

Abstract
The profiling of epigenetic marks like DNA methylation has become a central aspect of studies in evolution and ecology. Bisulphite sequencing is commonly used for assessing genome‐wide DNA methylation at single nucleotide resolution but these data can also provide information on genetic variants like single nucleotide polymorphisms (SNPs). However, bisulphite conversion causes unmethylated cytosines to appear as thymines, complicating the alignment and subsequent SNP calling. Several tools have been developed to overcome this challenge, but there is no independent evaluation of such tools for non‐model species, which often lack genomic references. Here, we used whole‐genome bisulphite sequencing (WGBS) data from four female great tits (Parus major) to evaluate the performance of seven tools for SNP calling from bisulphite sequencing data. We used SNPs from whole‐genome resequencing data of the same samples as baseline SNPs to assess common performance metrics like sensitivity, precision, and the number of true positive, false positive, and false negative SNPs for the full range of variant and genotype quality values. We found clear differences between the tools in either optimizing precision (bis‐snp), sensitivity (biscuit), or a compromise between both (all other tools). Overall, the choice of SNP caller strongly depends on which performance parameter should be maximized and whether ascertainment bias should be minimized to optimize downstream analysis, highlighting the need for studies that assess such differences.
Keywords: DNA methylation, great tit (Parus major), single nucleotide polymorphism, whole‐genome sequencing
1. INTRODUCTION
In recent years, epigenetic studies, in particular those linking DNA methylation to trait variation, have become an essential aspect of many key questions in ecology and evolution, such as the adaptation of natural populations to novel environments or mechanisms of nongenetic inheritance (Sepers et al., 2019; Stajic & Jansen, 2021; Verhoeven et al., 2016). Epigenetic modifications of the DNA sequence can affect the transcription of genes and consequently the expression of phenotypes (Suzuki & Bird, 2008). The most studied epigenetic modification is DNA methylation, which constitutes the addition of a methyl‐group to a cytosine within the DNA sequence. The methylation and demethylation of cytosines within the DNA sequence is a common event in eukaryotes making the methylome more dynamic than the nucleotide sequence (Laird, 2010), but it is unclear how independent variation in DNA methylation is from genetic variation (Guerrero‐Bosagna, 2020; Kilpinen et al., 2013). When both data on methylation status and data on genetic variants such as single nucleotide polymorphisms (SNPs) are available, we can identify the genetic variants that underly local and distant variation in DNA methylation (e.gDubin et al., 2015; Höglund et al., 2020). For this, bisulphite sequencing constitutes a promising approach as it provides information on the methylation status at single base pair resolution and, at the same time, can potentially be used for SNP calling. Whole‐genome bisulphite sequencing (WGBS) constitutes the current gold standard for methylation profiling and captures about 90% of the methylation events throughout the genome (Lister et al., 2009).
WGBS and other bisulphite sequencing techniques are based on a bisulphite treatment that converts unmethylated cytosines (Cs) into thymines (Ts) and as a consequence complicates SNP calling in several ways (Liu et al., 2012). First, the assumption of strand complementarity, which is made by all SNP calling algorithms, is violated as the two strands of bisulphite treated reads are not complementary at the unmethylated loci. Second, true C‐>T SNPs cannot necessarily be distinguished from bisulphite‐mediated C‐>T conversion and thus C‐>T SNPs might be misidentified as unmethylated cytosines. Given that almost 80% of SNPs at CpG sites are C‐>T substitutions (Tomso & Bell, 2003), this constitutes an important error source for SNP calling as well as methylation calling.
Whether C‐>T SNPs can be differentiated from unmethylated Cs depends on the protocol used for library preparation; directional bisulphite sequencing protocols are strand‐specific, which means that guanines (G) on the strand opposing a C are not affected by the bisulphite conversion (Krueger et al., 2012). Consequently, reads that map to a C can be used to quantify the methylation level of that C but yield no information on a potential C‐>T SNP, while reads that map to the other strand do not yield information of the methylation status of that C but can be used to identify the C‐>T SNPs, as an adenine (A) corresponds to a C‐>T SNPs while a G corresponds to a bisulphite‐mediated C‐>T conversion (Liu et al., 2012). This way, the directional bisulphite sequencing protocols can prevent the misidentification of C‐>T SNPs as unmethylated cytosines.
Tools for SNP calling from bisulphite sequencing data that implement solutions for bisulphite‐induced error sources are freely available and frequently used (Dubin et al., 2015; Gugger et al., 2016; Liew et al., 2020; Wang et al., 2020; Xu et al., 2019), but an independent and intensive evaluation of their performance using data from a non‐model species that often lack genomic references is not available. Here we evaluate the performance of seven tools for SNP calling from bisulphite sequencing data using WGBS and whole‐genome resequencing data of whole blood samples from four female great tits (Parus major). The great tit is an important model species for ecology and evolution (Gosler, 1993) and, more recently, has been used for molecular ecological studies. Currently the great tit has a reference genome, 650k SNP chip, and transcriptomes and methylomes for various tissues (Derks et al., 2016; Kim et al., 2018; Laine et al., 2016). Using SNPs called from whole‐genome resequencing data as baseline lists, we assessed common performance metrics such as precision and sensitivity, and the number of true positive, false positive and false negative SNPs of seven tools for SNP calling from bisulphite sequencing data. Overall, we found clear differences between the tools in performance metrics and potential for bisulphite‐induced ascertainment bias. Hence, the choice of SNP caller strongly depends on whether maximal precision, maximal sensitivity, or a compromise between both is required for optimized downstream analysis highlighting the need for studies that assess such differences.
2. MATERIALS AND METHODS
2.1. Samples used for sequencing
Here, we use whole blood samples of four female great tits for whole‐genome resequencing and WGBS. Whole blood constitutes mostly of erythrocytes (>90%, Verhulst et al., 2016), which are nucleated in avian species and hence are well suitable for isolation of genomic DNA. The four females were part of a genomic selection experiment for early and late timing of breeding (Gienapp et al., 2019; Verhagen et al., 2019) and were sequenced together with other samples from this experiment to test for genetic and epigenetic differentiation between the F3 generation of the selection experiment (unpublished data). Whole blood samples for sequencing were collected from females during their first year of breeding in 2017. Breeding pairs were housed in half‐open aviaries during the breeding season and repeatedly blood sampled from the jugular vein (up to 150 μl every two weeks). The experiment was performed under approval by the Animal Experimentation Committee of the Royal Academy of Sciences (DEC‐KNAW), Amsterdam, The Netherlands, protocol NIOO 14.10. Here, we selected WGBS data of two females from the early and two females from the late selection line of the F3 generation from four different families. DNA was extracted from whole blood samples taken closest to the first of June using FavorPrep DNA extraction kit (Bio‐Connect, The Netherlands) following the manufacturer's instructions.
2.2. Whole‐genome resequencing
Library preparation and sequencing of the four samples used in this study was performed by Novogene Company Limited (UK). The genomic DNA was randomly fragmented by sonication, after which DNA fragments were end polished, A‐tailed, ligated with the full‐length Illumina adapters, and followed by further PCR amplification with P5 and indexed P7 oligos. These PCR products as the final construction of the libraries were purified with AMPure XP system. Libraries were then checked for size distribution by Agilent 2100 Bioanalyser (Agilent Technologies) and quantified by real‐time PCR (to meet the criteria of 3 nM). Libraries were sequenced on one lane from both ends of the 150 bp fragments (i.e., paired‐end) using a NovaSeq 6000. See Table S1 for unique read counts per sample.
2.3. Whole‐genome bisulphite sequencing
The preparation and sequencing of whole‐genome bisulphite libraries of the four samples used in this study was performed by the Roy J. Carver Biotechnology Centre (University of Illinois at Urbana‐Champaign, USA) together with the other samples of the experiment (see above). Shotgun genomic libraries (with read length of 150 nucleotides) were prepared with the Hyper Library construction kit from kapa Biosystems (Roche) and treated with the EZ DNA Methylation Lightning kit from Zymo Research. Libraries were pooled, quantitated by qPCR and each pool was sequenced for 151 cycles from both ends of the fragments (i.e., paired‐end) on a S4 flow cell using a NovaSeq 6000. Samples selected for this study were sequenced on the same lane. Because WGBS data showed high duplication rates (Table S2), library preparation and sequencing were performed twice for all samples and data from both runs were merged. See Table S2 for unique read counts per sample and run.
2.4. Bioinformatic processing
We created the pipelines with snakemake v5.17.0 (Koster & Rahmann, 2012) and used R v4.0.1 (R Core Team, 2017) for additional scripts within the pipeline, data formatting, and graphical visualization. In addition to base R packages, we used dplyr v1.0.0 (Wickham et al., 2020), tidyr v1.1.0 (Wickham & Henry, 2020), stringr v1.4.0 (Wickham, 2019), ggplot2 v3.3.2 (Wickham, 2016), cowplot v1.1.0 (Wilke, 2020), rcolorbrewer v1.1.2 (Neuwirth, 2014), and r markdown v2.5 (Xie et al., 2018; Yihui et al., 2020). Software packages used within the snakemake pipelines were mostly built and managed with conda v4.8.4 (Anaconda Software Distribution, 2016). All pipelines and conda environments are publicly accessible on github (https://github.com/MLindner0/lindner_et_al‐2021‐mer‐snps_from_bs_data). For both the bioinformatic processing of the whole‐genome resequencing data and the WGBS data, we used the Parus major reference genome build (https://www.ncbi.nlm.nih.gov/assembly/GCF_001522545.3).
2.5. Bioinformatic processing of the whole‐genome resequencing data
We used SNPs called from the whole‐genome resequencing data as a baseline list of SNPs to evaluate SNPs called from the WGBS sequencing data. For the bioinformatic processing of whole‐genome resequencing data we followed the “GATK best practice” guidelines for model (Auwera et al., 2013) and non‐model species (https://evodify.com/gatk‐in‐non‐model‐organism/). Our snakemake pipeline included quality control, data trimming, alignment, recalibration of base‐quality‐scores, variant calling, and variant filtering and was executed such that samples were processed in parallel where applicable. Please note that in contrast to the GATK pipeline, in which SNPs and genotypes are called in separate steps, the tools for SNP calling from bisulphite sequencing data provide SNP and genotype calling in one step and hence we here refer to SNP calling as the calling of SNPs and genotypes.
For the initial quality control we used fastqc v0.11.9 (Andrew, 2010), fastq screen v0.11.1 (Wingett & Andrews, 2018), and multiqc v1.7 (Ewels et al., 2016) in default settings but allowed parallel processing of samples by fastqc. Results are presented in Table S1. We trimmed the data and removed adapters using trimgalore v0.6.5 (https://github.com/FelixKrueger/TrimGalore) in default settings for paired‐end data, but set a NovaSeq specific quality cutoff of 20 (by specifying ‐‐2coulor 20) accounting for NovaSeq specific over‐representation of Gs (poly‐G), and enabled the production of a fastqc output for the trimmed data. We completed the second quality control by running MultiQC for the trimmed data.
We prepared the Parus major reference genome for further processing of the trimmed whole‐genome resequencing data by building a BWA index using bwa v0.7.17 (Li & Durbin, 2010), a samtools fasta file index using samtools v1.3.1 (Li et al., 2009), and a sequence dictionary using picard v2.18.29 (https://github.com/broadinstitute/picard). We performed the alignment of paired‐end sequencing reads to the Parus major reference genome using BWA mem using eight threads per sample and adding sample‐specific read groups to the aligned reads. Alignments were coordinate‐sorted and deduplicated using Picard. We assessed the number of mapped reads, average coverage depth, and breadth of coverage using samtools (Table S3). The breadth of coverage was calculated as the number of bases with a minimum coverage of 10 divided by the bases within the great tit genome (i.e., genome length; calculated using bowtie2 (Langmead & Salzberg, 2012) “inspect”). We used a minimum coverage threshold of 10 throughout the manuscript and hence the breadth of coverage conditional on a minimum coverage of 10 will be most informative here. We performed base quality (BQ) score recalibration using gatk baserecalibrator and gatk applybqsr in default settings with 523,640 SNPs of 3344 great tits (415 males and 2929 females) derived from a high‐density SNP‐chip (Kim et al., 2018) as a list of known SNPs (da Silva et al., 2018; Verhagen et al., 2019). Finally, we removed reads mapping to the Z chromosome or mitochondrial DNA using samtools.
For SNP calling we used gatk v4.2.0 (DePristo et al., 2011; McKenna et al., 2010). We called the raw variants for each sample using the gatk haplotypecaller specifying “GVCF” as the mode for emitting reference confidence scores and “emit all confident sites” as the output mode. We combined sample‐specific variants using GATK CombineGVCFs and genotyped variants using GATK GenotypeGVCFs specifying a minimum phred‐scaled confidence threshold of 30 for genotyping of variants and a heterozygosity value of 0.003 following Hayes et al. (2020). We selected SNPs and visually inspected the quality of SNPs called (Figures S1 and S2) to determine appropriate filter thresholds. We hard‐filtered SNPs with mapping quality (MQ) smaller than 40.00, sequencing bias (in which one DNA strand is favoured over the other, SOR) larger than 4.00, variant confidence standardized by depth (QD) smaller than 2.00, strand bias (in support for REF vs. ALT allele calls, FS) larger than 60.00, Rank sum test for mapping qualities of REF versus ALT reads (MQRankSum) smaller than –12.50 or larger than 12.50, and rank sum of read position (i.e., are all SNPs located near the end of reads, ReadPosRankSum) smaller than –10.00 or larger than 10.00 following the “GATK best practice” guidelines for non‐model species (https://evodify.com/gatk‐in‐non‐model‐organism/). We set genotypes to “no call” for SNPs with sample‐specific coverage below 10 (5th percentile) and above 75 (99th percentile). We split the SNPs by sample and removed nonvariant sites to create sample‐specific baseline lists of SNPs.
2.6. Bioinformatic processing of the whole‐genome bisulphite sequencing data
We tested five tools for SNP calling and two tools that perform alignment and SNP calling (Table S4); bis‐snp v1.0.1 (Liu et al., 2012), biscuit v0.3.16 (https://github.com/zhou‐lab/biscuit), bs‐snper v1.0 (Gao et al., 2015), cgmaptools v0.1.2 (Guo et al., 2018), epidiverse‐snp pipeline v1.0 (Nunn et al., 2021), methylextract v1.9.1 (Barturen et al., 2014), and gembs v3.5.1 (Merkel et al., 2019). Our pipelines included quality control, data trimming, alignment, recalibration of base‐quality‐scores (for bis‐snp) or a double‐masking procedure (for epidiverse‐snp pipeline) of alignments, variant calling, and variant filtering.
For the initial quality control we used fastqc, fastq screen, and multiqc in default settings but allowed parallel processing of samples by fastqc. Results are presented in Table S2. We trimmed the data using trimgalore v0.6.5 (https://github.com/FelixKrueger/TrimGalore) in paired‐end mode and set a NovaSeq specific quality cutoff of 20 (by specifying ‐‐2coulor 20) accounting for NovaSeq specific overrepresentation of Gs (poly‐G). After trimming, we repeated the quality control with fastqc and multiqc.
To reduce aligner‐related variation between SNPs called, we performed alignments with bismark v0.22.3 (Krueger & Andrews, 2011) which utilizes bowtie2 where possible. For gembs and biscuit, that is, tools that include alignment and SNP calling, we used the tool‐specific aligner which utilize GEM3 (Marco‐Sola et al., 2012) and BWA‐mem (Li, 2013), respectively. All aligners were so called “three letter aligners”, but see Grehl et al. (2020) or Kunde‐Ramamoorthy et al. (2014) for explanation and comparison of different aligner types for bisulphite treated DNA.
We prepared the Parus major reference genome for the respective aligner; we bisulphite converted and indexed the reference genome for bismark alignments and indexed the reference genome for gembs alignments. We performed the alignments with bismark twice, using the new flag values (implemented since bismark v0.8.3, default) and using the old flag values which are required for SNP calling with cgmaptools. For the bismark alignments with new flag values we aligned the paired‐end reads with default settings but set the number of threads to eight. We used the percentage of CHH methylation from the bismark alignment reports to calculate the minimal bisulphite conversion efficiency as 100% ‐ %CHH methylation. For the bismark alignments with old flag values we additionally specified “‐‐old_flag” “‐‐no_dovetail”. We deduplicated the alignments using Bismark and added sample‐specific read groups to the aligned reads using Picard. We merged the alignments of the two sequencing runs for each sample using Picard and assessed the number of mapped reads, average coverage depth, and breadth of coverage using samtools. Finally, we removed reads mapping to the Z chromosome or mitochondrial DNA using samtools. For the biscuit alignments we aligned the paired‐end reads with default settings but set the number of threads to eight. Using Picard we deduplicated the alignments, added sample‐specific read groups to the aligned reads, and merged the alignments of the two sequencing runs for each sample. We assessed the number of mapped reads, average coverage depth, and breadth of coverage using samtools and finally removed reads mapping to the Z chromosome or mitochondrial DNA using samtools. gembs requires a metadata‐file that provides the connection between sample name and sequencing data files and a configuration with all pipeline parameters. All samples are processed in parallel for which we set the number of threads to 20. Alignment was performed with default settings for stranded libraries and included the merging of the alignments of the two sequencing runs for each sample. We assessed the number of mapped reads, average coverage depth, and breadth of coverage using samtools. In contrast to the other tools, duplicates were removed during SNP calling and SNPs located on the Z chromosome and mitochondrial DNA were removed after SNP calling using samtools.
We used the bismark alignments with the new flag values to call SNPs with bis‐snp, bs‐snper, epidiverse‐snp pipeline, and methylextract, Bismark alignments with the old flag values to call SNPs with cgmaptools, and tool‐specific alignments to call SNPs with gembs and biscuit. Prior to SNP calling with bis‐snp, we performed a BQ score recalibration of the alignments using default settings and 523,640 SNPs of 3344 great tits (415 males and 2929 females) derived from a high‐density SNP‐chip as a list of known SNPs (da Silva et al., 2018; Verhagen et al., 2019) (we used bis‐snp v0.82 for this step as the BQ score recalibration was not available in the newest release of bis‐snp). For the recalibration, we used the Bismark alignments prior to removal of reads mapping to the Z chromosome or mitochondrial DNA and removed those reads after BQ score recalibration. To aid comparison between tools and because not all non‐model species have a list of known SNPs, we performed SNP calling with bis‐snp from the recalibrated and nonrecalibrated alignments setting the maximal coverage to 1000 (default 250, for better calculation of the 99th percentile of coverage), heterozygosity to 0.003 following Hayes et al. (2020) and standard minimum confidence threshold for calling to 0 (to allow for a larger range of variant and genotype quality). We called SNPs with bs‐snper in default settings but setting the minimal and maximal coverage to 1 and 1000, respectively (for better calculation of the 99th percentile of coverage). We used nextflow v20.07.1 (Di Tommaso et al., 2017) to run the epidiverse‐snp pipeline in default settings but specifying the —“—variants” flag. We called SNPs with methylextract using the default setting for paired‐end reads, specified bismark‐specific new flag values, and set the minimal coverage to 1 (for better calculation of the 99th percentile of coverage). For SNP calling with cgmaptools we used the Bismark alignments with the old flag values and converted the alignments into atcgmaps and removed the overlap of read pairs. Using the atcgmaps and called SNPs with cgmaptools’ Bayesian and binomial wildcard strategy in default settings. In contrast to the previous five tools, biscuit and gembs provide a whole pipeline which involves alignment. Hence, tool‐specific alignments were used to call SNPs. For SNP calling with biscuit we used the default settings but specifying eight threads and that cytosines in overlapping read pairs must not be counted twice. For SNP calling with gembs we used the default settings for WGBS data but specified the removal of duplicates (which for all other tools is done prior to SNP calling).
We filtered the resulting lists of SNPs from different tools tested for depth such that all SNPs with depth lower than ten and higher than the tool‐specific 99th per centile of depth were removed (Figures S3–S11 and Table S5). Most of the parameters used for hard‐filtering of the SNPs called with GATK from the whole‐genome resequencing data (MQ, SOR, QD, FS, MQRankSum, ReadPosRankSum), were not given in the output files of the tools for SNP calling from bisulphite sequencing data. The only exception was MQ, which was provided in the methylextract output. Some of the tools (bis‐snp, biscuit, bs‐snper, gembs, and methylextract) provided the option to filter for BQ and/or MQ during the SNP calling, but as not all tools provided this option, we used the tool‐specific default settings. The output files of all tools tested for SNP calling provided values for the variant quality (QUAL) and most tools provided values for the genotype quality (GQ). These QUAL and GQ values, however, strongly varied between tools (Figure S3–S11 and Table S5).
2.7. Evaluation of SNP calls from whole‐genome bisulphite sequencing data
To evaluate the tools for SNP calling from bisulphite sequencing data, we compared the SNPs called with the seven different tools to baseline lists of true SNPs (i.e., SNPs called from whole‐genome resequencing data of the same samples) using rtgtools (Cleary et al., 2014). rtgtools provides common performance metrics such as the number of false positives (SNPs called that are not in the baseline list of true SNPs), false negatives (SNPs in the baseline list of true SNPs that are not called), true positives (SNPs called that are in the baseline list of true SNPs), precision, sensitivity, and f‐measure, which is the harmonic mean of precision and sensitivity. Precision was calculated as the number of SNPs called divided by the sum of the number of SNPs called and the number of false‐positive SNPs and sensitivity was calculated as the number of SNPs in the baseline lists of true SNPs divided by the sum of the number of SNPs in the baseline lists of true SNPs and the number of false‐negative SNPs called. Note that rtgtools operates on the level of local haplotypes such that a SNP (in diploid genome) is only considered a true positive SNP if both alleles of the genotype also match in order. Furthermore, we used rtgtools to estimate the performance metrics for each value of QUAL and GQ across the full range of the respective parameter to assess the effect of QUAL or GQ on the performance metrics. Please note that the range of QUAL and GQ values as well as the number of values within the respected parameter range differed between tools to such a degree that there is no QUAL or GQ value that is present within the respective parameter range of all tools tested.
3. RESULTS
3.1. Alignment statistics and bisulphite conversion efficiency
Mapping efficiency for the four samples ranged from 48%–53% for Bismark (irrespective of whether new or old flag values were used), from 96%–105% for biscuit, and from 186%–188% for gembs. Higher values for biscuit and gembs can be explained by multimapping (biscuit and gembs) and the presence of duplicated reads (gembs). In line with this, biscuit and especially gembs alignments also showed a higher average coverage depth than bismark; 23.89–29.93 for bismark, 39.07–48.34 for biscuit, and 80.31–97.01 for gembs. Breadth of coverage was overall high ranging from 78–89% for bismark, 87%–94% for biscuit, and 95%–97% for gembs. Alignment statistics including number of mapped reads, average coverage depth, and breadth of coverage are presented in Table S6. To ensure that bisulphite conversion was successful, we calculated the bisulphite conversion efficiency which was >99.1% for all samples and both sequencing runs (Table S7).
3.2. Evaluation of SNP calls from whole‐genome bisulphite sequencing data
Here, we assessed performance metrics of SNPs called with the seven tools tested for SNP calling from bisulphite sequencing data. The performance metrics were estimated for each value of QUAL and GQ across the full range of the respective parameter (Table S8). This allowed us to assess how the relationship between sensitivity and precision varied across parameter values of QUAL and GQ for the tools tested, which differs between tools. Ideally, tools would reach high precision and high sensitivity (i.e., located in the upper right quadrant of the plotting space in Figure 1). Most tools tested here show high precision (>0.8), but rather low levels of sensitivity (<0.6, Figure 1 and Figures S12 and S13). Especially bis‐snp showed a high precision (>0.9) but low sensitivity (<0.4, in particular when SNPs were called from the BQ score recalibrated alignments), while biscuit showed high sensitivity (up to almost 0.9) but comparably low precision (<0.8, Figure 1). Especially for the range of QUAL values (Figure 1a ), the other tools showed intermediate precision (i.e., higher than biscuit but lower than bis‐snp) and sensitivity (i.e., lower than biscuit but higher than bis‐snp).
FIGURE 1.
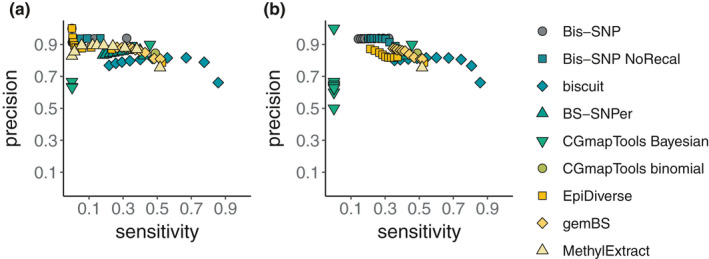
Relationship between precision and sensitivity for SNPs called from whole‐genome bisulphite sequencing data of one sample (F3_E_BD_27272) relative to a list of known SNPs derived from whole‐genome resequencing data of the same sample. Precision and sensitivity were calculated using rtgtools with (a) QUAL and (b) QG as score fields, which means that the performance metrics (here precision and sensitivity) were calculated across the full range of a parameter values for QUAL and GQ. Thus, the number of data points per tool, varies with the tool‐specific and parameter‐specific range of parameter values. If the parameter value is not given, the performance metrics are calculated for the full SNP list (resulting in one data point) and if the full range of a parameter value is longer than 20 values, we reduced the length of a parameter range to 20 equally spaced values across the full range of parameter values. Here, only one sample is displayed, but see Figures S12–13 for plots with all samples
While sensitivity and precision give a good indication of the performance of a tool, they provide little information on the magnitude of SNPs called. Thus, we assessed the number of true positive, false positive, and false negative SNPs called with the seven tools tested at the threshold value of QUAL that maximized the f‐measure, the sensitivity or the precision. The number of true positive, false positive, and false negative SNPs differed between the maximized performance metrics and there were clear tool‐specific patterns (Figure 2, Figures S14–S16 and Tables S9–S11). bis‐snp showed the lowest maximal f‐measure (0.42–0.51) and sensitivity (0.27–0.35) but highest maximal precision (0.89–0.95) based on a low number of true positive and false positive SNPs and high number of false negative SNPs. In contrast, biscuit showed the highest maximal f‐measure (0.77–0.82) and sensitivity (0.78–0.86) but lowest maximal precision (0.78–0.83) based on a high number of true positives and false positives SNPs and low number of false negative SNPs. The other five tools showed intermediate patterns and specifically cgmaptools, the epidiverse pipeline, gembs, and methylextract showed high maximal precision. For the epidiverse pipeline, gembs, and methylextract, however, high maximal precision was accompanied by low numbers of true positive SNPs (Figure 2c ). bs‐snper and cgmaptools showed slightly lower maximal f‐measure and sensitivity than the epidiverse pipeline, gembs, and methylextract based on a lower number of true positive and false positive SNPs and a higher number of false negative SNPs (Figure 2a,b ). Especially the Bayesian strategy with cgmaptools showed a low number of false positive SNPs, while showing a comparably high number of true positive SNPs irrespective of which performance metric is maximized.
FIGURE 2.
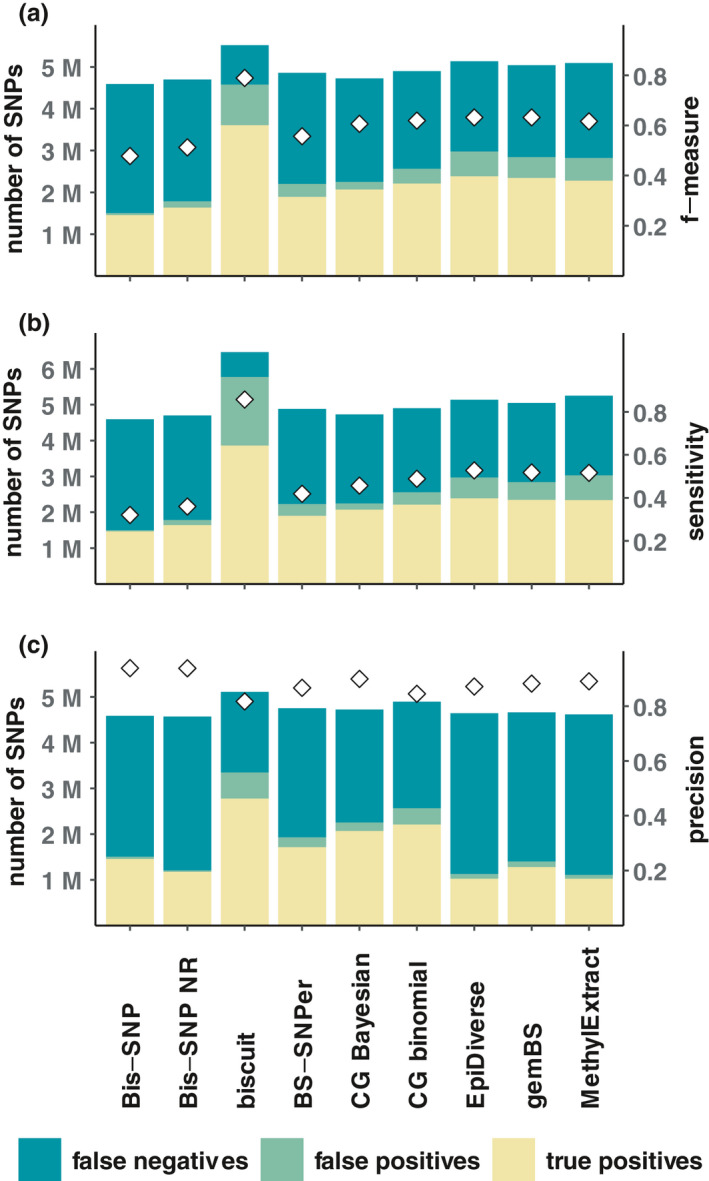
Number of false negative (teal), false positive (green), and true positive (yellow) SNPs called (bars and left y‐axis) with the different tools tested for SNP calling from bisulphite sequencing data for one sample (F3_E_BD_27272). Performance metrics are based on the evaluation with rtgtools and we here show the performance metrics for which the f‐measure (a), sensitivity (b), and (c) precision is maximized when using QUAL as score fields (white diamonds and right y‐axis). Note that the QUAL values for which f‐measure, sensitivity, or precision are maximized differ between tools and that precision is maximized on the condition that at least 1,000,000 SNPs were called. Here, only one sample is displayed, but see Figures S14–16 for plots with all samples
To understand whether substitution contexts affected by the bisulphite treatment (i.e., A‐>G, C‐>T, G‐>A, and C‐>T substitutions) are prone to bias during SNP calling, we visually inspected the tool‐specific distributions of false negative (Figure S17) and false positive SNPs (Figure 3) over substitution contexts relative to the distribution of baseline SNPs over substitution contexts. The tool‐specific distributions of false negative SNPs over substitution contexts closely followed the baseline distribution for all tools tested and hence did not indicate an enrichment of false negative SNPs for substitution contexts affected by the bisulphite treatment. In contrast, we found tool‐specific deviations between the distributions of false positive SNPs over substitution contexts and the distribution of baseline SNPs. There are four distinct patterns, (1) tool‐specific distributions of SNPs that roughly followed the distribution of baseline SNPs (binomial strategy with cgmaptools), (2) tool‐specific distributions of SNPs that constituted a mixture of the distribution of baseline SNPs and an uniform distribution (bis‐snp with BQ recalibration and the Bayesian strategy with cgmaptools), (3) tool‐specific distributions of SNPs that showed an enrichment for A‐>G and C‐>T substitutions relative to the baseline distribution of SNPs (bis‐snp without BQ recalibration, biscuit, epidiverse‐snp pipeline, gembs, and methylextract), and (4) tool‐specific distributions of SNPs that followed the distribution of baseline SNPs but show an enrichment for A‐>G, C‐>T, G‐>A, and C‐>T substitutions relative to the baseline distribution of SNPs (bs‐snper). To better understand these patterns, we inspected the tool‐specific distributions of false positive SNPs over substitution contexts but differentiated between homozygous and heterozygous SNPs (Figures S18–S21). Especially tools with false positive SNPs that showed an enrichment for A‐>G and C‐>T substitutions (3) or for A‐>G, C‐>T, G‐>A, and C‐>T substitutions (4) also showed a strong enrichment for heterozygous SNPs at these substitution contexts with a percentage of heterozygous SNPs of false positive SNPs up to 97.25% for bis‐snp without BQ recalibration, 92.94% for biscuit, 99.56% for bs‐snper, 72.35% for epidiverse‐snp pipeline, 97.70% for gembs, and 92.05% for methylextract (Table S12). This excess of heterozygous SNPs largely contributed to observed deviations between the tool‐specific distributions of false positive SNPs over substitution contexts and the distribution of baseline SNPs. The binomial strategy with cgmaptools also showed an enrichment of heterozygous SNPs for false positive SNPs at substitution contexts affected by the bisulphite treatment, but to a much smaller extent (up to 49.02%). bis‐snp with BQ recalibration and the Bayesian strategy with cgmaptools showed a strong enrichment of heterozygous SNPs (up to 92.12% for bis‐snp with BQ recalibration and up to 72.90% for the Bayesian strategy with gcmaptools), but not specifically for substitution contexts affected by the bisulphite treatment.
FIGURE 3.
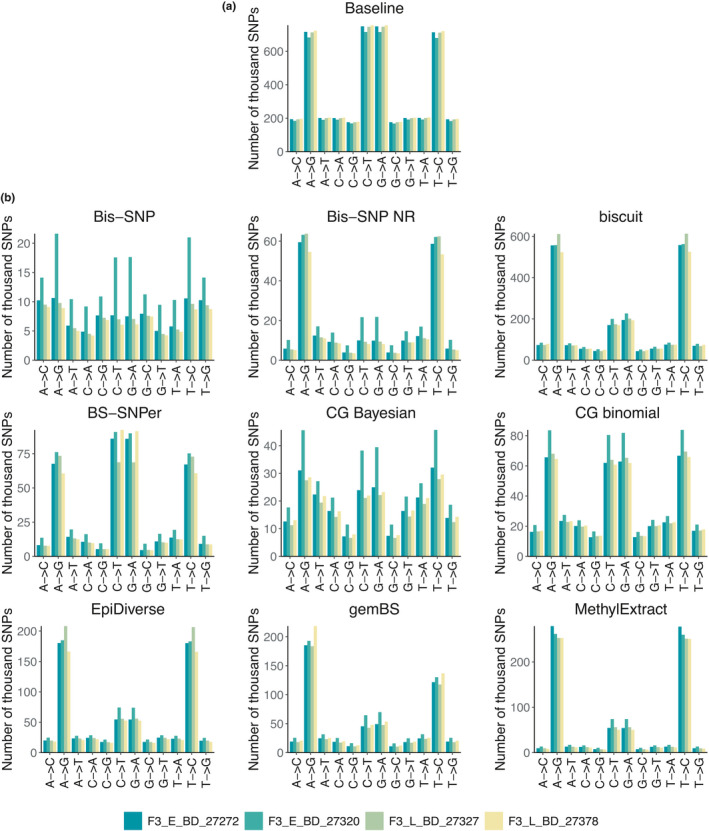
Distribution of SNPs over substitution contexts (alternative and reference allele) for the baseline list of true SNPs derived from whole‐genome resequencing data (a) and the tool‐specific lists of false positive SNPs (b). Samples are differentiated by colour (teal‐yellow) and plots in (b) have tool‐specific plot titles
4. DISCUSSION
The study of epigenetics and in particular DNA methylation has received much attention in ecology and evolution (Verhoeven et al., 2016) and bisulphite sequencing data used for DNA methylation profiling can also be utilized to detect genetic variants such as SNPs. When evaluating seven tools for SNP calling using WGBS and whole‐genome resequencing data of four whole blood samples from female great tits, we indeed found clear differences between the tools in performance metrics and the potential for bisulphite‐induced ascertainment bias. Overall, the choice of tools strongly depends on the downstream analysis, but for most applications the Bayesian strategy with cgmaptools or bis‐snp with BQ score recalibration will constitute the best choice (Table 1).
TABLE 1.
Number of true positive SNPs (low‐high), number of false positive SNPs (low‐high), potential for (bisulphite‐induced) ascertainment bias (low‐high), and additional requirements for SNP calling for the seven tools tested
| Tool name (strategy) | Number of true positive SNP | Number of false positive SNP | Potential for ascertainment bias | Requirement |
|---|---|---|---|---|
| bis‐snp (BQ score recalibration) | Low | Low | Medium | List of known SNPs |
| bis‐snp (no BQ score recalibration) | Low | Low | High | |
| biscuit | High | High | High | |
| bs‐snper | Medium | Medium | High | |
| cgmaptools (Bayesian strategy) | Medium | Low | Medium | |
| cgmaptools (binomial strategy) | Medium | Medium | Medium | |
| epidiverse | Medium | Medium | High | |
| methylextract | Medium | Medium | High | |
| gembs | Medium | Medium | High |
Abbreviation: BQ, base quality.
4.1. Evaluation of performance metrics
The clear differences in performance metrics between tools highlights that the choice of a tool for SNP calling has a clear impact on the resulting SNP list. Which tool is most suitable for a certain data set, however, strongly depends on the downstream analyses. In some analyses, where we care most about the individual SNPs rather than the combined effect of all SNPs, such as for genome‐wide association studies, we might want to maximize precision (or minimize the number of false positive SNPs) while caring less about the total number of SNPs called. In such a scenario bis‐snp and cgmaptools (specifically the Bayesian strategy) provide the optimal output. Albeit bis‐snp called fewer false positive SNPs than cgmaptools, it also called considerably fewer true positive SNPs and hence cgmaptools might constitute a better compromise between the number of false positive and the number of true positive SNPs. Furthermore, bis‐snp performs best when a list of known SNPs is available for the recalibration of BQ scores prior to SNP calling and hence might not be a good option for species or populations where this is not the case. When on the other hand we care most about the combined effect of all SNPs rather than the effect of individual SNPs, such as when inferring relatedness between individuals or F‐statistics between populations (but see 4.2 below), a tool that constitutes a compromise between precision and sensitivity and that reduces (bisulphite‐induced) potential for ascertainment bias might be the best choice. In such a scenario, cgmaptools constitutes the best choice followed by the epidiverse‐snp pipeline, gembs, methylextract and lastly bs‐snper. When focusing on the maximal f‐measure as an indication for a good compromise between precision and sensitivity, biscuit clearly has the lead, but also showed a considerably higher number of false positive SNPs compared to any other tool and a high potential for (bisulphite‐induced) ascertainment bias, which can negatively impact on downstream analysis.
4.2. Potential for bisulphite‐induced ascertainment bias
In addition to common performance metrics like sensitivity and precision, it is also important to assess potential bias on the allele frequency spectrum (AFS) of SNPs called from bisulphite sequencing data. The AFS constitutes a simple summary of the allele frequencies across loci in a population and is the basis of many estimates in population genetics. Deviations from the true AFS (i.e., ascertainment bias) can introduce strong bias in population genetic inferences potentially leading to wrong conclusions (Han et al., 2014).
Calling SNPs from bisulphite sequencing data potentially results in ascertainment bias at positions affected by the bisulphite conversion. Partially or unmethylated Cs are difficult to differentiate from true C‐>T SNPs (heterozygous or homozygous) potentially leading to an enrichment of false positive and false negative SNPs in such substitution contexts. Strand‐specificity can be used to avoid such misidentifications as Gs on the strand opposing Cs are not affected by the bisulphite conversion (Krueger et al., 2012). The tools, however, differ in how to make use of this information and avoid such misidentifications. bis‐snp and gembs use similar GATK‐based Bayesian inference models that consider C‐>T SNPs either as potential errors based on the BQ score or as a bisulphite conversion with the probability of observing a bisulphite conversion depending on the underlying methylation status and the bisulphite conversion error (Liu et al., 2012; Merkel et al., 2019). In addition, bis‐snp involves a GATK‐based and bisulphite sequencing adapted BQ score recalibration of the alignment prior to SNP calling to improve the Bayesian inference model (Liu et al., 2012). The epidiverse‐snp pipeline involves a double‐masking procedure of the alignment to facilitate SNP calling with conventional tools such as gatk or freebayes (Garrison & Marth, 2012; Nunn et al., 2021). The double‐masking procedure manipulates specific nucleotides and BQ scores of the alignment and, this way, imposes an indirect strand‐specificity on potential SNP calls to dissociate them from the effect of bisulphite conversion. cgmaptools provides two methods for SNP calling that are based on the introduction of wildcard genotypes (Guo et al., 2018). Due to the bisulphite treatment (conversion of C to T if C is unmethylated), the presence of Ts might indicate either Ts or Cs in the unconverted genome resulting in ambiguous genotypes. Wildcards are used to denote this ambiguity in predicted genotypes with Y referring to either T or C and R referring to either A or G. When both strands have high coverage, this ambiguity can be resolved and an exact genotype can be computed. cgmaptools provides two strategies that implement the wildcards; a Bayesian model and a binomial model. In the Bayesian model the posterior is noted as the product of the posteriors of each observed genotype and the genotype with the highest posterior from the exact genotype set and the wildcard genotype set is selected as the predicted genotype. In the binomial strategy, the genotype is predicted using a binomial distribution. methylextract relies on an approach in which positions with low BQ scores (indicative for sequencing errors) and reads with at least 90% of presumably unconverted cytosines in non‐CpG contexts (indicative for bisulphite conversion errors) are removed prior to SNP calling.
Due to our small sample size, we could not reliably infer the AFS and assess ascertainment bias. To get at least an indication of the potential for bisulphite‐induced ascertainment bias, we assessed whether the distributions of false negative and false positive SNPs over substitution contexts were biased towards substitution contexts affected by the bisulphite treatment when compared to the distribution of baseline SNPs over substitution contexts. Regardless of the tool used, we did not find strong deviations between the distribution of false negative SNPs and the distribution of baseline SNPs indicating that false negative SNPs are not biased towards substitution contexts affected by the bisulphite treatment. For false positive SNPs, however, we found substantial differences between tools in respect to deviations from the distribution of baseline SNPs. Interestingly, bs‐snper showed an enrichment for all four substitution contexts affected by the bisulphite treatment, while bis‐snp without BQ score recalibration, biscuit, the epidiverse‐snp pipeline, gembs, and methylextract only showed an enrichment of T‐>C and A‐>G SNPs (i.e., no enrichment for C‐>T and G‐>A SNPs). Furthermore, SNPs in these substitution contexts are strongly enriched for heterozygous SNPs and especially for biscuit, the epidiverse‐snp pipeline, gembs, and methylextract heterozygous SNPs seem to drive the enrichment of T‐>C and A‐>G SNPs. These findings might indicate that tools have difficulties to differentiate between partially methylated Cs and heterozygous SNPs in substitution contexts affected by the bisulphite treatment. Cs are not necessarily completely methylated or completely unmethylated at the tissue level, but can have intermediate methylation level, which means that only a part of the reads covering a C will be converted to Ts by the bisulphite treatment. Such partial methylation might be difficult to differentiate from heterozygous C‐>T SNPs. In directional libraries, however, the opposite strand is not affected by the bisulphite treatment and hence the position on the opposite strand is either an A (for C‐>T SNP) or a G (for bisulphite converted C) and in theory provides information to differentiate unmethylated or partially methylated Cs from homozygous or heterozygous SNPs (Liu et al., 2012). The high enrichment of T‐>C substitutions is more remarkable. As Ts are not expected to be affected by the bisulphite treatment (<1% bisulphite conversion efficiency for T; Holmes et al., 2014), these positions might constitute a homozygous T‐>C SNP that was partially methylated and hence wrongly genotyped as heterozygous T‐>C SNP.
In addition to bisulphite‐induced ascertainment bias, bs‐snper removed SNPs with low minor allele frequency (default 0.1, Gao et al., 2015), which will induce ascertainment bias in the AFS. Also, the general enrichment of false positive SNPs for heterozygous SNPs in all tools is likely to induce ascertainment bias in the AFS and bias downstream population genetic analyses. Our study, however, lacks the sample size needed to properly asses ascertainment bias in the AFS and hence future studies with much larger sample sizes are needed for assessing such bias for SNPs called from bisulphite sequencing data and the consequences for population genetic analyses.
4.3. Effect of aligner on SNP calling
We here used three different three letter aligners and previous studies have shown that there are differences between the aligners that are designed for bisulphite sequencing data in genomic coverage and quantitative accuracy (Grehl et al., 2020; Kunde‐Ramamoorthy et al., 2014). In general, the choice of aligner can also affect the accuracy of SNP calling and hence our findings are conditional on the aligners we used. While most tools use bismark alignments as input for SNP calling, biscuit and gembs are “whole‐pipeline‐tools” that utilize their own aligners. Consequently the comparison of biscuit and gembs with any other tool should be interpreted with caution, especially as the tool‐specific aligner of biscuit and gembs allow for multimapping and show a much higher mapping percentage than the bismark alignments. Here, we cannot explain whether the high number of false positive SNPs called with biscuit is associated to the biscuit SNP caller or to (the high mapping percentage of) the biscuit aligner. However, gembs alignments also had a high mapping percentage but showed a similar performance as the epidiverse‐snp pipeline and methylextract, indicating that a high mapping percentage does not per se increase the number of false positives SNPs.
4.4. Ecological and evolutionary applications for SNPs from WGBS data
Assessing to what extend genetic variation underlies variation in DNA methylation is of high scientific interest and has been investigated in a variety of species such as Arabidopsis thaliana (Dubin et al., 2015), maize (Xu et al., 2019), reef‐building corals (Liew et al., 2020), intercrosses between wild derived red junglefowl and domestic chickens (Höglund et al., 2020), and humans (Heyn et al., 2013). For example, in Arabidopsis thaliana variation in CHH methylation at transposons was strongly associated with genetic variants both in cis and trans (Dubin et al., 2015). In intercrosses between wild derived red junglefowl and domestic chickens over 46% of mapped trans quantitative trait loci for hypothalamus methylation were genotypically controlled by only five loci mainly associated with increased methylation in the red junglefowl genotype (Höglund et al., 2020). This large dependency of most DNA methylation variants on genetic variation also implies that more closely related individuals are more similar in their methylation patterns than unrelated individuals (Lea et al., 2017; van Oers et al., 2020; Viitaniemi et al., 2019). Depending on the experimental design, it therefore is important to infer and account for relatedness when analysing methylation data (e.g., Lindner et al., 2021).
5. CONCLUSION
Bisulphite sequencing offers the potential to analyse variation in both the genome and DNA methylation. However, the decision of which tools to use is crucial as the performance can be compromised by the bisulphite treatment and in turn affect downstream analyses. We found clear differences between the tools in performance metrics and the potential for bisulphite‐induced ascertainment bias and for most downstream analyses the Bayesian strategy with cgmaptools or bis‐snp with BQ score recalibration (if list of known SNPs is available) will constitute the best choice (Table 1). Our results highlight the need to assess the performance of tools to understand tool‐specific sources of bias and to choose a tool that optimizes the performance of SNP calling in respect to the downstream analysis. Lastly, our findings and pipelines will provide other molecular ecologists with a useful resource to choose appropriate tools for reliable SNP calling from bisulphite sequencing data of their own study systems.
AUTHOR CONTRIBUTIONS
Melanie Lindner, Fleur Gawehns, and Veronika N. Laine designed the analyses with input from Sebastiaan te Molder. Melanie Lindner conducted the analysis with advice from Veronika N. Laine and Fleur Gawehns, and Melanie Lindner wrote the manuscript with input from Veronika N. Laine, Fleur Gawehns, Kees van Oers, and Marcel E. Visser.
OPEN RESEARCH BADGES
This article has earned an Open Data Badge for making publicly available the digitally‐shareable data necessary to reproduce the reported results. The data is available at http://www.ncbi.nlm.nih.gov/bioproject/, https://doi.org/10.5061/dryad.ttdz08kzt and https://github.com/MLindner0/lindner_et_al‐2021‐mer‐snps_from_bs_data
Supporting information
Supplementary Material
Table S1
Supplementary Material
ACKNOWLEDGEMENTS
We thank Irene Verhagen for performing the selection line experiments, Koen Verhoeven, Bernice Sepers and other members of the NIOO‐KNAW “ecological epigenetics” theme group for useful discussions, Mattias de Hollander and Judith Risse for help with the bioinformatics, Christa Mateman and colleagues at NIOO‐KNAW for laboratory assistance, and the animal care takers at the NIOO‐KNAW for the care of the birds. We thank three anonymous reviewers and Alana Alexander for constructive comments that improved the manuscript. This work was supported by a European Research Council Advanced grant (ERC‐2013‐AdG 339092) to M.E.V.
Lindner, M. , Gawehns, F. , te Molder, S. , Visser, M. E. , van Oers, K. , & Laine, V. N. (2022). Performance of methods to detect genetic variants from bisulphite sequencing data in a non‐model species. Molecular Ecology Resources, 22, 834–846. 10.1111/1755-0998.13493
Contributor Information
Melanie Lindner, Email: M.Lindner@nioo.knaw.nl.
Veronika N. Laine, Email: veronika.laine@helsinki.fi.
DATA AVAILABILITY STATEMENT
Data used for this article have been made available in the NCBI BioProject database (http://www.ncbi.nlm.nih.gov/bioproject/) under BioProject PRJNA208335 (whole‐genome resequencing: SRR15365281‐SRR15365284, WGBS: SRR15410225‐ SRR15410232). SNP data have been made available on Dryad (https://doi.org/10.5061/dryad.ttdz08kzt). All code and pipelines can be accessed on gitHub (https://github.com/MLindner0/lindner_et_al‐2021‐mer‐snps_from_bs_data) and are presented in the Supplemental Code.
REFERENCES
- Anaconda Software Distribution (2016). Conda. Version 4.8.4, Anaconda. www.anaconda.com.
- Andrew, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. Retrieved from http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- Auwera, G. A. , Carneiro, M. O. , Hartl, C. , Poplin, R. , del Angel, G. , Levy‐Moonshine, A. , Jordan, T. , Shakir, K. , Roazen, D. , Thibault, J. , Banks, E. , Garimella, K. V. , Altshuler, D. , Gabriel, S. , & DePristo, M. A. (2013). From FastQ data to high‐confidence variant calls: The genome analysis toolkit best practices pipeline. Current Protocols in Bioinformatics, 43(1). 10.1002/0471250953.bi1110s43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow, D. P. , & Bartolomei, M. S. (2014). Genomic imprinting in mammals. Cold Spring Harbor Perspectives in Biology, 6(2), a018382. 10.1101/cshperspect.a018382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barturen, G. , Rueda, A. , Oliver, J. L. , & Hackenberg, M. (2014). MethylExtract: High‐quality methylation maps and SNV calling from whole genome bisulfite sequencing data. F1000Research 2, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary, J. G. , Braithwaite, R. , Gaastra, K. , Hilbush, B. S. , Inglis, S. , Irvine, S. A. , Jackson, A. , Littin, R. , Nohzadeh‐Malakshah, S. , Rathod, M. , Ware, D. , Trigg, L. , & De La Vega, F. M. (2014). Joint variant and De Novo mutation identification on pedigrees from high‐throughput sequencing data. Journal of Computational Biology, 21(6), 405–419. 10.1089/cmb.2014.0029 [DOI] [PubMed] [Google Scholar]
- da Silva, V. H. , Laine, V. N. , Bosse, M. , van Oers, K. , Dibbits, B. , Visser, M. E. , Crooijmans, R. P. M. A. , & Groenen, M. A. M. (2018). CNVs are associated with genomic architecture in a songbird. BMC Genomics, 19(1), 195. 10.1186/s12864-018-4577-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo, M. A. , Banks, E. , Poplin, R. E. , Garimella, K. V. , Maguire, J. R. , & Hartl, C. (2011). A framework for variation discovery and genotyping using next‐ generation DNA sequencing data. Nature Genetics, 43(5), 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derks, M. F. L. , Schachtschneider, K. M. , Madsen, O. , Schijlen, E. , Verhoeven, K. J. F. , & van Oers, K. (2016). Gene and transposable element methylation in great tit (Parus Major) brain and blood. BMC Genomics, 17(1), 332. 10.1186/s12864-016-2653-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Tommaso, P. , Chatzou, M. , Floden, E. W. , Barja, P. P. , Palumbo, E. , & Notredame, C. (2017). Nextflow enables reproducible computational workflows. Nature Biotechnology, 35(4), 316–319. 10.1038/nbt.3820 [DOI] [PubMed] [Google Scholar]
- Dubin, M. J. , Zhang, P. , Meng, D. , Remigereau, M.‐S. , Osborne, E. J. , Paolo Casale, F. , Drewe, P. , Kahles, A. , Jean, G. , Vilhjálmsson, B. , Jagoda, J. , Irez, S. , Voronin, V. , Song, Q. , Long, Q. , Rätsch, G. , Stegle, O. , Clark, R. M. , & Nordborg, M. (2015). DNA Methylation in arabidopsis has a genetic basis and shows evidence of local adaptation. Elife, 4(May), e05255. 10.7554/eLife.05255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewels, P. , Magnusson, M. , Lundin, S. , & Käller, M. (2016). MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics, 32(19), 3047–3048. 10.1093/bioinformatics/btw354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, S. , Zou, D. , Mao, L. , Liu, H. , Song, P. , Chen, Y. , Zhao, S. , Gao, C. , Li, X. , Gao, Z. , Fang, X. , Yang, H. , Ørntoft, T. F. , Sørensen, K. D. , & Bolund, L. (2015). BS‐SNPer: SNP calling in bisulfite‐seq data. Bioinformatics, August, btv507. 10.1093/bioinformatics/btv507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison, E. , & Marth, G. (2012). Haplotype‐Based Variant Detection from Short‐Read Sequencing. ArXiv:1207.3907 [q‐Bio], July. http://arxiv.org/abs/1207.3907.
- Gienapp, P. , Calus, M. P. L. , Laine, V. N. , & Visser, M. E. (2019). Genomic Selection on Breeding Time in a Wild Bird Population. Evolution Letters, 3(2), 142–151. 10.1002/evl3.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosler, A. (1993). The great tit. Hamlyn species guides. Hamlyn Limited. [Google Scholar]
- Grehl, C. , Wagner, M. , Lemnian, I. , Glaser, B. , & Grosse, I. (2020). Performance of mapping approaches for whole‐genome bisulfite sequencing data in crop plants. Frontiers in Plant Science, 11(February), 176. 10.3389/fpls.2020.00176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero‐Bosagna, C. (2020). From Epigenotype to new genotypes: Relevance of epigenetic mechanisms in the emergence of genomic evolutionary novelty. Seminars in Cell & Developmental Biology, 97(January), 86–92. 10.1016/j.semcdb.2019.07.006 [DOI] [PubMed] [Google Scholar]
- Gugger, P. F. , Fitz‐Gibbon, S. , PellEgrini, M. , & Sork, V. L. (2016). Species‐wide patterns of DNA methylation variation in Quercus Lobata and their association with climate gradients. Molecular Ecology, 25(8), 1665–1680. 10.1111/mec.13563 [DOI] [PubMed] [Google Scholar]
- Guo, W. , Zhu, P. , Pellegrini, M. , Zhang, M. Q. , Wang, X. , & Ni, Z. (2018). CGmapTools improves the precision of heterozygous SNV Calls and supports allele‐specific methylation detection and visualization in bisulfite‐sequencing data. Bioinformatics, 34(3), 381–387. 10.1093/bioinformatics/btx595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, E. , Sinsheimer, J. S. , & Novembre, J. (2014). Characterizing bias in population genetic inferences from low‐coverage sequencing data. Molecular Biology and Evolution, 31(3), 723–735. 10.1093/molbev/mst229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes, K. , Barton, H. J. , & Zeng, K. (2020). A study of Faster‐Z evolution in the great tit (Parus Major). Genome Biology and Evolution, 12(3), 210–222. 10.1093/gbe/evaa044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyn, H. , Moran, S. , Hernando‐Herraez, I. , Sayols, S. , Gomez, A. , Sandoval, J. , Monk, D. , Hata, K. , Marques‐Bonet, T. , Wang, L. , & Esteller, M. (2013). DNA methylation contributes to natural human variation. Genome Research, 23(9), 1363–1372. 10.1101/gr.154187.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höglund, A. , Henriksen, R. , Fogelholm, J. , Churcher, A. M. , Guerrero‐Bosagna, C. M. , Martinez‐Barrio, A. , Johnsson, M. , Jensen, P. , & Wright, D. (2020). The Methylation landscape and its role in domestication and gene regulation in the chicken. Nature Ecology & Evolution, 4(12), 1713–1724. 10.1038/s41559-020-01310-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes, E. E. , Jung, M. , Meller, S. , Leisse, A. , Sailer, V. , Zech, J. , Mengdehl, M. et al (2014). Performance evaluation of kits for bisulfite‐conversion of DNA from tissues, cell lines, FFPE tissues, aspirates, lavages, effusions, plasma, serum, and urine. PLoS One, 9(4), 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpinen, H. , Waszak, S. M. , Gschwind, A. R. , Raghav, S. K. , Witwicki, R. M. , Orioli, A. , Migliavacca, E. , Wiederkehr, M. , Gutierrez‐Arcelus, M. , Panousis, N. I. , Yurovsky, A. , Lappalainen, T. , Romano‐Palumbo, L. , Planchon, A. , Bielser, D. , Bryois, J. , Padioleau, I. , Udin, G. , Thurnheer, S. , … Dermitzakis, E. T. (2013). Coordinated effects of sequence variation on DNA binding, chromatin structure, and transcription. Science, 342(6159), 744–747. 10.1126/science.1242463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. M. , Santure, A. W. , Barton, H. J. , Quinn, J. L. , & Cole, E. F. , Great Tit HapMap Consortium , Visser, M. E. , Sheldon, B. C. , Groenen, M. A. M. , van Oers, K. , & Slate, J. (2018). A high‐density SNP chip for genotyping great tit (Parus Major) populations and its application to studying the genetic architecture of exploration behaviour. Molecular Ecology Resources 18 (4), 877–891. 10.1111/1755-0998.12778 [DOI] [PubMed] [Google Scholar]
- Koster, J. , & Rahmann, S. (2012). Snakemake–A scalable bioinformatics workflow engine. Bioinformatics, 28(19), 2520–2522. 10.1093/bioinformatics/bts480 [DOI] [PubMed] [Google Scholar]
- Krueger, F. , & Andrews, S. R. (2011). Bismark: A Flexible Aligner and Methylation Caller for Bisulfite‐Seq Applications. Bioinformatics, 27(11), 1571–1572. 10.1093/bioinformatics/btr167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger, F. , Kreck, B. , Franke, A. , & Andrews, S. R. (2012). DNA methylome analysis using short bisulfite sequencing data. Nature Methods, 9(2), 145–151. 10.1038/nmeth.1828 [DOI] [PubMed] [Google Scholar]
- Kunde‐Ramamoorthy, G. , Coarfa, C. , Laritsky, E. , Kessler, N. J. , Harris, R. A. , Xu, M. , Chen, R. , Shen, L. , Milosavljevic, A. , & Waterland, R. A. (2014). Comparison and quantitative verification of mapping algorithms for whole‐genome bisulfite sequencing. Nucleic Acids Research, 42(6), e43. 10.1093/nar/gkt1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine, V. N. , Gossmann, T. I. , Schachtschneider, K. M. , Garroway, C. J. , Madsen, O. , Verhoeven, K. J. F. , de Jager, V. , Megens, H.‐J. , Warren, W. C. , Minx, P. , Crooijmans, R. P. M. A. , Corcoran, P. , Sheldon, B. C. , Slate, J. , Zeng, K. , van Oers, K. , Visser, M. E. , & Groenen, M. A. M. (2016). Evolutionary signals of selection on cognition from the great tit genome and methylome. Nature Communications, 7(1), 10474. 10.1038/ncomms10474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird, P. W. (2010). Principles and challenges of genome‐wide DNA methylation analysis. Nature Reviews Genetics, 11(3), 191–203. 10.1038/nrg2732 [DOI] [PubMed] [Google Scholar]
- Langmead, B. , & Salzberg, S. L. (2012). Fast gapped‐read alignment with Bowtie 2. Nature Methods, 9(4), 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea, A. J. , Vilgalys, T. P. , Durst, P. A. P. , & Tung, J. (2017). Maximizing ecological and evolutionary insight in bisulfite sequencing data sets. Nature Ecology & Evolution, 1(8), 1074–1083. 10.1038/s41559-017-0229-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. ArXiv 1303.3997.
- Li, H. , & Durbin, R. (2010). Fast and accurate long‐read alignment with burrows‐wheeler transform. Bioinformatics, 26(5), 589–595. 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. , & Durbin, R. & 1000 Genome Project Data Processing Subgroup (2009). The sequence Alignment/Map format and SAMtools. Bioinformatics, 25(16), 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew, Y. J. , Howells, E. J. , Wang, X. , Michell, C. T. , Burt, J. A. , Idaghdour, Y. , & Aranda, M. (2020). Intergenerational epigenetic inheritance in reef‐building corals. Nature Climate Change, 10(3), 254–259. 10.1038/s41558-019-0687-2 [DOI] [Google Scholar]
- Lindner, M. , Laine, V. N. , Verhagen, I. , Viitaniemi, H. M. , Visser, M. E. , Oers, K. , & Husby, A. (2021). Rapid changes in DNA methylation associated with the initiation of reproduction in a small Songbird. Molecular Ecology. 10.1111/mec.15803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister, R. , Pelizzola, M. , Dowen, R. H. , Hawkins, R. D. , Hon, G. , Tonti‐Filippini, J. , Nery, J. R. , Lee, L. , Ye, Z. , Ngo, Q.‐M. , Edsall, L. , Antosiewicz‐Bourget, J. , Stewart, R. , Ruotti, V. , Millar, A. H. , Thomson, J. A. , Ren, B. , & Ecker, J. R. (2009). Human DNA methylomes at base resolution show widespread epigenomic differences. Nature, 462(7271), 315–322. 10.1038/nature08514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Siegmund, K. D. , Laird, P. W. , & Berman, B. P. (2012). Bis‐SNP: Combined DNA methylation and SNP calling for bisulfite‐seq data. Genome Biology, 13(7), R61. 10.1186/gb-2012-13-7-r61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco‐Sola, S. , Sammeth, M. , Guigó, R. , & Ribeca, P. (2012). The GEM mapper: Fast, accurate and versatile alignment by filtration. Nature Methods, 9(12), 1185–1188. 10.1038/nmeth.2221 [DOI] [PubMed] [Google Scholar]
- Marshall, H. , Jones, A. R. C. , Lonsdale, Z. N. , & Mallon, E. B. (2020). Bumblebee workers show differences in allele‐specific DNA methylation and allele‐specific expression. Genome Biology and Evolution, 12(8), 1471–1481. 10.1093/gbe/evaa132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. , Altshuler, D. , Gabriel, S. , Daly, M. , & DePristo, M. A. (2010). The genome analysis toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20(9), 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkel, A. , Fernández‐Callejo, M. , Casals, E. , Marco‐Sola, S. , Schuyler, R. , Gut, I. G. , & Heath, S. C. (2019). GemBS: High throughput processing for DNA methylation data from bisulfite sequencing. Bioinformatics, 35(5), 737–742. 10.1093/bioinformatics/bty690 [DOI] [PubMed] [Google Scholar]
- Neuwirth, E. (2014). RColorBrewer: ColorBrewer Palettes. R Package Version 1.1‐2. https://CRAN.R‐Project.Org/Package=RColorBrewer
- Nunn, A. , Otto, C. , Stadler, P. F. , & Langenberger, D. (2021). Manipulating base quality scores enables variant calling from bisulfite sequencing alignments using conventional bayesian approaches. BioRxiv, January, 2021.01.11.425926. 10.1101/2021.01.11.425926 [DOI] [PMC free article] [PubMed]
- R Core Team (2017). R: A language and environment for statistical computing. R Foundation for Statistical Computing. https://www.r‐project.org/ [Google Scholar]
- Sepers, B. , van den Heuvel, K. , Lindner, M. , Viitaniemi, H. , Husby, A. , & van Oers, K. (2019). Avian ecological epigenetics: Pitfalls and promises. Journal of Ornithology, 160(4), 1183–1203. 10.1007/s10336-019-01684-5 [DOI] [Google Scholar]
- Stajic, D. , & Jansen, L. E. T. (2021). Empirical evidence for epigenetic inheritance driving evolutionary adaptation. Philosophical Transactions of the Royal Society B: Biological Sciences 376 (1826): rstb.2020.0121, 20200121. 10.1098/rstb.2020.0121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, M. M. , & Bird, A. (2008). DNA methylation landscapes: Provocative insights from epigenomics. Nature Reviews Genetics, 9(6), 465–476. 10.1038/nrg2341 [DOI] [PubMed] [Google Scholar]
- Tomso, D. J. , & Bell, D. A. (2003). Sequence context at human single nucleotide polymorphisms: Overrepresentation of CpG dinucleotide at polymorphic sites and suppression of variation in CpG islands. Journal of Molecular Biology, 327(2), 303–308. [DOI] [PubMed] [Google Scholar]
- van Oers, K. , Sepers, B. , Sies, W. , Gawehns, F. , Verhoeven, K. J. F. , & Laine, V. N. (2020). Epigenetics of animal personality: DNA methylation cannot explain the heritability of exploratory behavior in a songbird. Integrative and Comparative Biology, 60(6), 1517–1530. 10.1093/icb/icaa138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhagen, I. , Gienapp, P. , Laine, V. N. , Grevenhof, E. M. , Mateman, A. C. , Oers, K. , & Visser, M. E. (2019). Genetic and phenotypic responses to genomic selection for timing of breeding in a wild songbird. Functional Ecology, 33(9), 1708–1721. 10.1111/1365-2435.13360 [DOI] [Google Scholar]
- Verhoeven, K. J. F. , von Holdt, B. M. , & Sork, V. L. (2016). Epigenetics in ecology and evolution: What we know and what we need to know. Molecular Ecology, 25(8), 1631–1638. 10.1111/mec.13617 [DOI] [PubMed] [Google Scholar]
- Verhulst, E. C. , Mateman, A. C. , Zwier, M. V. , Caro, S. P. , Verhoeven, K. J. F. , & van Oers, K. (2016). Evidence from pyrosequencing indicates that natural variation in animal personality is associated with DRD4 DNA methylation. Molecular Ecology, 25(8), 1801–1811. 10.1111/mec.13519 [DOI] [PubMed] [Google Scholar]
- Viitaniemi, H. M. , Verhagen, I. , Visser, M. E. , Honkela, A. , van Oers, K. , & Husby, A. (2019). Seasonal variation in genome‐wide DNA methylation patterns and the onset of seasonal timing of reproduction in great tits. Genome Biology and Evolution, 11(3), 970–983. 10.1093/gbe/evz044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Li, A. , Wang, W. , Que, H. , Zhang, G. , & Li, L. (2020). DNA methylation mediates differentiation in thermal responses of pacific oyster (Crassostrea Gigas) derived from different tidal levels. Heredity, 126(1), 10–22. 10.1038/s41437-020-0351-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham, H. (2016). Ggplot2: Elegant graphics for data analysis. Springer‐Verlag. [Google Scholar]
- Wickham, H. (2019). Stringr: Simple, consistent wrappers for common string operations. R Package Version 1.4.0. https://CRAN.R‐Project.Org/Package=stringr
- Wickham, H. , François, R. , Henry, L. , & Müller, K. (2020). Dplyr: A grammar of data manipulation. R Package Version 1.0.0. https://CRAN.R‐Project.Org/Package=dplyr
- Wickham, H. , & Henry, L. (2020). Tidyr: Tidy messy data. R Package Version 1.1.0. https://CRAN.R‐Project.Org/Package=tidyr
- Wilke, C. O. (2020). Cowplot: Streamlined plot theme and plot annotations for ‘Ggplot2.’ R Package Version 1.1.0. https://CRAN.R‐Project.Org/Package=cowplot
- Wingett, S. W. , & Andrews, S. (2018). FastQ Screen: A tool for multi‐genome mapping and quality control. F1000Research 7, 1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, J. , Chen, G. , Hermanson, P. J. , Xu, Q. , Sun, C. , Chen, W. , Kan, Q. , Li, M. , Crisp, P. A. , Yan, J. , Li, L. , Springer, N. M. , & Li, Q. (2019). Population‐level analysis reveals the widespread occurrence and phenotypic consequence of DNA methylation variation not tagged by genetic variation in maize. Genome Biology, 20(1), 243. 10.1186/s13059-019-1859-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Table S1
Supplementary Material
Data Availability Statement
Data used for this article have been made available in the NCBI BioProject database (http://www.ncbi.nlm.nih.gov/bioproject/) under BioProject PRJNA208335 (whole‐genome resequencing: SRR15365281‐SRR15365284, WGBS: SRR15410225‐ SRR15410232). SNP data have been made available on Dryad (https://doi.org/10.5061/dryad.ttdz08kzt). All code and pipelines can be accessed on gitHub (https://github.com/MLindner0/lindner_et_al‐2021‐mer‐snps_from_bs_data) and are presented in the Supplemental Code.