

Abstract
Aims
Cabotegravir is an integrase strand transfer inhibitor in clinical development as long‐acting (LA) injectable HIV preexposure prophylaxis.
Methods
This phase I study assessed pharmacokinetics of cabotegravir in plasma and anatomical sites associated with sexual HIV‐1 transmission after repeated oral and single intramuscular (IM) LA dosing in healthy adults. Following a 28‐day oral lead‐in period of cabotegravir 30 mg and a washout period of 14–42 days, participants were administered a single ultrasound‐guided gluteal IM cabotegravir LA 600‐mg injection. The study objective was to characterize cabotegravir concentrations in plasma, cervical, vaginal and rectal tissues, and cervicovaginal and rectal fluids and up to Week 12 after IM injection.
Results
Nineteen participants enrolled and 16 completed the study through Week 52. Cabotegravir was detected in plasma and all tissues and fluids. Median plasma cabotegravir concentrations exceeded the in vitro protein‐adjusted 90% maximal inhibitory concentration through Week 12. Median tissue‐ and fluid‐to‐plasma cabotegravir concentration ratios across all visits were 0.32 for rectal fluid and 0.08–0.16 for other tissues and fluids. Adjusted R 2 coefficients between cabotegravir concentrations in plasma and cervical, vaginal and rectal tissues were 0.78, 0.79 and 0.90, respectively. Injection‐site reactions were common (88% of participants) and were mostly grade 1 in intensity (82%). Two participants reported 11 non–drug‐related serious adverse events.
Conclusion
Concentrations of cabotegravir in tissues and fluids were proportional to plasma over time, with strong correlations between tissue and plasma concentrations. Cabotegravir LA tissue‐to‐plasma ratios may be important for understanding its use as preexposure prophylaxis.
Keywords: antiretrovirals, HIV/AIDS, pharmacokinetic–pharmacodynamic
What is already known about this subject
Long‐acting cabotegravir demonstrated superior efficacy vs. emtricitabine and tenofovir disoproxil fumarate as preexposure prophylaxis for HIV‐1 infection in 2 phase III studies and may be preferred by some individuals.
There are limited data in humans describing cabotegravir concentrations in anatomical sites related to sexual transmission of HIV‐1 following long‐acting injection.
What this study adds
After a single, long‐acting intramuscular 600‐mg cabotegravir dose, cervicovaginal and rectal tissue and fluid concentrations were lower but proportional to those in plasma; tissue and plasma cabotegravir concentrations were strongly correlated.
Tissue‐to‐plasma ratios may be important for evaluating the effectiveness of long‐acting agents as preexposure prophylaxis.
1. INTRODUCTION
Globally, 38 million people are living with HIV, and an estimated 1.7 million people were newly infected in 2019. 1 The HIV treatment 90–90–90 targets aiming to diagnose 90% of people with HIV infection, treat 90% of those diagnosed, and achieve viral suppression in 90% of people receiving treatment by 2020 were established to help end the AIDS epidemic. 2 Although maintaining an undetectable viral load is effective at preventing sexual transmission of HIV, consistent access and adherence to oral antiretroviral therapy, stable retention in care, and numerous barriers to daily lifelong oral therapy are significant challenges. 3 Multiple strategies to prevent HIV‐1 transmission have been evaluated, including preexposure prophylaxis (PrEP). 4 , 5 Two regimens approved for HIV‐1 PrEP are daily oral 2‐drug formulations of emtricitabine + tenofovir disoproxil fumarate and emtricitabine + tenofovir alafenamide. 6 , 7 The dapivirine vaginal ring for HIV‐1 PrEP in women is approved by the European Medicines Agency and prequalified by the World Health Organization. 8 , 9 However, global uptake of PrEP is low in individuals who would benefit from PrEP, with 590 000 people receiving at least 1 PrEP dose in 2019—falling far short of the goal of 3 000 000 by 2020. 10 Rate of PrEP uptake in the USA is low among African American men who have sex with men, despite the burden of HIV infection and likelihood of acquiring HIV being very high in this population. 11 , 12 , 13 Low rates of medication adherence to daily oral therapy reduce the effectiveness of PrEP. 14 , 15 Thus, PrEP with less‐frequent, parenteral administration could offer an alternative to daily oral dosing or event‐driven PrEP with improved compliance and longer duration of protection, which may be preferred by some people with high likelihood of acquiring HIV‐1. 16
Cabotegravir is an HIV integrase strand transfer inhibitor in late‐stage development as a long‐acting (LA) single agent for PrEP. 17 Cabotegravir is highly protein bound in plasma, with less than 1% free in circulation. 18 Preclinical studies in macaques demonstrated that cabotegravir LA administered as a single agent protected against simian–human immunodeficiency virus (SHIV) and simian immunodeficiency virus challenges, with plasma cabotegravir concentrations that can be attained in humans. 19 , 20 , 21 Plasma cabotegravir concentrations above the in vitro protein‐adjusted 90% maximal inhibitory concentration (PA‐IC90) provided protection in 97% of male macaques challenged intrarectally with SHIV (half‐maximal tissue culture infectious [TCID50] dose, 50) and >90% of medroxyprogesterone‐treated female macaques challenged intravaginally with SHIV (TCID50 dose, 300). 19 , 20 Female macaques had a 90% probability of protection from intravaginal simian immunodeficiency virus challenge (TCID50 dose, 1000) when plasma cabotegravir concentration was above 4× PA‐IC90. 21 Cabotegravir LA as PrEP was evaluated in the ECLAIR and HIV Prevention Trials Network (HPTN) 077 studies with dosing frequency of every 8 or 12 weeks to maintain target plasma cabotegravir levels above 1× PA‐IC90 (0.166 μg/mL) and above 4× PA‐IC90 (0.664 μg/mL) in 95 and 80% of study participants, respectively, based on concentrations affording protection in animal challenge studies. 22 , 23 In the ECLAIR study of men with a low likelihood of sexually acquiring HIV‐1, cabotegravir LA 800 mg intramuscularly (IM) every 12 weeks for 3 doses resulted in plasma cabotegravir levels below PA‐IC90 in 15–31% of participants. 22 These results were similar in men and women administered the same dosing schedule in the HPTN 077 study among persons with a low likelihood of acquiring HIV‐1, 23 confirming that dosing every 12 weeks may be insufficient. In a second cohort of the HPTN 077 trial, cabotegravir LA 600 mg IM injection every 8 weeks for 5 doses maintained plasma concentrations above 1× PA‐IC90 and above 4× PA‐IC90 pharmacokinetic (PK) targets up to 36 weeks in all participants, regardless of sex. 23
The HPTN 083 and 084 trials, 2 separate global, phase III, double‐blind, double‐dummy, noninferiority studies, were unblinded early at a predefined, interim analysis when cabotegravir 600 mg LA IM injections administered every 8 weeks demonstrated superior efficacy to daily oral emtricitabine + tenofovir disoproxil fumarate for PrEP in men who have sex with men and transgender women (HPTN 083) and cisgender women (HPTN 084) at high risk of acquiring HIV‐1 through sexual transmission. 17 , 24 Cabotegravir LA was generally well tolerated in phase II and III PrEP trials; therefore, it is a promising candidate for PrEP. 17 , 22 , 23 , 24 Cabotegravir plus rilpivirine LA dosed every month or every 2 months for HIV‐1 treatment is approved for maintenance of virological suppression in adults infected with HIV‐1.
Globally, sexual transmission is responsible for most new acquisitions of HIV, primarily through men who have sex with men and heterosexual contact. 1 , 12 Therefore, determining cabotegravir concentrations in anatomical sites associated with sexual transmission is important in the overall evaluation of cabotegravir LA as PrEP. Although cabotegravir concentrations associated with preclinical efficacy have been characterized in macaque challenge studies with SHIV, 19 , 20 , 25 cabotegravir concentration data in sites related to sexual HIV‐1 transmission following IM injection in humans are limited. The 114433 study evaluated cabotegravir concentrations following a lower 400‐mg IM dose (NCT01756131); however, anatomical sites were limited to plasma and tissues in a small sample size (n = 8). 26 Herein we report the PK of cabotegravir LA in tissues and fluids associated with sexual HIV‐1 transmission sites following repeat 30‐mg oral daily dosing and following a single 600‐mg IM dose administered under ultrasound guidance separated by a washout period between treatments.
2. METHODS
2.1. Design
This was a phase I, open‐label study of healthy adults that assessed PK of cabotegravir in plasma and anatomical tissues and secretions associated with sexual transmission of HIV‐1 following repeat oral and single LA dosing (NCT02478463). Participants received daily oral cabotegravir 30 mg for 28 days to assess safety and tolerability. Participants then underwent a washout period of 14–42 days to permit clinic scheduling of subsequent IM administration. Participants received a single, ultrasound‐guided, gluteal IM injection of cabotegravir LA 600 mg administered by a physician, with confirmation of depot injection location via magnetic resonance imaging at 1 of 2 study sites. A spinal needle ≥9 cm was used at both sites to ensure drug deposition took place within the gluteal muscle rather than deep subcutaneous tissue, which, at times, may occur using a standard 3.8‐cm needle. PK assessments were collected at the end of oral dosing and following injection through Week 12, with continued safety monitoring and quarterly PK sampling through study completion at 52 weeks postinjection. Cabotegravir LA 600 mg was selected because it is a clinically relevant dose predicted to achieve target PK concentrations in study participants associated with preclinical efficacy for prevention of SHIV infection in macaques. 19 , 23
The study was conducted at Johns Hopkins Hospital (Baltimore, MD, USA) and the University of Pittsburgh Medical Center (Pittsburgh, PA, USA) from 27 February 2017 to 25 July 2019, in accordance with International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use Good Clinical Practice and principles of the Declaration of Helsinki. Johns Hopkins Medicine Institutional Review Board (Baltimore, MD, USA) and Western Institutional Review Board (Puyallup, WA, USA) approved the study protocol and conduct. All participants provided written informed consent and could withdraw at any time.
2.2. Participants
Eligible participants were healthy men and women aged 18–55 years with body weight ≥40 kg and body mass index (BMI) between 18.5 and 35.0 kg/m2. Women were eligible if they were not pregnant or lactating and either of reproductive potential using a highly effective contraceptive method or not of reproductive potential (premenopausal with documented tubal ligation, bilateral tubal occlusion or bilateral oophorectomy). Women who consented to genital tract sampling but declined rectal sampling were also eligible. Exclusion criteria were related to medical history (e.g., history of seizure disorder, cardiovascular disease, liver disease) and diagnostic assessments, including positive test results for hepatitis B virus, hepatitis C virus, HIV or other sexually transmitted infections. Other exclusion criteria are summarized in the Supplemental Information.
2.3. Objectives and assessments
Primary study endpoint was cabotegravir concentrations following a single 600‐mg (3‐mL) IM gluteal injection in plasma and rectal tissue and fluid in men and women and cervical tissue and cervicovaginal fluid in women at Days 3 and 8 and Weeks 4, 8 and 12 and in vaginal tissue in women at Day 3 and Week 8 (Table S1). Secondary endpoints included cabotegravir concentration tissue‐ and fluid‐to‐plasma and tissue‐to‐fluid ratios; PK parameters of cabotegravir in plasma, tissues and fluids; tissue‐ and fluid‐to‐plasma area under the concentration–time curve (AUC) ratios; and safety parameters, including those observed through Week 52 (Supplemental Table S1).
PK sampling of plasma, cervical tissue, cervicovaginal fluid and rectal tissue and fluid occurred on Day 29 of the oral dosing period and on Days 3 and 8, and Weeks 4, 8 and 12 after IM injection. Vaginal tissue was collected on Day 3 and Week 8. Additional PK plasma samples were collected predose, 4 hours after IM injection on Day 1, and on Day 5 and Weeks 24, 36 and 52 after IM injection. Cervicovaginal and rectal fluids were collected via vaginal speculum and anoscopy, respectively, using polyethylene terephthalate swabs prior to cervical or vaginal and rectal biopsies, respectively. Collection of 3 vaginal and 2 cervical tissue samples via vaginal speculum and 3 rectal tissue samples via flexible sigmoidoscopy occurred at each biopsy. Vaginal tissue samples were collected first if both vaginal and cervical tissues were obtained at the same visit.
Cabotegravir concentration ratios for tissues and fluids to plasma and tissues to fluids were calculated for samples collected after repeated oral dosing and across all visits for samples collected after IM injection. Safety and tolerability were assessed by monitoring and recording adverse events (AEs), clinical laboratory assessments, electrocardiographic results, physical examination findings, and vital sign measurements. Blood samples were collected into potassium ethylenediaminetetraacetic acid tubes, centrifuged at 4°C to separate plasma, and stored at ≤−80°C until analysis.
Plasma, tissue, and fluid cabotegravir concentrations were analysed by the Clinical Pharmacology Analytical Laboratory at Johns Hopkins University Bayview Medical Center (Baltimore, MD, USA) via liquid chromatography with tandem mass spectrometry (LC–MS/MS). Assays were validated in accordance with the US Food and Drug Administration's Bioanalytical Method Validation Guidance for Industry. 27 , 28 The plasma assay was described previously. 29 For cabotegravir measurement in tissue, biopsies were homogenized in 70% methanol, subjected to solid‐phase extraction, and analysed via LC–MS/MS on an API 5000 mass spectrometer (SCIEX, Redwood City, CA, USA). For cabotegravir measurement in fluid, vaginal and rectal fluids were extracted from a polyethylene terephthalate swab and analysed via LC–MS/MS using an API 5000 mass spectrometer. Assay lower limits of quantification for plasma, tissues and fluids were 0.025 μg/mL, 0.05 ng/sample and 0.0625 ng/swab, respectively. Tissue and fluid concentrations were normalized to net swab or tissue weight and expressed in μg/mL, assuming a density of 1 g/mL. Data were acquired and quantified using Analyst 1.6 (SCIEX).
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20. 30
2.4. Data analyses
Cabotegravir PK parameters were estimated from concentration–time data using noncompartmental methods with Phoenix WinNonlin ≥6.3 (Certara, Princeton, NJ, USA). Relationship between plasma and tissue or fluid cabotegravir concentrations at matched timepoints was graphically assessed with log–log linear regression using R software (R Foundation, Boston, MA, USA). PK parameters were summarized using descriptive statistics. Log‐transformed maximum observed concentration (Cmax), apparent terminal phase half‐life (t1/2) and AUC from time 0 to infinity (AUC0‐∞), the last quantifiable time point (AUC0‐t), Week 4 (AUC0‐Wk4), Week 8 (AUC0‐Wk8) and Week 12 (AUC0‐Wk12) were analysed by analysis of covariance using mixed‐linear models with sex as a fixed effect and BMI as a continuous covariate. All ratios were individually calculated for each participant. Safety results were summarized using descriptive statistics.
3. RESULTS
3.1. Study population and baseline characteristics
Of 29 individuals screened, 19 were enrolled and 16 (84%) completed 52 weeks of follow‐up. Of those enrolled, mean age was 33 years, mean BMI was 27 kg/m2; 53% were women (Table 1).
TABLE 1.
Baseline demographics
| Parameter | Participants in overall population (n = 19) | Participants in evaluable pharmacokinetic population (n = 15) a |
|---|---|---|
| Age (y) | ||
| Mean (SD) | 33.3 (9.1) | 30.1 (7.0) |
| Range | 22.0–53.0 | 22.0–46.0 |
| Sex, n (%) | ||
| Female | 10 (53) | 7 (47) |
| Male | 9 (47) | 8 (53) |
| Body mass index (kg/m2) | ||
| Mean (SD) | 27.2 (3.3) | 26.9 (2.9) |
| Range, min‐max | 21.4–33.1 | 21.4–31.1 |
| Height, mean (SD), cm | 171.1 (10.2) | 171.8 (9.0) |
| Weight, mean (SD), kg | 79.4 (11.5) | 79.1 (9.0) |
| Race and ethnicity, n (%) | ||
| Not Hispanic and not Latino | 19 (100) | 15 (100) |
| Asian | 1 (5) | 1 (7) |
| Black and African American | 6 (32) | 5 (33) |
| White | 12 (63) | 9 (60) |
SD, standard deviation.
Four participants did not have evaluable pharmacokinetic data: 2 withdrew before receiving intramuscular injection; 1 had injection maladministration into the retroperitoneal cavity; and 1 was hospitalized for non–study drug‐related serotonin syndrome.
Prior to receiving IM injection, 2 participants withdrew at investigator discretion due to missed study visits or lost to follow‐up. A third participant withdrew consent on Day 282 following IM injection due to relocation. Two participants did not undergo PK tissue sampling because of IM injection maladministration into the retroperitoneal cavity (n = 1) and hospitalization for non–study‐drug‐related serotonin syndrome (n = 1).
3.2. PK
Cabotegravir concentrations were evaluated in plasma, tissues and fluids following repeat oral dosing and after a single IM injection (Figure 1; Table 2). Plasma cabotegravir concentrations were ≥4× PA‐IC90 in 100% of participants after repeat oral dosing and by Day 3 to Week 4 following a single IM 600‐mg injection (Table 3). Median cabotegravir plasma concentrations were ≥4× PA‐IC90 (0.664 μg/mL) at Week 8 and remained above PA‐IC90 (0.166 μg/mL) at Week 12 following IM injection. Following a single 600‐mg IM injection, 100 and 60% of participants had plasma cabotegravir concentrations above PA‐IC90 at Weeks 8 and 12, respectively. Plasma cabotegravir concentrations were detected in 2 women among 17 participants (12%) at ≥48 weeks postinjection (BMI, 28.0 and 30.4 kg/m2, respectively; Figure S1). Fourteen (82%) participants had no measurable plasma cabotegravir concentrations after Week 24; of these, 6 (43%) were women (median [range] BMI, 29.0 [24.1–33.1] kg/m2) and 8 (57%) were men (median [range] BMI, 25.5 [21.4–29.2] kg/m2).
FIGURE 1.
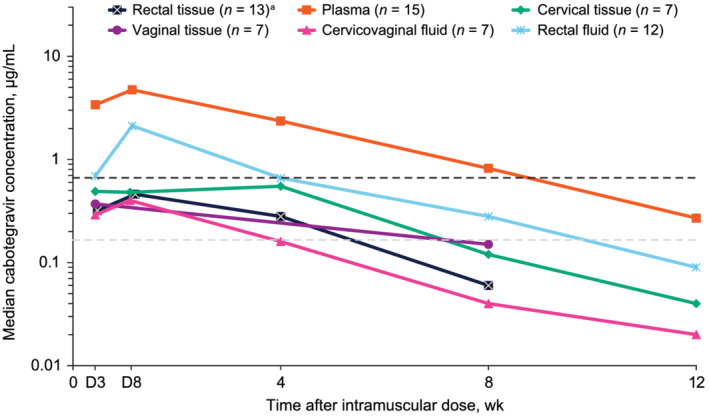
Median cabotegravir concentration–time profiles presented by matrix following a single, ultrasound‐guided cabotegravir LA 600‐mg intramuscular gluteal injection. Black and grey dashed lines indicate 4× PA‐IC90 = 0.664 μg/mL and PA‐IC90 = 0.166 μg/mL, respectively. Plasma LLOQ = 0.025 μg/mL; fluid LLOQ = 0.0000625 μg/mL; tissue LLOQ = 0.00005 μg/mL. D, day; LA, long‐acting; LLOQ, lower limit of quantification; PA‐IC90, in vitro protein‐adjusted 90% maximal inhibitory concentration. a Median rectal tissue concentration at Week 12 was less than tissue LLOQ
TABLE 2.
Median (range) cabotegravir concentrations (μg/mL) in plasma, tissues and fluids after oral dosing and intramuscular injection
| Visit | Median (range) a | |||||
|---|---|---|---|---|---|---|
| Plasma (n = 15) | Cervical tissue (n = 7) | Vaginal tissue (n = 7) | Cervicovaginal fluid (n = 7) | Rectal tissue (n = 13) | Rectal fluid (n = 12) | |
| Oral 30 mg | ||||||
| Day 28 | 5.98 (2.19–8.78) | 1.10 (0.56–2.12) | 0.54 (0.22–1.56) | 0.45 (0.16–2.75) | 0.59 (0.20–1.17) | 3.36 (0.91–22.70) |
| Intramuscular 600 mg | ||||||
| Day 3 | 3.39 (0.72–6.9) | 0.49 (0.19–1.77) | 0.37 (0.06–1.46) | 0.29 (0.04–2.63) | 0.32 (0.05–0.67) | 0.69 (0.00–6.63) |
| Day 8 | 4.74 (1.01–11.2) | 0.48 (0.12–2.54) | — | 0.40 (0.17–1.86) | 0.46 (0.11–0.95) | 2.12 (0.77–9.6) |
| Week 4 | 2.36 (0.80–5.12) | 0.55 (0.11–0.77) | — | 0.16 (0.02–1.10) | 0.28 (0.10–0.45) | 0.66 (0.04–21.4) |
| Week 8 | 0.82 (0.33–3.10) | 0.12 (<LLOQ to 0.41) | 0.15 (<LLOQ to 0.40) | 0.04 (0.02–0.43) | 0.06 (0.00–0.31) | 0.28 (0.03–0.77) |
| Week 12 | 0.27 (0.04–1.31) | 0.04 (<LLOQ to 0.18) | — | 0.02 (<LLOQ to 0.50) | <LLOQ (<LLOQ to 0.17) | 0.09 (0.01–6.12) |
LLOQ, lower limit of quantification.
LLOQs were 0.025, 0.0000625 and 0.000050 μg/mL for plasma, fluids and tissues, respectively.
TABLE 3.
Proportion of participants with cabotegravir concentrations >PA‐IC90 and ≥4× PA‐IC90 in plasma, tissues and fluids after repeat oral dosing and after a single 600‐mg intramuscular injection across sexes
| Visit | Proportion, % a | |||||
|---|---|---|---|---|---|---|
| Plasma (n = 15) | Cervical tissue (n = 7) | Vaginal tissue (n = 7) | Cervicovaginal fluid (n = 7) | Rectal tissue (n = 13) | Rectal fluid (n = 12) | |
| Oral 30 mg | ||||||
| Day 28 | ||||||
| >PA‐IC90 | 100 | 100 | 100 | 86 | 100 | 100 |
| ≥4× PA‐IC90 | 100 | 71 | 29 | 43 | 23 | 100 |
| IM 600 mg | ||||||
| Day 3 | ||||||
| >PA‐IC90 | 100 | 100 | 71 | 86 | 69 | 92 |
| ≥4× PA‐IC90 | 100 | 43 | 29 | 29 | 8 | 50 |
| Day 8 | ||||||
| > PA‐IC90 | 100 | 86 | — | 100 | 92 | 100 |
| ≥4× PA‐IC90 | 100 | 43 | — | 29 | 39 | 100 |
| Week 4 | ||||||
| >PA‐IC90 | 100 | 71 | — | 43 | 77 | 83 |
| ≥4× PA‐IC90 | 100 | 29 | — | 14 | 0 | 50 |
| Week 8 | ||||||
| >PA‐IC90 | 100 | 43 | 43 | 14 | 23 | 67 |
| ≥4 × PA‐IC90 | 53 | 0 | 0 | 0 | 0 | 8 |
| Week 12 | ||||||
| >PA‐IC90 | 60 | 14 | — | 14 | 8 | 33 |
| ≥4 × PA‐IC90 | 20 | 0 | — | 0 | 0 | 17 |
IM, intramuscular; PA‐IC90, in vitro protein‐adjusted 90% maximal inhibitory concentration.
PA‐IC90 = 0.166 μg/mL and 4× PA‐IC90 = 0.664 μg/mL.
Following repeat oral dosing at Day 28, median cabotegravir concentrations were ≥4× PA‐IC90 in cervical tissue and rectal fluid and more than PA‐IC90 in vaginal tissue, rectal tissue and cervicovaginal fluid (Table 2). Cabotegravir concentrations were above PA‐IC90 in 100 and 86% of participants in all tissues and cervicovaginal fluid, respectively, after oral dosing (Table 3). Following a single 600‐mg IM injection, median cabotegravir concentrations were greater than PA‐IC90 in cervical and rectal tissues through Week 4 and rectal fluid through Week 8 (Figure 1; Table 2). Median cabotegravir concentrations in all tissues and cervicovaginal fluid were below PA‐IC90 at Week 8 after IM injection. Following IM injection, cabotegravir concentrations were above PA‐IC90 in 77, 83, 71 and 43% of participants in rectal tissue, rectal fluid, cervical tissue and cervicovaginal fluid, respectively, at Week 4 and 43% of participants in vaginal tissue at Week 8 (Table 3). Cabotegravir concentrations in cervical and vaginal tissues were higher than in cervicovaginal fluid after oral dosing and at Week 8 following IM injection (Table 2). Median cabotegravir concentrations for the entire study population in rectal tissue at Week 12 were below the lower limit of quantification because >50% of participants had non‐quantifiable values.
Median cabotegravir fluid‐ and tissue‐to‐plasma concentration ratios were highest for rectal fluid and cervical tissue, respectively, on Day 28 following repeat oral dosing (Table 4). Despite absolute tissue and fluid cabotegravir concentrations being higher following repeat oral dosing than after IM injection, median cabotegravir fluid‐ and tissue‐to‐plasma concentration ratios were proportionally similar between dosing regimens. One participant had an increased rectal fluid‐to‐plasma concentration ratio of 10.4 following repeat oral dosing because of a high rectal fluid cabotegravir concentration (22.7 μg/mL). Similarly, median cabotegravir fluid‐ and tissue‐to‐plasma concentration ratios were also highest for rectal fluid and cervical and vaginal tissues, respectively, across all visits following IM injection.
TABLE 4.
Median cabotegravir tissue‐ and fluid‐to‐plasma ratios after oral dosing and IM injection across all visits vs. 114433 study results
| Study | Treatment | Ratio (range) | ||||
|---|---|---|---|---|---|---|
| Cervical tissue to plasma (n = 7) | Vaginal tissue to plasma (n = 7) | Cervicovaginal fluid to plasma (n = 7) | Rectal tissue to plasma (n = 13) | Rectal fluid to plasma (n = 12) | ||
| 201767 (present) a | Oral 30 mg | 0.18 (0.11–0.25) | 0.14 (0.03–0.18) | 0.13 (0.02–0.36) | 0.10 (0.07–0.17) | 0.45 (0.15–10.40) |
| IM 600 mg | 0.14 (<LLOQ‐0.31) | 0.16 (<LLOQ‐0.34) | 0.08 (0.01–0.44) | 0.09 (<LLOQ‐0.16) | 0.32 (<LLOQ‐11.38) | |
| Cervical tissue to plasma (n = 4 each) | Vaginal tissue to plasma (n = 4 each) | Rectal tissue to plasma (n = 4 each) | ||||
| 11443326 (previous) | IM 400 mg (unsplit) b | 0.20 (<LLOQ‐0.40) | 0.28 (<LLOQ‐0.70) | — | <LLOQ (<LLOQ‐0.10) | — |
| IM 400 mg (split) c | 0.16 (<LLOQ‐0.40) | 0.19 (<LLOQ‐0.70) | — | 0.08 (<LLOQ‐0.20) | — | |
IM, intramuscular; LLOQ, lower limit of quantification.
LLOQs were 0.025, 0.0000625, and 0.000050 μg/mL, for plasma, tissues, and fluids, respectively.
Administered as a single injection. Samples were collected on Weeks 2 and 8.
Administered as 2 200‐mg injections. Samples were collected on Weeks 4 and 12.
Median cabotegravir cervical tissue‐ and vaginal tissue‐to‐cervicovaginal fluid concentration ratios were >1 after oral dosing and at Week 8 after IM injection (Table S2). By contrast, cabotegravir concentrations in cervical tissue were lower than those observed in cervicovaginal fluid at Week 12. Cabotegravir concentrations were also lower in rectal tissue than rectal fluid after oral dosing and all time points after IM injection. Adjusted R 2 coefficients from log–log linear regression of plasma and time‐matched tissue and fluid cabotegravir concentrations following a single IM injection were 0.78, 0.79 and 0.90 for cervical, vaginal and rectal tissues, respectively, and 0.60 and 0.44 for cervicovaginal and rectal fluids, respectively (Figure 2).
FIGURE 2.
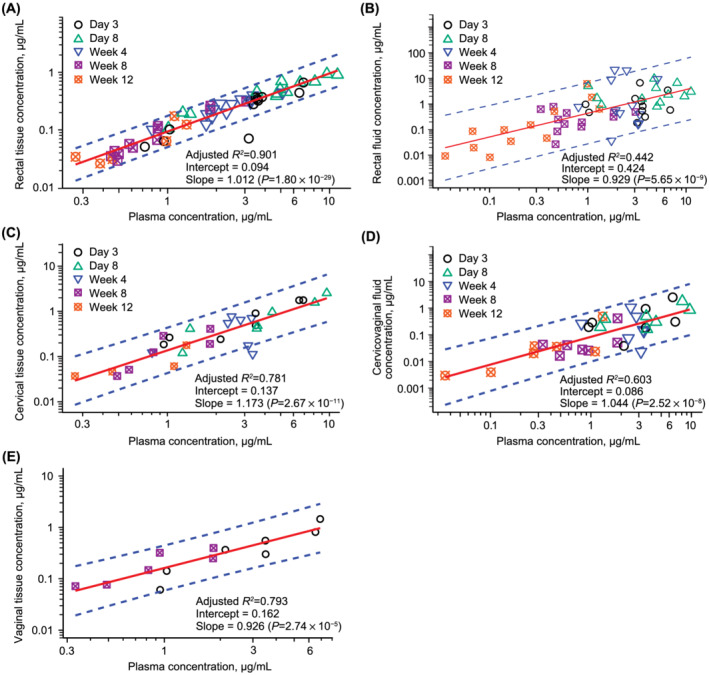
Cabotegravir concentration following cabotegravir 600 mg IM injection in plasma vs. time matched samples from (A) rectal tissue, (B) rectal fluid, (C) cervical tissue, (D) cervicovaginal fluid, and (E) vaginal tissue. Log–log linear regression assessed the relationship between plasma and time‐matched tissue and fluid concentrations. IM, intramuscular
Peak cabotegravir concentrations were observed at a median time of maximum observed concentration (tmax) of 7 days following injection in all matrices (Tables 5 and 6). Compared with participants receiving IM injection into the gluteal muscle, the participant with injection maladministration into the retroperitoneal cavity had decreased plasma PK values for Cmax, AUC0‐Wk4, AUC0‐Wk8 and AUC0‐Wk12 (1.26 μg/mL, 736 μg*h/mL, 1513 μg*h/mL and 2130 μg*h/mL, respectively); this participant had longer overall exposure to plasma cabotegravir, demonstrating increased values for tmax (21 days), t1/2 (124 days), AUC0‐t (4697 μg*h/mL) and AUC0‐∞ (5502 μg*h/mL). Rectal fluid had higher exposures (Cmax and AUC0‐Wk12) and a shorter t1/2 vs. cervical tissue, rectal tissue and cervicovaginal fluid.
TABLE 5.
Summary of plasma PK parameters by sex after IM cabotegravir injection
| PK parameter | Geometric mean (%CVb), range a | Geometric LS mean | Geometric LS mean (90% CI) | |
|---|---|---|---|---|
| Overall (n = 15) | Men (n = 8) | Women (n = 7) | Women vs. men | |
| Cmax, μg/mL b | 5.04 (62), 1.2–11.2 | 6.26 | 3.94 | 0.63 (0.34–1.15) |
| tmax, median (range), d | 6.9 (3.9–52.2) | — | — | — |
| AUC0‐Wk4, μg*h/mL | 2141 (59), 650–5063 | 2285 | 1990 | 0.87 (0.47–1.62) |
| AUC0‐Wk8, μg*h/mL | 3214 (40), 1106–6372 | 3342 | 3073 | 0.92 (0.60–1.42) |
| AUC0‐Wk12, μg*h/mL | 3639 (36), 1442–6555 | 3710 | 3570 | 0.96 (0.64–1.44) |
| AUC0‐t, μg*h/mL | 3992 (25), 2811–6547 | — | — | — |
| AUC0‐∞, μg*h/mL | 4172 (24), 2839‐6597 c | 3829 | 4547 | 1.19 (0.91–1.54) |
| t1/2, d | 19.1 (81), 9.1–148.6 c | 14.6 | 25.1 | 1.72 (0.78–3.79) |
| KALA, h | 0.0014 (97) c | — | — | — |
| Time > PA‐IC90, dd | 78.3 (46) | 66.5 | 94.3 | — |
AUC0‐Wk4, area under the concentration–time curve from time 0 to Week 4; AUC0‐Wk8, area under the concentration–time curve from time 0 to Week 8; AUC0‐Wk12, area under the concentration–time curve from time 0 to Week 12; AUC0‐t, area under the concentration–time curve from time 0 to the last quantifiable time point; AUC0‐∞, area under the concentration–time curve from time 0 to infinity; CI, confidence interval; Cmax, maximum observed concentration; %CVb, geometric coefficient of variation; IM, intramuscular; KA, absorption rate constant; LA, long acting; LS, least squares; PA‐IC90, in vitro protein‐adjusted 90% maximal inhibitory concentration; PK, pharmacokinetics; t1/2, apparent terminal phase half‐life; tmax, time to first occurrence of maximum observed concentration.
Except where noted for tmax.
The lower limit of quantification was 0.025 μg/mL.
n = 14. dPA‐IC90 = 0.166 μg/mL.
TABLE 6.
Tissue and fluid PK parameters after IM cabotegravir injection
| Geometric mean, %CVb a | Cervical tissue (n = 7) | Cervicovaginal fluid (n = 7) | Rectal tissue (n = 13) | Rectal fluid (n = 12) |
|---|---|---|---|---|
| Cmax, μg/mL b | 0.81 (105) | 0.55 (118) | 0.50 (47) | 3.27 (172) |
| Ratio to plasma Cmax (95% CI) | 0.20 (0.16–0.25) | 0.13 (0.07–0.26) | 0.10 (0.08–0.11) | 0.62 (0.31–1.25) |
| tmax, median (range), d | 7.0 (1.9–30.0) | 7.0 (1.9–84.0) | 7.0 (6.9–52.2) | 7.0 (6.9–80.0) |
| AUC0‐Wk4, μg*h/mL | 277 (104) | 203 (126) | 206 (57) | 1170 (192) |
| Ratio to plasma AUC0‐Wk4 (95% CI) | 0.16 (0.09–0.26) | 0.10 (0.04–0.25) | 0.10 (0.09–0.11) | 0.55 (0.23–1.28) |
| AUC0‐Wk8, μg*h/mL | 365 (85) | 282 (125) | 287 (39) | 1576 (203) |
| Ratio to plasma AUC0‐Wk8 (95% CI) | 0.15 (0.07–0.33) | 0.09 (0.04–0.23) | 0.11 (0.09–0.13) | 0.49 (0.21–1.11) |
| AUC0‐Wk12, μg*h/mL | 350 (79) | 381 (108) | 324 (55) | 1345 (138) |
| Ratio to plasma AUC0‐Wk12 (95% CI) | 0.12 (0.04–0.38) | 0.09 (0.02–0.33) | 0.11 (0.09–0.14) | 0.39 (0.14–1.07) |
| AUC0‐t, μg*h/mL | 523 (77) | 324 (121) | 348 (36) | 1841 (194) |
| AUC0‐∞, μg*h/mL c | 696 (NA) | 399 (100) | 292 (35) | 850 (43) |
| t1/2, d c | 15.14 (NA) | 14.83 (79) | 23.65 (23) | 12.77 (4) |
| Time > PA‐IC90, d d | 18.7 (190) | 10.8 (203) | 24.9 (75) e | 33.1 (140) f |
AUC0‐Wk4, area under the concentration–time curve from time 0 to Week 4; AUC0‐Wk8, area under the concentration–time curve from time 0 to Week 8; AUC0‐Wk12, area under the concentration–time curve from time 0 to Week 12; AUC0‐t, area under the concentration–time curve from time 0 to the last quantifiable time point; AUC0‐∞, area under the concentration–time curve from time 0 to infinity; CI, confidence interval; Cmax, maximum observed concentration; %CVb, geometric coefficient of variation; IM, intramuscular; KA, absorption rate constant; LA, long acting; NA, not available; PA‐IC90, in vitro protein‐adjusted 90% maximal inhibitory concentration; PK, pharmacokinetics; t1/2, apparent terminal phase half‐life; tmax, time to first occurrence of maximum observed concentration.
Except where noted for tmax and ratios to plasma parameters.
Lower limits of quantification were 0.025, 0.0000625 and 0.000050 μg/mL for plasma, fluids and tissues, respectively.
n = 14.
PA‐IC90 = 0.166 μg/mL.
n = 11.
n = 10.
Sex‐based comparison of evaluable cabotegravir plasma PK parameters was performed using a mixed‐effect linear model, with sex as a fixed effect and BMI as a continuous covariate. Differences in plasma cabotegravir exposures varied between men and women, with an increased Cmax (6.26 vs. 3.94 μg/mL) and decreased t1/2 (14.6 vs. 25.1 days) in men vs. women, respectively (Table 5).
3.3. Safety
Eighty‐six AEs were reported; all participants experienced at least 1 AE. Ten participants reported AEs during the oral lead‐in phase; none were considered drug‐related (Table 7). Seventeen participants reported AEs following IM injection. Excluding injection‐site reactions (ISRs), the most frequently reported AEs were depression, headache, insomnia, palpitations and pyrexia (two participants for each event [12%]). Most AEs were grade 1 (60%) or 2 (33%) in intensity, and none led to study withdrawal. Drug‐related AEs determined by the investigator were reported by 15 participants following IM injection, including ISRs of pain (14 participants [82%]), erythema (two participants [12%]), and induration, pruritus, muscle cramp and swelling (one participant for each event [6%]). Most drug‐related ISR AEs were of grade 1 intensity; all resolved (median duration, 6 d) and none led to study withdrawal. Two participants withdrew early prior to receiving IM injection based on investigator discretion due to missed study visits and loss to follow‐up. No obvious trends in clinical laboratory abnormalities were noted.
TABLE 7.
Summary of AEs
| Preferred term, n (%) | Oral lead‐in cabotegravir 30 mg once daily (n = 19) | Cabotegravir long‐acting intramuscular injection 600 mg (n = 17) |
|---|---|---|
| Total AE a | 10 (53) | 17 (100) |
| Injection‐site pain | — | 15 (88) |
| Viral gastroenteritis | 3 (16) | 0 |
| Increased blood glucose | 2 (11) | 0 |
| Depression | 0 | 2 (12) |
| Headache | 0 | 2 (12) |
| Injection‐site erythema | — | 2 (12) |
| Insomnia | 0 | 2 (12) |
| Palpitations | 0 | 2 (12) |
| Pyrexia | 0 | 2 (12) |
| Fatigue | 1 (5) | 1 (6) |
| Drug‐related AE | 0 | 15 (88) |
| Injection‐site pain | — | 14 (82) |
| Injection‐site erythema | — | 2 (12) |
| Gait disturbance | 0 | 1 (6) |
| Injection‐site induration | — | 1 (6) |
| Injection‐site pruritus | — | 1 (6) |
| Injection‐site reaction | — | 1 (6) |
| Injection‐site swelling | — | 1 (6) |
| Insomnia | 0 | 1 (6) |
| Myalgia | 0 | 1 (6) |
AE, adverse event.
AEs reported in >1 participant.
Two participants reported a total of 11 serious AEs (SAEs) during the study. No deaths occurred during the study. One participant with a personal history of recurrent pregnancy loss reported pregnancy at her Week 52 visit and experienced a suspected spontaneous abortion no more than 2 days later. The second participant developed serotonin syndrome 9 weeks following injection that was related to 4 serotonergic drugs that the participant concomitantly received during the study. Due to subsequent prolonged hospitalization and consequent complications resulting in additional hospital‐related SAEs, including upper extremity deep venous thrombosis, extensive intraparenchymal haemorrhage after anticoagulation for deep venous thrombosis, aphasia, dysphagia and urinary tract infection with leucocytosis, the participant was withdrawn from the study but monitored in long‐term follow‐up up to Week 52. Two additional SAEs reported by this participant were related to 2 subsequent hospitalizations, including a brief hospitalization for dyspnoea and subsequent hospitalization for post‐traumatic stress disorder. After discharge, adjustments were made to the participant's antianxiety medications; the participant subsequently returned to the approximate baseline level of psychological functioning that existed prior to the first SAE being reported. All SAEs resolved except for intracerebral haemorrhage, which was still resolving at the last clinical follow‐up visit. No SAEs reported in any study participant were considered to be study drug related.
4. DISCUSSION
After a single ultrasound‐guided IM injection of cabotegravir 600 mg, drug concentrations were quantifiable in plasma and in most tissue and fluid samples over 12 weeks postinjection from anatomical sites associated with sexual HIV‐1 acquisition. Median plasma cabotegravir concentrations remained ≥4× PA‐IC90 through Week 8, a clinical threshold associated with efficacy in study participants with HIV infection in phase II and III treatment trials and expected to be associated with efficacy when using the identical regimen for HIV‐1 PrEP, and above PA‐IC90 through Week 12. All participants had plasma cabotegravir concentrations that were ≥4× PA‐IC90 beginning as early as Day 3 through Week 4 and above PA‐IC90 through Week 8. Plasma cabotegravir concentrations were detectable in 12% of participants 52 weeks after the last injection, a finding similar to those observed in men following the final injection in ECLAIR (17%) and 52–60 weeks following the final injection in men in HPTN 077 (23%). 22 , 29 However, most participants in this study had no measurable plasma cabotegravir concentrations 24 weeks after injection (82%), similar to what was observed after the final injection in ECLAIR (81%). In the HPTN 077 study, cabotegravir LA 600 mg every 8 weeks yielded plasma cabotegravir trough concentrations at or above 1× and 4× PA‐IC90 targets in 95 and 80% of participants, respectively, for 8 weeks after the last injection; therefore, dosing every 8 weeks is likely to be sufficient to provide coverage for most participants regardless of absorption kinetics. 23
Of the anatomical sites sampled, median cabotegravir concentrations were highest in rectal fluid, followed by vaginal tissue, cervical tissue, rectal tissue and, lastly, cervicovaginal fluid. Peak concentrations occurred at approximately 7 days in all tissues and fluids. Median cabotegravir concentrations following a single 600‐mg IM injection were greater than PA‐IC90 in cervical and rectal tissues through Week 4 and in rectal fluid through Week 8. Median cabotegravir concentrations were less than PA‐IC90 in all tissue and fluid samples at Week 12. At Week 4, 71 and 77% of participants had cabotegravir concentrations above PA‐IC90 in cervical and rectal tissue, respectively, consistent with a prior study that demonstrated lower absolute tissue concentrations relative to plasma. 26 At Week 12, only a small proportion of participants had cabotegravir concentrations above PA‐IC90 in cervical and rectal tissue and cervicovaginal fluid. Median concentration–time profiles in plasma, tissues and fluids had comparable slopes. Strong correlations (adjusted R 2 > 0.75) were observed between plasma and time‐matched cervical, vaginal and rectal tissue cabotegravir concentrations. By contrast, correlations were weaker between plasma and cervicovaginal fluid (adjusted R 2 = 0.60) and rectal fluid (adjusted R 2 = 0.44), probably due to increased variability in fluid concentration between participants and possibly from variability in active and passive transport of drug from cells into fluid. Rectal fluid cabotegravir concentrations at Week 4 in 2 participants (1 man, 1 woman) were >30× median concentration (0.66 μg/mL), potentially contributing to its lower correlation coefficient. The slope of the log–log linear regression for all tissues and fluids was approximately 1, indicating a direct correlation with plasma concentrations. Overall, cabotegravir concentrations were the most variable in rectal fluid, followed by plasma and cervicovaginal fluid, with tissues demonstrating much lower variability between participants.
Distribution of cabotegravir into tissues and fluids was similar between oral dosing and IM injection. Furthermore, tissue‐to‐plasma ratios after a single 600‐mg IM injection were similar to those observed following cabotegravir 400‐mg IM split or nonsplit injections in the 114433 study (Table 4). 26 Taken together, these results suggest that distribution of cabotegravir into tissues associated with HIV‐1 sexual transmission sites remains consistent and declines in a parallel fashion, regardless of dose, dose splitting or administration. Tissue‐to‐plasma ratios following a single cabotegravir 600‐mg injection were generally low, with values ≤0.16 across all tissues. Cabotegravir 600 mg every 8 weeks starting 4 weeks after an initial loading injection demonstrated efficacy for PrEP in HPTN 083 and HPTN 084, 17 , 24 suggesting that cabotegravir concentrations in tissues, plasma or both, similar to those observed here sufficiently confer a high rate of protection. Because HPTN 083 and HPTN 084 did not involve compartmentalized collections, direct comparisons to this study can be made for plasma alone. Understanding cabotegravir PK and pharmacodynamics within genital tract compartments in study participants would assist in interpreting PrEP effectiveness.
Cabotegravir LA exhibits absorption‐limited flip‐flop kinetics. 31 Plasma cabotegravir PK parameters were consistent with results from previous studies. 22 , 23 Plasma PK exposures (Cmax and AUC0‐Wk12) were similar to those following cabotegravir 800 mg IM split injection in ECLAIR despite the lower dose and nonsplit injection in the present study. 22 Geometric mean of the cabotegravir LA absorption constant observed in participants with higher absorption rate was similar between the current study (0.0014) and ECLAIR (0.0011). 32 The higher Cmax, which is reflective of absorption rate, and lower t1/2 observed in men vs. women in the present study was consistent with differing sex‐specific PK parameter results observed in HPTN 077. 23 , 33 Cabotegravir LA for PrEP demonstrated superior efficacy among men and transgender women in HPTN 083 and cisgender women in HPTN 084 using an every‐8‐week dosing regimen of the identical dose used in this study, indicating that the observed sex‐specific PK differences are unlikely to impact the rate of protection. 17 , 24 It is unknown whether ultrasound‐guided IM delivery of cabotegravir using a longer needle to ensure deep gluteal IM injection in this study, rather than using standard 3.8‐ or 5.1‐cm needle lengths with typical free‐hand IM gluteal injection in clinical studies, might have impacted the rate of drug absorption from the injection site. Achieving true IM injection is challenging, even with direct ultrasound guidance. However, all participants maintained plasma cabotegravir concentrations ≥4× PA‐IC90 through 4 weeks following a single 600‐mg IM injection. A loading dose strategy with every‐8‐week dosing, in which participants receive initial loading injections at Weeks 0 and 4 before maintenance injections every 8 weeks starting at Week 12, achieved cabotegravir trough concentrations >4× PA‐IC90 in 95% of participants in a phase IIIb HIV‐1 treatment study and would be expected when using this identical regimen for HIV‐1 PrEP. 34 One participant who received an injection into the retroperitoneal cavity exhibited plasma exposures that continued to decrease through Week 12 postinjection but were higher overall, with a prolonged t1/2 compared with participants who received injections into the gluteal muscle. However, in routine settings, it is unlikely that using a shorter needle (generally 3.8 cm) to administer an IM injection, which is recommended in clinical practice, would reach the depth required for inadvertent drug deposition into the retroperitoneal cavity. Data are limited regarding cabotegravir LA PK following confirmed injection maladministration.
This study has some limitations. The sample size was small and was further reduced by 3 withdrawals and 2 participants who did not undergo tissue PK sampling. The study population was limited to a small overall range of BMI (18.5–35.0 kg/m2) to reduce PK variability and better permit interpretation of the results. Thus, extrapolation of these results to individuals with higher BMIs may be difficult. Cabotegravir concentrations in tissues and fluids could be overestimated due to possible blood contamination while collecting samples. Tissue cabotegravir concentrations were evaluated after tissue homogenization and may not be reflective of total tissue concentrations. Because cabotegravir protein binding data are only available for plasma, the tissue and fluid cabotegravir concentration ratios are not adjusted for protein binding. These data reflect cabotegravir PK in plasma, tissues and fluids following an ultrasound‐guided injection, which may differ when administered as a free‐hand gluteal injection without imaging guidance as intended in the clinic.
Following a single IM LA injection, cabotegravir was detected in tissues and fluids of anatomical sites associated with sexual HIV‐1 transmission. Tissue and fluid cabotegravir concentrations were proportional to plasma over time, and time‐matched tissue concentrations were strongly correlated with plasma concentrations. Plasma PK parameters and tissue‐to‐plasma ratios were similar to those observed in previous studies. Given the apparently sufficient distribution of cabotegravir into mucosal tissues and fluids related to sexual HIV‐1 transmission, this study provides data to inform the conduct and interpretation of future cabotegravir PrEP studies.
COMPETING INTERESTS
J.S.S., K.B., K.H. and S.L.F. are employees of and own stock in GlaxoSmithKline. E.D.W. received institutional research funding from ViiV Healthcare and GlaxoSmithKline. S.E. has nothing to disclose. E.F. received grants from ViiV Healthcare and GlaxoSmithKline. S.R. received grants from GlaxoSmithKline, Merck and Gilead and consultant fees from Novimab. M.A.M. received grants from ViiV Healthcare and GlaxoSmithKline and the National Institutes of Health and research support from Gilead and Merck. R.D.A., W.S. and P.P. are employees of ViiV Healthcare and own stock in GlaxoSmithKline. Y.L. is an employee of Precision Biosciences and owns stock in GlaxoSmithKline. C.H. received grants from ViiV Healthcare, GlaxoSmithKline, Merck, Gilead and the National Institutes of Health; personal fees from Merck, ViiV Healthcare and GlaxoSmithKline; and nonfinancial support from Gilead. D.M. was an employee of ViiV Healthcare and may have owned stock in GlaxoSmithKline at the time of the study.
CONTRIBUTORS
All authors have: made substantial contributions to conception and design, or acquisition of data, or analysis and interpretation of data; been involved in drafting the manuscript or revising it critically for important intellectual content; given final approval of the version to be published; have participated sufficiently in the work to take public responsibility for appropriate portions of the content; and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
STUDY PI DECLARATION
The authors confirm that the PI for this paper is Craig Hendrix, MD and that he had direct clinical responsibility for patients.
ETHICS APPROVAL
The study was conducted at Johns Hopkins Hospital (Baltimore, MD, USA) and the University of Pittsburgh Medical Center (Pittsburgh, PA, USA) from 27 February 2017 to 25 July 2019 in accordance with the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use Good Clinical Practice and the principles of the Declaration of Helsinki. Johns Hopkins Medicine Institutional Review Board (Baltimore, MD, USA) and Western Institutional Review Board (Puyallup, WA, USA) approved the study protocol and conduct.
PATIENT CONSENT
All participants provided written informed consent and could withdraw from the study at any time.
CLINICAL TRIAL REGISTRATION
5.
Supporting information
TABLE S1 Study primary and secondary endpoints
TABLE S2 Median (range) cabotegravir tissue‐to‐fluid ratios after oral dosing and IM injection
FIGURE S1 Plasma cabotegravir concentration–time profiles for each participant after oral cabotegravir dosing and after a single cabotegravir LA 600‐mg intramuscular gluteal injection. Plasma cabotegravir concentration is in blue or red for women and green for men. Plasma cabotegravir concentration after intramuscular injection is in red for the woman with maladministration into the retroperitoneal cavity. Black and grey dashed lines indicate 4× PA‐IC90 = 0.664 μg/mL and PA‐IC90 = 0.166 μg/mL, respectively. Dark blue dashed line indicates a plasma lower limit of quantification of 0.025 μg/mL. LA, long‐acting; PA‐IC90, in vitro protein‐adjusted 90% maximal inhibitory concentration. aCabotegravir concentration at 24 hours after administration of oral cabotegravir for 28 days.
ACKNOWLEDGEMENT
The authors would like to acknowledge Stephen Piscitelli, PharmD and Elizabeth Gould for their contributions to study design and conduct. Editorial assistance was provided under the direction of the authors by Megan Schmidt, PhD, and Sherri Damlo, ELS, MedThink SciCom, and funded by ViiV Healthcare. This study was sponsored by ViiV Healthcare.
Shaik JS, Weld ED, Edick S, et al. Multicompartmental pharmacokinetic evaluation of long‐acting cabotegravir in healthy adults for HIV preexposure prophylaxis. Br J Clin Pharmacol. 2022;88(4):1667-1678. 10.1111/bcp.14980
Jafar Sadik Shaik and Ethel D. Weld are joint first authors and contributed equally to the manuscript.
Funding information ViiV Healthcare
DATA AVAILABILITY STATEMENT
Anonymized individual participant data and study documents can be requested for further research from www.clinicalstudydatarequest.com.
REFERENCES
- 1. UNAIDS UNAIDS Data 2020. www.unaids.org/en/resources/documents/2020/unaids-data. Accessed August 18, 2020.
- 2. UNAIDS 90–90‐90: An Ambitious Treatment Target to Help End the AIDS Epidemic. www.unaids.org/sites/default/files/media_asset/90-90-90_en.pdf. Accessed August 18, 2020.
- 3. Eisinger RW, Dieffenbach CW, Fauci AS. HIV viral load and transmissibility of HIV infection: undetectable equals untransmittable. JAMA. 2019;321(5):451‐452. [DOI] [PubMed] [Google Scholar]
- 4. Baeten JM, Haberer JE, Liu AY, Sista N. Preexposure prophylaxis for HIV prevention: where have we been and where are we going? J Acquir Immune Defic Syndr. 2013;63(Supplement 2):S122‐S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. AIDSinfo The Basics of HIV Prevention. https://aidsinfo.nih.gov/understanding-hiv-aids/fact-sheets/20/48/the-basics-of-hiv-prevention. Accessed July 29, 2020.
- 6. Descovy [package insert]. Foster City, CA: Gilead Sciences, Inc; 2020. [Google Scholar]
- 7. Truvada [package insert]. Foster City, CA: Gilead Sciences, Inc; 2020. [Google Scholar]
- 8. Dapivirine vaginal ring [summary of product characteristics]. Malmo, Sweden: QPharma AB; 2020. [Google Scholar]
- 9. World Health Organization Prequalification Unit 19th Invitation to Manufacturers and Suppliers of Medicinal Products for HIV Infections and Related Diseases to Submit an Expression of Interest (EOI) for Product Evaluation to the WHO Prequalification Unit – Medicines Team. https://extranet.who.int/pqweb/news/new-invitation-expression-interest-eoi-hiv-infection-and-related-diseases-medicines-published-0. Accessed January 5, 2020.
- 10. UNAIDS 2020 Global AIDS Update Report. www.unaids.org/en/resources/documents/2020/global-aids-report. Accessed July 23, 2020.
- 11. Finlayson T, Cha S, Xia M, et al. Changes in HIV preexposure prophylaxis awareness and use among men who have sex with men ‐ 20 urban areas, 2014 and 2017. MMWR Morb Mortal Wkly Rep. 2019;68(27):597‐603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. UNAIDS The Gap Report. www.unaids.org/en/resources/documents/2014/20140716_UNAIDS_gap_report. Accessed October 6, 2020.
- 13. Hess KL, Hu X, Lansky A, Mermin J, Hall HI. Lifetime risk of a diagnosis of HIV infection in the United States. Ann Epidemiol. 2017;27(4):238‐243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Haberer JE, Bangsberg DR, Baeten JM, et al. Defining success with HIV pre‐exposure prophylaxis: a prevention‐effective adherence paradigm. Aids. 2015;29(11):1277‐1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Van Damme L, Corneli A, Ahmed K, et al. Preexposure prophylaxis for HIV infection among African women. N Engl J Med. 2012;367(5):411‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Molina JM, Capitant C, Spire B, et al. On‐demand preexposure prophylaxis in men at high risk for HIV‐1 infection. N Engl J Med. 2015;373(23):2237‐2246. [DOI] [PubMed] [Google Scholar]
- 17. Landovitz RJ, Donnell D, Clement M, et al. HPTN 083 final results: pre‐exposure prophylaxis containing long‐acting injectable cabotegravir is safe and highly effective for cisgender men and transgender women who have sex with men. AIDS 2020; July 6–10, 2020; Virtual.
- 18. Spreen W, Min S, Ford SL, et al. Pharmacokinetics, safety, and monotherapy antiviral activity of GSK1265744, an HIV integrase strand transfer inhibitor. HIV Clin Trials. 2013;14(5):192‐203. [DOI] [PubMed] [Google Scholar]
- 19. Andrews CD, Spreen WR, Mohri H, et al. Long‐acting integrase inhibitor protects macaques from intrarectal simian/human immunodeficiency virus. Science. 2014;343(6175):1151‐1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Andrews CD, Yueh YL, Spreen WR, et al. A long‐acting integrase inhibitor protects female macaques from repeated high‐dose intravaginal SHIV challenge. Sci Transl Med. 2015;7: 270ra274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Spreen W, Lowry A, Pal R, et al. Correlation of in vivo cabotegravir concentration & prevention of SIV in macaques. CROI 2015; February 23–26, 2015; Seattle, WA.
- 22. Markowitz M, Frank I, Grant RM, et al. Safety and tolerability of long‐acting cabotegravir injections in HIV‐uninfected men (ECLAIR): a multicentre, double‐blind, randomised, placebo‐controlled, phase 2a trial. Lancet HIV. 2017;4(8):e331‐e340. [DOI] [PubMed] [Google Scholar]
- 23. Landovitz RJ, Li S, Grinsztejn B, et al. Safety, tolerability, and pharmacokinetics of long‐acting injectable cabotegravir in low‐risk HIV‐uninfected individuals: HPTN 077, a phase 2a randomized controlled trial. PLoS Med. 2018;15(11):e1002690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Delany‐Moretlwe S, Hughes JP, Bock P, et al. Long acting injectable cabotegravir is safe and effective in preventing HIV infection in cisgender women: results from HPTN 084. HIV Research for Prevention; January 27–28 and February 3–4, 2021; Virtual.
- 25. Radzio J, Spreen W, Yueh YL, et al. The long‐acting integrase inhibitor GSK744 protects macaques from repeated intravaginal SHIV challenge. Sci Transl Med. 2015;7: 270ra275 [DOI] [PubMed] [Google Scholar]
- 26. Spreen W, Ford SL, Chen S, et al. GSK1265744 Pharmacokinetics in plasma and tissue after single‐dose long‐acting injectable administration in healthy subjects. J Acquir Immune Defic Syndr. 2014;67(5):481‐486. [DOI] [PubMed] [Google Scholar]
- 27. US Department of Health and Human Services , US Food and Drug Administration , Center for Drug Evaluation and Research (CDER) , Center for Veterinary Medicine (CVM) . Guidance for Industry: Bioanalytical Method Validation. Silver Spring, MD: DHHS; 2018. [Google Scholar]
- 28. DiFrancesco R, Tooley K, Rosenkranz SL, et al. Clinical pharmacology quality assurance for HIV and related infectious diseases research. Clin Pharmacol Ther. 2013;93(6):479‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Landovitz RJ, Li S, Eron JJ Jr, et al. Tail‐phase safety, tolerability, and pharmacokinetics of long‐acting injectable cabotegravir in HIV‐uninfected adults: a secondary analysis of the HPTN 077 trial. Lancet HIV. 2020;7(7):e472‐e481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alexander SPH, Kelly E, Mathie A, et al. The concise guide to pharmacology 2019/20: introduction and other protein targets. Br J Pharmacol. 2019;176(suppl 1):S1‐S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Spreen WR, Margolis DA, Pottage JC Jr. Long‐acting injectable antiretrovirals for HIV treatment and prevention. Curr Opin HIV AIDS. 2013;8(6):565‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ford S, Stancil BS, Markowitz M, et al. ECLAIR study of cabotegravir (CAB) LA injections: characterization of safety and PK during the "PK tail" phase. HIV Research for Prevention; October 17‐21, 2016; Chicago, IL
- 33. Landovitz RJ, Li S, Grinsztejn B, et al. Tail‐phase safety, tolerability and pharmacokinetics of long‐acting injectable cabotegravir in HIV‐uninfected individuals. HIV Research for Prevention; October 24, 2018; Madrid, Spain
- 34. Overton ET, Richmond G, Rizzardini G, et al. Long‐acting cabotegravir and rilpivirine dosed every 2 months in adults with HIV‐1 infection (ATLAS‐2M), 48‐week results: a randomised, multicentre, open‐label, phase 3b, non‐inferiority study. Lancet. 2020;396(10267):1994‐2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 Study primary and secondary endpoints
TABLE S2 Median (range) cabotegravir tissue‐to‐fluid ratios after oral dosing and IM injection
FIGURE S1 Plasma cabotegravir concentration–time profiles for each participant after oral cabotegravir dosing and after a single cabotegravir LA 600‐mg intramuscular gluteal injection. Plasma cabotegravir concentration is in blue or red for women and green for men. Plasma cabotegravir concentration after intramuscular injection is in red for the woman with maladministration into the retroperitoneal cavity. Black and grey dashed lines indicate 4× PA‐IC90 = 0.664 μg/mL and PA‐IC90 = 0.166 μg/mL, respectively. Dark blue dashed line indicates a plasma lower limit of quantification of 0.025 μg/mL. LA, long‐acting; PA‐IC90, in vitro protein‐adjusted 90% maximal inhibitory concentration. aCabotegravir concentration at 24 hours after administration of oral cabotegravir for 28 days.
Data Availability Statement
Anonymized individual participant data and study documents can be requested for further research from www.clinicalstudydatarequest.com.