

Abstract
In the adult hippocampus, synaptic plasticity is important for information processing, learning, and memory encoding. Astrocytes, the most common glial cells, play a pivotal role in the regulation of hippocampal synaptic plasticity. While astrocytes were initially described as a homogenous cell population, emerging evidence indicates that in the adult hippocampus, astrocytes are highly heterogeneous and can differentially respond to changes in neuronal activity in a subregion‐dependent manner to actively modulate synaptic plasticity. In this review, we summarize how local neuronal activity changes regulate the interactions between astrocytes and synapses, either by modulating the secretion of gliotransmitters and synaptogenic proteins or via contact‐mediated signaling pathways. In turn, these specific responses induced in astrocytes mediate the interactions between astrocytes and neurons, thus shaping synaptic communication in the adult hippocampus. Importantly, the activation of astrocytic signaling is required for memory performance including memory acquisition and recall. Meanwhile, the dysregulation of this signaling can cause hippocampal circuit dysfunction in pathological conditions, resulting in cognitive impairment and neurodegeneration. Indeed, reactive astrocytes, which have dysregulated signaling associated with memory, are induced in the brains of patients with Alzheimer's disease (AD) and transgenic mouse model of AD. Emerging technologies that can precisely manipulate and monitor astrocytic signaling in vivo enable the examination of the specific actions of astrocytes in response to neuronal activity changes as well as how they modulate synaptic connections and circuit activity. Such findings will clarify the roles of astrocytes in hippocampal synaptic plasticity and memory in health and disease.
Keywords: Alzheimer's disease, hippocampal circuit, learning and memory, neurodegeneration, neuronal activity, synapse, synaptic dysfunction, synaptic plasticity
Astrocytes regulate hippocampal synaptic plasticity in an activity‐ and circuit‐dependent manner. In this review, we discuss the molecular and cellular bases of such region‐specific regulation of astrocyte–synapse interactions in the adult hippocampal circuitry. Importantly, the activation of astrocytic signaling is important for hippocampal synaptic homeostasis and thus memory processes. Meanwhile, dysregulation of this signaling can result in hippocampal circuit dysfunction and cognitive impairment during the progression of Alzheimer's disease.
Abbreviations
- AAV
adeno‐associated virus
- AD
Alzheimer's disease
- AMPAR
AMPA receptor
- APC
anaphase‐promoting complex
- ApoE
apolipoprotein E
- ATP
adenosine triphosphate
- Aβ
beta‐amyloid
- Best‐1
Bestrophin‐1
- CA
cornu ammonis
- CHRDL1
chordin‐like 1
- CRAC
calcium release-activated calcium
- DG
dentate gyrus
- EAAT
excitatory amino acid transporter
- EPSCs
excitatory postsynaptic currents
- FPKM
fragments per kilobase million
- GPCR
G protein-coupled receptor
- IL-33
interleukin 33
- LTD
long-term depression
- LTP
long-term potentiation
- mGluRs
metabotropic glutamate receptors
- NFIA
nuclear factor I‐A
- NMDAR
NMDA receptor
- Nptx1
neuronal pentraxin-1
- P2Y12R
P2Y12 receptor
- SNARE
soluble N-ethylmaleimide-sensitive factor attachment protein receptor
- SPARCL1
SPARC-like protein 1
- TNFα
tumor necrosis factor alpha
- TREK-1
TWIK-related potassium channel 1
- TSP
thrombospondin
- TTX
tetrodotoxin
Introduction
Throughout life, synapses remain plastic by altering their structure and strength—a process termed ‘synaptic plasticity.’ Such synaptic adaptions in neuronal circuits are critically dependent on experience‐driven neuronal activity changes [1, 2]. Synaptic plasticity has multiple forms including Hebbian plasticity and homeostatic plasticity [1, 2, 3]. Hebbian plasticity is a positive feedback mechanism that facilitates the reinforcement of synaptic connections and includes two major forms: long‐term potentiation (LTP) and long‐term depression (LTD) [4]. Meanwhile, homeostatic plasticity (also known as ‘synaptic scaling’) is a negative feedback mechanism whereby neurons counteract excessive excitation or inhibition in response to prolonged neuronal activity changes [5, 6]. Different forms of synaptic plasticity interact to facilitate the network functions of the adult brain. Meanwhile, synaptic plasticity impairment is implicated in several neurological and cognitive disorders, notably Alzheimer's disease (AD) [7, 8].
The coordination of cellular events in different forms of synaptic plasticity not only depends on the bidirectional communication between pre‐ and postsynaptic neurons but also on the interactions between neurons and their enveloping glia [9]. Notably, astrocytes, the most abundant glial cells in the central nervous system, integrate into the neuronal circuitry via their intricate processes and interact with pre‐ and postsynaptic neurons to form tripartite structures [10, 11]. Through their interactions with synapses, astrocytes monitor changes in synaptic activity and modify the structures and functions of those synapses accordingly, thereby shaping specific circuits—for example, the hippocampal circuit during learning and memory [9, 12, 13]. Concordantly, the disruption of such astrocyte–synapse interactions in the adult mouse brain leads to impaired synaptic plasticity in the hippocampal circuits, which is considered a key contributor to the synaptic dysfunctions and memory deficits in AD [14, 15, 16, 17].
The hippocampus, which comprises the cornu ammonis 1 (CA1), CA2, CA3, and dentate gyrus (DG) subregions, is important for information encoding as well as memory storage and retrieval. In particular, during sensory experience, CA1 pyramidal neurons receive excitatory synaptic inputs from CA3 pyramidal neurons via the Schaffer collateral pathway or from layer III pyramidal neurons in the entorhinal cortex via the perforant pathway [18]. This sensory information is subsequently processed and integrated by the hippocampal CA1 pyramidal neurons and eventually exits the hippocampus to other brain regions including the subiculum, perirhinal cortex, prefrontal cortex, and amygdala [19, 20]. As the CA1 microcircuit is a major output of the hippocampus, its activity is precisely controlled by the synapse‐interacting astrocytes, which is essential for the storage and retrieval of most hippocampus‐dependent memories [21, 22]. However, the mechanisms underlying such astrocyte‐mediated synaptic remodeling in the adult hippocampal circuits remain unclear.
Here, we review how astrocytes interact with synapses to regulate neuronal activity‐dependent synaptic plasticity in the adult hippocampus. First, we discuss how astrocytes specifically respond to the neuronal activity changes in adult hippocampal circuits. We then discuss how the astrocyte‐derived signals mediate activity‐dependent hippocampal synaptic plasticity. Next, we summarize recent studies that link astrocytic signaling with hippocampus‐dependent cognitive processes. Finally, we discuss how astrocyte–synapse interactions are altered as well as their roles in pathological conditions, particularly AD.
Astrocytes regulate hippocampal synaptic functions in a neuronal activity‐ and circuit‐dependent manner
Astrocytes actively integrate into hippocampal circuits through their neuronal activity‐dependent interactions with synapses—either by regulating the probability of presynaptic release or through the modulation of postsynaptic structures and functions.
Regulation of gliotransmitters and their receptors
Notably, astrocytes express multiple ion channels, neurotransmitter, and neuromodulator receptors, enabling them to sense changes in neuronal activity. Sensing such neuronal activity changes subsequently triggers astrocytes to secrete various factors, such as gliotransmitters and synaptogenic proteins, to act on neurons [23, 24, 25, 26]. The release of gliotransmitters including glutamate, ATP (adenosine triphosphate), and d‐serine [a co‐agonist of NMDA receptors (NMDARs)] from astrocytes is mediated through two major mechanisms: calcium‐dependent exocytosis from storage organelles and direct release from cytosol via membrane ion channels [27, 28]. Specifically, neuronal activity‐dependent vesicular exocytosis of gliotransmitters requires the actions of various neurotransmitter receptors, vesicular transporters, and SNARE (soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor) complexes [25, 28, 29, 30]. Meanwhile, the activity‐induced gliotransmitter flux across the plasma membrane of astrocytes is critically dependent on the opening of distinct gliotransmitter‐permeable ion channels [27].
Through such activity‐dependent secretion of gliotransmitters, astrocytes actively regulate synaptic transmission and plasticity in the adult hippocampus [28, 31, 32, 33]. In particular, glutamate release by astrocytes in hippocampal subregions is differentially regulated, which subsequently modulates synaptic transmission in the hippocampal neurons in a circuit‐specific manner. For example, in the DG, glutamate exocytosis processes are activated through the neuronal activity‐dependent stimulation of purinergic P2Y1 receptors on astrocytes, which in turn increases the probability of neurotransmitter release in surrounding neurons via the activation of presynaptic NMDAR 2B subunits [34]. On the other hand, in the hippocampal CA3–CA1 synapses, such astrocytic glutamate exocytosis‐facilitated neurotransmitter release is dependent on another mechanism, which requires the activation of Ca2+ and SNARE proteins in astrocytes and group I mGluRs (metabotropic glutamate receptors) in neurons [35]. Besides conventional vesicular exocytosis, hippocampal CA1 astrocytes directly release glutamate via glutamate‐permeable ion channels including the two‐pore‐domain potassium channel TREK‐1 (TWIK‐related potassium channel 1) and the calcium‐activated anion channel Best‐1 (Bestrophin‐1) [27, 36, 37]. Upon neuronal activity‐stimulated activation of specific G protein‐coupled receptors (GPCRs) in CA1 astrocytes, TREK‐1 and Best‐1 channels regulate two distinct modes of astrocytic glutamate release. Specifically, upon the activation of Gi‐coupled GPCR, TREK‐1, which is expressed in the cell bodies and processes of astrocytes, facilitates rapid glutamate release and initiates neuronal mGluR signaling to impact synaptic transmission [36]. Meanwhile, the activation of the Gq family of GPCRs stimulates the action of Best‐1, which is localized at the microdomains of astrocytes near synapses, to mediate calcium‐dependent slow glutamate release and activate synaptic NMDARs [36, 37]. These findings suggest that hippocampal astrocytes employ multiple mechanisms to regulate glutamate release in response to dynamic neuronal activity changes at specific timescales, subcellular locations, and synaptic receptors. Such neuronal activity‐dependent regulation of the interaction between astrocytes and synapses enables hippocampal neurons to specifically modulate their activity levels in the adult hippocampal circuitry in response to distinct physiological stimuli.
Moreover, depending on the basal or sustained neuronal activity, astrocytes differentially regulate the secretion of ATP to regulate the synaptic efficacy of hippocampal CA3–CA1 synapses via the activation of distinct adenosine receptor subtypes [25, 38, 39, 40, 41]. In response to a minimal stimulus of a single presynaptic fiber, astrocytic mGluR5‐mediated ATP secretion activates adenosine A2A receptors, which subsequently enhance basal synaptic transmission of CA3–CA1 excitatory synapses [39]. Meanwhile, in response to a strong tetanic stimulus, ATP is readily released by astrocytes to activate both adenosine A2A and A1 receptors. Studies using specific pharmacological inhibitors of different adenosine receptors revealed that A2A receptors mediate sustained potentiation of excitatory synapses whereas adenosine A1 receptors regulate synaptic depression [38, 41, 42]. Moreover, the excitatory A2A and inhibitory A1 receptors are enriched at different synaptic compartments in excitatory hippocampal neurons: A2A receptors are enriched at both the pre‐ and postsynaptic terminals, whereas A1 receptors are exclusively localized at presynaptic terminals [43]. Thus, hippocampal astrocytes release ATP to activate distinct adenosine receptors and specifically regulate the pre‐ and postsynaptic signaling and functions of CA3–CA1 synapses. Through the coordinated regulation of A2A and A1 receptor‐mediated signaling pathways, hippocampal astrocytes can process different forms of neuronal activity (i.e., basal or sustained) and in turn actively modulate the capability of specific synapses to express synaptic plasticity [25, 38]. Accordingly, the neuronal activity‐dependent regulation of ATP and its converted product adenosine has been implicated in the regulation of synaptic plasticity (i.e., LTP) in the adult hippocampus [44, 45, 46]. Specifically, the ATP released by astrocytes facilitates the recruitment and activation of its neuronal receptors in hippocampal excitatory synapses, which in turn regulates the abundance of postsynaptic AMPA receptors (AMPARs) to modulate synaptic strength via several calcium‐dependent signaling pathways [47, 48]. Furthermore, a very recent study revealed that the release of ATP by astrocytes in the adult hippocampus also actively regulates the activity of CA1 pyramidal neurons by stimulating their surrounding GABAergic interneurons, which involves the activation of CRAC (calcium release‐activated calcium) channels encoded by ORAI1 and STIM1 [30]. Accordingly, in response to increased neuronal activity, astrocytic CRAC channels induce the cellular calcium signals upon the activation of metabotropic purinergic and protease‐activated receptors, which further triggers the secretion of ATP to act on CA1 interneurons, thus modulating the activity of hippocampal CA1 circuitry [30]. Collectively, these results suggest that astrocytes play an essential role in maintaining hippocampal network activity by specifically regulating the secretion of gliotransmitters. Through such mechanisms, astrocytes can set a suitable threshold for the induction of activity‐dependent synaptic plasticity in different subregions of the adult hippocampus in response to experience [38, 49].
Regulation of synaptogenic proteins
To regulate neuronal activity‐dependent hippocampal synaptic plasticity, astrocytes send synaptogenic signals to neurons to rearrange the structures and modulate the functions of excitatory synapses. The extracellular matrix proteins TSP‐1 and TSP‐2 (thrombospondin 1 and 2, respectively) were the first astrocytic factors shown to be important for synaptogenesis in hippocampal neurons [50, 51, 52]. Astrocytes secrete TSPs to promote the formation of structural glutamatergic synapses in cultured hippocampal cells via the activation of the neuronal gabapentin receptor α2δ1 [51] and the adhesion molecule neuroligin‐1 [52]. Moreover, in response to neuronal activity changes, hippocampal astrocytes secrete several cytokines that act on neurons and regulate their synaptic structures. One of the best characterized cytokines is TNF‐α (tumor necrosis factor alpha). Accordingly, pharmacological deprivation of neuronal activity in cultured hippocampal cells by the sodium channel blocker tetrodotoxin (TTX) increases the expression and secretion of TNFα, which subsequently modulates the homeostatic plasticity of both excitatory and inhibitory neurons [53, 54]. Accordingly, one study prepared mixed cultures of neurons and astrocytes from either TNFα‐knockout or wild‐type hippocampi and demonstrated that astrocytic TNFα is required for such TTX‐stimulated increase of excitatory synaptic transmission [53].
Specifically, such astrocyte‐derived TNFα acts on postsynaptic TNF receptors to increase AMPARs at existing excitatory synapses, thus actively regulating homeostatic plasticity to rebalance network activity [53, 55, 56]. The action of astrocyte‐derived TNFα on such postsynaptic receptor trafficking is dependent on the activation of several kinases including p38 and PI3K [57]. Further in vivo studies on germline TNFα‐knockout mice corroborate the role of TNFα in homeostatic adaptation in the cortical circuitry during sensory deprivation, which is mediated by the regulation of excitatory synapses in layer V pyramidal cells [58, 59]. Nonetheless, the precise mechanisms underlying these activity‐dependent, TNFα‐mediated postsynaptic changes in the intact hippocampal circuitry are unclear.
Our recent study demonstrates that another cytokine, IL‐33 (interleukin 33), is selectively regulated in the astrocytes in the hippocampal CA1 subregion to maintain network homeostasis during homeostatic synaptic plasticity (Fig. 1) [60]. IL‐33 acting as an alarmin is released from damaged cells to maintain tissue homeostasis [61]. Prolonged suppression of neuronal activity in CA1 excitatory pyramidal neurons either by TTX administration to hippocampal slices or by optogenetic stimulation of adult mouse hippocampi increases the local expression and release of IL‐33 in neighboring astrocytes. Astrocyte‐secreted IL‐33 subsequently serves as a negative feedback signal to increase excitatory synaptic transmission to maintain hippocampal network homeostasis during homeostatic synaptic plasticity [60]. Of note, blockade of the global neuronal activity of hippocampal slices by TTX can induce IL‐33 protein only in CA1 astrocytes [60], suggesting that CA1 astrocytes distinctly respond to neuronal activity changes to perform their synaptic plasticity‐related functions.
Fig. 1.
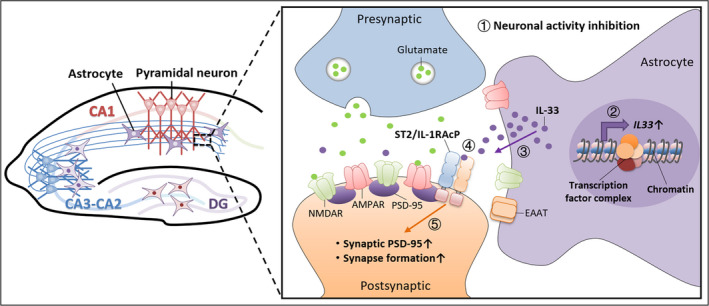
Astrocyte‐derived IL‐33 regulates homeostatic synaptic plasticity at hippocampal CA3–CA1 excitatory synapses in a negative feedback manner. In response to the inhibition of hippocampal neuronal activity (1), astrocytes in the CA1 stratum radiatum region undergo transcriptional reprogramming to increase IL‐33 expression (2). The synthesized IL‐33 is then secreted from astrocytes (3) and binds to the neuronal postsynaptic receptor complex ST2/IL‐1RAcP (4), which further promotes the synaptic accumulation of PSD‐95 as well as subsequent synaptogenesis in CA1 pyramidal neurons (5). AMPAR, AMPA receptor; CA, cornu ammonis; DG,dentate gyrus; EAAT, excitatory amino acid transporter; NMDAR, NMDA receptor.
A recent single‐cell RNA sequencing study showed that hippocampal astrocytes exhibit a unique, region‐specific molecular signature [62]. Characterizing the astrocytic transcriptional program in response to neuronal activity blockade will help uncover how the activity‐dependent regulation of astrocytic functions potentiates synaptic transmission and synaptic plasticity. Of note, genome‐wide transcriptome profiling of neuronal cultures treated with TTX revealed that long‐term activity blockade leads to the activation of a transcriptional program in neurons during homeostatic synaptic plasticity [63]. In that study, among the upregulated genes identified in neurons after TTX administration, Nptx1 (neuronal pentraxin‐1) in particular was shown to be important for synaptic upscaling by enhancing the clustering of AMPARs. While our RNA sequencing analysis revealed that in addition to increasing the expression of TNFα and IL‐33, neuronal activity blockade of cultured hippocampal cells by TTX increases a panel of genes encoding astrocyte‐secreted proteins including two synaptogenic factors—Sparcl1 and Chrdl1—which are well known to regulate synaptic development (Table 1) [60, 64, 65, 66]. SPARCL1 (SPARC‐like protein 1, also known as Hevin) acts as an astrocytic signal to promote the formation of structural glutamatergic synapses via their interactions with neuronal adhesion molecules [65, 67, 68]. Meanwhile, astrocyte‐secreted CHRDL1 (chordin‐like 1) regulates the maturation of excitatory synapses through the upregulation of the GluA2‐containing AMPARs at synapses [64, 66]. Thus, neuronal activity blockade may stimulate transcriptional reprogramming in hippocampal neurons and astrocytes in a subregion‐specific manner. Further single‐cell transcriptome profiling of hippocampal astrocytes would elucidate their functional specialization in the homeostatic upregulation of synaptic strength.
Table 1.
Neuronal activity blockade increases the expression of specific astrocyte‐secreted synaptogenic factors [60]. RNA sequencing analysis was performed on mixed hippocampal neuron–glia cultures after treatment with TTX (1 μm) or vehicle (Con) for 24 h. FPKM, fragments per kilobase million.
| Gene name | Log2 fold change | Adjusted P value | Mean_FPKM_Con | Mean_FPKM_TTX | Mean_Count_Con | Mean_Count_TTX |
|---|---|---|---|---|---|---|
| Il33 | 0.56 | 4.70E‐30 | 19.30 | 28.73 | 3164 | 4895 |
| Tnf | 2.23 | 2.20E‐04 | N/A | N/A | 5 | 23 |
| Sparcl1 | 0.23 | 5.62E‐12 | 1059.67 | 1252.64 | 188 307 | 233 055 |
| Chrdl1 | 0.28 | 3.55E‐05 | 6.60 | 8.10 | 1811 | 2311 |
To understand how the transcriptional program of hippocampal astrocytes is activated upon neuronal activity blockade, it is critical to identify which transcription factors control this program. Accordingly, various neuronal activity‐regulated transcription factors in cortical neurons, such as SRF, CREB, MEF2A, and MEF2D, as well as their roles in transcription have been well characterized [63, 69]. However, the specific transcription factor(s) that controls the region‐specific functions of astrocytes has yet to be investigated. While distinct transcription factors have been identified in astrocytes in different brain regions, no enriched transcription factors have been identified in the hippocampus [70]. Nevertheless, a recent study elegantly demonstrates that the transcription factor NFIA (nuclear factor I‐A) is important for astrocyte–synapse interactions and functions in the hippocampus [71]. Interestingly, brain‐wide genetic deletion of the NFIA gene specifically in adult astrocytes alters the morphology, physiology, and gene expression signatures of astrocytes only in the hippocampus; almost no such changes are observed in other brain regions. NFIA exerts its actions in this selective control of the properties of hippocampal astrocytes through its region‐specific DNA‐binding capacity and gene regulation. This NFIA‐mediated transcriptional mechanism is required for the dynamic astrocyte–synapse interactions and the subsequent induction of synaptic plasticity in the adult hippocampus. Moreover, transcriptional reprogramming is regulated by dynamic chromatin modulation: the regulation of chromatin accessibility at gene regulatory regions and the alteration of their binding affinity to transcription factors ultimately lead to gene regulation [72]. Indeed, emerging studies have identified that specific chromatin regulators are involved in activity‐dependent gene regulation and synaptic scaling [73, 74]. Accordingly, further characterization of epigenomic profiling will uncover the epigenetic and transcriptional control molecules that function in hippocampal astrocytes during synaptic scaling.
In addition to the neuronal activity‐dependent regulation of IL‐33 in CA1 astrocytes, its specific action in CA1 hippocampal synaptic scaling is attributed to the selective expression and activation of IL‐33 receptors in hippocampal CA1 neurons. Accordingly, in vivo two‐photon imaging of CA1 pyramidal neurons revealed that IL‐33 administration promotes excitatory synapse formation in young adult mice. Such signaling is mediated through the activation of the synaptosomally enriched IL‐33 neuronal receptor complex, ST2/IL‐1RAcP, followed by the phosphorylation‐dependent synaptic accumulation of PSD‐95 and subsequent recruitment of AMPARs (Fig. 1). Meanwhile, conditional knockout of IL‐33 in CA1 astrocytes locally decreases the number of excitatory synapses [60, 75, 76]. In addition, IL‐33 signaling can regulate the number of excitatory synapses by promoting microglial synapse engulfment during circuit development [77] or enhancing synaptic plasticity in the DG and promoting memory consolidation through the microglial engulfment of the extracellular matrix [78]. Nevertheless, it remains to be investigated whether IL‐33 can regulate CA1 synapse formation by modulating the clearance functions of microglia during homeostatic synaptic plasticity.
Regulation of contact‐mediated signaling
Besides the selective secretion of astrocyte‐derived factors and activation of their cognate receptors at synapses, astrocytes can regulate synaptic plasticity via their direct contacts with neurons. Accordingly, astrocytic processes encapsulate synapses, enabling astrocytes to communicate with neurons. The neurexin/neuroligin and ephrin/Eph receptor‐mediated contacts between astrocytic processes and synaptic terminals have been reported to regulate astrocyte‐mediated synaptic plasticity [79, 80, 81, 82, 83, 84]. Of note, the deletion of Nrxn1 (neurexin‐1) in astrocytes but not in neurons suppresses AMPAR‐mediated excitatory postsynaptic currents (EPSCs) in CA1 pyramidal neurons of acute hippocampal slices, suggesting that astrocytic Nrxn1 interacts with neuronal neuroligins to regulate excitatory synaptic transmission through the modulation of AMPAR response [84]. Meanwhile, ephrins expressed by astrocytes regulate dendritic spine formation and elimination as well as synaptic plasticity through the activation of neuronal Eph receptors (Fig. 2) [80, 82, 85, 86]. For example, astrocyte‐specific deletion of ephrin‐B1 in mice enhances the formation of dendritic spines in hippocampal CA1 neurons induced by learning, while ephrin‐B1 overexpression in astrocytes using an adeno‐associated virus (AAV) approach abolishes dendritic spine formation in activated hippocampal neurons during memory recall following fear conditioning. Thus, such astrocytic ephrin‐B1 signaling might compete with presynaptic ephrin‐B1 to act on postsynaptic neurons and trigger the neuronal activity‐dependent elimination of EphB receptor‐containing excitatory synapses [82, 83]. These lines of evidence suggest that the astrocyte‐mediated ephrin‐B1 contact signaling negatively regulates excitatory synapse formation in neurons during learning and memory. This raises the intriguing possibility that local changes in synaptic activity might regulate the expression of ephrin‐B1 in certain astrocytes and in turn control the formation and removal of excitatory synapses on specific neurons or dendrites during learning; such a process potentially underlies memory encoding and consolidation in the adult hippocampus. Interestingly, the activation of CA1 neurons enhances the astrocytic coverage of these CA3–CA1 synapses, which subsequently regulates synaptic transmission [39, 87]. As such, these contact‐mediated astrocyte–neuron signals can specifically regulate the neuronal activity‐dependent synaptic responses in the hippocampal CA3–CA1 circuit.
Fig. 2.
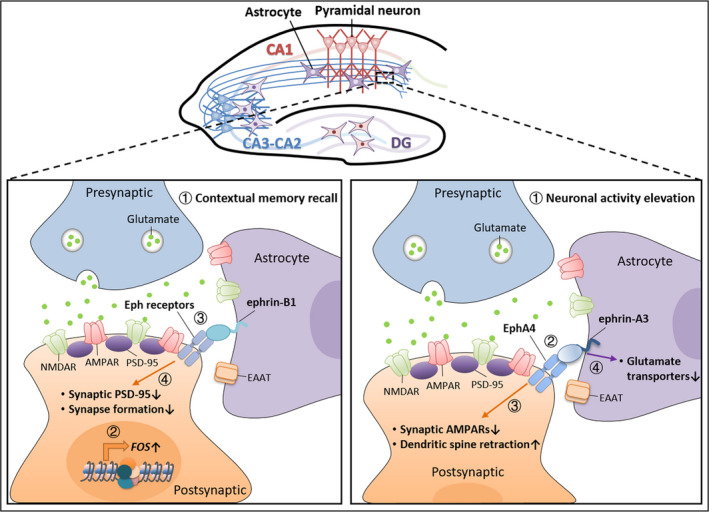
Astrocytic ephrin signaling regulates neuronal activity‐dependent hippocampal CA3–CA1 synaptic responses in a feedback regulatory manner. Lower left panel: A negative regulatory role of astrocytic ephrin‐B1 in the control of synaptogenesis in the CA3–CA1 circuit during learning and memory. During contextual memory recall (1), dendritic spines form in the activated c‐Fos‐expressing CA1 pyramidal neurons (2). The astrocytic ephrin‐B1 forward signaling is subsequently activated in these neurons (3), which inhibits such learning‐induced synaptic PSD‐95 targeting and formation of dendritic spines (4). Lower right panel: Neuron–astrocyte communication via EphA4/ephrin A3 mediates activity‐dependent hippocampal synaptic plasticity. Upon chronic elevation of neuronal activity (1), the activation of neuronal EphA4 forward signaling via astrocytic ephrin A3 (2) causes reduction of synaptic AMPARs and the retraction of dendritic spines (3) during homeostatic plasticity. Meanwhile, the activation of ephrin A3 reverse signaling in astrocytes by postsynaptic EphA4 (2) might decrease the expression of glutamate transporters in astrocytes (4), which further impacts astrocytic functions and the LTP induction at CA3–CA1 synapses. AMPAR, AMPA receptor; CA, cornu ammonis; DG, dentate gyrus; EAAT, excitatory amino acid transporter; NMDAR, NMDA receptor.
In addition, the interaction between astrocytic ephrins and EphA4 in postsynaptic CA1 neurons is important for hippocampal synaptic plasticity (Fig. 2) [80, 85]. Of note, postsynaptic EphA4 signaling induces hippocampal LTP and homeostatic downregulation of excitatory synaptic strength in a neuronal activity‐dependent manner [86]. Accordingly, the ephrin/Eph receptors coordinate multiple signaling pathways to regulate the structure and functions of excitatory synapses, including actin cytoskeleton regulators, glutamate transporters, and the ubiquitin–proteasome system [86, 88, 89]. For example, the activation of postsynaptic EphA4 signaling stimulates dendritic spine retraction, which involves astrocyte–neuron interaction: ephrins in astrocytes bind to postsynaptic EphA4 to initiate downstream Rho guanine nucleotide exchange factor ephexin‐1/RhoA GTPase‐dependent actin dynamics [88]. Moreover, neuron–astrocyte communication via EphA4/ephrin A3 plays an important role in hippocampal LTP. Accordingly, in either EphA4‐ or ephrinA3‐knockout mice, the induction of LTP at hippocampal CA3–CA1 synapses is impaired and accompanied by increased expression and enhanced activity of glial glutamate transporters in the hippocampal CA1 stratum radiatum subregion. Pharmacological inhibition of glial glutamate transporters in these knockout mice rescues such LTP defects [80, 85]. These findings suggest that in response to neuronal activity changes, neuron–astrocyte EphA4/ephrinA3 signaling is activated to inhibit the glial glutamate uptake process near the CA3–CA1 excitatory synapses via the regulation of glial glutamate transporters; thus, such neuron–astrocyte communication facilitates the activity‐induced synaptic transmission and plasticity in the adult hippocampal circuit. Furthermore, postsynaptic EphA4 signaling regulates synaptic strength during hippocampal homeostatic plasticity through the degradation of AMPAR subunits via the ubiquitin ligase APC (anaphase‐promoting complex)‐dependent pathway. Meanwhile, knockdown of neuronal EphA4 abolishes the decrease in excitatory synaptic transmission induced by chronically elevated neuronal activity [86]. Taken together, astrocytic ephrin–neuronal Eph interaction is an important mechanism that integrates different forms of activity‐dependent synaptic plasticity in the hippocampus.
Regulation of lipid homeostasis
Lipid metabolic crosstalk between astrocytes and synapses is essential for neuronal activity‐dependent synaptic modulation and functioning [90, 91]. Of note, astrocyte‐derived ApoE (apolipoprotein E) is a key molecule for this lipid‐mediated astrocyte–synapse communication during neuronal activity changes [92, 93]. During enhanced neuronal activity, the activation of hippocampal neurons leads to the buildup and secretion of fatty acids via small dense carriers. Meanwhile, nearby astrocytes reuptake these excess fatty acids from neurons via an ApoE‐associated mechanism to protect hyperactive neurons against fatty acid toxicity [94]. Through this process, astrocytes might store energy in the form of lipid droplets to protect against hyperactivity‐induced synaptic toxicity, which is important for maintaining hippocampal functions. Moreover, lipoprotein‐enriched astrocytes are essential for the activity‐dependent remodeling of synaptic structures; this is because the synaptogenesis requires massive cholesterol production and delivery, which are critically dependent on ApoE‐containing lipoproteins in astrocytes [65, 95]. Accordingly, a very recent study revealed that a distinct class of microRNAs in astrocyte‐derived ApoE particles specifically inhibits the neuronal cholesterol biosynthesis pathways upon their uptake by nearby neurons; this consequently promotes histone acetylation in these neurons, which initiates the transcription of certain genes. Of note, such epigenetic modification facilitated by the treatment of ApoE particles is required for the expression of multiple neuronal immediate early genes during the learning process, which subsequently enhances memory consolidation in mice [96]. Meanwhile, depriving hippocampal astrocytes of ApoE abolishes the neuronal histone acetylation, leading to the inhibition of the transcriptional activation of neuronal activity‐induced genes in hippocampal neurons; this ultimately results in synaptic dysfunctions and memory deficits in adult mice [96, 97, 98].
Regulation of indirect astrocyte–synapse interactions
Astrocytes maintain the functioning of hippocampal circuits not only by directly interacting with synapses but also by communicating with other neural cells including microglia and endothelial cells. Interestingly, microglia can regulate neuronal activity through their response to ATP [99]. Specifically, in response to increased neuronal activity, neurons and astrocytes secrete ATP, which triggers microglial recruitment to the activated synapses in the adult striatum [99]. Microglia then convert the secreted ATP into adenosine; the subsequent binding of adenosine to the neuronal adenosine A1 receptors inhibits further synaptic transmission by limiting presynaptic neurotransmitter release and suppressing postsynaptic responses, thus protecting the brain against excessive activation [99, 100]. Concomitantly, conditional knockout of microglia P2Y12 receptors (P2Y12Rs) in the adult hippocampus induces abnormally elevated neuronal activity in CA1 neurons, suggesting that microglia directly regulate CA1 neuronal excitability via their P2Y12R‐mediated responses to ATP in the hippocampal circuitry [101]. Given that neuronal activation in the adult hippocampus is associated with local neuronal and astrocytic ATP secretion [32, 38, 39, 99], astrocytes might communicate with microglia via ATP to modulate neuronal activity levels to maintain hippocampal network homeostasis. Such a feedback mechanism demonstrates the wide range of crosstalk among astrocytes, microglia, and neurons during learning‐induced neuronal activity changes.
In addition, astrocytes interact with vascular endothelial cells via their perivascular end‐feet to form the blood–brain barrier [102, 103, 104, 105]. In response to changes in neuronal activity, elevated astrocytic Ca2+ leads to the secretion of several vasodilators, which facilitate activity‐induced microcirculation. Through this mechanism, astrocytes serve as exchange sites for ions, metabolites, and energy substrates from the blood to the brain, thereby maintaining the activity and functioning of the hippocampal circuitry [103, 104]. RNA sequencing analysis revealed that the activation or inhibition of glutamatergic neurons via chemogenetic approaches significantly regulates the expression of genes associated with blood–brain barrier efflux transport and circadian rhythms in brain endothelial cells [106]. Given the close anatomical proximity between endothelial cells and perivascular astrocytes at the blood–brain barrier, astrocytes probably sense changes in neuronal activity and send signals to endothelial cells to induce such activity‐dependent transcriptional changes. Hence, astrocytes are likely the key mediators that induce the neuronal activity‐dependent changes in microglia and endothelial cells, which in turn act on neurons to control the activity and functioning of the adult hippocampal circuitry.
Subregion‐specific regulation of astrocyte–synapse interactions in the adult hippocampal circuits
Heterogeneity of hippocampal astrocytes
The astrocyte–synapse interactions in the adult hippocampus are highly dynamic, and specific astrocytic signaling is regulated only in certain hippocampal subregions or circuits. The heterogeneity of such astrocytic responses is attributed to their distinct activity‐dependent morphological and transcriptional changes. Accordingly, astrocytes within different hippocampal subregions exhibit distinct transcriptomic signatures, which are regulated by specific transcription factors such as NFIA as well as the modulation of dynamic chromatin modifications. Such regulation subsequently contributes to the distinct differential functional responses of hippocampal astrocytes upon neuronal activity changes [62, 107]. Moreover, compared to CA3 astrocytes, the processes of hippocampal CA1 astrocytes have closer contacts with excitatory synapses [25]. Therefore, astrocytes in the CA1 and CA3 subregions respond differently to neuronal activity: a single synaptic stimulation is sufficient to stimulate a calcium signal in CA1 stratum radiatum astrocytes [39, 108], whereas a stronger stimulus (e.g., a tense burst of action potential) is required to activate CA3 stratum lucidum astrocytes [109]. These distinct features of hippocampal astrocytes might lead to the differential expression of specific factors in response to neuronal activity changes. Even in the CA1 subregion, astrocyte–synapse interactions only occur in some defined synapses and astrocytic processes [87, 110], suggesting that astrocytes can sense neuronal activity changes in a synapse‐specific manner. Thus, such distinct regulation of astrocytic responses might explain how astrocytes regulate specific learning‐related structural rearrangements in the hippocampal CA3–CA1 synapses. Further analysis of the functional, morphological, and transcriptomic properties of different hippocampal astrocytes at the single‐cell level will advance our understanding of the activity‐dependent, region‐specific interactions between astrocytes and synapses in the hippocampal circuits.
CA1 astrocytes in memory
The importance of astrocytes in long‐term synaptic plasticity and normal memory performance is well established. One key finding is that the interruption of astrocytic activity impairs memory responses [13, 111]. Of note, studies using chemogenetic GPCR platforms have shown that astrocyte activation is sufficient to enhance synaptic plasticity and improve memory performance. Accordingly, Gq‐stimulated activation of hippocampal astrocytes in the CA1 subregion is sufficient to potentiate synaptic transmission at CA3–CA1 excitatory synapses and consequently enhances recent memory acquisition in rodents when coupled with learning [112]. Meanwhile, modulating the activity of CA1 astrocytes by Gi activation, which mimics their response to GABAergic stimuli [113], selectively impairs remote (but not recent) memory recall by affecting the functional connectivity of CA1 to the anterior cingulate cortex (a frontal cortical region) [111]. This finding suggests that CA1 astrocytic activity is important for the coordinated activities of the hippocampus and cortex during remote memory acquisition. Given that the activation of different GPCRs (i.e., Gq and Gi) in hippocampal astrocytes is associated with distinct modes of glutamate release via astrocytic ion channels [36], these chemogenetically stimulated astrocytes might regulate hippocampal synaptic transmission and plasticity via glutamate‐dependent mechanisms during such experience‐driven memory processes. Hence, astrocytes specifically modulate different aspects of hippocampus‐dependent memory processes in the adult brain.
Dysregulation of astrocyte–synapse signaling in Alzheimer's disease
Synaptic dysfunction is believed to be a key mechanism that leads to hippocampus‐dependent memory impairment in AD along with the deposition of misfolded beta‐amyloid (Aβ) peptides and the formation of neurofibrillary tau tangles. Studies involving transcriptomic analysis have identified reactive astrocyte populations involved in synaptic pathology during AD progression [114, 115, 116, 117, 118, 119, 120], namely a neurotoxic reactive astrocyte population in AD, termed ‘A1 astrocytes’ [16, 121, 122]. More recently, a single‐nucleus RNA sequencing study of hippocampal cells identified a new and unique astrocyte population in the AD transgenic model mice (5xFAD), termed ‘disease‐associated astrocytes’ [123]. The signature genes of these disease‐associated astrocytes are associated with specific pathways including metabolic pathways (i.e., lipid and cholesterol pathways), inflammatory responses, and complement cascades [123]. Of note, our RNA sequencing results revealed that 67 of 239 disease‐associated astrocyte signature genes are highly regulated by neuronal activity in cultured hippocampal cells [60, 123]. Specifically, 10 of these genes encode secreted proteins: Lgals1, Timp2, Cst3, Igfbp5, Mmp16, Smpdl3a, Agt, Itm2b, Serpina3n, and Vwa1 (Table 2) [124]. This disease‐associated astrocyte population also exhibits reduced expression of some important genes involved in neuronal support and communication, such as Slc7a10 and Trpm3 [121, 123], which might further contribute to the dysregulated astrocyte–synapse interactions in AD. Concomitantly, patients with mild cognitive impairment (who have an increased risk of developing AD) exhibit aberrant excitatory neuronal activity in the hippocampus [125, 126], which might cause the observed changes in astrocyte signatures in the early stage of AD. As such, it will be interesting to determine whether the dysregulation of activity‐dependent astrocyte–synapse interactions contributes to hippocampal synaptic pathology in AD.
Table 2.
RNA sequencing analysis of disease‐associated astrocyte signature genes that encode secreted factors regulated by neuronal activity [60, 123]. FPKM, fragments per kilobase million.
| Gene name | Log2 fold change | Adjusted P value | Mean_FPKM_Con | Mean_FPKM_TTX | Mean_Count_Con | Mean_Count_TTX |
|---|---|---|---|---|---|---|
| Lgals1 | 0.78 | 4.18E‐42 | 37.15 | 64.39 | 1253 | 2262 |
| Timp2 | 0.59 | 1.74E‐59 | 209.14 | 318.25 | 23 071 | 36 745 |
| Cst3 | 0.56 | 8.27E‐43 | 3109.66 | 4636.78 | 150 318 | 233 906 |
| Igfbp5 | 0.48 | 1.25E‐07 | 105.91 | 149.30 | 11 548 | 16 873 |
| Mmp16 | 0.45 | 2.32E‐40 | 13.67 | 18.87 | 3210 | 4646 |
| Smpdl3a | 0.43 | 2.04E‐25 | 44.46 | 60.57 | 4915 | 6998 |
| Agt | 0.40 | 1.73E‐05 | 69.59 | 92.49 | 9020 | 12 453 |
| Itm2b | 0.39 | 2.55E‐55 | 608.37 | 806.41 | 71 250 | 98 791 |
| Serpina3n | −1.04 | 4.12E‐05 | 22.90 | 11.28 | 3093 | 1625 |
| Vwa1 | −1.70 | 7.64E‐111 | 11.49 | 3.55 | 1779 | 582 |
Dysregulation of complement protein C3 signaling
Studies involving transcriptomic analysis have revealed that the expression of complement protein C3 is elevated in disease‐associated astrocytes in AD transgenic model mice (including APP/TTA and PS2APP mice) and patients with AD [16, 121, 127]. Specifically, astrocyte‐secreted C3 disrupts dendritic morphology and network functioning in the adult mouse hippocampus via the postsynaptic C3 receptor during AD progression [16]. Moreover, during development, C3 is recognized by the microglial C3 receptor, which is required for microglia‐mediated synaptic elimination, termed ‘synaptic pruning’ [128, 129]. Hence, the dysregulated expression of complement molecules in disease‐associated astrocytes might be responsible for the aberrant synaptic pruning by microglia during AD progression, which would partly explain the synaptic loss and cognitive decline observed in AD [127, 130, 131]. Accordingly, inhibiting such abnormal C3 signaling by administration of C3 receptor antagonists into APP/TTA transgenic mice improves cognitive performance [16].
Dysregulation of ApoE4 signaling
APOE is another top candidate gene identified in disease‐associated astrocytes. APOE has three major alleles in the human brain: APOE‐ɛ2, APOE‐ɛ3, and APOE‐ɛ4. The APOE‐ɛ4 variant is a well‐known genetic risk factor for AD that reportedly increases the risk of AD by 3–15 times compared to noncarriers [116, 117]. Other than coding variants, we also identified several noncoding variants that reside in regions near APOE that confer APOE‐independent AD risk and potentially modify brain APOE expression [132]. In AD, ApoE is thought to play an important role related to Aβ clearance: ApoE4 significantly attenuates Aβ clearance compared to that with ApoE3 or ApoE2, thus accelerating the amyloid pathology. Indeed, different APOE alleles also differentially control the rate of astrocyte‐mediated synaptic pruning in the brain: knock‐in of APOE‐ɛ2 and APOE‐ɛ4, respectively, potentiate and prevent efficient synapse phagocytosis by astrocytes in vivo through the regulation of the phagocytic capacity of astrocytes [133]. A very recent study on induced pluripotent stem cell‐derived organoids from patients with AD also supports the notion that ApoE4 is involved in the synaptic loss and neurodegeneration observed in AD, while conversion of ApoE4 to ApoE3 in these organoids would attenuate the AD‐related phenotypes [134].
Dysregulation of IL‐33/ST2 signaling
Human genetic mapping has identified additional astrocyte‐enriched genes in AD [114, 115, 116, 117, 118, 119, 120]. For example, IL‐33/ST2 signaling is closely associated with AD progression: patients with AD exhibit decreased transcript levels of IL‐33 in the brain [114], while levels of soluble ST2 (the IL‐33 decoy receptor) are elevated in patients with mild cognitive impairment [135]. Given the importance of astrocytic IL‐33 signaling in neuronal activity‐dependent excitatory synapse formation [60], a decrease in such IL‐33/ST2 signaling is likely to impair activity‐induced synaptic adaption, causing imbalanced synaptic activity in AD. Concordantly, replenishing IL‐33 ameliorates synaptic plasticity deficits and cognitive impairment in the APP/PS1 transgenic mouse model of AD [135].
Dysregulation of TSP‐1 signaling
Besides IL‐33, a panel of astrocyte‐secreted synaptogenic factors is also dysregulated in AD [123, 136, 137]. For example, TSP‐1, a matrix protein synthesized and released by astrocytes that is well known as a regulator of synaptogenesis [50], exhibits reduced expression in the brains of both AD model mice (i.e., Tg2576 and 5XFAD mice) and patients with AD [137]. Accordingly, administration of TSP‐1 into the brains of these AD model mice rescues synaptic deficits in AD, including decreased dendritic spine density and reduced synaptic activity in hippocampal neurons, through the activation of the TSP‐1 neuronal receptor α2δ1 [137]. Concordantly, a more recent study revealed a protective role of TSP‐1 in Aβ‐induced mitochondrial damage in hippocampal cells [138]. Given that synaptic deficits in AD are closely associated with mitochondrial disruptions [139], the impaired astrocytic TSP‐1 signaling observed in AD might contribute to the synaptic pathology of AD not only because of its important roles in astrocyte–synapse interactions but also because of its involvement in balancing mitochondrial dynamics.
Conclusion and perspectives
Here, we have summarized the literature showing how astrocytes respond to changes in experience and regulate neuronal activity‐dependent synaptic plasticity in the hippocampus as well as the literature elucidating how altered astrocyte–synapse interactions contribute to neurodegenerative diseases. Further studies are warranted to define the molecular mechanisms underlying the neuronal activity‐dependent interactions between astrocytes and neurons in the hippocampus—for example, single‐cell transcriptome profiling of hippocampal astrocytes. In addition, two‐photon imaging and optogenetic approaches in live animals might help link the dynamic changes in hippocampal astrocytes with the experience‐driven synaptic changes in specific hippocampal circuits. Furthermore, the involvement of astrocytes in information processing during hippocampus‐dependent memory is only beginning to be unraveled. With the development of better technologies and experimental protocols, future studies will advance our understanding of the astrocyte‐specific responses during different learning and cognitive processes.
In addition, emerging studies indicate that astrocytic dysfunctions might contribute to the synaptic failure and impaired synaptic plasticity in neurodegenerative diseases. Therefore, a better understanding of the dysregulation of the neuronal activity‐dependent astrocyte–synapse interactions in the hippocampus during neurodegeneration and the resultant imbalanced network stability will provide insights into the pathological mechanisms underlying the synaptic dysfunctions in these diseases.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
YW, AKYF, and NYI all contributed to the drafting and editing of the manuscript.
Acknowledgements
We apologize to the researchers whose studies could not be discussed or cited because of space limitations. We thank Ka‐Chun Lok for preparing the figures as well as members of the Ip Laboratory for many helpful discussions. Work performed in the Ip Laboratory was supported in part by the Research Grants Council of Hong Kong (16149616, 16102717, and 16102019; Collaborative Research Fund C6027‐19GF), the National Key R&D Program of China (2017YFE0190000 and 2018YFE0203600), the Area of Excellence Scheme of the University Grants Committee (AoE/M‐604/16), the Theme‐based Research Scheme (T13‐605/18W), the Innovation and Technology Commission (ITCPD/17‐9), the Guangdong Provincial Key S&T Program (2018B030336001), and the Shenzhen Knowledge Innovation Program (JCYJ20180507183642005 and JCYJ20170413173717055).
References
- 1. Magee JC & Grienberger C (2020) Synaptic plasticity forms and functions. Annu Rev Neurosci 43, 95–117. [DOI] [PubMed] [Google Scholar]
- 2. Citri A & Malenka RC (2008) Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology 33, 18–41. [DOI] [PubMed] [Google Scholar]
- 3. Bannerman DM, Sprengel R, Sanderson DJ, McHugh SB, Rawlins JN, Monyer H & Seeburg PH (2014) Hippocampal synaptic plasticity, spatial memory and anxiety. Nat Rev Neurosci 15, 181–192. [DOI] [PubMed] [Google Scholar]
- 4. Malenka RC & Bear MF (2004) LTP and LTD: an embarrassment of riches. Neuron 44, 5–21. [DOI] [PubMed] [Google Scholar]
- 5. Turrigiano GG (2008) The self‐tuning neuron: synaptic scaling of excitatory synapses. Cell 135, 422–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pozo K & Goda Y (2010) Unraveling mechanisms of homeostatic synaptic plasticity. Neuron 66, 337–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen Y, Fu AKY & Ip NY (2019) Synaptic dysfunction in Alzheimer's disease: mechanisms and therapeutic strategies. Pharmacol Ther 195, 186–198. [DOI] [PubMed] [Google Scholar]
- 8. Martella G, Bonsi P, Johnson SW & Quartarone A (2018) Synaptic plasticity changes: hallmark for neurological and psychiatric disorders. Neural Plast 2018, 9230704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Allen NJ & Lyons DA (2018) Glia as architects of central nervous system formation and function. Science 362, 181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Perea G, Navarrete M & Araque A (2009) Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci 32, 421–431. [DOI] [PubMed] [Google Scholar]
- 11. Perez‐Alvarez A, Navarrete M, Covelo A, Martin ED & Araque A (2014) Structural and functional plasticity of astrocyte processes and dendritic spine interactions. J Neurosci 34, 12738–12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Allen NJ (2014) Astrocyte regulation of synaptic behavior. Annu Rev Cell Dev Biol 30, 439–463. [DOI] [PubMed] [Google Scholar]
- 13. Santello M, Toni N & Volterra A (2019) Astrocyte function from information processing to cognition and cognitive impairment. Nat Neurosci 22, 154–166. [DOI] [PubMed] [Google Scholar]
- 14. Lee E & Chung WS (2019) Glial control of synapse number in healthy and diseased brain. Front Cell Neurosci 13, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gonzalez‐Reyes RE, Nava‐Mesa MO, Vargas‐Sanchez K, Ariza‐Salamanca D & Mora‐Munoz L (2017) Involvement of astrocytes in Alzheimer's disease from a neuroinflammatory and oxidative stress perspective. Front Mol Neurosci 10, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lian H, Yang L, Cole A, Sun L, Chiang AC, Fowler SW, Shim DJ, Rodriguez‐Rivera J, Taglialatela G, Jankowsky JL et al. (2015) NFkappaB‐activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer's disease. Neuron 85, 101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Henstridge CM, Tzioras M & Paolicelli RC (2019) Glial contribution to excitatory and inhibitory synapse loss in neurodegeneration. Front Cell Neurosci 13, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Basu J & Siegelbaum SA (2015) The corticohippocampal circuit, synaptic plasticity, and memory. Cold Spring Harb Perspect Biol 7, a021733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tonegawa S & McHugh TJ (2008) The ins and outs of hippocampal circuits. Neuron 57, 175–177. [DOI] [PubMed] [Google Scholar]
- 20. van Strien NM, Cappaert NL & Witter MP (2009) The anatomy of memory: an interactive overview of the parahippocampal‐hippocampal network. Nat Rev Neurosci 10, 272–282. [DOI] [PubMed] [Google Scholar]
- 21. Volpe BT, Davis HP, Towle A & Dunlap WP (1992) Loss of hippocampal CA1 pyramidal neurons correlates with memory impairment in rats with ischemic or neurotoxin lesions. Behav Neurosci 106, 457–464. [DOI] [PubMed] [Google Scholar]
- 22. Bartsch T, Schonfeld R, Muller FJ, Alfke K, Leplow B, Aldenhoff J, Deuschl G & Koch JM (2010) Focal lesions of human hippocampal CA1 neurons in transient global amnesia impair place memory. Science 328, 1412–1415. [DOI] [PubMed] [Google Scholar]
- 23. Allen NJ & Eroglu C (2017) Cell biology of astrocyte‐synapse interactions. Neuron 96, 697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Araque A, Carmignoto G, Haydon PG, Oliet SH, Robitaille R & Volterra A (2014) Gliotransmitters travel in time and space. Neuron 81, 728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dallerac G, Zapata J & Rouach N (2018) Versatile control of synaptic circuits by astrocytes: where, when and how? Nat Rev Neurosci 19, 729–743. [DOI] [PubMed] [Google Scholar]
- 26. Habas A, Hahn J, Wang X & Margeta M (2013) Neuronal activity regulates astrocytic Nrf2 signaling. Proc Natl Acad Sci USA 110, 18291–18296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Olsen ML, Khakh BS, Skatchkov SN, Zhou M, Lee CJ & Rouach N (2015) New insights on astrocyte ion channels: critical for homeostasis and neuron‐glia signaling. J Neurosci 35, 13827–13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sahlender DA, Savtchouk I & Volterra A (2014) What do we know about gliotransmitter release from astrocytes? Philos Trans R Soc Lond B Biol Sci 369, 20130592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Harada K, Kamiya T & Tsuboi T (2015) Gliotransmitter release from astrocytes: functional, developmental, and pathological implications in the brain. Front Neurosci 9, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Toth AB, Hori K, Novakovic MM, Bernstein NG, Lambot L & Prakriya M (2019) CRAC channels regulate astrocyte Ca(2+) signaling and gliotransmitter release to modulate hippocampal GABAergic transmission. Sci Signal 12, eaaw5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Covelo A & Araque A (2018) Neuronal activity determines distinct gliotransmitter release from a single astrocyte. Elife 7, e32237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, Jiang ZL, Wu CP, Poo MM & Duan S (2003) ATP released by astrocytes mediates glutamatergic activity‐dependent heterosynaptic suppression. Neuron 40, 971–982. [DOI] [PubMed] [Google Scholar]
- 33. Parpura V & Haydon PG (2000) Physiological astrocytic calcium levels stimulate glutamate release to modulate adjacent neurons. Proc Natl Acad Sci USA 97, 8629–8634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jourdain P, Bergersen LH, Bhaukaurally K, Bezzi P, Santello M, Domercq M, Matute C, Tonello F, Gundersen V & Volterra A (2007) Glutamate exocytosis from astrocytes controls synaptic strength. Nat Neurosci 10, 331–339. [DOI] [PubMed] [Google Scholar]
- 35. Perea G & Araque A (2007) Astrocytes potentiate transmitter release at single hippocampal synapses. Science 317, 1083–1086. [DOI] [PubMed] [Google Scholar]
- 36. Woo DH, Han KS, Shim JW, Yoon BE, Kim E, Bae JY, Oh SJ, Hwang EM, Marmorstein AD, Bae YC et al. (2012) TREK‐1 and Best1 channels mediate fast and slow glutamate release in astrocytes upon GPCR activation. Cell 151, 25–40. [DOI] [PubMed] [Google Scholar]
- 37. Park H, Han KS, Oh SJ, Jo S, Woo J, Yoon BE & Lee CJ (2013) High glutamate permeability and distal localization of Best1 channel in CA1 hippocampal astrocyte. Mol Brain 6, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pascual O, Casper KB, Kubera C, Zhang J, Revilla‐Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K & Haydon PG (2005) Astrocytic purinergic signaling coordinates synaptic networks. Science 310, 113–116. [DOI] [PubMed] [Google Scholar]
- 39. Panatier A, Vallee J, Haber M, Murai KK, Lacaille JC & Robitaille R (2011) Astrocytes are endogenous regulators of basal transmission at central synapses. Cell 146, 785–798. [DOI] [PubMed] [Google Scholar]
- 40. Serrano A, Haddjeri N, Lacaille JC & Robitaille R (2006) GABAergic network activation of glial cells underlies hippocampal heterosynaptic depression. J Neurosci 26, 5370–5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fontinha BM, Delgado‐Garcia JM, Madronal N, Ribeiro JA, Sebastiao AM & Gruart A (2009) Adenosine A(2A) receptor modulation of hippocampal CA3‐CA1 synapse plasticity during associative learning in behaving mice. Neuropsychopharmacology 34, 1865–1874. [DOI] [PubMed] [Google Scholar]
- 42. Todd KJ, Darabid H & Robitaille R (2010) Perisynaptic glia discriminate patterns of motor nerve activity and influence plasticity at the neuromuscular junction. J Neurosci 30, 11870–11882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rebola N, Lujan R, Cunha RA & Mulle C (2008) Adenosine A2A receptors are essential for long‐term potentiation of NMDA‐EPSCs at hippocampal mossy fiber synapses. Neuron 57, 121–134. [DOI] [PubMed] [Google Scholar]
- 44. Henneberger C, Papouin T, Oliet SH & Rusakov DA (2010) Long‐term potentiation depends on release of D‐serine from astrocytes. Nature 463, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Boue‐Grabot E & Pankratov Y (2017) Modulation of central synapses by astrocyte‐released ATP and postsynaptic P2X receptors. Neural Plast 2017, 9454275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yamazaki Y & Fujii S (2015) Extracellular ATP modulates synaptic plasticity induced by activation of metabotropic glutamate receptors in the hippocampus. Biomed Res 36, 1–9. [DOI] [PubMed] [Google Scholar]
- 47. Pougnet JT, Toulme E, Martinez A, Choquet D, Hosy E & Boue‐Grabot E (2014) ATP P2X receptors downregulate AMPA receptor trafficking and postsynaptic efficacy in hippocampal neurons. Neuron 83, 417–430. [DOI] [PubMed] [Google Scholar]
- 48. Pougnet JT, Compans B, Martinez A, Choquet D, Hosy E & Boue‐Grabot E (2016) P2X‐mediated AMPA receptor internalization and synaptic depression is controlled by two CaMKII phosphorylation sites on GluA1 in hippocampal neurons. Sci Rep 6, 31836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bonansco C, Couve A, Perea G, Ferradas CA, Roncagliolo M & Fuenzalida M (2011) Glutamate released spontaneously from astrocytes sets the threshold for synaptic plasticity. Eur J Neurosci 33, 1483–1492. [DOI] [PubMed] [Google Scholar]
- 50. Christopherson KS, Ullian EM, Stokes CCA, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P & Barres BA (2005) Thrombospondins are astrocyte‐secreted proteins that promote CNS synaptogenesis. Cell 120, 421–433. [DOI] [PubMed] [Google Scholar]
- 51. Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD et al. (2009) Gabapentin receptor alpha 2 delta‐1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139, 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xu JY, Xiao N & Xia J (2010) Thrombospondin 1 accelerates synaptogenesis in hippocampal neurons through neuroligin 1. Nat Neurosci 13, 22–24. [DOI] [PubMed] [Google Scholar]
- 53. Stellwagen D & Malenka RC (2006) Synaptic scaling mediated by glial TNF‐alpha. Nature 440, 1054–1059. [DOI] [PubMed] [Google Scholar]
- 54. Pribiag H & Stellwagen D (2013) TNF‐alpha downregulates inhibitory neurotransmission through protein phosphatase 1‐dependent trafficking of GABA(A) receptors. J Neurosci 33, 15879–15893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS & Malenka RC (2002) Control of synaptic strength by glial TNFalpha. Science 295, 2282–2285. [DOI] [PubMed] [Google Scholar]
- 56. Stellwagen D, Beattie EC, Seo JY & Malenka RC (2005) Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor‐alpha. J Neurosci 25, 3219–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Heir R & Stellwagen D (2020) TNF‐mediated homeostatic synaptic plasticity: from in vitro to in vivo models. Front Cell Neurosci 14, 565841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kaneko M, Stellwagen D, Malenka RC & Stryker MP (2008) Tumor necrosis factor‐alpha mediates one component of competitive, experience‐dependent plasticity in developing visual cortex. Neuron 58, 673–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Greenhill SD, Ranson A & Fox K (2015) Hebbian and homeostatic plasticity mechanisms in regular spiking and intrinsic bursting cells of cortical layer 5. Neuron 88, 539–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang Y, Fu W‐Y, Cheung K, Hung K‐W, Chen C, Geng H, Yung W‐H, Qu JY, Fu AKY & Ip NY (2021) Astrocyte‐secreted IL‐33 mediates homeostatic synaptic plasticity in the adult hippocampus. Proc Natl Acad Sci USA 118, e2020810118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liew FY, Girard JP & Turnquist HR (2016) Interleukin‐33 in health and disease. Nat Rev Immunol 16, 676–689. [DOI] [PubMed] [Google Scholar]
- 62. Batiuk MY, Martirosyan A, Wahis J, de Vin F, Marneffe C, Kusserow C, Koeppen J, Viana JF, Oliveira JF, Voet T et al. (2020) Identification of region‐specific astrocyte subtypes at single cell resolution. Nat Commun 11, 1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schaukowitch K, Reese AL, Kim SK, Kilaru G, Joo JY, Kavalali ET & Kim TK (2017) An intrinsic transcriptional program underlying synaptic scaling during activity suppression. Cell Rep 18, 1512–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Allen NJ, Bennett ML, Foo LC, Wang GX, Chakraborty C, Smith SJ & Barres BA (2012) Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 486, 410–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kucukdereli H, Allen NJ, Lee AT, Feng A, Ozlu MI, Conatser LM, Chakraborty C, Workman G, Weaver M, Sage EH et al. (2011) Control of excitatory CNS synaptogenesis by astrocyte‐secreted proteins Hevin and SPARC. Proc Natl Acad Sci USA 108, E440–E449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Blanco‐Suarez E, Liu TF, Kopelevich A & Allen NJ (2018) Astrocyte‐secreted chordin‐like 1 drives synapse maturation and limits plasticity by increasing synaptic GluA2 AMPA receptors. Neuron 100, 1116–1132 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chung WS, Allen NJ & Eroglu C (2015) Astrocytes control synapse formation, function, and elimination. Cold Spring Harb Perspect Biol 7, a020370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Singh SK, Stogsdill JA, Pulimood NS, Dingsdale H, Kim YH, Pilaz LJ, Kim IH, Manhaes AC, Rodrigues WS Jr, Pamukcu A et al. (2016) Astrocytes assemble thalamocortical synapses by bridging NRX1alpha and NL1 via Hevin. Cell 164, 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Flavell SW & Greenberg ME (2008) Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu Rev Neurosci 31, 563–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lozzi B, Huang TW, Sardar D, Huang AY & Deneen B (2020) Regionally distinct astrocytes display unique transcription factor profiles in the adult brain. Front Neurosci 14, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Huang AY, Woo J, Sardar D, Lozzi B, Bosquez Huerta NA, Lin CJ, Felice D, Jain A, Paulucci‐Holthauzen A & Deneen B (2020) Region‐specific transcriptional control of astrocyte function oversees local circuit activities. Neuron 106, 992–1008 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Klemm SL, Shipony Z & Greenleaf WJ (2019) Chromatin accessibility and the regulatory epigenome. Nat Rev Genet 20, 207–220. [DOI] [PubMed] [Google Scholar]
- 73. Garay PM, Chen A, Tsukahara T, Rodriguez Diaz JC, Kohen R, Althaus JC, Wallner MA, Giger RJ, Jones KS, Sutton MA et al. (2020) RAI1 regulates activity‐dependent nascent transcription and synaptic scaling. Cell Rep 32, 108002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mao WJ, Salzberg AC, Uchigashima M, Hasegawa Y, Hock H, Watanabe M, Akbarian S, Kawasawa YI & Futai K (2018) Activity‐induced regulation of synaptic strength through the chromatin reader L3mbtl1. Cell Rep 23, 3209–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kim MJ, Futai K, Jo J, Hayashi Y, Cho K & Sheng M (2007) Synaptic accumulation of PSD‐95 and synaptic function regulated by phosphorylation of serine‐295 of PSD‐95. Neuron 56, 488–502. [DOI] [PubMed] [Google Scholar]
- 76. Kim E & Sheng M (2004) PDZ domain proteins of synapses. Nat Rev Neurosci 5, 771–781. [DOI] [PubMed] [Google Scholar]
- 77. Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, Liddelow SA, Nguyen PT, Nakao‐Inoue H, Dorman LC et al. (2018) Astrocyte‐derived interleukin‐33 promotes microglial synapse engulfment and neural circuit development. Science 359, 1269–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nguyen PT, Dorman LC, Pan S, Vainchtein ID, Han RT, Nakao‐Inoue H, Taloma SE, Barron JJ, Molofsky AB, Kheirbek MA et al. (2020) Microglial remodeling of the extracellular matrix promotes synapse plasticity. Cell 182, 388–403 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Baldwin KT & Eroglu C (2017) Molecular mechanisms of astrocyte‐induced synaptogenesis. Curr Opin Neurobiol 45, 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Carmona MA, Murai KK, Wang L, Roberts AJ & Pasquale EB (2009) Glial ephrin‐A3 regulates hippocampal dendritic spine morphology and glutamate transport. Proc Natl Acad Sci USA 106, 12524–12529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Garrett AM & Weiner JA (2009) Control of CNS synapse development by {gamma}‐protocadherin‐mediated astrocyte‐neuron contact. J Neurosci 29, 11723–11731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Koeppen J, Nguyen AQ, Nikolakopoulou AM, Garcia M, Hanna S, Woodruff S, Figueroa Z, Obenaus A & Ethell IM (2018) Functional consequences of synapse remodeling following astrocyte‐specific regulation of Ephrin‐B1 in the adult hippocampus. J Neurosci 38, 5710–5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nguyen AQ, Koeppen J, Woodruff S, Mina K, Figueroa Z & Ethell IM (2020) Astrocytic Ephrin‐B1 controls synapse formation in the hippocampus during learning and memory. Front Synaptic Neurosci 12, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Trotter JH, Dargaei Z, Wöhr M, Liakath‐Ali K, Raju K, Essayan‐Perez S, Nabet A, Liu X & Südhof TC (2020) Astrocytic neurexin‐1 orchestrates functional synapse assembly. bioRxiv 2020.08.21.262097 [PREPRINT]. [Google Scholar]
- 85. Filosa A, Paixao S, Honsek SD, Carmona MA, Becker L, Feddersen B, Gaitanos L, Rudhard Y, Schoepfer R, Klopstock T et al. (2009) Neuron‐glia communication via EphA4/ephrin‐A3 modulates LTP through glial glutamate transport. Nat Neurosci 12, 1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Fu AK, Hung KW, Fu WY, Shen C, Chen Y, Xia J, Lai KO & Ip NY (2011) APC(Cdh1) mediates EphA4‐dependent downregulation of AMPA receptors in homeostatic plasticity. Nat Neurosci 14, 181–189. [DOI] [PubMed] [Google Scholar]
- 87. Bernardinelli Y, Randall J, Janett E, Nikonenko I, Konig S, Jones EV, Flores CE, Murai KK, Bochet CG, Holtmaat A et al. (2014) Activity‐dependent structural plasticity of perisynaptic astrocytic domains promotes excitatory synapse stability. Curr Biol 24, 1679–1688. [DOI] [PubMed] [Google Scholar]
- 88. Fu WY, Chen Y, Sahin M, Zhao XS, Shi L, Bikoff JB, Lai KO, Yung WH, Fu AK, Greenberg ME et al. (2007) Cdk5 regulates EphA4‐mediated dendritic spine retraction through an ephexin 1‐dependent mechanism. Nat Neurosci 10, 67–76. [DOI] [PubMed] [Google Scholar]
- 89. Chen Y, Fu AK & Ip NY (2012) Eph receptors at synapses: implications in neurodegenerative diseases. Cell Signal 24, 606–611. [DOI] [PubMed] [Google Scholar]
- 90. Barber CN & Raben DM (2019) Lipid metabolism crosstalk in the brain: glia and neurons. Front Cell Neurosci 13, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Van Deijk ALF, Camargo N, Timmerman J, Heistek T, Brouwers JF, Mogavero F, Mansvelder HD, Smit AB & Verheijen MHG (2017) Astrocyte lipid metabolism is critical for synapse development and function in vivo. Glia 65, 670–682. [DOI] [PubMed] [Google Scholar]
- 92. Poirier J, Miron J, Picard C, Gormley P, Theroux L, Breitner J & Dea D (2014) Apolipoprotein E and lipid homeostasis in the etiology and treatment of sporadic Alzheimer's disease. Neurobiol Aging 35 (Suppl 2), S3–S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lane RM & Farlow MR (2005) Lipid homeostasis and apolipoprotein E in the development and progression of Alzheimer's disease. J Lipid Res 46, 949–968. [DOI] [PubMed] [Google Scholar]
- 94. Ioannou MS, Jackson J, Sheu SH, Chang CL, Weigel AV, Liu H, Pasolli HA, Xu CS, Pang S, Matthies D et al. (2019) Neuron‐astrocyte metabolic coupling protects against activity‐induced fatty acid toxicity. Cell 177, 1522–1535 e14. [DOI] [PubMed] [Google Scholar]
- 95. Mauch DH, Nagler K, Schumacher S, Goritz C, Muller EC, Otto A & Pfrieger FW (2001) CNS synaptogenesis promoted by glia‐derived cholesterol. Science 294, 1354–1357. [DOI] [PubMed] [Google Scholar]
- 96. Li X, Zhang J, Li D, He C, He K, Xue T, Wan L, Zhang C & Liu Q (2021) Astrocytic ApoE reprograms neuronal cholesterol metabolism and histone‐acetylation‐mediated memory. Neuron 109, 957–970.e8. [DOI] [PubMed] [Google Scholar]
- 97. Lane‐Donovan C, Wong WM, Durakoglugil MS, Wasser CR, Jiang S, Xian X & Herz J (2016) Genetic restoration of plasma ApoE improves cognition and partially restores synaptic defects in ApoE‐deficient mice. J Neurosci 36, 10141–10150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Masliah E, Mallory M, Ge N, Alford M, Veinbergs I & Roses AD (1995) Neurodegeneration in the central nervous system of apoE‐deficient mice. Exp Neurol 136, 107–122. [DOI] [PubMed] [Google Scholar]
- 99. Badimon A, Strasburger HJ, Ayata P, Chen X, Nair A, Ikegami A, Hwang P, Chan AT, Graves SM, Uweru JO et al. (2020) Negative feedback control of neuronal activity by microglia. Nature 586, 417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Dunwiddie TV & Masino SA (2001) The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci 24, 31–55. [DOI] [PubMed] [Google Scholar]
- 101. Peng J, Liu Y, Umpierre AD, Xie M, Tian D‐S, Richardson JR & Wu L‐J (2019) Microglial P2Y12 receptor regulates ventral hippocampal CA1 neuronal excitability and innate fear in mice. Mol Brain 12, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Nortley R & Attwell D (2017) Control of brain energy supply by astrocytes. Curr Opin Neurobiol 47, 80–85. [DOI] [PubMed] [Google Scholar]
- 103. Patel DC, Tewari BP, Chaunsali L & Sontheimer H (2019) Neuron‐glia interactions in the pathophysiology of epilepsy. Nat Rev Neurosci 20, 282–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Belanger M, Allaman I & Magistretti PJ (2011) Brain energy metabolism: focus on astrocyte‐neuron metabolic cooperation. Cell Metab 14, 724–738. [DOI] [PubMed] [Google Scholar]
- 105. Abbott NJ (2002) Astrocyte‐endothelial interactions and blood‐brain barrier permeability. J Anat 200, 629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Pulido RS, Munji RN, Chan TC, Quirk CR, Weiner GA, Weger BD, Rossi MJ, Elmsaouri S, Malfavon M, Deng A et al. (2020) Neuronal activity regulates blood‐brain barrier efflux transport through endothelial circadian genes. Neuron 108, 937–952 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Miller SJ (2018) Astrocyte heterogeneity in the adult central nervous system. Front Cell Neurosci 12, 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Bernardinelli Y, Salmon C, Jones EV, Farmer WT, Stellwagen D & Murai KK (2011) Astrocytes display complex and localized calcium responses to single‐neuron stimulation in the hippocampus. J Neurosci 31, 8905–8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Haustein MD, Kracun S, Lu XH, Shih T, Jackson‐Weaver O, Tong X, Xu J, Yang XW, O'Dell TJ, Marvin JS et al. (2014) Conditions and constraints for astrocyte calcium signaling in the hippocampal mossy fiber pathway. Neuron 82, 413–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Di Castro MA, Chuquet J, Liaudet N, Bhaukaurally K, Santello M, Bouvier D, Tiret P & Volterra A (2011) Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat Neurosci 14, 1276–1284. [DOI] [PubMed] [Google Scholar]
- 111. Kol A, Adamsky A, Groysman M, Kreisel T, London M & Goshen I (2020) Astrocytes contribute to remote memory formation by modulating hippocampal‐cortical communication during learning. Nat Neurosci 23, 1229–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Adamsky A, Kol A, Kreisel T, Doron A, Ozeri‐Engelhard N, Melcer T, Refaeli R, Horn H, Regev L, Groysman M et al. (2018) Astrocytic activation generates de novo neuronal potentiation and memory enhancement. Cell 174, 59–71 e14. [DOI] [PubMed] [Google Scholar]
- 113. Durkee CA, Covelo A, Lines J, Kofuji P, Aguilar J & Araque A (2019) Gi/o protein‐coupled receptors inhibit neurons but activate astrocytes and stimulate gliotransmission. Glia 67, 1076–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Chapuis J, Hot D, Hansmannel F, Kerdraon O, Ferreira S, Hubans C, Maurage CA, Huot L, Bensemain F, Laumet G et al. (2009) Transcriptomic and genetic studies identify IL‐33 as a candidate gene for Alzheimer's disease. Mol Psychiatr 14, 1004–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Yu JT, Song JH, Wang ND, Wu ZC, Zhang Q, Zhang N, Zhang W, Xuan SY & Tan L (2012) Implication of IL‐33 gene polymorphism in Chinese patients with Alzheimer's disease. Neurobiol Aging 33, 1014 e11‐4. [DOI] [PubMed] [Google Scholar]
- 116. Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL & Pericak‐Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261, 921–923. [DOI] [PubMed] [Google Scholar]
- 117. Lange KL, Bondi MW, Salmon DP, Galasko D, Delis DC, Thomas RG & Thal LJ (2002) Decline in verbal memory during preclinical Alzheimer's disease: examination of the effect of APOE genotype. J Int Neuropsychol Soc 8, 943–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Korvatska O, Leverenz JB, Jayadev S, McMillan P, Kurtz I, Guo X, Rumbaugh M, Matsushita M, Girirajan S, Dorschner MO et al. (2015) R47H variant of TREM2 associated With Alzheimer disease in a large late‐onset family: clinical, genetic, and neuropathological study. JAMA Neurol 72, 920–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Schlepckow K, Kleinberger G, Fukumori A, Feederle R, Lichtenthaler SF, Steiner H & Haass C (2017) An Alzheimer‐associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO Mol Med 9, 1356–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Chung WS, Welsh CA, Barres BA & Stevens B (2015) Do glia drive synaptic and cognitive impairment in disease? Nat Neurosci 18, 1539–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC et al. (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Yun SP, Kam TI, Panicker N, Kim S, Oh Y, Park JS, Kwon SH, Park YJ, Karuppagounder SS, Park H et al. (2018) Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson's disease. Nat Med 24, 931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Habib N, McCabe C, Medina S, Varshavsky M, Kitsberg D, Dvir‐Szternfeld R, Green G, Dionne D, Nguyen L, Marshall JL et al. (2020) Disease‐associated astrocytes in Alzheimer's disease and aging. Nat Neurosci 23, 701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Meinken J, Walker G, Cooper CR & Min XJ (2015) MetazSecKB: the human and animal secretome and subcellular proteome knowledgebase. Database (Oxford) 2015, bav077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Dickerson BC, Salat DH, Greve DN, Chua EF, Rand‐Giovannetti E, Rentz DM, Bertram L, Mullin K, Tanzi RE, Blacker D et al. (2005) Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology 65, 404–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Yassa MA, Stark SM, Bakker A, Albert MS, Gallagher M & Stark CE (2010) High‐resolution structural and functional MRI of hippocampal CA3 and dentate gyrus in patients with amnestic mild cognitive impairment. NeuroImage 51, 1242–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Wu T, Dejanovic B, Gandham VD, Gogineni A, Edmonds R, Schauer S, Srinivasan K, Huntley MA, Wang Y, Wang TM et al. (2019) Complement C3 is activated in human AD brain and is required for neurodegeneration in mouse models of amyloidosis and tauopathy. Cell Rep 28, 2111–2123 e6. [DOI] [PubMed] [Google Scholar]
- 128. Stephan AH, Barres BA & Stevens B (2012) The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci 35, 369–389. [DOI] [PubMed] [Google Scholar]
- 129. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA & Stevens B (2012) Microglia sculpt postnatal neural circuits in an activity and complement‐dependent manner. Neuron 74, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Hong S, Beja‐Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA et al. (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Luchena C, Zuazo‐Ibarra J, Alberdi E, Matute C & Capetillo‐Zarate E (2018) Contribution of neurons and glial cells to complement‐mediated synapse removal during development, aging and in Alzheimer's disease. Mediators Inflamm 2018, 2530414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Zhou X, Chen Y, Mok KY, Kwok TCY, Mok VCT, Guo Q, Ip FC, Chen Y, Mullapudi N, Neuroimaging AD et al. (2019) Non‐coding variability at the APOE locus contributes to the Alzheimer's risk. Nat Commun 10, 3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Chung WS, Verghese PB, Chakraborty C, Joung J, Hyman BT, Ulrich JD, Holtzman DM & Barres BA (2016) Novel allele‐dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc Natl Acad Sci USA 113, 10186–10191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Zhao J, Fu Y, Yamazaki Y, Ren Y, Davis MD, Liu CC, Lu W, Wang X, Chen K, Cherukuri Y et al. (2020) APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer's disease patient iPSC‐derived cerebral organoids. Nat Commun 11, 5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Fu AK, Hung KW, Yuen MY, Zhou X, Mak DS, Chan IC, Cheung TH, Zhang B, Fu WY, Liew FY et al. (2016) IL‐33 ameliorates Alzheimer's disease‐like pathology and cognitive decline. Proc Natl Acad Sci USA 113, E2705–E2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Jha MK, Kim JH, Song GJ, Lee WH, Lee IK, Lee HW, An SSA, Kim S & Suk K (2018) Functional dissection of astrocyte‐secreted proteins: implications in brain health and diseases. Prog Neurogibol 162, 37–69. [DOI] [PubMed] [Google Scholar]
- 137. Son SM, Nam DW, Cha MY, Kim KH, Byun J, Ryu H & Mook‐Jung I (2015) Thrombospondin‐1 prevents amyloid beta‐mediated synaptic pathology in Alzheimer's disease. Neurobiol Aging 36, 3214–3227. [DOI] [PubMed] [Google Scholar]
- 138. Kang S, Byun J, Son SM & Mook‐Jung I (2018) Thrombospondin‐1 protects against Abeta‐induced mitochondrial fragmentation and dysfunction in hippocampal cells. Cell Death Discov 4, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Reddy PH & Beal MF (2008) Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer's disease. Trends Mol Med 14, 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]