1169–1178. [DOI] [PubMed] [Google Scholar]
- 112. Yamaguchi K, Itoh Y, Yokomizo C, Nishimura T, Niimi T, Umemura A, Fujii H, Okanoue T & Yoshikawa T (2011) Blockade of IL‐6 signaling exacerbates liver injury and suppresses antiapoptotic gene expression in methionine choline‐deficient diet‐fed db/db mice. Lab Invest 91, 609–618. [DOI] [PubMed] [Google Scholar]
- 113. Mohlenberg M, Terczynska‐Dyla E, Thomsen KL, George J, Eslam M, Gronbaek H & Hartmann R (2019) The role of IFN in the development of NAFLD and NASH. Cytokine 124, 154519. [DOI] [PubMed] [Google Scholar]
- 114. Luo XY, Takahara T, Kawai K, Fujino M, Sugiyama T, Tsuneyama K, Tsukada K, Nakae S, Zhong L & Li XK (2013) IFN‐gamma deficiency attenuates hepatic inflammation and fibrosis in a steatohepatitis model induced by a methionine‐ and choline‐deficient high‐fat diet. Am J Physiol Gastrointest Liver Physiol 305, G891–G899. [DOI] [PubMed] [Google Scholar]
- 115. Tan‐Garcia A, Lai F, Sheng Yeong JP, Irac SE, Ng PY, Msallam R, Tatt Lim JC, Wai LE, Tham CYL, Choo SP et al. (2020) Liver fibrosis and CD206(+) macrophage accumulation are suppressed by anti‐GM‐CSF therapy. JHEP Rep 2, 100062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Cintra DE, Pauli JR, Araujo EP, Moraes JC, de Souza CT, Milanski M, Morari J, Gambero A, Saad MJ & Velloso LA (2008) Interleukin‐10 is a protective factor against diet‐induced insulin resistance in liver. J Hepatol 48, 628–637. [DOI] [PubMed] [Google Scholar]
- 117. Kawai T & Akira S (2007) Signaling to NF‐kappaB by Toll‐like receptors. Trends Mol Med 13, 460–469. [DOI] [PubMed] [Google Scholar]
- 118. Baker RG, Hayden MS & Ghosh S (2011) NF‐kappaB, inflammation, and metabolic disease. Cell Metab 13, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Liu CP, Zhang X, Tan QL, Xu WX, Zhou CY, Luo M, Li X, Huang RY & Zeng X (2017) NF‐kappaB pathways are involved in M1 polarization of RAW 264.7 macrophage by polyporus polysaccharide in the tumor microenvironment. PLoS One 12, e0188317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Robert O, Boujedidi H, Bigorgne A, Ferrere G, Voican CS, Vettorazzi S, Tuckermann JP, Bouchet‐Delbos L, Tran T, Hemon P et al. (2016) Decreased expression of the glucocorticoid receptor‐GILZ pathway in Kupffer cells promotes liver inflammation in obese mice. J Hepatol 64, 916–924. [DOI] [PubMed] [Google Scholar]
- 121. Jin K, Liu Y, Shi Y, Zhang H, Sun Y, Zhangyuan G, Wang F, Yu W, Wang J, Tao X et al. (2020) PTPROt aggravates inflammation by enhancing NF‐kappaB activation in liver macrophages during nonalcoholic steatohepatitis. Theranostics 10, 5290–5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Ji Z, He L, Regev A & Struhl K (2019) Inflammatory regulatory network mediated by the joint action of NF‐kB, STAT3, and AP‐1 factors is involved in many human cancers. Proc Natl Acad Sci USA 116, 9453–9462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Solinas G, Vilcu C, Neels JG, Bandyopadhyay GK, Luo JL, Naugler W, Grivennikov S, Wynshaw‐Boris A, Scadeng M, Olefsky JM et al. (2007) JNK1 in hematopoietically derived cells contributes to diet‐induced inflammation and insulin resistance without affecting obesity. Cell Metab 6, 386–397. [DOI] [PubMed] [Google Scholar]
- 124. Hao J, Hu Y, Li Y, Zhou Q & Lv X (2017) Involvement of JNK signaling in IL4‐induced M2 macrophage polarization. Exp Cell Res 357, 155–162. [DOI] [PubMed] [Google Scholar]
- 125. Schulien I, Hockenjos B, Schmitt‐Graeff A, Perdekamp MG, Follo M, Thimme R & Hasselblatt P (2019) The transcription factor c‐Jun/AP‐1 promotes liver fibrosis during non‐alcoholic steatohepatitis by regulating Osteopontin expression. Cell Death Differ 26, 1688–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Zhang X, Fan L, Wu J, Xu H, Leung WY, Fu K, Wu J, Liu K, Man K, Yang X et al. (2019) Macrophage p38alpha promotes nutritional steatohepatitis through M1 polarization. J Hepatol 71, 163–174. [DOI] [PubMed] [Google Scholar]
- 127. Fleetwood AJ, Dinh H, Cook AD, Hertzog PJ & Hamilton JA (2009) GM‐CSF‐ and M‐CSF‐dependent macrophage phenotypes display differential dependence on type I interferon signaling. J Leukoc Biol 86, 411–421. [DOI] [PubMed] [Google Scholar]
- 128. Tarassishin L, Suh HS & Lee SC (2011) Interferon regulatory factor 3 plays an anti‐inflammatory role in microglia by activating the PI3K/Akt pathway. J Neuroinflammation 8, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Lin R, Heylbroeck C, Genin P, Pitha PM & Hiscott J (1999) Essential role of interferon regulatory factor 3 in direct activation of RANTES chemokine transcription. Mol Cell Biol 19, 959–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Fleetwood AJ, Lawrence T, Hamilton JA & Cook AD (2007) Granulocyte‐macrophage colony‐stimulating factor (CSF) and macrophage CSF‐dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J Immunol 178, 5245–5252. [DOI] [PubMed] [Google Scholar]
- 131. Qiao JT, Cui C, Qing L, Wang LS, He TY, Yan F, Liu FQ, Shen YH, Hou XG & Chen L (2018) Activation of the STING‐IRF3 pathway promotes hepatocyte inflammation, apoptosis and induces metabolic disorders in nonalcoholic fatty liver disease. Metabolism 81, 13–24. [DOI] [PubMed] [Google Scholar]
- 132. Luo X, Li H, Ma L, Zhou J, Guo X, Woo SL, Pei Y, Knight LR, Deveau M, Chen Y et al. (2018) Expression of STING is increased in liver tissues from patients with NAFLD and promotes macrophage‐mediated hepatic inflammation and fibrosis in mice. Gastroenterology 155, 1971–1984.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Tanaka T, Murakami K, Bando Y & Yoshida S (2015) Interferon regulatory factor 7 participates in the M1‐like microglial polarization switch. Glia 63, 595–610. [DOI] [PubMed] [Google Scholar]
- 134. Pinilla‐Vera M, Xiong Z, Zhao Y, Zhao J, Donahoe MP, Barge S, Horne WT, Kolls JK, McVerry BJ, Birukova A et al. (2016) Full spectrum of LPS activation in alveolar macrophages of healthy volunteers by whole transcriptomic profiling. PLoS One 11, e0159329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Andrilenas KK, Ramlall V, Kurland J, Leung B, Harbaugh AG & Siggers T (2018) DNA‐binding landscape of IRF3, IRF5 and IRF7 dimers: implications for dimer‐specific gene regulation. Nucleic Acids Res 46, 2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ghazarian M, Revelo XS, Nøhr MK, Luck H, Zeng K, Lei H, Tsai S, Schroer SA, Park YJ, Chng MHY et al. (2017) Type I interferon responses drive intrahepatic T cells to promote metabolic syndrome. Sci Immunol 2, eaai7616. doi: 10.1126/sciimmunol.aai7616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Kim SY, Choi YJ, Joung SM, Lee BH, Jung YS & Lee JY (2010) Hypoxic stress up‐regulates the expression of Toll‐like receptor 4 in macrophages via hypoxia‐inducible factor. Immunology 129, 516–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Wang T, Liu H, Lian G, Zhang SY, Wang X & Jiang C (2017) HIF1alpha‐induced glycolysis metabolism is essential to the activation of inflammatory macrophages. Mediators Inflamm. 2017, 9029327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V et al. (2003) HIF‐1alpha is essential for myeloid cell‐mediated inflammation. Cell 112, 645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Werno C, Menrad H, Weigert A, Dehne N, Goerdt S, Schledzewski K, Kzhyshkowska J & Brune B (2010) Knockout of HIF‐1alpha in tumor‐associated macrophages enhances M2 polarization and attenuates their pro‐angiogenic responses. Carcinogenesis 31, 1863–1872. [DOI] [PubMed] [Google Scholar]
- 141. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG & Karin M (2008) NF‐kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF‐1alpha. Nature 453, 807–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Jantsch J, Wiese M, Schodel J, Castiglione K, Glasner J, Kolbe S, Mole D, Schleicher U, Eckardt KU, Hensel M et al. (2011) Toll‐like receptor activation and hypoxia use distinct signaling pathways to stabilize hypoxia‐inducible factor 1alpha (HIF1A) and result in differential HIF1A‐dependent gene expression. J Leukoc Biol 90, 551–562. [DOI] [PubMed] [Google Scholar]
- 143. Wang X, de Carvalho Ribeiro M, Iracheta‐Vellve A, Lowe P, Ambade A, Satishchandran A, Bukong T, Catalano D, Kodys K & Szabo G (2019) Macrophage‐specific hypoxia‐inducible factor‐1alpha contributes to impaired autophagic flux in nonalcoholic steatohepatitis. Hepatology 69, 545–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Wang XA, Zhang R, She ZG, Zhang XF, Jiang DS, Wang T, Gao L, Deng W, Zhang SM, Zhu LH et al. (2014) Interferon regulatory factor 3 constrains IKKbeta/NF‐kappaB signaling to alleviate hepatic steatosis and insulin resistance. Hepatology 59, 870–885. [DOI] [PubMed] [Google Scholar]
- 145. Copple BL, Kaska S & Wentling C (2012) Hypoxia‐inducible factor activation in myeloid cells contributes to the development of liver fibrosis in cholestatic mice. J Pharmacol Exp Ther 341, 307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Mantena SK, Vaughn DP, Andringa KK, Eccleston HB, King AL, Abrams GA, Doeller JE, Kraus DW, Darley‐Usmar VM & Bailey SM (2009) High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochem J 417, 183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Chistiakov DA, Myasoedova VA, Revin VV, Orekhov AN & Bobryshev YV (2018) The impact of interferon‐regulatory factors to macrophage differentiation and polarization into M1 and M2. Immunobiology 223, 101–111. [DOI] [PubMed] [Google Scholar]
- 148. Krausgruber T, Saliba D, Ryzhakov G, Lanfrancotti A, Blazek K & Udalova IA (2010) IRF5 is required for late‐phase TNF secretion by human dendritic cells. Blood 115, 4421–4430. [DOI] [PubMed] [Google Scholar]
- 149. Dalmas E, Toubal A, Alzaid F, Blazek K, Eames HL, Lebozec K, Pini M, Hainault I, Montastier E, Denis RG et al. (2015) Irf5 deficiency in macrophages promotes beneficial adipose tissue expansion and insulin sensitivity during obesity. Nat Med 21, 610–618. [DOI] [PubMed] [Google Scholar]
- 150. Liu J, Cao S, Herman LM & Ma X (2003) Differential regulation of interleukin (IL)‐12 p35 and p40 gene expression and interferon (IFN)‐gamma‐primed IL‐12 production by IFN regulatory factor 1. J Exp Med 198, 1265–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Xu H, Zhu J, Smith S, Foldi J, Zhao B, Chung AY, Outtz H, Kitajewski J, Shi C, Weber S et al. (2012) Notch‐RBP‐J signaling regulates the transcription factor IRF8 to promote inflammatory macrophage polarization. Nat Immunol 13, 642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Langlais D, Barreiro LB & Gros P (2016) The macrophage IRF8/IRF1 regulome is required for protection against infections and is associated with chronic inflammation. J Exp Med 213, 585–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Honma K, Udono H, Kohno T, Yamamoto K, Ogawa A, Takemori T, Kumatori A, Suzuki S, Matsuyama T & Yui K (2005) Interferon regulatory factor 4 negatively regulates the production of proinflammatory cytokines by macrophages in response to LPS. Proc Natl Acad Sci USA 102, 16001–16006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, Miyake T, Matsushita K, Okazaki T, Saitoh T et al. (2010) The Jmjd3‐Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol 11, 936–944. [DOI] [PubMed] [Google Scholar]
- 155. Negishi H, Ohba Y, Yanai H, Takaoka A, Honma K, Yui K, Matsuyama T, Taniguchi T & Honda K (2005) Negative regulation of Toll‐like‐receptor signaling by IRF‐4. Proc Natl Acad Sci USA 102, 15989–15994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Loh CY, Arya A, Naema AF, Wong WF, Sethi G & Looi CY (2019) Signal transducer and activator of transcription (STATs) proteins in cancer and inflammation: functions and therapeutic implication. Front Oncol 9, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Kovarik P, Stoiber D, Novy M & Decker T (1998) Stat1 combines signals derived from IFN‐gamma and LPS receptors during macrophage activation. EMBO J 17, 3660–3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, Kaplan DH, Riley JK, Greenlund AC, Campbell D et al. (1996) Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK‐STAT signaling pathway. Cell 84, 431–442. [DOI] [PubMed] [Google Scholar]
- 159. Hong HJ, Lee JW, Park SS, Kang YJ, Chang SY, Kim KM, Kim JO, Murthy KK, Payne JS, Yoon SK et al. (2000) A humanized anti–4‐1BB monoclonal antibody suppresses antigen‐induced humoral immune response in nonhuman primates. J Immunother 23, 613–621. [DOI] [PubMed] [Google Scholar]
- 160. Lawrence T & Natoli G (2011) Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol 11, 750–761. [DOI] [PubMed] [Google Scholar]
- 161. Dabritz J, Weinhage T, Varga G, Wirth T, Walscheid K, Brockhausen A, Schwarzmaier D, Bruckner M, Ross M, Bettenworth D et al. (2015) Reprogramming of monocytes by GM‐CSF contributes to regulatory immune functions during intestinal inflammation. J Immunol 194, 2424–2438. [DOI] [PubMed] [Google Scholar]
- 162. Szanto A, Balint BL, Nagy ZS, Barta E, Dezso B, Pap A, Szeles L, Poliska S, Oros M, Evans RM et al. (2010) STAT6 transcription factor is a facilitator of the nuclear receptor PPARgamma‐regulated gene expression in macrophages and dendritic cells. Immunity 33, 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I & Akira S (1999) Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity 10, 39–49. [DOI] [PubMed] [Google Scholar]
- 164. Lang R, Patel D, Morris JJ, Rutschman RL & Murray PJ (2002) Shaping gene expression in activated and resting primary macrophages by IL‐10. J Immunol 169, 2253–2263. [DOI] [PubMed] [Google Scholar]
- 165. Yin Z, Ma T, Lin Y, Lu X, Zhang C, Chen S & Jian Z (2018) IL‐6/STAT3 pathway intermediates M1/M2 macrophage polarization during the development of hepatocellular carcinoma. J Cell Biochem 119, 9419–9432. [DOI] [PubMed] [Google Scholar]
- 166. Chawla A, Repa JJ, Evans RM & Mangelsdorf DJ (2001) Nuclear receptors and lipid physiology: opening the X‐files. Science 294, 1866–1870. [DOI] [PubMed] [Google Scholar]
- 167. Evans RM & Mangelsdorf DJ (2014) Nuclear receptors, RXR, and the big bang. Cell 157, 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, Karlsson A, Al‐Lazikani B, Hersey A, Oprea TI et al. (2017) A comprehensive map of molecular drug targets. Nat Rev Drug Discov 16, 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Xiao Y, Kim M, Lazar MA (2020) Nuclear receptors and transcriptional regulation in non‐alcoholic fatty liver disease. Mol Metab 101119. doi: 10.1016/j.molmet.2020.101119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Saponaro F, Sestito S, Runfola M, Rapposelli S & Chiellini G (2020) Selective thyroid hormone receptor‐beta (TRbeta) agonists: new perspectives for the treatment of metabolic and neurodegenerative disorders. Front Med (Lausanne) 7, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Yen PM (2001) Physiological and molecular basis of thyroid hormone action. Physiol Rev 81, 1097–1142. [DOI] [PubMed] [Google Scholar]
- 172. Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y, Soderstrom M, Glass CK et al. (1995) Ligand‐independent repression by the thyroid hormone receptor mediated by a nuclear receptor co‐repressor. Nature 377, 397–404. [DOI] [PubMed] [Google Scholar]
- 173. Xu L, Glass CK & Rosenfeld MG (1999) Coactivator and corepressor complexes in nuclear receptor function. Curr Opin Genet Dev 9, 140–147. [DOI] [PubMed] [Google Scholar]
- 174. Radenne A, Akpa M, Martel C, Sawadogo S, Mauvoisin D & Mounier C (2008) Hepatic regulation of fatty acid synthase by insulin and T3: evidence for T3 genomic and nongenomic actions. Am J Physiol Endocrinol Metab 295, E884–E894. [DOI] [PubMed] [Google Scholar]
- 175. Hashimoto K, Yamada M, Matsumoto S, Monden T, Satoh T & Mori M (2006) Mouse sterol response element binding protein‐1c gene expression is negatively regulated by thyroid hormone. Endocrinology 147, 4292–4302. [DOI] [PubMed] [Google Scholar]
- 176. De Vito P, Incerpi S, Pedersen JZ, Luly P, Davis FB & Davis PJ (2011) Thyroid hormones as modulators of immune activities at the cellular level. Thyroid 21, 879–890. [DOI] [PubMed] [Google Scholar]
- 177. Perrotta C, Buldorini M, Assi E, Cazzato D, De Palma C, Clementi E & Cervia D (2014) The thyroid hormone triiodothyronine controls macrophage maturation and functions: protective role during inflammation. Am J Pathol 184, 230–247. [DOI] [PubMed] [Google Scholar]
- 178. Ortega E, Forner MA, Garcia JJ, Rodriguez AB & Barriga C (1999) Enhanced chemotaxis of macrophages by strenuous exercise in trained mice: thyroid hormones as possible mediators. Mol Cell Biochem 201, 41–47. [DOI] [PubMed] [Google Scholar]
- 179. van der Spek AH, Fliers E & Boelen A (2017) Thyroid hormone metabolism in innate immune cells. J Endocrinol 232, R67–R81. [DOI] [PubMed] [Google Scholar]
- 180. Chen Y, Sjolinder M, Wang X, Altenbacher G, Hagner M, Berglund P, Gao Y, Lu T, Jonsson AB & Sjolinder H (2012) Thyroid hormone enhances nitric oxide‐mediated bacterial clearance and promotes survival after meningococcal infection. PLoS One 7, e41445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Forner MA, Barriga C & Ortega E (1985) (1996) Exercise‐induced stimulation of murine macrophage phagocytosis may be mediated by thyroxine. J Appl Physiol 80, 899–903. [DOI] [PubMed] [Google Scholar]
- 182. Billon C, Canaple L, Fleury S, Deloire A, Beylot M, Dombrowicz D, Del Carmine P, Samarut J & Gauthier K (2014) TRalpha protects against atherosclerosis in male mice: identification of a novel anti‐inflammatory property for TRalpha in mice. Endocrinology 155, 2735–2745. [DOI] [PubMed] [Google Scholar]
- 183. Tapia G, Pepper I, Smok G & Videla LA (1997) Kupffer cell function in thyroid hormone‐induced liver oxidative stress in the rat. Free Radic Res 26, 267–279. [DOI] [PubMed] [Google Scholar]
- 184. Valencia C, Cornejo P, Romanque P, Tapia G, Varela P, Videla LA & Fernandez V (2004) Effects of acute lindane intoxication and thyroid hormone administration in relation to nuclear factor‐kappaB activation, tumor necrosis factor‐alpha expression, and Kupffer cell function in the rat. Toxicol Lett 148, 21–28. [DOI] [PubMed] [Google Scholar]
- 185. Tapia G, Fernandez V, Pino C, Ardiles R & Videla LA (2006) The acute‐phase response of the liver in relation to thyroid hormone‐induced redox signaling. Free Radic Biol Med 40, 1628–1635. [DOI] [PubMed] [Google Scholar]
- 186. Fernandez V, Tapia G, Varela P & Videla LA (2005) Redox regulation of thyroid hormone‐induced Kupffer cell‐dependent IkappaB‐alpha phosphorylation in relation to inducible nitric oxide synthase expression. Free Radic Res 39, 411–418. [DOI] [PubMed] [Google Scholar]
- 187. Contreras‐Jurado C, Alonso‐Merino E, Saiz‐Ladera C, Valino AJ, Regadera J, Alemany S & Aranda A (2016) The thyroid hormone receptors inhibit hepatic interleukin‐6 signaling during endotoxemia. Sci Rep 6, 30990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Vaitkus JA, Farrar JS & Celi FS (2015) Thyroid hormone mediated modulation of energy expenditure. Int J Mol Sci 16, 16158–16175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Gullberg H, Rudling M, Salto C, Forrest D, Angelin B & Vennstrom B (2002) Requirement for thyroid hormone receptor beta in T3 regulation of cholesterol metabolism in mice. Mol Endocrinol 16, 1767–1777. [DOI] [PubMed] [Google Scholar]
- 190. Mantovani A, Nascimbeni F, Lonardo A, Zoppini G, Bonora E, Mantzoros CS & Targher G (2018) Association between primary hypothyroidism and nonalcoholic fatty liver disease: a systematic review and meta‐analysis. Thyroid 28, 1270–1284. [DOI] [PubMed] [Google Scholar]
- 191. Sinha RA, Bruinstroop E, Singh BK & Yen PM (2019) Nonalcoholic fatty liver disease and hypercholesterolemia: roles of thyroid hormones, metabolites, and agonists. Thyroid 29, 1173–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Accorroni A, Saponaro F & Zucchi R (2016) Tissue thyroid hormones and thyronamines. Heart Fail Rev 21, 373–390. [DOI] [PubMed] [Google Scholar]
- 193. Gullberg H, Rudling M, Forrest D, Angelin B & Vennstrom B (2000) Thyroid hormone receptor beta‐deficient mice show complete loss of the normal cholesterol 7alpha‐hydroxylase (CYP7A) response to thyroid hormone but display enhanced resistance to dietary cholesterol. Mol Endocrinol 14, 1739–1749. [DOI] [PubMed] [Google Scholar]
- 194. Johansson C, Vennstrom B & Thoren P (1998) Evidence that decreased heart rate in thyroid hormone receptor‐alpha1‐deficient mice is an intrinsic defect. Am J Physiol 275, R640–R646. [DOI] [PubMed] [Google Scholar]
- 195. Araki O, Ying H, Zhu XG, Willingham MC & Cheng SY (2009) Distinct dysregulation of lipid metabolism by unliganded thyroid hormone receptor isoforms. Mol Endocrinol 23, 308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Zucchi R (2020) Thyroid hormone analogues: an update. Thyroid 30, 1099–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Cable EE, Finn PD, Stebbins JW, Hou J, Ito BR, van Poelje PD, Linemeyer DL & Erion MD (2009) Reduction of hepatic steatosis in rats and mice after treatment with a liver‐targeted thyroid hormone receptor agonist. Hepatology 49, 407–417. [DOI] [PubMed] [Google Scholar]
- 198. Erion MD, Cable EE, Ito BR, Jiang H, Fujitaki JM, Finn PD, Zhang BH, Hou J, Boyer SH, van Poelje PD et al. (2007) Targeting thyroid hormone receptor‐beta agonists to the liver reduces cholesterol and triglycerides and improves the therapeutic index. Proc Natl Acad Sci USA 104, 15490–15495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Hartley MD, Banerji T, Tagge IJ, Kirkemo LL, Chaudhary P, Calkins E, Galipeau D, Shokat MD, DeBell MJ, Van Leuven S et al. (2019) Myelin repair stimulated by CNS‐selective thyroid hormone action. JCI Insight 4. doi: 10.1172/jci.insight.126329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Derosa G, Sahebkar A & Maffioli P (2018) The role of various peroxisome proliferator‐activated receptors and their ligands in clinical practice. J Cell Physiol 233, 153–161. [DOI] [PubMed] [Google Scholar]
- 201. Francque S, Szabo G, Abdelmalek MF, Byrne CD, Cusi K, Dufour JF, Roden M, Sacks F & Tacke F (2021) Nonalcoholic steatohepatitis: the role of peroxisome proliferator‐activated receptors. Nat Rev Gastroenterol Hepatol 18, 24–39. [DOI] [PubMed] [Google Scholar]
- 202. Dreyer C, Krey G, Keller H, Givel F, Helftenbein G & Wahli W (1992) Control of the peroxisomal beta‐oxidation pathway by a novel family of nuclear hormone receptors. Cell 68, 879–887. [DOI] [PubMed] [Google Scholar]
- 203. Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, Grimaldi PA, Kadowaki T, Lazar MA, O'Rahilly S et al. (2006) International Union of Pharmacology. LXI. Peroxisome proliferator‐activated receptors. Pharmacol Rev 58, 726–741. [DOI] [PubMed] [Google Scholar]
- 204. Wanders RJ & Waterham HR (2006) Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem 75, 295–332. [DOI] [PubMed] [Google Scholar]
- 205. Kersten S & Stienstra R (2017) The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie 136, 75–84. [DOI] [PubMed] [Google Scholar]
- 206. Xu HE, Lambert MH, Montana VG, Plunket KD, Moore LB, Collins JL, Oplinger JA, Kliewer SA, Gampe RT Jr, McKee DD et al. (2001) Structural determinants of ligand binding selectivity between the peroxisome proliferator‐activated receptors. Proc Natl Acad Sci USA 98, 13919–13924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Braissant O, Foufelle F, Scotto C, Dauca M & Wahli W (1996) Differential expression of peroxisome proliferator‐activated receptors (PPARs): tissue distribution of PPAR‐alpha, ‐beta, and ‐gamma in the adult rat. Endocrinology 137, 354–366. [DOI] [PubMed] [Google Scholar]
- 208. Zardi EM, Navarini L, Sambataro G, Piccinni P, Sambataro FM, Spina C & Dobrina A (2013) Hepatic PPARs: their role in liver physiology, fibrosis and treatment. Curr Med Chem 20, 3370–3396. [DOI] [PubMed] [Google Scholar]
- 209. Chakravarthy MV, Lodhi IJ, Yin L, Malapaka RR, Xu HE, Turk J & Semenkovich CF (2009) Identification of a physiologically relevant endogenous ligand for PPARalpha in liver. Cell 138, 476–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Pawlak M, Bauge E, Bourguet W, De Bosscher K, Lalloyer F, Tailleux A, Lebherz C, Lefebvre P & Staels B (2014) The transrepressive activity of peroxisome proliferator‐activated receptor alpha is necessary and sufficient to prevent liver fibrosis in mice. Hepatology 60, 1593–1606. [DOI] [PubMed] [Google Scholar]
- 211. Bougarne N, Paumelle R, Caron S, Hennuyer N, Mansouri R, Gervois P, Staels B, Haegeman G & De Bosscher K (2009) PPARalpha blocks glucocorticoid receptor alpha‐mediated transactivation but cooperates with the activated glucocorticoid receptor alpha for transrepression on NF‐kappaB. Proc Natl Acad Sci USA 106, 7397–7402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Guan D, Xiong Y, Borck PC, Jang C, Doulias PT, Papazyan R, Fang B, Jiang C, Zhang Y, Briggs ER et al. (2018) Diet‐induced circadian enhancer remodeling synchronizes opposing hepatic lipid metabolic processes. Cell 174, 831–842.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Chen L & Yang G (2014) PPARs integrate the mammalian Clock and energy metabolism. PPAR Res 2014, 653017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Gervois P, Vu‐Dac N, Kleemann R, Kockx M, Dubois G, Laine B, Kosykh V, Fruchart JC, Kooistra T & Staels B (2001) Negative regulation of human fibrinogen gene expression by peroxisome proliferator‐activated receptor alpha agonists via inhibition of CCAAT box/enhancer‐binding protein beta. J Biol Chem 276, 33471–33477. [DOI] [PubMed] [Google Scholar]
- 215. Bougarne N, Weyers B, Desmet SJ, Deckers J, Ray DW, Staels B & De Bosscher K (2018) Molecular actions of PPARalpha in lipid metabolism and inflammation. Endocr Rev 39, 760–802. [DOI] [PubMed] [Google Scholar]
- 216. Patsouris D, Reddy JK, Muller M & Kersten S (2006) Peroxisome proliferator‐activated receptor alpha mediates the effects of high‐fat diet on hepatic gene expression. Endocrinology 147, 1508–1516. [DOI] [PubMed] [Google Scholar]
- 217. Montagner A, Polizzi A, Fouche E, Ducheix S, Lippi Y, Lasserre F, Barquissau V, Regnier M, Lukowicz C, Benhamed F et al. (2016) Liver PPARalpha is crucial for whole‐body fatty acid homeostasis and is protective against NAFLD. Gut 65, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Botta M, Audano M, Sahebkar A, Sirtori C, Mitro N, Ruscica M (2018) PPAR agonists and metabolic syndrome: an established role?. Int J Mol Sci 19, 1197. doi: 10.3390/ijms19041197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Pawlak M, Lefebvre P & Staels B (2015) Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non‐alcoholic fatty liver disease. J Hepatol 62, 720–733. [DOI] [PubMed] [Google Scholar]
- 220. Rakhshandehroo M, Hooiveld G, Muller M & Kersten S (2009) Comparative analysis of gene regulation by the transcription factor PPARalpha between mouse and human. PLoS One 4, e6796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Francque S, Verrijken A, Caron S, Prawitt J, Paumelle R, Derudas B, Lefebvre P, Taskinen MR, Van Hul W, Mertens I et al. (2015) PPARalpha gene expression correlates with severity and histological treatment response in patients with non‐alcoholic steatohepatitis. J Hepatol 63, 164–173. [DOI] [PubMed] [Google Scholar]
- 222. Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E & Fruchart JC (1998) Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 98, 2088–2093. [DOI] [PubMed] [Google Scholar]
- 223. Rosenson RS (2008) Fenofibrate: treatment of hyperlipidemia and beyond. Expert Rev Cardiovasc Ther 6, 1319–1330. [DOI] [PubMed] [Google Scholar]
- 224. Belfort R, Berria R, Cornell J & Cusi K (2010) Fenofibrate reduces systemic inflammation markers independent of its effects on lipid and glucose metabolism in patients with the metabolic syndrome. J Clin Endocrinol Metab 95, 829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Fabbrini E, Mohammed BS, Korenblat KM, Magkos F, McCrea J, Patterson BW & Klein S (2010) Effect of fenofibrate and niacin on intrahepatic triglyceride content, very low‐density lipoprotein kinetics, and insulin action in obese subjects with nonalcoholic fatty liver disease. J Clin Endocrinol Metab 95, 2727–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Laurin J, Lindor KD, Crippin JS, Gossard A, Gores GJ, Ludwig J, Rakela J & McGill DB (1996) Ursodeoxycholic acid or clofibrate in the treatment of non‐alcohol‐induced steatohepatitis: a pilot study. Hepatology 23, 1464–1467. [DOI] [PubMed] [Google Scholar]
- 227. Fernandez‐Miranda C, Perez‐Carreras M, Colina F, Lopez‐Alonso G, Vargas C & Solis‐Herruzo JA (2008) A pilot trial of fenofibrate for the treatment of non‐alcoholic fatty liver disease. Dig Liver Dis 40, 200–205. [DOI] [PubMed] [Google Scholar]
- 228. Basaranoglu M, Acbay O & Sonsuz A (1999) A controlled trial of gemfibrozil in the treatment of patients with nonalcoholic steatohepatitis. J Hepatol 31, 384. [DOI] [PubMed] [Google Scholar]
- 229. Araki E, Yamashita S, Arai H, Yokote K, Satoh J, Inoguchi T, Nakamura J, Maegawa H, Yoshioka N, Tanizawa Y et al. (2019) Efficacy and safety of pemafibrate in people with type 2 diabetes and elevated triglyceride levels: 52‐week data from the PROVIDE study. Diabetes Obes Metab 21, 1737–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Yokote K, Yamashita S, Arai H, Araki E, Suganami H, Ishibashi S & on behalf of the K‐Study Group (2019) Long‐term efficacy and safety of pemafibrate, a novel selective peroxisome proliferator‐activated receptor‐alpha modulator (SPPARMalpha), in dyslipidemic patients with renal impairment. Int J Mol Sci 20, 706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, Romero‐Gomez M, Boursier J, Abdelmalek M, Caldwell S et al. (2016) Elafibranor, an agonist of the peroxisome proliferator‐activated receptor‐alpha and ‐delta, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 150, 1147–1159.e5. [DOI] [PubMed] [Google Scholar]
- 232. Cariou B, Hanf R, Lambert‐Porcheron S, Zair Y, Sauvinet V, Noel B, Flet L, Vidal H, Staels B & Laville M (2013) Dual peroxisome proliferator‐activated receptor alpha/delta agonist GFT505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care 36, 2923–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. ClinicalTrials.gov [Internet]. National Library of Medicine (US), Bethesda, MD. 2000 Feb 29. Identifier NCT02704403, P. S. t. E. t. E. a. S. o. & https://clinicaltrials.gov/ct2/show/study/NCT02704403, c. F. A. f. in [Google Scholar]
- 234. Eichenbaum N & Lavin H (2020) GENFIT: Announces Results from Interim Analysis of RESOLVE‐IT Phase 3 Trial of Elafibranor in Adults with NASH and Fibrosis, pp. 1–5.GENFIT, Loos. [Google Scholar]
- 235. Schattenberg JM, Pares A, Kowdley KV, Heneghan MA, Caldwell S, Pratt D, Bonder A, Hirschfield GM, Levy C, Vierling J et al. (2021) A randomized placebo‐controlled trial of elafibranor in patients with primary biliary cholangitis and incomplete response to UDCA. J Hepatol. doi: 10.1016/j.jhep.2021.01.013 [DOI] [PubMed] [Google Scholar]
- 236. Tailleux A, Wouters K & Staels B (2012) Roles of PPARs in NAFLD: potential therapeutic targets. Biochim Biophys Acta 1821, 809–818. [DOI] [PubMed] [Google Scholar]
- 237. Sanderson LM, Boekschoten MV, Desvergne B, Muller M & Kersten S (2010) Transcriptional profiling reveals divergent roles of PPARalpha and PPARbeta/delta in regulation of gene expression in mouse liver. Physiol Genomics 41, 42–52. [DOI] [PubMed] [Google Scholar]
- 238. Liu S, Hatano B, Zhao M, Yen CC, Kang K, Reilly SM, Gangl MR, Gorgun C, Balschi JA, Ntambi JM et al. (2011) Role of peroxisome proliferator‐activated receptor delta}/{beta in hepatic metabolic regulation. J Biol Chem 286, 1237–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Liu S, Brown JD, Stanya KJ, Homan E, Leidl M, Inouye K, Bhargava P, Gangl MR, Dai L, Hatano B et al. (2013) A diurnal serum lipid integrates hepatic lipogenesis and peripheral fatty acid use. Nature 502, 550–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Lefere S & Tacke F (2019) Macrophages in obesity and non‐alcoholic fatty liver disease: crosstalk with metabolism. JHEP Rep 1, 30–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Fan Y, Wang Y, Tang Z, Zhang H, Qin X, Zhu Y, Guan Y, Wang X, Staels B, Chien S et al. (2008) Suppression of pro‐inflammatory adhesion molecules by PPAR‐delta in human vascular endothelial cells. Arterioscler Thromb Vasc Biol 28, 315–321. [DOI] [PubMed] [Google Scholar]
- 242. Kilgore KS & Billin AN (2008) PPARbeta/delta ligands as modulators of the inflammatory response. Curr Opin Investig Drugs 9, 463–469. [PubMed] [Google Scholar]
- 243. Ham SA, Hwang JS, Yoo T, Lee WJ, Paek KS, Oh JW, Park CK, Kim JH, Do JT, Kim JH et al. (2015) Ligand‐activated PPARdelta upregulates alpha‐smooth muscle actin expression in human dermal fibroblasts: a potential role for PPARdelta in wound healing. J Dermatol Sci 80, 186–195. [DOI] [PubMed] [Google Scholar]
- 244. Park JR, Ahn JH, Jung MH, Koh JS, Park Y, Hwang SJ, Jeong YH, Kwak CH, Lee YS, Seo HG et al. (2016) Effects of peroxisome proliferator‐activated receptor‐delta agonist on cardiac healing after myocardial infarction. PLoS One 11, e0148510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Iwaisako K, Haimerl M, Paik YH, Taura K, Kodama Y, Sirlin C, Yu E, Yu RT, Downes M, Evans RM et al. (2012) Protection from liver fibrosis by a peroxisome proliferator‐activated receptor delta agonist. Proc Natl Acad Sci USA 109, E1369–E1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Choi YJ, Roberts BK, Wang X, Geaney JC, Naim S, Wojnoonski K, Karpf DB & Krauss RM (2012) Effects of the PPAR‐delta agonist MBX‐8025 on atherogenic dyslipidemia. Atherosclerosis 220, 470–476. [DOI] [PubMed] [Google Scholar]
- 247. Pott S, Kamrani NK, Bourque G, Pettersson S & Liu ET (2012) PPARG binding landscapes in macrophages suggest a genome‐wide contribution of PU.1 to divergent PPARG binding in human and mouse. PLoS One 7, e48102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Lefterova MI, Steger DJ, Zhuo D, Qatanani M, Mullican SE, Tuteja G, Manduchi E, Grant GR & Lazar MA (2010) Cell‐specific determinants of peroxisome proliferator‐activated receptor gamma function in adipocytes and macrophages. Mol Cell Biol 30, 2078–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Vidal‐Puig A, Jimenez‐Linan M, Lowell BB, Hamann A, Hu E, Spiegelman B, Flier JS & Moller DE (1996) Regulation of PPAR gamma gene expression by nutrition and obesity in rodents. J Clin Invest 97, 2553–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Moran‐Salvador E, Lopez‐Parra M, Garcia‐Alonso V, Titos E, Martinez‐Clemente M, Gonzalez‐Periz A, Lopez‐Vicario C, Barak Y, Arroyo V & Claria J (2011) Role for PPARgamma in obesity‐induced hepatic steatosis as determined by hepatocyte‐ and macrophage‐specific conditional knockouts. FASEB J 25, 2538–2550. [DOI] [PubMed] [Google Scholar]
- 251. Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, Ward JM, Brewer B Jr, Reitman ML & Gonzalez FJ (2003) Liver‐specific disruption of PPARgamma in leptin‐deficient mice improves fatty liver but aggravates diabetic phenotypes. J Clin Invest 111, 737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Skat‐Rordam J, Hojland Ipsen D, Lykkesfeldt J & Tveden‐Nyborg P (2019) A role of peroxisome proliferator‐activated receptor gamma in non‐alcoholic fatty liver disease. Basic Clin Pharmacol Toxicol 124, 528–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Ricote M & Glass CK (2007) PPARs and molecular mechanisms of transrepression. Biochim Biophys Acta 1771, 926–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG & Glass CK (2005) A SUMOylation‐dependent pathway mediates transrepression of inflammatory response genes by PPAR‐gamma. Nature 437, 759–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Zizzo G & Cohen PL (2015) The PPAR‐gamma antagonist GW9662 elicits differentiation of M2c‐like cells and upregulation of the MerTK/Gas6 axis: a key role for PPAR‐gamma in human macrophage polarization. J Inflamm (Lond) 12, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Miyahara T, Schrum L, Rippe R, Xiong S, Yee HF Jr, Motomura K, Anania FA, Willson TM & Tsukamoto H (2000) Peroxisome proliferator‐activated receptors and hepatic stellate cell activation. J Biol Chem 275, 35715–35722. [DOI] [PubMed] [Google Scholar]
- 257. Liu X, Xu J, Rosenthal S, Zhang LJ, McCubbin R, Meshgin N, Shang L, Koyama Y, Ma HY, Sharma S et al. (2020) Identification of lineage‐specific transcription factors that prevent activation of hepatic stellate cells and promote fibrosis resolution. Gastroenterology 158, 1728–1744.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Liss KH & Finck BN (2017) PPARs and nonalcoholic fatty liver disease. Biochimie 136, 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Ratziu V, Giral P, Jacqueminet S, Charlotte F, Hartemann–Heurtier A, Serfaty L, Podevin P, Lacorte JM, Bernhardt C, Bruckert E et al. (2008) Rosiglitazone for nonalcoholic steatohepatitis: one‐year results of the randomized placebo‐controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 135, 100–110. [DOI] [PubMed] [Google Scholar]
- 260. Pettinelli P & Videla LA (2011) Up‐regulation of PPAR‐gamma mRNA expression in the liver of obese patients: an additional reinforcing lipogenic mechanism to SREBP‐1c induction. J Clin Endocrinol Metab 96, 1424–1430. [DOI] [PubMed] [Google Scholar]
- 261. Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, Balas B, Gastaldelli A, Tio F, Pulcini J et al. (2006) A placebo‐controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med 355, 2297–2307. [DOI] [PubMed] [Google Scholar]
- 262. Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz‐Lopez C, Tio F, Hardies J, Darland C, Musi N et al. (2016) Long‐term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: a randomized trial. Ann Intern Med 165, 305–315. [DOI] [PubMed] [Google Scholar]
- 263. Harrison SA, Alkhouri N, Davison BA, Sanyal A, Edwards C, Colca JR, Lee BH, Loomba R, Cusi K, Kolterman O et al. (2020) Insulin sensitizer MSDC‐0602K in non‐alcoholic steatohepatitis: a randomized, double‐blind, placebo‐controlled phase IIb study. J Hepatol 72, 613–626. [DOI] [PubMed] [Google Scholar]
- 264. Wilding JP (2012) PPAR agonists for the treatment of cardiovascular disease in patients with diabetes. Diabetes Obes Metab 14, 973–982. [DOI] [PubMed] [Google Scholar]
- 265. Jani RH, Pai V, Jha P, Jariwala G, Mukhopadhyay S, Bhansali A & Joshi S (2014) A multicenter, prospective, randomized, double‐blind study to evaluate the safety and efficacy of Saroglitazar 2 and 4 mg compared with placebo in type 2 diabetes mellitus patients having hypertriglyceridemia not controlled with atorvastatin therapy (PRESS VI). Diabetes Technol Ther 16, 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Jain MR, Giri SR, Bhoi B, Trivedi C, Rath A, Rathod R, Ranvir R, Kadam S, Patel H, Swain P et al. (2018) Dual PPARalpha/gamma agonist saroglitazar improves liver histopathology and biochemistry in experimental NASH models. Liver Int 38, 1084–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Wettstein G, Luccarini JM, Poekes L, Faye P, Kupkowski F, Adarbes V, Defrene E, Estivalet C, Gawronski X, Jantzen I et al. (2017) The new‐generation pan‐peroxisome proliferator‐activated receptor agonist IVA337 protects the liver from metabolic disorders and fibrosis. Hepatol Commun 1, 524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Boubia B, Poupardin O, Barth M, Binet J, Peralba P, Mounier L, Jacquier E, Gauthier E, Lepais V, Chatar M et al. (2018) Design, synthesis, and evaluation of a novel series of indole sulfonamide peroxisome proliferator activated receptor (PPAR) alpha/gamma/delta triple activators: discovery of lanifibranor, a new antifibrotic clinical candidate. J Med Chem 61, 2246–2265. [DOI] [PubMed] [Google Scholar]
- 269. Ruzehaji N, Frantz C, Ponsoye M, Avouac J, Pezet S, Guilbert T, Luccarini JM, Broqua P, Junien JL & Allanore Y (2016) Pan PPAR agonist IVA337 is effective in prevention and treatment of experimental skin fibrosis. Ann Rheum Dis 75, 2175–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Avouac J, Konstantinova I, Guignabert C, Pezet S, Sadoine J, Guilbert T, Cauvet A, Tu L, Luccarini JM, Junien JL et al. (2017) Pan‐PPAR agonist IVA337 is effective in experimental lung fibrosis and pulmonary hypertension. Ann Rheum Dis 76, 1931–1940. [DOI] [PubMed] [Google Scholar]
- 271. Lefere S, Puengel T, Hundertmark J, Penners C, Frank AK, Guillot A, de Muynck K, Heymann F, Adarbes V, Defrene E et al. (2020) Differential effects of selective‐ and pan‐PPAR agonists on experimental steatohepatitis and hepatic macrophages. J Hepatol 73, 757–770. [DOI] [PubMed] [Google Scholar]
- 272. Cren F (2020) Inventiva's lanifibranor meets the primary and key secondary endpoints in the Phase IIb NATIVE clinical trial in non‐alcoholic steatohepatitis (NASH). Inventiva Pharma. [Google Scholar]
- 273. Aguilar‐Olivos NE, Carrillo‐Cordova D, Oria‐Hernandez J, Sanchez‐Valle V, Ponciano‐Rodriguez G, Ramirez‐Jaramillo M, Chable‐Montero F, Chavez‐Tapia NC, Uribe M & Mendez‐Sanchez N (2015) The nuclear receptor FXR, but not LXR, up‐regulates bile acid transporter expression in non‐alcoholic fatty liver disease. Ann Hepatol 14, 487–493. [PubMed] [Google Scholar]
- 274. Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, Stimmel JB, Willson TM, Zavacki AM, Moore DD et al. (1999) Bile acids: natural ligands for an orphan nuclear receptor. Science 284, 1365–1368. [DOI] [PubMed] [Google Scholar]
- 275. Wang H, Chen J, Hollister K, Sowers LC & Forman BM (1999) Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell 3, 543–553. [DOI] [PubMed] [Google Scholar]
- 276. Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ & Shan B (1999) Identification of a nuclear receptor for bile acids. Science 284, 1362–1365. [DOI] [PubMed] [Google Scholar]
- 277. Stofan M & Guo GL (2020) Bile acids and FXR: novel targets for liver diseases. Front Med (Lausanne) 7, 544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Laffitte BA, Kast HR, Nguyen CM, Zavacki AM, Moore DD & Edwards PA (2000) Identification of the DNA binding specificity and potential target genes for the farnesoid X‐activated receptor. J Biol Chem 275, 10638–10647. [DOI] [PubMed] [Google Scholar]
- 279. Chong HK, Infante AM, Seo YK, Jeon TI, Zhang Y, Edwards PA, Xie X & Osborne TF (2010) Genome‐wide interrogation of hepatic FXR reveals an asymmetric IR‐1 motif and synergy with LRH‐1. Nucleic Acids Res 38, 6007–6017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Chong HK, Biesinger J, Seo YK, Xie X & Osborne TF (2012) Genome‐wide analysis of hepatic LRH‐1 reveals a promoter binding preference and suggests a role in regulating genes of lipid metabolism in concert with FXR. BMC Genom 13, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Li L, Zhang Q, Peng J, Jiang C, Zhang Y, Shen L, Dong J, Wang Y & Jiang Y (2015) Activation of farnesoid X receptor downregulates monocyte chemoattractant protein‐1 in murine macrophage. Biochem Biophys Res Commun 467, 841–846. [DOI] [PubMed] [Google Scholar]
- 282. Claudel T, Sturm E, Duez H, Torra IP, Sirvent A, Kosykh V, Fruchart JC, Dallongeville J, Hum DW, Kuipers F et al. (2002) Bile acid‐activated nuclear receptor FXR suppresses apolipoprotein A‐I transcription via a negative FXR response element. J Clin Invest 109, 961–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Barbier O, Torra IP, Sirvent A, Claudel T, Blanquart C, Duran‐Sandoval D, Kuipers F, Kosykh V, Fruchart JC & Staels B (2003) FXR induces the UGT2B4 enzyme in hepatocytes: a potential mechanism of negative feedback control of FXR activity. Gastroenterology 124, 1926–1940. [DOI] [PubMed] [Google Scholar]
- 284. Chennamsetty I, Claudel T, Kostner KM, Baghdasaryan A, Kratky D, Levak‐Frank S, Frank S, Gonzalez FJ, Trauner M & Kostner GM (2011) Farnesoid X receptor represses hepatic human APOA gene expression. J Clin Invest 121, 3724–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Kim I, Ahn SH, Inagaki T, Choi M, Ito S, Guo GL, Kliewer SA & Gonzalez FJ (2007) Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res 48, 2664–2672. [DOI] [PubMed] [Google Scholar]
- 286. Plass JR, Mol O, Heegsma J, Geuken M, Faber KN, Jansen PL & Muller M (2002) Farnesoid X receptor and bile salts are involved in transcriptional regulation of the gene encoding the human bile salt export pump. Hepatology 35, 589–596. [DOI] [PubMed] [Google Scholar]
- 287. Landrier JF, Eloranta JJ, Vavricka SR & Kullak‐Ublick GA (2006) The nuclear receptor for bile acids, FXR, transactivates human organic solute transporter‐alpha and ‐beta genes. Am J Physiol Gastrointest Liver Physiol 290, G476–G485. [DOI] [PubMed] [Google Scholar]
- 288. Zollner G, Wagner M, Moustafa T, Fickert P, Silbert D, Gumhold J, Fuchsbichler A, Halilbasic E, Denk H, Marschall HU et al. (2006) Coordinated induction of bile acid detoxification and alternative elimination in mice: role of FXR‐regulated organic solute transporter‐alpha/beta in the adaptive response to bile acids. Am J Physiol Gastrointest Liver Physiol 290, G923–G932. [DOI] [PubMed] [Google Scholar]
- 289. Denson LA, Sturm E, Echevarria W, Zimmerman TL, Makishima M, Mangelsdorf DJ & Karpen SJ (2001) The orphan nuclear receptor, shp, mediates bile acid‐induced inhibition of the rat bile acid transporter, ntcp. Gastroenterology 121, 140–147. [DOI] [PubMed] [Google Scholar]
- 290. Wang L, Lee YK, Bundman D, Han Y, Thevananther S, Kim CS, Chua SS, Wei P, Heyman RA, Karin M et al. (2002) Redundant pathways for negative feedback regulation of bile acid production. Dev Cell 2, 721–731. [DOI] [PubMed] [Google Scholar]
- 291. Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, Moore DD & Auwerx J (2004) Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP‐1c. J Clin Invest 113, 1408–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Schoenfield LJ & Lachin JM (1981) Chenodiol (chenodeoxycholic acid) for dissolution of gallstones: the National Cooperative Gallstone Study. A controlled trial of efficacy and safety. Ann Intern Med 95, 257–282. [DOI] [PubMed] [Google Scholar]
- 293. Prieur X, Coste H & Rodriguez JC (2003) The human apolipoprotein AV gene is regulated by peroxisome proliferator‐activated receptor‐alpha and contains a novel farnesoid X‐activated receptor response element. J Biol Chem 278, 25468–25480. [DOI] [PubMed] [Google Scholar]
- 294. Kast HR, Nguyen CM, Sinal CJ, Jones SA, Laffitte BA, Reue K, Gonzalez FJ, Willson TM & Edwards PA (2001) Farnesoid X‐activated receptor induces apolipoprotein C‐II transcription: a molecular mechanism linking plasma triglyceride levels to bile acids. Mol Endocrinol 15, 1720–1728. [DOI] [PubMed] [Google Scholar]
- 295. Claudel T, Inoue Y, Barbier O, Duran‐Sandoval D, Kosykh V, Fruchart J, Fruchart JC, Gonzalez FJ & Staels B (2003) Farnesoid X receptor agonists suppress hepatic apolipoprotein CIII expression. Gastroenterology 125, 544–555. [DOI] [PubMed] [Google Scholar]
- 296. Pineda Torra I, Claudel T, Duval C, Kosykh V, Fruchart JC & Staels B (2003) Bile acids induce the expression of the human peroxisome proliferator‐activated receptor alpha gene via activation of the farnesoid X receptor. Mol Endocrinol 17, 259–272. [DOI] [PubMed] [Google Scholar]
- 297. Thomas AM, Hart SN, Kong B, Fang J, Zhong XB & Guo GL (2010) Genome‐wide tissue‐specific farnesoid X receptor binding in mouse liver and intestine. Hepatology 51, 1410–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Zhan L, Liu HX, Fang Y, Kong B, He Y, Zhong XB, Fang J, Wan YJ & Guo GL (2014) Genome‐wide binding and transcriptome analysis of human farnesoid X receptor in primary human hepatocytes. PLoS One 9, e105930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Ma K, Saha PK, Chan L & Moore DD (2006) Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest 116, 1102–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Zhang Y, Lee FY, Barrera G, Lee H, Vales C, Gonzalez FJ, Willson TM & Edwards PA (2006) Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci USA 103, 1006–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Wang YD, Chen WD, Wang M, Yu D, Forman BM & Huang W (2008) Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 48, 1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Zhang S, Wang J, Liu Q & Harnish DC (2009) Farnesoid X receptor agonist WAY‐362450 attenuates liver inflammation and fibrosis in murine model of non‐alcoholic steatohepatitis. J Hepatol 51, 380–388. [DOI] [PubMed] [Google Scholar]
- 303. Fiorucci S & Distrutti E (2015) Bile acid‐activated receptors, intestinal microbiota, and the treatment of metabolic disorders. Trends Mol Med 21, 702–714. [DOI] [PubMed] [Google Scholar]
- 304. Campbell C, McKenney PT, Konstantinovsky D, Isaeva OI, Schizas M, Verter J, Mai C, Jin WB, Guo CJ, Violante S et al. (2020) Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 581, 475–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Hang S, Paik D, Yao L, Kim E, Trinath J, Lu J, Ha S, Nelson BN, Kelly SP, Wu L et al. (2019) Bile acid metabolites control TH17 and Treg cell differentiation. Nature 576, 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Vavassori P, Mencarelli A, Renga B, Distrutti E & Fiorucci S (2009) The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol 183, 6251–6261. [DOI] [PubMed] [Google Scholar]
- 307. Renga B, Mencarelli A, Cipriani S, D'Amore C, Carino A, Bruno A, Francisci D, Zampella A, Distrutti E & Fiorucci S (2013) The bile acid sensor FXR is required for immune‐regulatory activities of TLR‐9 in intestinal inflammation. PLoS One 8, e54472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Wildenberg ME & van den Brink GR (2011) FXR activation inhibits inflammation and preserves the intestinal barrier in IBD. Gut 60, 432–433. [DOI] [PubMed] [Google Scholar]
- 309. Mencarelli A, Renga B, Migliorati M, Cipriani S, Distrutti E, Santucci L & Fiorucci S (2009) The bile acid sensor farnesoid X receptor is a modulator of liver immunity in a rodent model of acute hepatitis. J Immunol 183, 6657–6666. [DOI] [PubMed] [Google Scholar]
- 310. McMahan RH, Wang XX, Cheng LL, Krisko T, Smith M, El Kasmi K, Pruzanski M, Adorini L, Golden‐Mason L, Levi M et al. (2013) Bile acid receptor activation modulates hepatic monocyte activity and improves nonalcoholic fatty liver disease. J Biol Chem 288, 11761–11770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Carino A, Cipriani S, Marchiano S, Biagioli M, Santorelli C, Donini A, Zampella A, Monti MC & Fiorucci S (2017) BAR502, a dual FXR and GPBAR1 agonist, promotes browning of white adipose tissue and reverses liver steatosis and fibrosis. Sci Rep 7, 42801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Hogenauer K, Arista L, Schmiedeberg N, Werner G, Jaksche H, Bouhelal R, Nguyen DG, Bhat BG, Raad L, Rauld C et al. (2014) G‐protein‐coupled bile acid receptor 1 (GPBAR1, TGR5) agonists reduce the production of proinflammatory cytokines and stabilize the alternative macrophage phenotype. J Med Chem 57, 10343–10354. [DOI] [PubMed] [Google Scholar]
- 313. Fiorucci S, Rizzo G, Antonelli E, Renga B, Mencarelli A, Riccardi L, Morelli A, Pruzanski M & Pellicciari R (2005) Cross‐talk between farnesoid‐X‐receptor (FXR) and peroxisome proliferator‐activated receptor gamma contributes to the antifibrotic activity of FXR ligands in rodent models of liver cirrhosis. J Pharmacol Exp Ther 315, 58–68. [DOI] [PubMed] [Google Scholar]
- 314. Renga B, Mencarelli A, Migliorati M, Cipriani S, D'Amore C, Distrutti E & Fiorucci S (2011) SHP‐dependent and ‐independent induction of peroxisome proliferator‐activated receptor‐gamma by the bile acid sensor farnesoid X receptor counter‐regulates the pro‐inflammatory phenotype of liver myofibroblasts. Inflamm Res 60, 577–587. [DOI] [PubMed] [Google Scholar]
- 315. Chapman RW & Lynch KD (2020) Obeticholic acid‐a new therapy in PBC and NASH. Br Med Bull 133, 95–104. [DOI] [PubMed] [Google Scholar]
- 316. Neuschwander‐Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, Chalasani N, Dasarathy S, Diehl AM, Hameed B et al. (2015) Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet 385, 956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Hameed B, Terrault NA, Gill RM, Loomba R, Chalasani N, Hoofnagle JH, Van Natta ML & Nash CRN (2018) Clinical and metabolic effects associated with weight changes and obeticholic acid in non‐alcoholic steatohepatitis. Aliment Pharmacol Ther 47, 645–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Ratziu V, Sanyal AJ, Loomba R, Rinella M, Harrison S, Anstee QM, Goodman Z, Bedossa P, MacConell L, Shringarpure R et al. (2019) REGENERATE: Design of a pivotal, randomised, phase 3 study evaluating the safety and efficacy of obeticholic acid in patients with fibrosis due to nonalcoholic steatohepatitis. Contemp Clin Trials 84, 105803. [DOI] [PubMed] [Google Scholar]
- 319.(2020) Intercept's NASH hopes dashed. Nat Biotechnol 38, 911. [DOI] [PubMed] [Google Scholar]
- 320. Trauner M, Gulamhusein A, Hameed B, Caldwell S, Shiffman ML, Landis C, Eksteen B, Agarwal K, Muir A, Rushbrook S et al. (2019) The nonsteroidal farnesoid X receptor agonist cilofexor (GS‐9674) improves markers of cholestasis and liver injury in patients with primary sclerosing cholangitis. Hepatology 70, 788–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Patel K, Harrison SA, Elkhashab M, Trotter JF, Herring R, Rojter SE, Kayali Z, Wong VW, Greenbloom S, Jayakumar S et al. (2020) Cilofexor, a nonsteroidal FXR agonist, in patients with noncirrhotic NASH: a phase 2 randomized controlled trial. Hepatology 72, 58–71. [DOI] [PubMed] [Google Scholar]
- 322. Hernandez ED, Zheng L, Kim Y, Fang B, Liu B, Valdez RA, Dietrich WF, Rucker PV, Chianelli D, Schmeits J et al. (2019) Tropifexor‐mediated abrogation of steatohepatitis and fibrosis is associated with the antioxidative gene expression profile in rodents. Hepatol Commun 3, 1085–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Orliaguet L, Dalmas E, Drareni K, Venteclef N & Alzaid F (2020) Mechanisms of macrophage polarization in insulin signaling and sensitivity. Front Endocrinol (Lausanne) 11, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Eaton RP, Allen RC & Schade DS (1983) Hepatic removal of insulin in normal man: dose response to endogenous insulin secretion. J Clin Endocrinol Metab 56, 1294–1300. [DOI] [PubMed] [Google Scholar]
- 325. Polonsky KS & Rubenstein AH (1984) C‐peptide as a measure of the secretion and hepatic extraction of insulin. Pitfalls and limitations. Diabetes 33, 486–494. [DOI] [PubMed] [Google Scholar]
- 326. Song SH, McIntyre SS, Shah H, Veldhuis JD, Hayes PC & Butler PC (2000) Direct measurement of pulsatile insulin secretion from the portal vein in human subjects. J Clin Endocrinol Metab 85, 4491–4499. [DOI] [PubMed] [Google Scholar]
- 327. Ieronymaki E, Daskalaki MG, Lyroni K & Tsatsanis C (2019) Insulin signaling and insulin resistance facilitate trained immunity in macrophages through metabolic and epigenetic changes. Front Immunol 10, 1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Artyomov MN & Van den Bossche J (2020) Immunometabolism in the Single‐Cell Era. Cell Metab 32, 710–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Bedossa P, Poitou C, Veyrie N, Bouillot JL, Basdevant A, Paradis V, Tordjman J & Clement K (2012) Histopathological algorithm and scoring system for evaluation of liver lesions in morbidly obese patients. Hepatology 56, 1751–1759. [DOI] [PubMed] [Google Scholar]
- 330. Poynard T, Peta V, Deckmyn O, Pais R, Ngo Y, Charlotte F, Ngo A, Munteanu M, Imbert‐Bismut F, Monneret D et al. (2020) Performance of liver biomarkers, in patients at risk of nonalcoholic steato‐hepatitis, according to presence of type‐2 diabetes. Eur J Gastroenterol Hepatol 32, 998–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. López‐Sánchez G, Dóminguez‐Pérez M, Uribe M, Chávez‐Tapia NC, Nuño‐Lámbarri N (2021) Non‐alcoholic fatty liver disease and microRNAs expression, how it affects the development and progression of the disease. Ann Hepatol 21, 100212. doi: 10.1016/j.aohep.2020.04.012 [DOI] [PubMed] [Google Scholar]
- 332. Sanyal AJ (2019) Past, present and future perspectives in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 16, 377–386. [DOI] [PubMed] [Google Scholar]
- 333. Holan V & Minowada J (1992) Selective enhancement of interleukin 1 beta production in myelomonocytic cell lines by insulin and its related cytokines. Immunol Lett 34, 243–247. [DOI] [PubMed] [Google Scholar]
- 334. Iida KT, Shimano H, Kawakami Y, Sone H, Toyoshima H, Suzuki S, Asano T, Okuda Y & Yamada N (2001) Insulin up‐regulates tumor necrosis factor‐alpha production in macrophages through an extracellular‐regulated kinase‐dependent pathway. J Biol Chem 276, 32531–32537. [DOI] [PubMed] [Google Scholar]
- 335. Mauer J, Chaurasia B, Plum L, Quast T, Hampel B, Bluher M, Kolanus W, Kahn CR & Bruning JC (2010) Myeloid cell‐restricted insulin receptor deficiency protects against obesity‐induced inflammation and systemic insulin resistance. PLoS Genet 6, e1000938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Park YM, Kashyap SR, Major JA & Silverstein RL (2012) Insulin promotes macrophage foam cell formation: potential implications in diabetes‐related atherosclerosis. Lab Invest 92, 1171–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337. Rotllan N, Chamorro‐Jorganes A, Araldi E, Wanschel AC, Aryal B, Aranda JF, Goedeke L, Salerno AG, Ramirez CM, Sessa WC et al. (2015) Hematopoietic Akt2 deficiency attenuates the progression of atherosclerosis. FASEB J 29, 597–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Dror E, Dalmas E, Meier DT, Wueest S, Thevenet J, Thienel C, Timper K, Nordmann TM, Traub S, Schulze F et al. (2017) Postprandial macrophage‐derived IL‐1beta stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat Immunol 18, 283–292. [DOI] [PubMed] [Google Scholar]
- 339. Tessaro FHG, Ayala TS, Nolasco EL, Bella LM & Martins JO (2017) Insulin influences LPS‐induced TNF‐alpha and IL‐6 release through distinct pathways in mouse macrophages from different compartments. Cell Physiol Biochem 42, 2093–2104. [DOI] [PubMed] [Google Scholar]
- 340. Ieronymaki E, Theodorakis EM, Lyroni K, Vergadi E, Lagoudaki E, Al‐Qahtani A, Aznaourova M, Neofotistou‐Themeli E, Eliopoulos AG, Vaporidi K et al. (2019) Insulin resistance in macrophages alters their metabolism and promotes an M2‐like phenotype. J Immunol 202, 1786–1797. [DOI] [PubMed] [Google Scholar]
- 341. Costa Rosa LF, Safi DA, Cury Y & Curi R (1996) The effect of insulin on macrophage metabolism and function. Cell Biochem Funct 14, 33–42. [DOI] [PubMed] [Google Scholar]
- 342. Dandona P, Aljada A, Mohanty P, Ghanim H, Hamouda W, Assian E & Ahmad S (2001) Insulin inhibits intranuclear nuclear factor kappaB and stimulates IkappaB in mononuclear cells in obese subjects: evidence for an anti‐inflammatory effect? J Clin Endocrinol Metab 86, 3257–3265. [DOI] [PubMed] [Google Scholar]
- 343. Iida KT, Suzuki H, Sone H, Shimano H, Toyoshima H, Yatoh S, Asano T, Okuda Y & Yamada N (2002) Insulin inhibits apoptosis of macrophage cell line, THP‐1 cells, via phosphatidylinositol‐3‐kinase‐dependent pathway. Arterioscler Thromb Vasc Biol 22, 380–386. [DOI] [PubMed] [Google Scholar]
- 344. Leffler M, Hrach T, Stuerzl M, Horch RE, Herndon DN & Jeschke MG (2007) Insulin attenuates apoptosis and exerts anti‐inflammatory effects in endotoxemic human macrophages. J Surg Res 143, 398–406. [DOI] [PubMed] [Google Scholar]
- 345. Martins JO, Ferracini M, Ravanelli N, Landgraf RG & Jancar S (2008) Insulin suppresses LPS‐induced iNOS and COX‐2 expression and NF‐kappaB activation in alveolar macrophages. Cell Physiol Biochem 22, 279–286. [DOI] [PubMed] [Google Scholar]
- 346. Senokuchi T, Liang CP, Seimon TA, Han S, Matsumoto M, Banks AS, Paik JH, DePinho RA, Accili D, Tabas I et al. (2008) Forkhead transcription factors (FoxOs) promote apoptosis of insulin‐resistant macrophages during cholesterol‐induced endoplasmic reticulum stress. Diabetes 57, 2967–2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347. Cuschieri J, Bulger E, Grinsell R, Garcia I & Maier RV (2008) Insulin regulates macrophage activation through activin A. Shock 29, 285–290. [DOI] [PubMed] [Google Scholar]
- 348. Su D, Coudriet GM, Hyun Kim D, Lu Y, Perdomo G, Qu S, Slusher S, Tse HM, Piganelli J, Giannoukakis N et al. (2009) FoxO1 links insulin resistance to proinflammatory cytokine IL‐1beta production in macrophages. Diabetes 58, 2624–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349. Mita T, Goto H, Azuma K, Jin WL, Nomiyama T, Fujitani Y, Hirose T, Kawamori R & Watada H (2010) Impact of insulin resistance on enhanced monocyte adhesion to endothelial cells and atherosclerogenesis independent of LDL cholesterol level. Biochem Biophys Res Commun 395, 477–483. [DOI] [PubMed] [Google Scholar]
- 350. Mita T, Azuma K, Goto H, Jin WL, Arakawa M, Nomiyama T, Suzuki R, Kubota N, Tobe K, Kadowaki T et al. (2011) IRS‐2 deficiency in macrophages promotes their accumulation in the vascular wall. Biochem Biophys Res Commun 415, 545–550. [DOI] [PubMed] [Google Scholar]
- 351. Liang CP, Han S, Li G, Tabas I & Tall AR (2012) Impaired MEK signaling and SERCA expression promote ER stress and apoptosis in insulin‐resistant macrophages and are reversed by exenatide treatment. Diabetes 61, 2609–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Yan H, Ma Y, Li Y, Zheng X, Lv P, Zhang Y, Li J, Ma M, Zhang L, Li C et al. (2016) Insulin inhibits inflammation and promotes atherosclerotic plaque stability via PI3K‐Akt pathway activation. Immunol Lett 170, 7–14. [DOI] [PubMed] [Google Scholar]
- 353. Reardon CA, Lingaraju A, Schoenfelt KQ, Zhou G, Cui C, Jacobs‐El H, Babenko I, Hoofnagle A, Czyz D, Shuman H et al. (2018) Obesity and insulin resistance promote atherosclerosis through an IFNgamma‐regulated macrophage protein network. Cell Rep 23, 3021–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Pal S, Nath P, Das D, Hajra S & Maitra S (2018) Cross‐talk between insulin signalling and LPS responses in mouse macrophages. Mol Cell Endocrinol 476, 57–69. [DOI] [PubMed] [Google Scholar]
- 355. Yu T, Gao M, Yang P, Pei Q, Liu D, Wang D, Zhang X & Liu Y (2017) Topical insulin accelerates cutaneous wound healing in insulin‐resistant diabetic rats. Am J Transl Res 9, 4682–4693. [PMC free article] [PubMed] [Google Scholar]
- 356. Kubota T, Inoue M, Kubota N, Takamoto I, Mineyama T, Iwayama K, Tokuyama K, Moroi M, Ueki K, Yamauchi T et al. (2018) Downregulation of macrophage Irs2 by hyperinsulinemia impairs IL‐4‐indeuced M2a‐subtype macrophage activation in obesity. Nat Commun 9, 4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357. Yu T, Gao M, Yang P, Liu D, Wang D, Song F, Zhang X & Liu Y (2019) Insulin promotes macrophage phenotype transition through PI3K/Akt and PPAR‐gamma signaling during diabetic wound healing. J Cell Physiol 234, 4217–4231. [DOI] [PubMed] [Google Scholar]
- 358. Yang P, Wang X, Wang D, Shi Y, Zhang M, Yu T, Liu D, Gao M, Zhang X, Liu Y (2020) Topical insulin application accelerates diabetic wound healing by promoting anti‐inflammatory macrophage polarization. J Cell Sci 133, jcs235838. doi: 10.1242/jcs.235838 [DOI] [PubMed] [Google Scholar]