

Abstract
Chronic exposure to hexavalent chromium compounds [Cr(VI)] is associated with an increased risk of cancers, but the molecular mechanisms remain to be elucidated. In this study, we found that CXCL5 levels in peripheral blood monocytes (PBMCs) and plasma from workers with occupational exposure to Cr(VI) were dramatically upregulated compared to non-exposure healthy subjects, and plasma C-X-C Motif Chemokine Ligand 5 (CXCL5) CXCL5 levels were positively correlated with Cr concentrations in subjects’ toenails. Zinc chromate exposed mice showed higher levels of CXCL5 and its receptor CXCR2 in lung tissues, and in PBMCs. Similar CXCL5 upregulation was evident in Cr(VI)-induced transformed (Cr-T) cells with long-term Cr(VI) treatment. Mechanistic studies showed that elevated CXCL5 expression levels were regulated by Cr(VI)-induced histone modifications and DNA hypomethylation, and that the c-Myc/p300 complex was a key upstream regulator of histone H3 acetylation. CXCL5 overexpression promoted Cr (VI)-induced the epithelial to mesenchyme transition (EMT) by upregulating zinc finger E-box binding homeobox 1 (ZEB1) to promote tumor development. Our findings identify a novel mechanism by which CXCL5 is upregulated and promotes EMT and carcinogenesis upon chronic Cr(VI) exposure. Our work also implies that CXCL5 mRNA and protein levels will elevate in PBMCs and serum after occupational Cr(VI) exposure, which may be a potential target and biomarker for cancer prevention and health surveillance among populations exposed to Cr(VI).
Keywords: Epigenetic regulation, CXCL5, Cr(VI) exposure, Carcinogenesis, EMT
Graphical Abstract
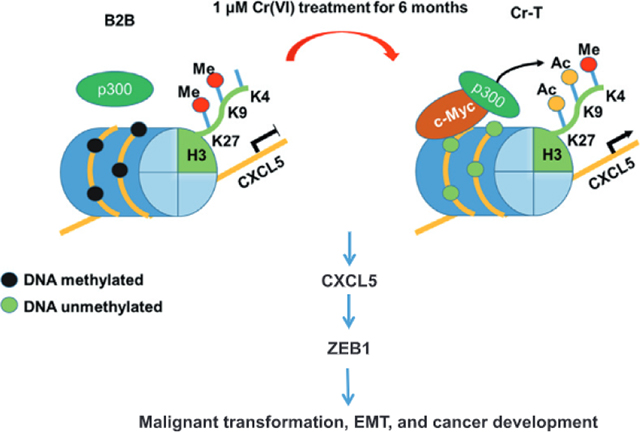
1. Introduction
Hexavalent chromium compounds [Cr(VI)] is recognized as a class I human carcinogen. However, chromium (Cr)-containing compounds are extensively used in various industries for plating, tanning leather, and preserving wood etc. (Xie et al., 2017), where people can be exposed to chromium by breathing chromium dust or fumes which is the major route of chromium exposure. Epidemiological studies identified a high risk of lung cancer associated with Cr(VI) occupational exposure (Proctor et al., 2016). In addition, non-occupational exposures to particulate chromates are also on the rise due to industrial waste disposal, concrete pavement and other sources (De Flora et al., 1997). Cr (VI) compounds are also present in cigarette smoke, automobile emissions, landfills, and water. There is currently no other substitute for chromium. Therefore, it becomes an urgent need to unveil molecular mechanisms of Cr(VI)-induced lung cancer and pinpoint potential target(s) for monitoring lung cancer or other respiratory diseases caused by occupational chromium exposure.
CXCL5 belongs to a superfamily of small, inflammatory proteins that drive multiple processes relevant to tumor progression including angiogenesis, cell proliferation, and leukocyte recruitment through certain G-protein-coupled receptors (Stillie et al., 2009). Accumulating evidence implicates increased expression of CXCL5 in various kinds of neoplastic transformation including gastric, prostate (Park et al., 2007), and pancreatic cancers (Li et al., 2011). Up to now, studies of CXCL5 in cancer research mainly focused on the effect of neutrophil recruitment and cell metastasis (Disteldorf et al., 2015). However, little information is known about whether CXCL5 plays a role in Cr(VI)-induced malignant transformation.
Cr(VI) generally induces carcinogenesis via several mechanisms, including genome instability (GI), abnormal epigenetic changes, alterations of signaling transduction and so on. Under physiological conditions, Cr(VI) can cross the cell membrane through non-specific phosphate/sulfate anionic transporters. Once Cr(VI) gets inside the cell, it can be reduced to insoluble Cr(III), which interacts with DNA and forms DNA-Cr adducts to induce DNA damage and GI. Moreover, the reduction of Cr(VI) produces toxic levels of ROS that can be lethal (Sedman et al., 2006). Cr(VI) is also linked to abnormal epigenetic alterations. For example, the DNA promoter region of human mutant L homolog 1 (hMLHl), the DNA mismatch repair gene, was hypermethylated in 63% Cr(VI)-related lung cancers, which caused a high frequency of replication error (Takahashi et al., 2005). In addition, Cr(VI) impacts histone modifications. It cross-links chromatin to histone deacetylase 1-DNA methyltransferase 1 (HDAC1-DNMT1) complexes and inhibits histone marks of transactivation including phosphorylation of histone H3 Ser-10, trimethylation of H3 Lys-4, and various acetylation marks in histones H3 and H4, which inhibits RNA polymerase II recruitment and represses expression of certain genes (Schnekenburger et al., 2007). The epigenetic alterations upon long-term Cr(VI) exposure remain to be elucidated for further understanding Cr(VI)-induced carcinogenesis.
We have found that C-X-C Motif Chemokine Ligand 5 (CXCL5 or ENA-78) was the most upregulated gene in peripheral blood mononuclear cells (PBMCs) samples taken from subjects occupationally exposed to Cr(VI) using whole genome expression array analysis. In the present study, our human subjects, animal and cell models confirmed that CXCL5 was upregulated at hazardous levels of Cr(VI) exposure. Cr(VI)-induced CXCL5 expression by inducing epigenetic shifts, including histone H3 acetylation and DNA hypometylation, which in turn accelerated EMT and carcinogenesis.
2. Materials and methods
2.1. Human subject samples
The human subject samples with 200–300 have been collected and stored in the Tissue Bank of Nanjing Medical University over several years. These samples were collected using the cross-sectional study of recruited workers from two chrome-electroplating plants in Changzhou City, China. In-person interviews collected standard information about age, gender, occupational history, medical history, and smoking. Regular smokers (>1 cigarette per day, for at least one year) and workers with a history of less than three years Cr (VI) exposure were excluded from the study. Workers from a mechanical manufacturing plant without any Cr(VI) exposure were included as a control group (n = 49). These samples were deidentified with the codes without personal information. The 54 samples with Cr(VI) exposure and 50 nonexposure samples were randomly obtained from the tissue bank using the code number. The human subject information including names, age and other personal information was not known to the investigators.
2.2. Measurement of Cr concentrations in the ambient occupational environment, and worker blood and toenails
Ambient Cr concentrations were determined according to GBZ/T 160.7 (National Occupational Health Standards of the People’s Republic of China). Briefly, air samplers fitted with a 0.8 μm pore size filter were set up to sample air at a rate of 1 L/min on site. After 15 min, the filters were collected and incubated in 5 mL digestion buffer per filter at 160 °C until the buffer evaporated. Appropriate volumes of nitric acid (0.16 M) were used to dissolve any residue and set the final volume to 10 mL. The samples’ absorption at a wavelength of 357.9 nm was measured by an atomic absorption spectrophotometer (iCE™ 3400, Thermo Scientific). Personal air-samplers were worn by workers during their entire shift.
Blood Cr concentrations were measured as described previously (Schnekenburger et al., 2007). Briefly, whole-blood samples obtained from Tissue Bank of Nanjing Medical University were gently homogenized and then 4 mL of ashing acid (HNO3: HC1O4 = 20:1) was added per 0.5 mL of sample. For digestion, samples were heated to 180–200 °C by an electric-hot plate. Extracts were diluted with 1% HN03 to an appropriate protein concentration. The internal standard solutions were rhodium and indium at a concentration of 1 μg/L and 0.1 μg/L, respectively. After calibration, blood Cr concentrations were determined by atomic absorption spectroscopy (iCE™ 3400, Thermo Scientific).
Chromium concentrations in toenails were measured as described before (O’Brien et al., 2019). In brief, 0.1 g of toenail clippings were collected and dried in an oven set to 80 °C. Dried samples were ground before incubation in digesting buffer (nitric acid: hydrogen chloride = 9: 1). A coupled plasma mass spectromctty analysis was conducted using atomic absorption spectroscopy (iCE™ 3400, Thermo Scientific).
2.3. Cell lines and reagents
BEAS-2B (B2B) cells were obtained from American Type Culture Collection and cultured in Dulbecco’s Modified Eagle Medium (Gibco, Grand Island, NY, USA) with 10% FBS. B2B cells were treated with 1 μM Potassium dichromate (Sigma-Aldrich, USA) for 6 months and analyzed by colony formation and tumor-formation assays to confirm the malignantly transformed status. These Cr(VI)-induced transformed cells were named Cr-T cells. Passage-matched B2B cells were used as parental control cells. Trichostatin A (TSA), C646, GSK343, CPI-455, BIX01294, A196, and Sirtinol were purchased from Selleck Chemicals (Houston, TX, USA). Potassium dichromatc was purchased from Sigma (St. Louis, MO, USA). CXCL5 was purchased from R&D Systems (Minneapolis, MN, USA).
2.4. Immunoblotting and immunoprecipitation
Cells were lysed in RIPA Lysis and Extraction Buffer supplemented with protease inhibitors (Life Technologies, USA) on ice for 30 min as previously described (Ge et al., 2018). Samples were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF membranes. Membranes were blocked with 5% non-fat milk and incubated with primary antibodies at 4 °C overnight. Immunoreactive signal was visualized by appropriate horseradish pcroxidase-conjugated secondary antibodies and the Super Signal West Pico Chemiluminescent Substrate Kit (Thermo Scientific, MA, USA).
Co-IP Kits from Thermo Scientific™ were used to perform immunoprecipitation assays following the instruction. Briefly, the extraction of proteins from cultured cells were incubated with antibodies. After overnight incubation at 4 °C, magnetic protein A/G-agarose beads were added. After 3 h, the beads were then washed by washing buffer for 4 times. Immunocomplexes were eluted by elution buffer and followed to analyze by immunoblotting assay with indicated antibodies.
Antibodies used are listed in Supplementary Table 2.
2.5. RNA isolation and quantitative real-time PCR (qRT-PCR)
Total RNAs were extracted by TRIzol reagent (Life Technologies, USA) from cultured cells or tissues. Real-time PCR was performed to test the expression of specific genes using SYBR™ Green PCR Master Mix (Life Technologies, USA) on QuantStudio 3 and 5 Real-Time PCR Systems. All primers were from Integrated DNA Technologies (Coralville, IA) and are listed in Supplementary Table 1.
2.6. Dual-luciferase reporter assay
CXCL5 promoter regions were cloned into the Sac I and Hind III restriction enzyme sites of the pGL3 vector (Promega, WI, USA). Plasmids containing the CXCL5 fragments and renilla luciferase-thymidine kinase (pRL-TK) plasmid were transfected into the B2B and Cr-T cell lines, respectively, using jetPRIME. Firefly luciferase activity was confirmed using a dual luciferase assay kit (Promega, WI, USA) 24 h after transfection.
2.7. DNA methylation analysis
Methylation-specific PCR (MSP) and bisulfate-sequencing PCR (BSP) were used to evaluate the methylation status of the CXCL5 promoter as previously described (Xu et al., 2013). Genomic DNAs were modified with sodium bisulfite using the EpiTect Kit from Qiagen (Germantown, MD, USA) and the levels of CXCL5 methylation in B2B and Cr-T cells were assessed.
2.8. Methylated DNA immunoprecipitation (MeDIP)
The SimpleDIP™ Methylated DNA IP (MeDIP) Kit from Cell Signaling Technology (Danvers, MA, USA) was used to detect methylation status of CXCL5 promoter region. Genomic DNA extracted from B2B and Cr-T cells was sonicated to generate 500 bp fragments that were quantified using the NanoDrop™ 2000/2000c Spectrophotometers. Sheared genomic DNA fragments were incubated with a Rabbit mAb to 5-Methylcytosine (5-mC; D3S2Z) overnight at 4 °C with rotation. The complex was then ligated with ChIP-Grade Protein G Magnetic Beads for 2 h at room temperature; and DNA fragments were subsequently eluted from the beads. After purification, samples were quantified by qPCR. The final results were calculated using the Percent Input Method and the equation shown below. With this method, signals were displayed as a percent ratio of the total input of genomic DNA. Percent INPUT = 10% × 2(C[T] 10%Input Sample − C[T] IP Sample).
2.9. Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed using the Simple ChIP Plus Enzymatic Chromatin IP Kit (Agarose Beads; Cell Signaling Technology) following the protocol as previously described (Ge et al., 2018). Fold enrichment was analyzed according to C[T] value and the following equation: Percent Input = 2% × 2(C[T] 2%Input Sample − C[T] IP Sample).
2.10. Wound healing assay
Cell monolayers at 95% confluency, cultured in 6-well plates, were scratched using 200 μL tips to form gaps, and washed with PBS. Wounds (gaps) were photographed at different time points and analyzed for the migration distance of cells in three different areas per wound.
2.11. Cell migration assay
Cell migration was determined using 24-well chambers with an 8 μm pore (Coming, USA). Cells (5 × 104/well) were seeded in the upper well of the chamber in 100 μL DMEM without FBS. The bottom chamber contained 500 μL DMEM supplemented with 10% FBS to stimulate cell migration. After incubation for 24 h, a cotton swab was used to remove nonmigrated cells, and 3% paraformaldehyde was used to fix the bottom cells, which were stained with 0.1% crystal violet and later photographed in 3 independents 10 × fields per well. Membranes were air-dried, and fixed cells were eluted for 15 min at room temperature with 33% acetic acid decolorization (200 μL/well). The solution was transferred to 96-well plates, and the absorbance value was read at 570 nm using a microplate reader (BMG Labtech, Germany).
2.12. Soft agar colony formation and CCK-8 cell proliferation assay
For soft agar colony formation assays, the wells of a 6-well plate were coated with 1% soft agar. Cells mixed with 1% soft agar were overlayed on this coating. A layer of complete medium was maintained over the upper layer of agar, to prevent desiccation. 100 μL of medium was added twice weekly. Cells were stained with 200 μL of nitroblue tetrazolium chloride solution per well and incubated overnight at 37 °C, then photographed and counted. To confirm the effect of increased expression of CXCL5 on proliferation, we plated 2000 cells per well in 96-well plates. The cell proliferation rate was determined using CCK8 kit (Dojindo Laboratories, Kumamoto, Japan) and the absorption value of 570 nm was measured at indicated time points.
2.13. Immunofluorescence
B2B and Cr-T cells with different treatment as indicated were fixed with 37% paraformaldehyde for 30 min, permeabilized with 0.5% Triton X-100 in PBS at room temperature, and blocked by 1% bovine serum albumin (BSA) in PBST. For immunostaining, cell preparations were probed with antibodies against E-Cadhcrin and Vimentin at 4 °C, then incubated with fluorescence isothiocyanate (FITC)-labeled goat anti-rabbit secondary antibody (Santa Cruz) and tetramethylrhodamine isothiocyanate (TRITC)-labeled goat anti-mouse secondary antibody (Santa Cruz). Cells were incubated with 10 μM Hoechst 33342 for 10 min to locate nuclei. Images were taken under a fluorescence microscope (ZEISS, Germany).
2.14. Intranasal instillation of Cr(VI) and immunohistochemical (IHC) staining
BALB/cJ mice at 6–8 weeks old from the Jackson Laboratory (Bar Harbor, ME, USA) were acclimatized for 12 days before exposure. Zinc chromate [ZnCrO44Zn(OH)2] suspension in saline solution at 50 μg/50 μL/mouse or 50 μL of saline was Intranasally instilled once a week for 12 weeks, as we described before (Wang et al., 2022). After mice were euthanized, blood and lung tissues were collected for further examination. PBMCs were separated using Flcoll-Paque (ThermoScientific, Rockford, IL). Partial lung tissues were snap-frozen for RT-qPCR assay to detect CXCL5 and CXCR5 expression. Partial lung tissues were fixed and embedded using formalin. The animal experiments were performed following the protocols and procedures approved by the Institutional Animal Care and Use Ethics Committee at Thomas Jefferson University. IHC staining was performed to detect CXCL5 and its receptor CXCR2. CXCL5 (BS-2549R from Bioss) and its receptor CXCR2 (ab14935 from abeam) antibodies were used for IHC staining.
2.15. ELISA
Plasma and cell culture supernatants were collected and levels of human CXCL5 were assessed using the Human CXCL5 SimpleStep ELISA® Kit (ab212163, Abeam, USA).
2.16. In vivo study using orthotopic lung cancer xenograft model
Six-week-old BALB/c nude mice were purchased from The Jackson laboratory (Bar Harbor, Maine, USA) and fed under special pathogen-free conditions for one week. Animal experimental procedures were consistent with the Care and Use of Laboratory Animals Guide and approved by the Institutional Animal Care and Use Ethics Committee at Thomas Jefferson University. Cells (4 × 106) resuspended in 50 μL of DMEM basal medium plus 50 μL of Matrigel were injected into the lung of a nude mouse. The injection site was on the left anterior axillary line between the third and fourth ribs. Tumor growth was visualized weekly in anesthetized mice by intraperitoneal injection of D-luciferin (150 mg/kg) followed by imaging under the IVIS Illumina System (Caliper Life Sciences). Five weeks after implantation, tumors were harvested from sacrificed mice.
2.17. Statistical analysis
The data were shown as means ± SEM from at least three independent experiments. A two-tailed Student’s unpaired t-test was used to compare experimental and control groups. Pearson Correlation was used for correlation analysis. Differences were considered significant at P < 0.05. GraphPad Prism 8.0 (https://www.graphpad.com/) was used for graph-drawing. For the field samples and clinical lung adenocarcinoma (LUAD) samples in Fig. 1 and Fig. 9, all the data were conformed to the normal distribution.
Fig. 1.
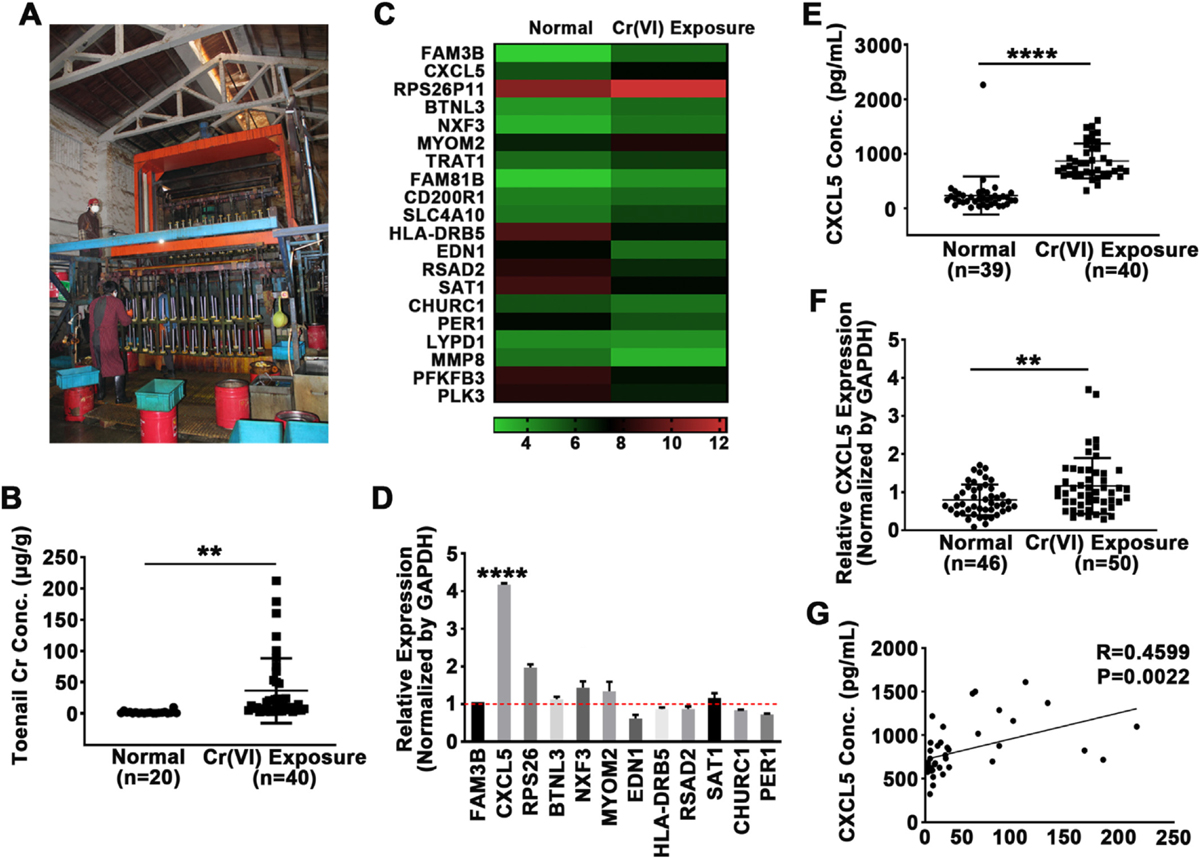
CXCL5 was upregulated in blood samples of Cr(VI)-exposed workers. (A) Scene of industrial chromate-plating plants, the sampling sites. (B) Chromium (Cr) concentrations in toenails from Cr(VI)-exposed workers (n = 40) and control subjects (n = 20) were analyzed by atomic absorption spectroscopy. (C) Heatmap of mRNA microarray analysis of peripheral blood mononuclear cells (PBMCs) that were isolated from blood samples of three Cr(VI)-exposed workers and three healthy subjects. PBMCs from each group were pooled and RNA was extracted for transcriptome array. (D) Quantitative real-time PCR (qRT-PCR) data of 12 genes identified as significantly differentially expressed by microarray in Cr(VI)-exposed workers and healthy subjects. GAPDH was used as internal control and normalized to the values of healthy controls. (E) CXCL5 protein levels in the plasma of Cr(VI)-exposed workers (n = 40) and control subjects (n = 39) were determined by ELISA. (F) CXCL5 mRNA levels in PBMCs from Cr(VI)-exposed workers (n = 50) and control subjects (n = 46) were determined by qRT-PCR, GAPDH was used as internal control. (G) Pearson correlation analysis between CXCL5 protein expression in plasma and the concentration of Cr(VI) in toenails of Cr(VI)-exposed workers (n = 42). ** and ****, p < 0.01 and p < 0.0001 compared to control group.
Fig. 9.
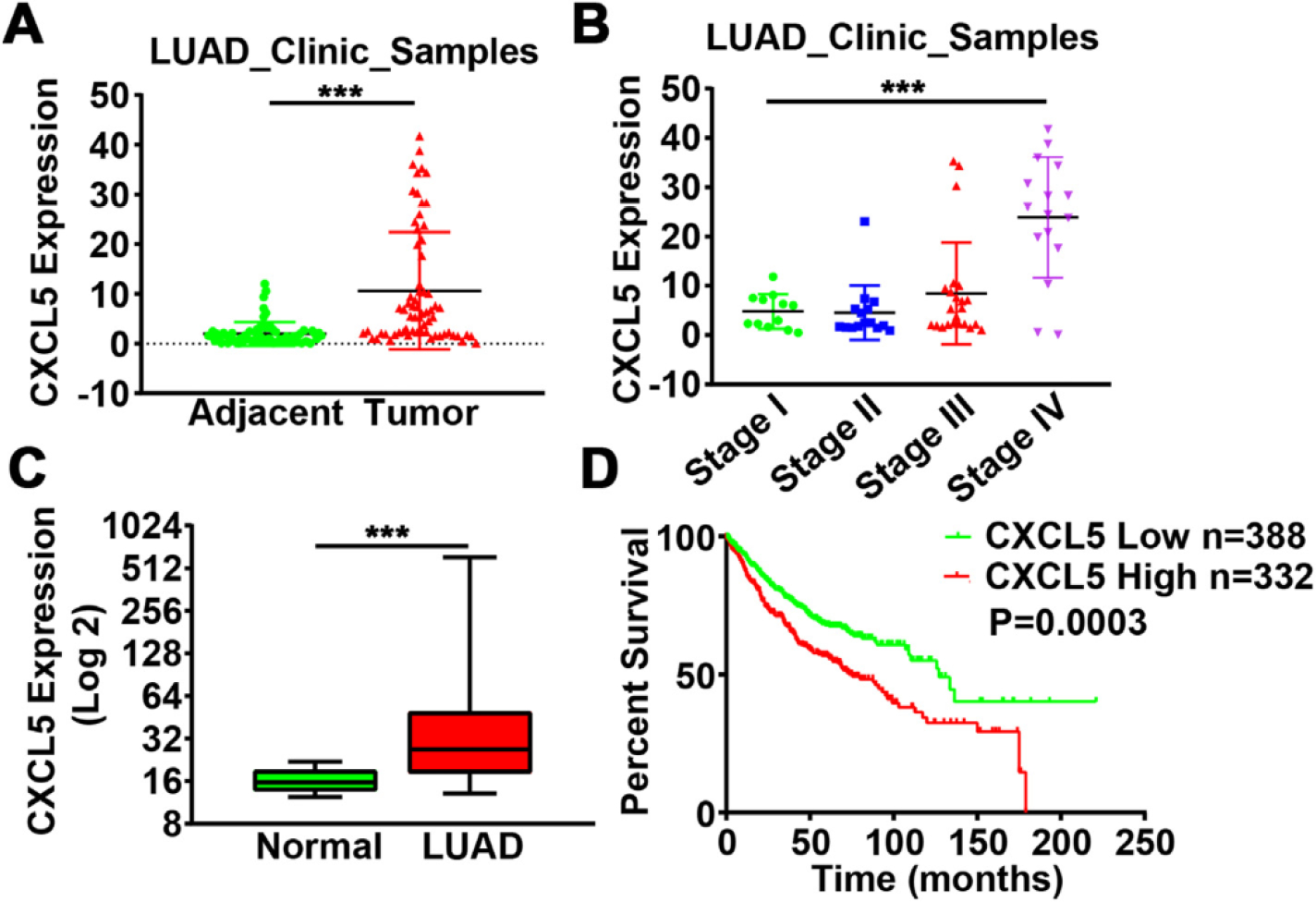
Elevated expression of CXCL5 in lung adenocarcinoma correlated with a poor prognosis. (A) CXCL5 expression levels in 66 pairs of lung adenocarcinoma tissues (LAUD) and adjacent normal tissues. ***, p < 0.001 compared to adjacent normal tissues. (B) CXCL5 expression in tissues from lung adenocarcinoma patients, categorized into different TNM stages. ***, p < 0.001 compared to Stage I. (C) CXCL5 expression levels in normal lung and adenocarcinoma tissues (LUAD) from Metabolic Gene Rapid Visualizer (http://merav.wi.mit.edu/). ***, p < 0.001 compared to normal tissues. (D) Kaplan-Meier plot of the overall survival of patients with lung adenocarcinoma with high or low expression of CXCL5 (Affy ID = 215101_s_at) from Kaplan-Meier plotter (http://kmplot.com/analysis/).
3. Results and discussion
3.1. Ambient Cr, blood Cr, and toenail Cr upon long-term occupational Cr(VI) exposure
Chromium is a widely used industrial raw material due to its strength and resistance to corrosion and extreme high temperature. However, Cr (VI) also induces respiratory diseases including lung cancer by generating reactive oxygen species, DNA damage, and chronic inflammation (Hou et al., 2012; Nickens et al., 2010). The increasing use and waste emission of chromium, and the threats to human health caused by both occupational and non-occupational exposures have recently garnered vigilance, eliciting calls to explore the intrinsic mechanisms of Cr(VI)-induced respiratory diseases.
We collected large amounts of field samples including blood and toenails from industrial chromate-plating plants, depicted in Fig. 1A. Characteristics of the study participants were presented in Table 1, showing no significant difference in terms of age, gender, and years of employment between the exposure and control groups. Field study and subsequent lab tests showed that the 8 h-time-weighted average (TWA) of ambient chromium concentrations in the electroplating plants ranged from 0.016 to 0.02 mg/m3 (Table 2), which is below the permissible limit for exposure (0.05 mg/m3, China). Similarly, personal air samplers also showed that exposure complied with the national regulation standard (Table 3). The Cr concentration in blood was measured by atomic absorption spectroscopy; and it did not differ between Cr(VI) exposure workers and control subjects (data not shown), suggesting that blood Cr is rapidly metabolized and it’s not a good indicator of Cr(VI) exposure. Toenails grow relatively slowly and in isolation from the body’s metabolic activities, so their Cr concentration was expected to reflect whole-body intake and exposure during the past one to one and a half year. Metabolically inactive tissues such as nails in biomonitoring are widely used in research, especially in the study of many trace elements accumulated in the body (Mehra and Juneja, 2005). Unlike blood samples which reflect concentrations only transiently, nails can provide a continuous record of trace element concentrations of the body. Moreover, nails are easily sampled, transported, and stored. Thus, despite some well-known shortcomings (e.g., the potential for contamination), this method is first choice for many clinical studies (Wilhelm et al., 1991). Here we tested Cr concentrations in toenails of 40 occupational workers who exposed to Cr(VI) for more than three years and 20 healthy controls using atomic absorption spectroscopy. The average Cr concentration in toenails from the exposed group was 36.3 μg/g compared with 1.5 μg/g in the control group, confirming Cr(VI)-exposed workers have higher concentrations of Cr in their toenails (Fig. 1B). Thus, toenail Cr concentration detection might be more reliable than blood Cr concentration to trance long-term Cr(VI) exposure in occupational population.
Table 1.
Characteristics of Cr(VI)-exposed workers and control subjects.
| Characteristic | Cr(VI)-exposed workers | Control subjects | P value |
|---|---|---|---|
|
| |||
| No. of subjects | 35 (25–49)a | 33 (25–53)a | 0.527 |
| Age (years) | |||
| Gender | |||
| Male | 29 | 25 | 0.549 |
| Female | 25 | 24 | |
Median (interquartile range).
Table 2.
Ambient total Cr concentration at different work sites measured by area air samplings (mg/m3) using two methods.
| Area of sampling | Sample number | GBZ/T 160.7a |
NIOSH 7703b |
||
|---|---|---|---|---|---|
| Mean | Range | Mean | Range | ||
|
| |||||
| Products loading area after electroplating | 6 | <0.010 | <0.010 | 0.0019 | 0.0016–0.0023 |
| Electroplating tanks | 6 | 0.027 | 0.026–0.028 | 0.0276 | 0.0241–0.0286 |
| Administrative offices | 12 | <0.010 | <0.010 | 0.0012 | 0.0007–0.0022 |
GBZ/T 160.7: Methods for determination of chromium and its compounds in the air of workplace according to “National occupational health standards of the People’s Republic of China”.
NIOSH 7703: Methods for determination of chromium and its compounds in the air of workplace according to “The National Institute for Occupational Safety and Health (NIOSH)”.
Table 3.
Total Cr concentration among Cr(VI)-exposed workers measured by personal air samplers (n = 7).
| Workers | Exposure time (min) | Inhaled volume (L) | ConcentrationTWAa (mg/m3) |
|---|---|---|---|
|
| |||
| A | 392 | 392 | 0.0104 |
| B | 383 | 766 | 0.0160 |
| C | 384 | 1152 | 0.0201 |
| D | 377 | 1508 | 0.0214 |
| E | 369 | 1845 | 0.0149 |
| F | 375 | 2250 | 0.0220 |
| G | 415 | 2905 | 0.0147 |
TWA: 8 h-time-weighted-average.
3.2. CXCL5 was upregulated in blood samples of workers with occupational exposure to Cr(VI)
CXCL5 enhances the malignant phenotype in lung cancer, breast cancer and other types of cancer (Begley et al., 2008; Hsu et al., 2013; Kawamura et al., 2012; Li et al., 2011; Miyazaki et al., 2006; Zhou et al., 2012). In addition, CXCL5 plays an oncogenic role in colorectal cancer and promotes tumor angiogenesis by increasing FOXD1 expression levels (Chen et al., 2019). CXCR2, the receptor of CXCL5, is highly expressed in both lung cancer cells and stromal cells to increase tumor inflammation, metastasis and angiogenesis. CXCL5 mainly drives CXCR2-mediated gene cluster expression (Arenberg et al., 1998; Saintigny et al., 2013). Also, bone marrow cells can secrete CXCL5, which is sensed by breast cancer cells via CXCR2, recruiting breast cancer cells to metastasize to bone (Romero-Moreno et al., 2019). Despite that CXCL5 is considered an oncogene in certain malignant transform models, the role and mechanism of CXCL5 in metal-induced carcinogenesis have never been demonstrated. To identify molecular changes caused by Cr (VI) exposure, gene expression in PBMCs samples from the exposed group was compared to that of the control group using whole genome expression array analysis. To reduce the dimension of array results, the data were filtered, and the top twenty differentially expressed genes sorted in two groups (Fig. 1C). The expression changes were verified by qRT-PCR, showing that among these twenty genes, CXCL5 was the most significantly upregulated one (Fig. 1D). Protein concentrations of CXCL5 in plasma and levels of CXCL5 mRNA in PBMCs were determined by ELISA assay and qRT-PCR, respectively. The average plasma concentration of CXCL5 protein was 6-fold higher in the Cr(VI)-exposed group compared to controls (Fig. IE). Similarly, CXCL5 mRNA expression was higher in PMBCs from Cr(VI)-exposed workers (Fig. IF), suggesting that Cr(VI) exposure elevates levels of CXCL5 expression in humans. The positive correlation between the toenail Cr concentration and the expression level of CXCL5 was confirmed by Pearson correlation analysis (R = 0.4599, p = 0.0022, Fig. 1G). Collectively, these results suggest that Cr (VI) exposure is associated with higher expression levels of CXCL5, both in plasma and PBMCs samples.
3.3. Intranasal instillation of Cr(VI) for 12 weeks promoted CXCL5 expression in BALB/cJ mice
The data from occupational Cr(VI)-exposed workers demonstrated that CXCL5 expression was upregulated in response to long-term Cr(VI) exposure. To further verify the correlation of CXCL5 and Cr(VI) exposure in vivo, BALB/cJ mice were exposed to zinc chromate [ZnCrO44Zn(OH)2] suspension in saline solution at 50 μg/50 μL/mouse once a week for 12 weeks by intranasal administration. Mice intranasally instilled saline alone were used as control. The PBMCs from blood samples and lung tissues were collected after the mice were euthanized. Chronic Cr(VI) exposure led to an aberrant remodeling of the alveolar epithelial cells and exaggerated accumulation of extracellular matrix components in the interstitial. IHC staining results showed that CXCL5 and CXCR2 expression dramatically increased in lung tissues from Cr(VI)-exposed mice compared to control mice (Fig. 2A). Consistent with the above results, CXCL5 and CXCR2 mRNA levels in lung tissues were higher after Cr(VI) exposure (Fig. 2B). Meanwhile, the CXCL5 levels in PBMCs of Cr(VI)-exposed mice was also significantly increased (Fig. 2C). Thus, our data solidly provided the evidence that long-term Cr(VI) exposure enhances CXCL5 expression, and higher levels of CXCL5 in PBMCs from blood samples of both human and mice might be a potential biomarker for chronic Cr(VI) exposure.
Fig. 2.
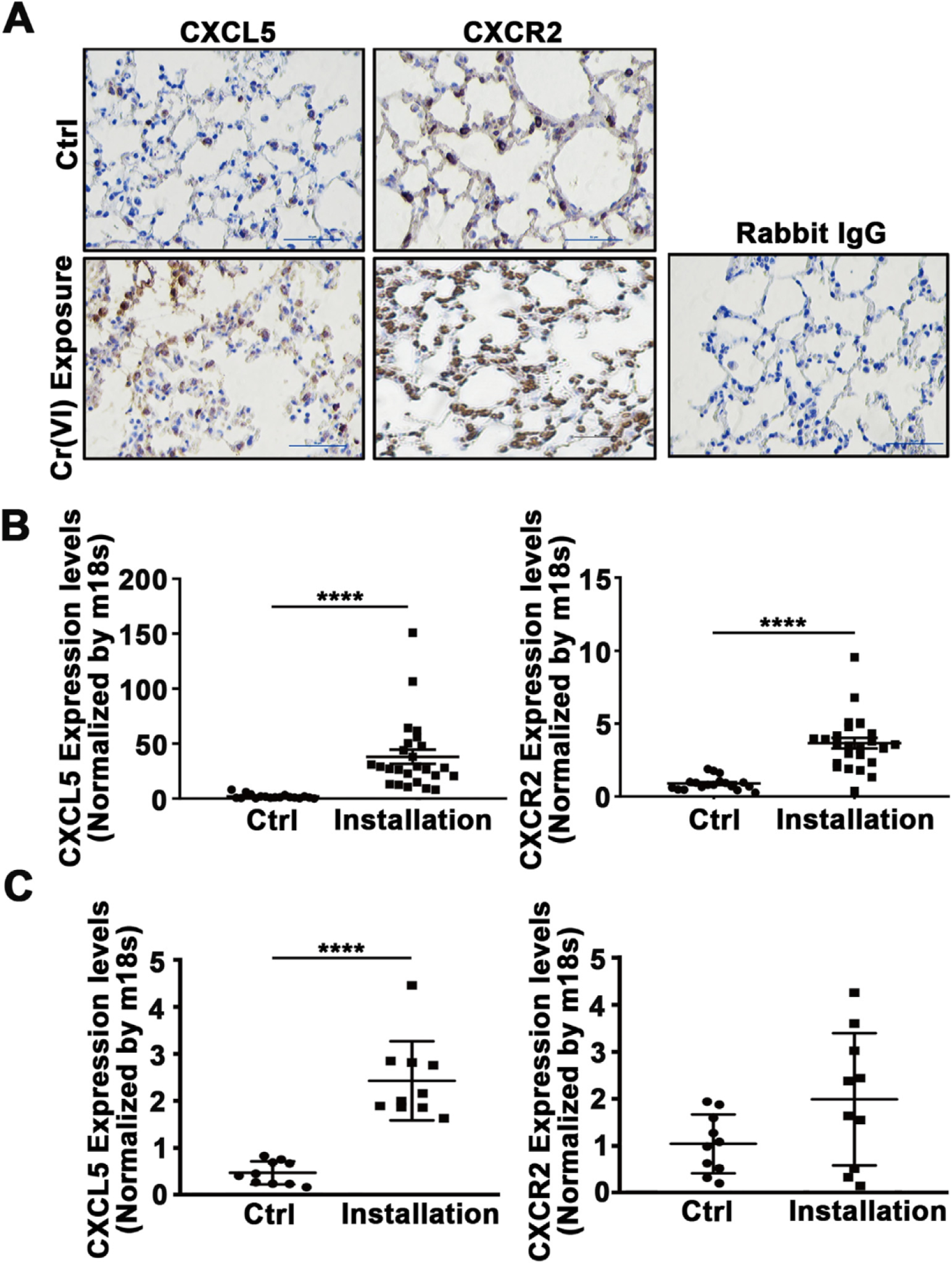
Intranasal instillation of Cr(VI) for 12 weeks promoted CXCL5 expression in BALB/cJ mice. BALB/cJ mice were treated intranasally with zinc chromate (50 μg/50 μL/ mouse) or saline solution (vehicle for the control group) once a week. After 12 weeks, mice were euthanized, and blood and lung tissues were collected for further examination. PBMCs were separated using Ficoll-Paque. (A) Lung tissues were fixed and embedded using formalin. Immunohistochemical (IHC) staining was performed to detect CXCL5 and its receptor CXCR2. Magnification: 200×. (B) Lung tissues were used to extract RNAs. The expression levels of CXCL5 and CXCR2 was determined using RT-qPCR. (C) PBMCs were isolated from blood samples of mice, the expression of CXCL5 and CXCR2 was analyzed using RT-qPCR as above. ****, p < 0.0001 compared to control group without Cr(VI) intranasal instillation.
3.4. Chronic Cr(VI) treatment increased CXCL5 expression in human bronchial epithelial cells
To verify the results showing upregulated CXCL5 upon Cr(VI) exposure from field samples and animal models, CXCL5 levels were studied in three cell lines exposed to Cr(VI), including human fetal lung fibroblast 1 (HFL1), human umbilical vein endothelial cells (HUVEC), and BEAS-2B (B2B) cells. In both HFL1 and B2B cells, acute Cr(VI) treatment induced CXCL5 expression, with B2B cells being more sensitive to Cr(VI) exposure (Fig. 3A), indicating that bronchial epithelial cells, the major type of cell from which lung cancer origins, are more sensitive upon Cr(VI) treatment. Next, B2B cells were treated with 1 μM Cr(VI) for 0, 3, 4, 5 and 6 months to model chronic exposure. CXCL5 mRNA expression was significantly induced by Cr(VI) exposure for 3 months, and reached highest level for six months’ exposure (Fig. 3B). Consistent with the qRT-PCR results, the cell supernatants contained significantly higher levels of CXCL5 protein after 3 months of treatment (Fig. 3C). Recent studies have reported that CXCL5 is highly expressed in human non-small cell lung cancer cells and it contributes to promote cancer cell proliferation and migration (Wang et al., 2018; Wu et al., 2017). In agreement with these studies, we found that several lung adenocarcinoma cell lines expressed higher CXCL5 mRNA levels than the immortalized normal human bronchial epithelial B2B cell line (Supplementary Fig. 1). Collectively, our findings show Cr(VI) can induce CXCL5 expression in human bronchial epithelial cells.
Fig. 3.
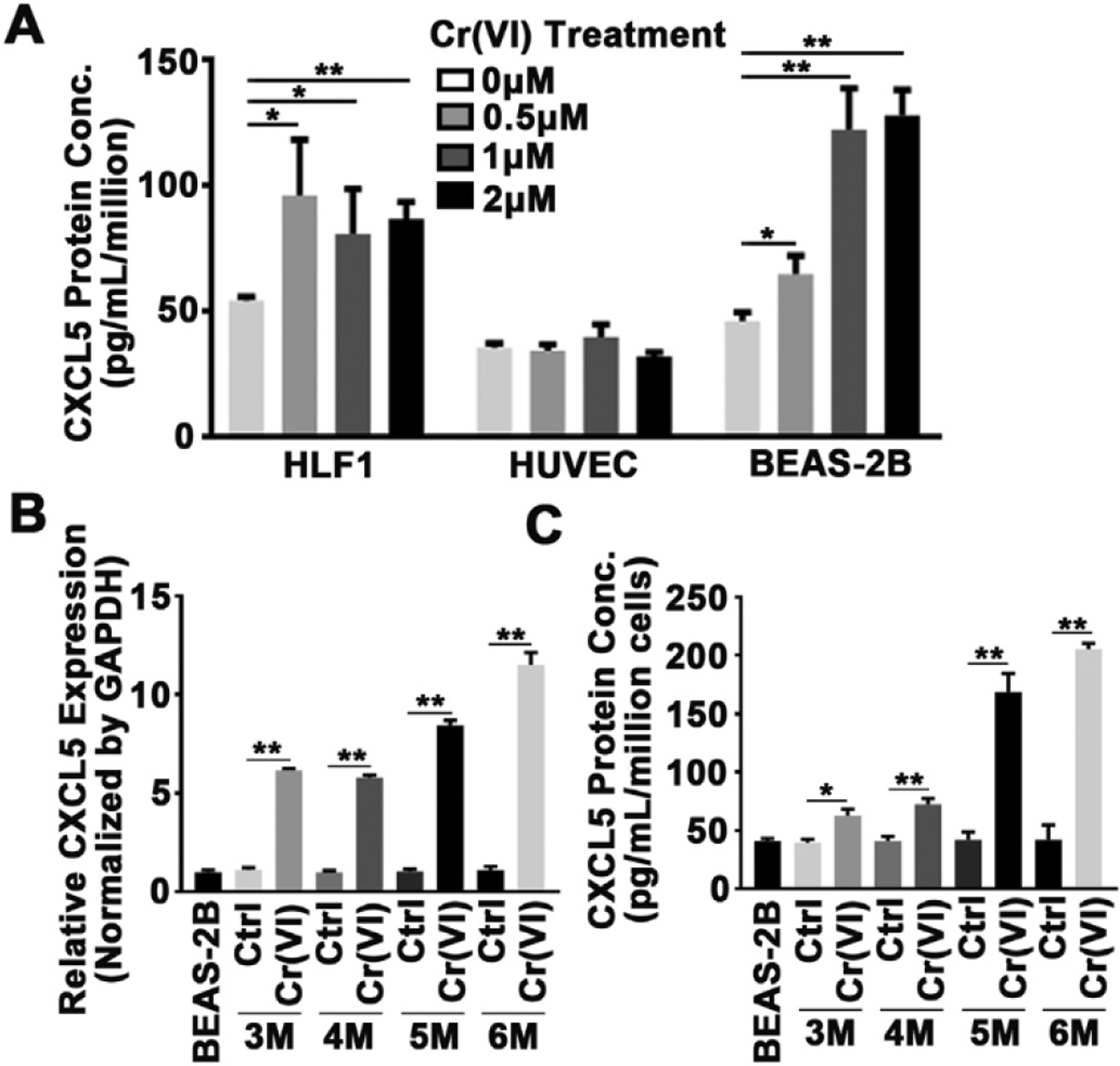
Chronic Cr(VI) treatment increased CXCL5 expression in human bronchial epithelial cells. (A) CXCL5 protein expression levels in human lung fibroblasts (HLF), human umbilical vein endothelial (HUVEC) cells, and immortalized bronchial epithelial cells (B2B) treated with different concentrations of Cr(VI) for 72 h were determined by ELISA. * and **, p < 0.05 and p < 0.01 compared to control group without Cr(VI) treatment. (B) The expression of CXCL5 mRNA in cells treated with Cr (VI) for indicated time points was analyzed using qRT-PCR. (C) CXCL5 protein in supernatant of cells treated as above was detected by ELISA. P values <0.05 (5.0E-2) means the results are statistically significant. * and **, p < 0.05 and p < 0.01 compared to parental B2B cells.
3.5. Cr(VI) exposure induced histone H3 acetylation at the CXCL5 promoter region in human bronchial epithelial cells
Epigenetic gene regulation can be controlled by histone modifications, such as acetylation and methylation, which affect chromatin packaging. To determine whether epigenetics factors control CXCL5 gene expression changes during Cr(VI) exposure, we examined data sets from the UCSC Genome Browser (http://genome.ucsc.edu/) and the ENCODE database (https://www.encodeproject.org/) to check CXCL5 histone modification levels according to the ChIP-Seq data. These databases suggested that epigenetic marks H3K27ac and H3K4me3 were enriched within 1000 bp upstream of the CXCL5 transcription start site (TSS), linking this region to CSCL5 transcriptional regulation (Fig. 4A). To evaluate whether this region contains open chromatin, we further analyzed the data from DNase-seq, a high-resolution technique for mapping active gene regulatory elements across the genome that uses deoxyribonuclease (DNase) to hydrolyze exposed phosphodiester linkages in the DNA backbone (Wang et al., 2012). DNase-seq identified open chromatin with high DNase activity around the same region containing modified histone H3, suggesting the promoter region of CXCL5 has a crucial function in regulating its expression (Fig. 4A).
Fig. 4.
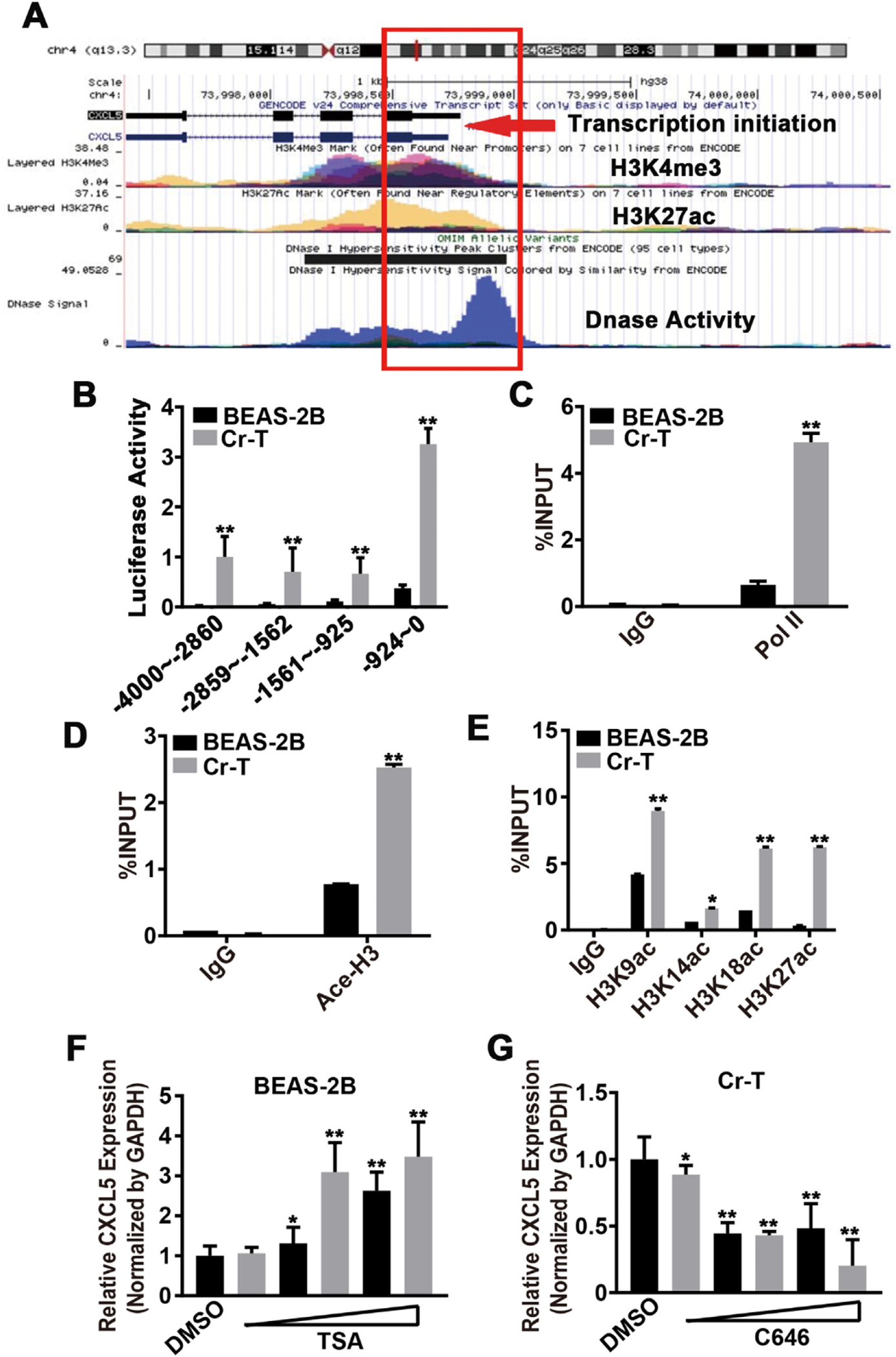
Cr(VI) exposure induced histone H3 acetylation at the CXCL5 promoter region in human bronchial epithelial cells. (A) Tracks from the UCSC Genome Browser (http://genome.ucsc.edu/) and ENCODE database (https://www.encodeproject.org/) depicting H3K27Ac, H3K4Me3, and DNase activity at the promoter region of CXCL5. Red box outlines the regulation fragment. Red arrow indicates the transcription initiation site. (B) Luciferase reporter plasmids expressing different regions from −4000 to 0 before the transcription start site (TSS) of CXCL5 were constructed. Luciferase activities in B2B and Cr-T cells that were transfected with four different luciferase reporter plasmids were measured by a luciferase assay 24 h after transfection. (C) The enrichment of RNA polymerase II at the most active area (−924–0) within the CXCL5 promotor region in B2B and Cr-T cells was determined using chromatin immunoprecipitation (ChIP) assays. IgG was used as a negative control. IgG or Pol II binding was shown as a percentage of the total input. (D) Total Acetyl-Histone H3 in the CXCL5 promoter region from −924–0 bp in B2B and Cr-T cells using ChIP assay. (E) The acetylation sites K9, K14, KI 8 and K27 on Histone H3 in the CXCL5 promoter region (−924–0) in B2B and Cr-T cells using ChIP assay. Data were presented as means ± SEM of triplicates. * and **, p < 0.05 and p < 0.01 compared to parental B2B cells. (F and G) B2B and Cr-T cells were treated with different doses of TSA (0,0.5, 1, 2.5, 5, 7.5 μM) and C646 (0, 20, 40, 60, 80,100 μM), respectively. The expression of CXCL5 and GAPDH was tested by qRT-PCR. Data were presented as means ± SEM of triplicates. * and **, p < 0.05 and p < 0.01 compared to DMSO solvent control group.
Next, fragments of the CXCL5 promoter (ranging from −4000 to 0 bp from the TSS) were used to make luciferase reporter constructs. In both parental B2B cells and Cr-T cells, CXCL5 transcriptional activation was upregulated using luciferase reporter plasmids containing the four promoter fragments, particularly the region from −924 bp to 0 bp, in agreement with the database predictions (Fig. 4B). Also, Pol II binding area was enriched in the promoter region from −924 bp to 0 bp and was more pronounced in Cr-T cells compared to the parental B2B cells (Fig. 4C). To confirm the predicted increase in histone marks in this region, acetyl-Histone H3 was determined using a chromatin immunoprecipitation (ChIP) assay. Cr-T cells showed more acetyl-Histone H3 in the −924 to 0 bp of CXCL5 promoter region compared to B2B cells (Fig. 4D). Moreover, in Cr-T cells, the CXCL5 promoter was more strongly marked by histone modifications associated with transcriptional activation (i.e., H3K9ac, H3K14ac, H3K18ac, H3K27ac, H3K4, H3K36me3, and H3K79me2), whereas the histone modifications of transcriptional repression (H3K9 and H3K27) were weaker (Fig. 4E, Supplementary Fig. 2A and B). These findings indicate that the chromatin structure of the CXCL5 promoter region in Cr-T cells is favorable for transcriptional activation.
To identify the type of histone modification(s) that drives CXCL5 transcriptional activation, B2B and Cr-T cells were treated with a variety of agents: C646 (a HAT inhibitor/p300 inhibitor), trichostatin A (TSA, a histone deacetylases [HDACs] inhibitor), GSK343 (an EZH2 inhibitor that blocks H3K27me3), CPI-455 (a KDM5 inhibitor that increases H3K4me3), BIX01294 (a G9a histone methyltransferase inhibitor that blocks H3K9me2 and H3K9me3), A196 (a SUV420H1 and SUV420H2 inhibitor), and Sirtinol (a SIRT1 and SIRT2 inhibitor). TSA, a potent and specific inhibitor of HDACs, increased CXCL5 expression in parental B2B cells by enhancing histone H3 acetylation (Fig. 4F, Supplementary Fig. IC), whereas C646, a novel CBP/p300-specific inhibitor of histone acetyltransferase (HAT), suppressed CXCL5 in Cr-T cells by inhibiting histone H3 acetylation (Fig. 4G, Supplementary Fig. 2C). In contrast, GSK343, CPI-455, BIX01294, A196, and Sirtinol did not change the expression levels of CXCL5 (Supplementary Fig. 2D). These results demonstrate that histone acetylation is likely one of the key factors affecting the Cr (VI)-induced CXCL5 upregulation.
3.6. c-Myc upregulation increased CXCL5 expression by recruiting p300 and enhancing histone H3 acetylation in Cr-T cells
c-Myc, a proto-oncogene, is dysfunctional in over 50% of human cancers and is associated with a poor prognosis (Dang, 2012). It is a crucial transcription factor that associates with promoters enriched for euchromatic markers. It acts as the master regulator of many signaling pathways including cell growth, differentiation, and apoptosis (Dang, 2012) via recruitment of histone acetylases (Dang et al., 2006). c-Myc interacts with histone modifying enzymes to form a transcription complex that induces chromatin changes such as hyperacetylation of histones (Amente et al., 2011). To determine which factors influence the hyperacetylation of CXCL5, the transcription factors enriched at the CXCL5 promoter region were assessed by ChIP-seq data from GSE31477 dataset The results showed highly enriched c-Myc in A549 lung cancer cell line (Fig. 5A), and there was a c-Myc-specific cis-acting element in promoter region of CXCL5 by analyzing the JASPAR databases (Fig. 5B), implicating that c-Myc promote CXCL5 transcriptional activation by binding to the CXCL5 promoter. C-Myc usually recruits p300 to format a transcriptional complex that activates transcription of downstream genes via histone acetylation (Tikhanovich et al., 2017). Next, to test whether c-Myc and its co-activator p300 are involved in mediating CXCL5 upregulation upon Cr(VI) exposure through increased histone acetylation, the expression levels of c-Myc and p300 were determined in B2B and Cr-T cells. As expected, Cr-T cells expressed much higher level of c-Myc, but p300 expression was equivalent by immunoblotting assay (Fig. 5D). Moreover, forced overexpression or silencing of c-Myc and p300 in B2B or Cr-T cells consequently upregulated or downregulated CXCL5 mRNA expression levels, respectively (Fig. 5C). Since c-Myc and CBP/p300 synergistically activate gene transcription via cooperative binding and chromatin acetylation (Cho et al., 2015; Vervoorts ct al., 2003), ChIP assays were used to analyze binding at the CXCL5 promoter. We found that c-Myc and p300 were enriched at the CXCL5 promoter region in Cr-T cells compared to parental B2B cells (Fig. 5E). Co-immunoprecipitation assays verified the c-Myc/p300 interaction, showing an increased association in Cr-T cells than that in B2B cells, further suggesting that c-Myc activation is pivotal for recruiting p300 to the CXCL5 promoter region in Cr-T cells (Fig. 5F). Finally, H3 acetylation status was assessed in B2B cells overexpressing or Cr-T cells knocking down c-Myc and p300 using ChIP assay. The results showed that overexpression of c-Myc or p300 in parental B2B cells promoted histone H3 acetylation, whereas knockdown of c-Myc or p300 in Cr-T cells markedly decreased total acetylation levels of histone H3, indicating that the c-Myc/p300 transcription complex promotes CXCL5 transcription by hyperacetylating histone H3 at its promoter region (Fig. 5G). Overall, these results confirm that c-Myc is activated by long-term Cr (VI) exposure, and then specifically binds to the CXCL5 promoter region. Once bound to the CXCL5 promoter, c-Myc recruits p300 to form a transcription complex that eventually drives CXCL5 transcription through enhancing histone H3 acetylation.
Fig. 5.
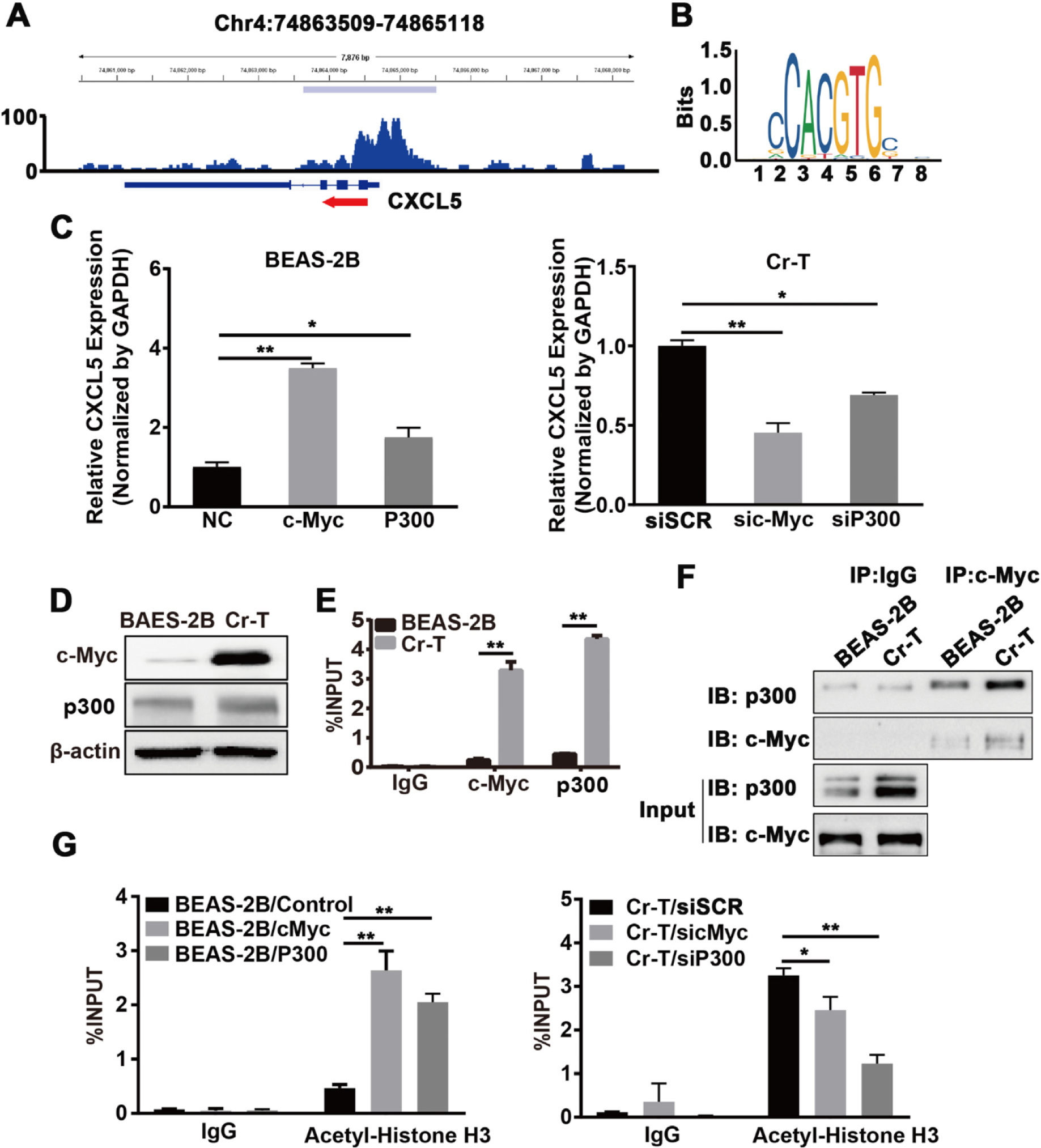
C-Myc upregulation increased CXCL5 expression by recruiting p300 and enhancing histone H3 acetylation in Cr-T cells. (A) The GSE31477 GEO dataset was visualized and peaks were called using the Integrated Genomics Viewer (IGV) to predict the binding sites and enrichment intensity of c-Myc in the promoter region of CXCL5. (B) The potential binding motif of c-Myc in the CXCL5 promoter was identified using JASPAR (http://jaspar.genereg.net). (C) B2B cells transfected with pcDNA3 empty vector, pcDNA3-c-Myc, or pcDNA3-p300 (left panel) or Cr-T cells transfected with an siRNA scrambled control (siSCR), an siRNA targeting c-Myc (sic-Myc) or an siRNA targeting p300 (sip300, right panel) were harvested 72 h post transfection for analysis of CXCL5 and GAPDH expression by qRT-PCR. * and **, p < 0.05 and p < 0.01 compared to empty negative control (NC) or siSCR group. (D) c-Myc and p300 protein expression in parental B2B and Cr-T cells using immunoblotting, β-actin was used as a loading control. (E) ChIP assay of the CXCL5 promoter from B2B or Cr-T cells using antibodies against IgG, c-Myc or p300. **, p < 0.01 compared to B2B cells. (F) Co-immunoprecipitation (Co-IP) analysis of whole cell lysates from parental B2B and Cr-T cells using IgG, c-Myc, and p300 antibodies for IP and blotting. 5% of protein input depicted in the bottom panels (G) Histone H3 acetylation in B2B cells overexpressing c-Myc or p300 (as in E) and Cr-T cells silencing c-Myc or p300 was detected using ChIP assay. Data were presented as means ± SEM of three independent experiments. * and **, p < 0.05 and p < 0.01 compared to empty vector control or siSCR group.
3.7. DNA hypomethylation contributed to CXCL5 upregulation in Cr-T cells
Previous studies have shown that DNA methylation and histone modification may show crosstalk (Cedar and Bergman, 2009). In lung cancer, CXCL5 might be regulated by abnormal DNA methylation, which is one of the main mechanisms of Cr(VI)-induced cell malignant transformation (Hu et al., 2018; Lou et al., 2013; Saintigny et al., 2013). DNA methylation of CpG islands typically occurs in conjunction with histone modifications, so we identified two CpG islands in the CXCL5 gene promoter and coding regions using Methprimer 2.0 (http://www.urogene.org/methprimer2/) (Fig. 6A). Meanwhile, the TCGA database showed that CXCL5 promoter methylation status negatively correlated with CXCL5 expression level in lung squamous and adenocarcinoma tissues (Fig. 6B), suggesting DNA methylation status of the CXCL5 CpG islands may be an important determinant of CXCL5 expression. To test this, methylation specific PCR (MSP) was performed to test the DNA methylation status of two CpG islands above. The results showed that parental B2B cells possessed more methylated CXCL5 CpG islands than Cr-T cells (Fig. 6C). To quantitatively evaluate the methylation levels of CXCL5, we performed a MeDIP coupled with qPCR assay in parental B2B and Cr-T cells to test the methylation levels of two CpG islands using 5-mC antibody. The results showed that Cr-T cells were less methylated at CXCL5 CpG islands than parental B2B cells (Fig. 6D). Bisulfite sequencing analysis provided further evidence that the average methylation levels of the two CpG islands in the CXCL5 promoter region was lower in Cr-T cells (39.06% and 55.88%) than those in parental B2B cells (81.26% and 85.29%) (Fig. 6E). To test whether DNA methylation controls CXCL5 expression, parental B2B cells and Cr-T cells were treated with 5-aza-2’-deoxycytidine (5-Aza-dC), a demethylation agent. 5-Aza-dC dramatically induced CXCL5 expression in parental B2B cells, but not in Cr-T cells, suggesting that DNA hypomethylation contributing to CXCL5 upregulation in Cr-T cells (Fig. 6F and Supplementary Fig. 3). To further determine whether the hypomethylation was due to the activity of DNA methyltransferases (DNMTs), ChIP assays were performed to detect DNMT1, DNMT3a and DNMT3b enrichment binding to CXCL5 promoter. Only DNMT1 was enriched in Cr-T cells, suggesting it might regulate DNA methylation at the CXCL5 promoter (Fig. 6G). Indeed, DNMT1 knockdown in parental B2B cells or DNMT1 overexpression in Cr-T cells increased or suppressed CXCL5 expression, respectively (Fig. 6H), validating that underexpression of DNMT1 in Cr-T cells drives CXCL5 upregulation.
Fig. 6.
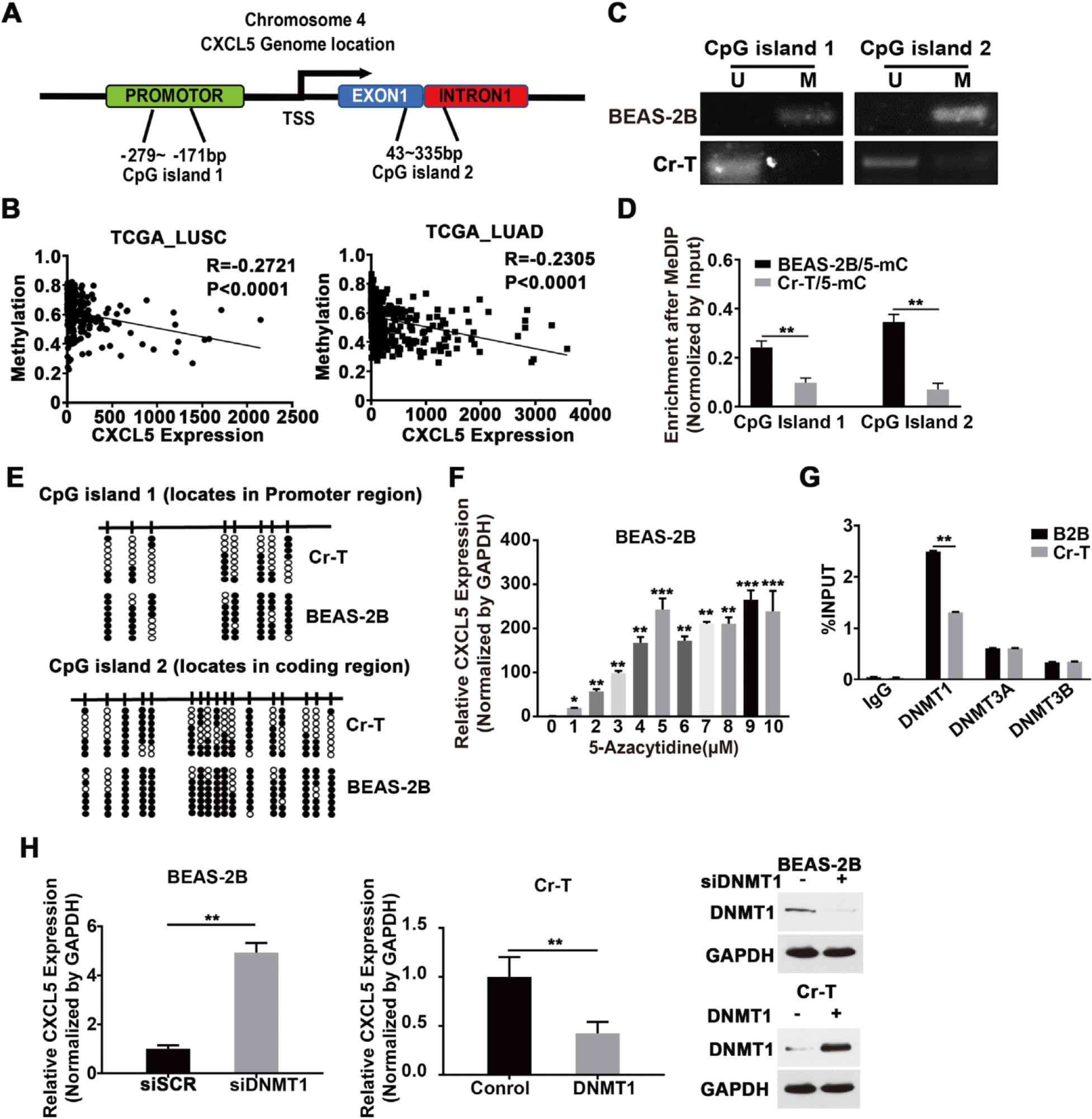
DNA hypomethylation contributed to CXCL5 upregulation in Cr-T cells. (A) Predicted CpG islands in the CXCL5 promoter region and coding region, as determined using MethPrimer 2.0 (http://www.urogene.org/methprimer2/). (B) Correlation between the methylation status of CXCL5 promoter and CXCL5 expression in lung cancer. Datasets were obtained from the MethHC database (https://methhc.mbc.nctu.edu) and TCGA database (https://portal.gdc.cancer.gov). (C) Methylation specific PCR (MSP) analyses of the CXCL5 gene promoter in parental B2B and Cr-T cells. U, unmethylated status; M, methylated status. (D) Methylated DNA immunoprecipitation (MeDIP) coupled with quantitative polymerase chain reaction (qPCR) using a 5-Melhylcytosine (5-mC) antibody in parental B2B and Cr-T cells. **, p < 0.01 compared to B2B cells. (E) Bisulfite sequencing of CpG islands in the CXCL5 promoter region in parental B2B and Cr-T cells. The CpG sites are indicated by vertical ticks. Each row represents a single clone for each individual genomic. Open and filled circles represent unmethylated and methylated CpG sites, respectively. (F) B2B cells that were treated with different doses of 5-Aza-dC for 5 days and the expression of CXCL5 was analyzed by qRT-PCR. GAPDH was used as a loading control. Data were normalized to control group. *. ** and p < 0.05, p < 0.01 and p < 0.001 compared to control without 5-Aza-dC treatment. (G) ChIP assays of DNMT1, DNMT3a and DNMT3b binding to the CXCL5 promotor, in B2B and Cr-T cells. **, p < 0.01 compared to B2B cells. (H) B2B cells were transfected with a control siRNA (siSCR) or an siRNA targeting DNMT1 (siDNMT1) (left panel), or Cr-T cells were transfected with an empty vector or one encoding DNMT1 (middle panel). The overexpression or knockdown of DNMT1 was verified using immunoblotting assay (right panel). Cells were harvested 72 h after transfection and the expression of CXCL5 and GAPDH was analyzed by qRT-PCR. Data were presented as means ± SEM of three independent experiments. **, p < 0.01 compared to siSCR or empty vector control group.
3.8. CXCL5 played a critical role in Cr(VI)-induced oncogenic transformation and carcinogenesis
In vitro, cell transformation assays (CTAs) are considered an ethical proxy for evaluating the carcinogenic potential of chemicals and are frequently used as an initial screening method for carcinogens. BALB/3 T3 cell is one of the most notable CTA models and a useful tool by virtue of easily recognized morphological and biological changes after transformation such as soft agar colony formation ability, short transformation cycle, and low incidence of spontaneous transformation (Kakunaga, 1973; Mascolo et al., 2010; Matthews et al., 1993). To test whether CXCL5 mediates Cr(VI)-induced malignant cell transformation, BALB/3T3 cells were treated with Cr(VI) for two weeks, with or without recombinant human CXCL5 protein. CXCL5 treatment accelerated Cr(VI)-induced colony formation of BALB/3T3 cells, suggesting a critical role for CXCL5 in oncogenic transformation (Fig. 7A). The effect of CXCL5 on cell anchorage-independent-growth and cell proliferation was tested via CRISPR/Cas9-mediated genomic ablation of CXCL5 in Cr-T cells using immunoblotting assays (Fig. 7B). Also, a B2B cell line constitutively expressing CXCL5 was established by lentiviral transduction. Colony-formation assays showed that loss of CXCL5 significantly decreased the ability of Cr-T cells to form colonies to about 20% of wild type control group (Fig. 7C). Parental B2B cells overexpressing CXCL5 grew more rapidly, and Cr-T cells silencing CXCL5 grew more slowly (Fig. 7D). More interestingly, in functional assays, morphological changes were easily observed in B2B cells that constitutively overexpressed CXCL5 and in CXCL5-knockout Cr-T cells: cells deficient for CXCL5 displayed a rounded or cobblestone shape, while cells upregulating CXCL5 displayed a spindle shape, suggesting that CXCL5 is likely involved in the epithelial-mesenchymal transition (EMT; Fig. 7E). The results above indicate CXCL5 indeed enhances Cr (VI)-induced malignant transformation and cell proliferation. To verify the role of CXCL5 in tumor growth in vivo, we orthotopically implanted CXCL5 wild type or CXCL5 knockout (KO) Cr-T-Luc cells into lungs of nude mice and monitored tumor growth using noninvasive bioluminescence imaging for five weeks. Mice that received CXCL5 KO cells had smaller tumor size, less tumor weight, and better overall survival compared to mice that received wild type Cr-T cells (Fig. 7F–I). The expression levels of CXCL5 mRNA and protein in xenografts were verified to confirm the deletion of CXCL5 in Cr-T cells (Fig. 7J and K). These results demonstrate that CXCL5 is critical in Cr(VI)-mediated malignant cell transformation and tumor development.
Fig. 7.
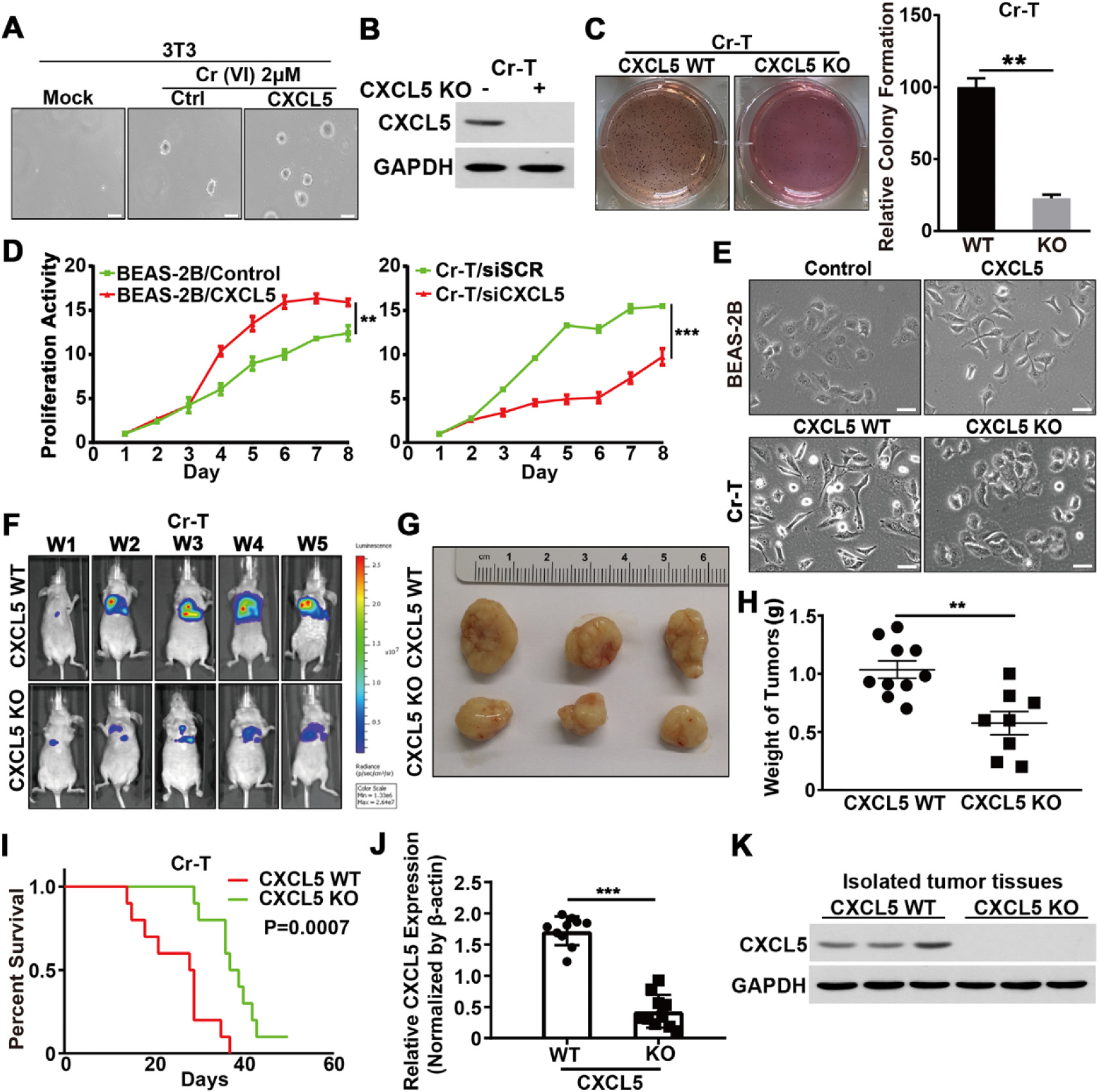
CXCL5 played a critical role in Cr(VI)-induced oncogenic transformation and carcinogenesis. (A) Soft agar assay of BALB/3 T3 cells that were cultured with or without Cr (VI)(2 μM) and CXCL5 (100 ng/mL) for 6 weeks. Images of colonies were taken 2 weeks after seeding. (B) Immunoblotting was used to confirm the levels of CRISPR/Cas9-mediated CXCL5 deletion. (C) Soft agar assay of wild-type (WT) Cr-T cells or Cr-T cells in which stable knockout (KO) of CXCL5 was achieved using CRISPR/Cas9-mediated deletion (left). Right panel depicts the relative number of colonies formed for each cell line. (D) B2B cells were infected with a lend virus stably expressing an empty vector CXCL5, respectively. MTT assay was conducted to determine relative proliferation rates of B2B cells (left). Cr-T cells were transfected with a control siRNA (siSCR) and an siRNA targeting CXCL5, respectively. The MTT assay determined the relative proliferation rates of Cr-T cells (right). (E) B2B and Cr-T cells were treated as above in Fig. 7C and D, and morphological changes were tracked by microscopy. Scale bar = 100 μM (× 400 magnification). (F) CXCL5 wild type and CXCL5 KO Cr-T cells (4 × 106) stably expressing the firefly luciferase were resuspended in 50 μL of DMEM basal medium plus 50 μL of Matrigel and injected into the lung of a nude mouse (n = 10/group). Bioluminescence imaging was performed at post-implantation time points in mice. (G) Representative tumors from CXCL5 wild type and CXCL5 KO Cr-T cells. (H) After the mice were euthanized, xenografts were trimmed out and tumor weight was measured. (I) Kaplan-Meier survival curves of mice that received CXCL5 wild type and CXCL5 KO Cr-T cells for tumor growth assay. (J) CXCL5 levels in the tumor tissues were detected using RT-qPCR. (K) The expression levels of CXCL5 and GAPDH in xenografts. Data were presented as means ± SEM of three independent experiments. ** and ***, p < 0.01 and p < 0.001 compared to CXCL5 wild type group.
3.9. CXCL5/ZEB1 promoted the EMT during Cr (VI)-induced oncogenic transformation
The EMT is a biological process in which epithelial cells differentiate into motile mesenchymal cells. The EMT is associated with stem cell behavior, wound healing, fibrosis, and cancer progression (Lamouillc et al., 2014). EMT can be tracked via changes in several key molecular markers, including loss of E-cadherin and gain of N-cadherin and vimentin. The functional loss of E-cadherin impairs cell polarity and tissue organization (Larsen et al., 2016), while the gain of N-cadherin and N-cadherin-mediated interactions between cancer and stromal cells heighten invasive potential (Nakajima et al., 2004). Vimentin, the cytoskeletal component responsible for maintaining cell integrity and resilience, enables cancer cells to pass through the junctional space between endothelial cells of blood vessels (Goldman et al., 1996).
Both short- and long-term Cr(VI) exposure can induce an EMT in B2B cells; and Cr(VI)-induccd transformed BEAS-2B cells show greater invasion and migration abilities (Ding et al., 2013; Pratheeshkumar et al., 2016). CXCL5 can also promote EMT processes in colorectal cancer (Zhao et al., 2017); however, the function of CXCL5 in Cr(VI)-induced EMT remains unknown. We found that Cr(VI) exposure triggered morphology changes and CXCL5-mediated cell malignant transformation. Thus, we tested whether B2B cells undergo EMT upon chronic Cr(VI) exposure and whether CXCL5 was required during this process by determining EMT markers. Compared to B2B cells, N-cadherin and vimentin were upregulated, whereas E-cadherin was downregulated in Cr-T cells, indicating increased EMT in Cr-T cells. Numerous studies have shown that the EMT process is regulated by some EMT-related transcription factors including zinc finger E-box binding homeobox 1 (ZEB1) (Haensel et al., 2019; Larsen et al., 2016). The transcriptional repressor ZEB1 is crucial in triggering the EMT during organ fibrosis and cancer cell metastasis by inhibiting the transcriptional activities of E-cadherin (Larsen et al., 2016). We found that ZEB1 expression was higher in Cr-T cells compared to parental B2B cells (data not shown here). Recombinant CXCL5 treatment upregulated the expression of N-cadherin, vimentin, and ZEB1 and downregulated the expression of E-cadherin in parental B2B cells, further suggesting CXCL5 induced an EMT (Fig. SA, left panel). In contrast, decreasing CXCL5 expression by siRNA in Cr-T cells suppressed signs of the EMT (Fig. 8A, right panel). The relationship of CXCL5 and ZEB1 in lung adenocarcinoma tissues was assessed using the TCGA database. Consistent with our in vitro data, CXCL5 and ZEB1 were in positive correlation (Fig. 8B). The CXCL5/ZEB1 regulatory axis was also confirmed in lung cancer cell lines (Fig. 8C). Next, we demonstrated that ZEB1 was required for CXCL5-induccd EMT in B2B cells, whereas forced expression of ZEB1 recovered CXCL5 KO-suppressed EMT in Cr-T cells (Fig. 8D and E), confirming that CXCL5 promotes EMT through its downstream target ZEB1. Accordingly, immunofluorescence staining results showed that ZEB1 is key mediator for CXCL5-induced alterations of vimentin and E-cadherin (Fig. 8F), indicating the importance of CXCL5/ZEB1 regulatory axis in the EMT. Moreover, Transwell migration and wound healing assays showed B2B cells overexpressing CXCL5 had increased migration ability, whereas this effect was abrogated when ZEB1 was knocked down. Conversely, in Cr-T cells, CXCL5 deficiency inhibited cell migration, and the effect was reversed by overexpression of ZEB1 (Fig. 8G, Supplementary Fig. 4A). Similar results were obtained for migration assay using lung adenocarcinoma A549 and H2199 cells (Supplementary Fig. 4B). Collectively, our results demonstrate that the CXCL5/ZEB1 axis plays an important role in driving a Cr(VI)-induced EMT.
Fig. 8.
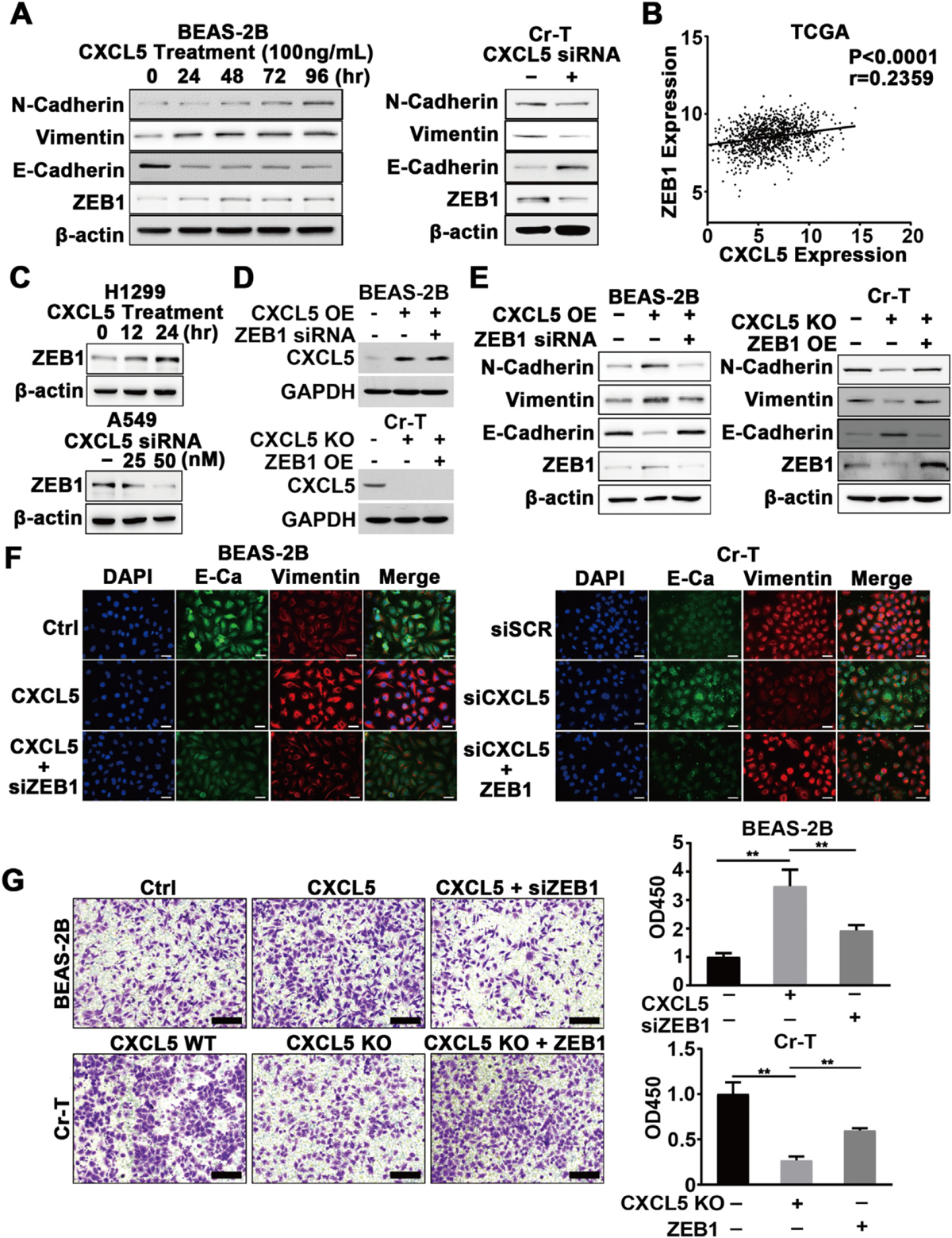
CXCL5/ZEB1 promoted the EMT during Cr (VI)-induced oncogenic transformation. (A) B2B cells were treated with 100 ng/mL of recombinant human CXCL5 for the indicated time points, the expression of N-Cadherin, Vimentin, E-Cadherin, and ZEB1 was determined using immunoblotting assay (left panel). Cr-T cells were transfected with control siRNA or siRNA targeting CXCL5 (siCXCL5) for 72 h, the proteins were measured using immunoblotting assay as above (right panel), β-actin was used as a loading control. (B) Correlation analysis of the TCGA dataset to assess expression of CXCL5 and ZEB1 in lung adenocarcinoma tissue. (C) H1299 cells were treated with 100 ng/mL of recombinant human CXCL5 for different time points, ZEB1 and β-actin expression was tested by immunoblotting assay. A549 cells were transfected with control siRNA (siSCR), or 25 nM and 50 nM of siRNA targeting CXCL5 (CXCL5 siRNA) for 72 h. ZEB1 and β-actin expression was detected as above. (D) B2B cells stably overexpressing empty vector control or CXCL5 (CXCL5 OE) were transfected with control siRNA (siSCR) or siRNA against ZEB1 (ZEB1 siRNA). Cr-T cells expressing wild type CXCL5 (CXCL5 WT) or CXCL5 knockout (CXCL5 KO) were transfected with pCMV6 (negative control) or pCMV6-ZEB1 to overexpress ZEB1 (ZEB OE). The expression levels of CXCL5 and GAPDH were determined using immunoblotting assay. (E) Cell lysates were obtained 72 h after transfection, and the expression levels of N-Cadherin, Vimentin, E-Cadherin, ZEB1 and β-actin were determined using immunoblotting assay. (F) B2B cells were treated with PBS or CXCL5 and transfected siSCR or siZEB1, as indicated. Cr-T cells were transfected with siSCR or siCXCL5 and concurrently ZEB1 was overexpressed. Fluorescent pictures were taken after immunofluorescent staining of E-cadherin (green) and Vimentin (red) in cells treated as above. DAPI was used for nuclei staining. (G) Cells treated as above were subjected to a cell migration assay (left panel). Cells were eluted by acetic acid and their absorbance was measured using microplate reader. Right graph depicts quantitation of relative absorbance. Data were presented as means ± SEM of three independent experiments. **, p < 0.01 compared to CXCL5 + siZEB1 or CXCL5 KO + ZEB1 group.
3.10. Elevated expression of CXCL5 in lung adenocarcinoma correlated with a poor prognosis
To evaluate our results in a clinical setting, we analyzed tumor tissues of lung adenocarcinoma from Tissue Bank of Nanjing Medical University and confirmed CXCL5 levels were upregulated in lung adenocarcinoma tissues compared with adjacent normal tissues (Fig. 9A). In addition, CXCL5 expression levels also positively correlated with clinical TNM stage of the tumors, with much higher expression of CXCL5 observed in stage IV of disease (Fig. 9B). Moreover, the Metabolic Gene Rapid Visualizer (http://merav.wi.mit.edu/)(Shaul et al., 2016), a tool for comparing gene expression, was used to summarize and normalize GEO datasets to evaluate CXCL5 expression levels in lung cancer. Consistent with our clinical data, the samples from GEO datasets showed higher expression of CXCL5 in lung cancers compared to normal tissues (Fig. 9C). Finally, we analyzed the relationship between overall survival and CXCL5 levels by Kaplan-Meier plotter (http://kmplot.com/analysis/). Among the available data of all lung adenocarcinoma patients (n = 720), high CXCL5 expression was associated with poor survival rate (Fig. 9D). These results indicate that increased CXCL5 expression is closely related with cancer progression and a poor prognosis, especially in lung adenocarcinoma.
4. Conclusions
Cr(VI) is a well-known carcinogen which can cause EMT and abnormal epigenetic alterations. In this study, we found that non-smoking workers chronically exposed to Cr(VI) showed higher CXCL5 levels in PBMCs and plasma from blood samples, which positively correlate with Cr concentration in their toenails. Cr (VI) upregulated CXCL5 by promoting c-Myc/p300-mediated histone H3 acetylation and suppressing DNMT1-mediated DNA methylation. In turn, the elevated expression of CXCL5 promoted Cr (VI)-induced malignant transformation, accelerated EMT and promoted cell proliferation and migration. CXCL5 played an important role in tumor growth. Taken together, our findings uncover the epigenetic regulatory mechanism of CXCL5 upregulation by which Cr(VI) induces lung cancer, and suggest that circulating CXCL5 may be a potential target molecule for clinical screening of long-term Cr(VI) exposure-related diseases, including lung cancer.
Supplementary Material
HIGHLIGHTS.
CXCL5 is induced by chronic Cr(VI) exposure in human, animal and cell models.
Histone H3 acetylation leads to CXCL5 upregulation upon Cr(VI) exposure.
DNA hypomethylation is involved in Cr (VI)-induced CXCL5 upregulation.
CXCL5 induces transformation, EMT and cancer development by chronic Cr(VI) exposure.
CXCL5 promotes EMT through ZEB1 upregulation.
Acknowledgements
This work was supported by the National Institutes of Health grants (nos. R01ES027901, R01CA232587, R01ES033197, R01CA263506 and K02ES029119), American Cancer Society Research Scholar (no. NEC-129306), and Commonwealth University Research Enhancement Program grant with the Pennsylvania Department of Health (SAP#4100088563).
Abbreviations
- 5-Aza-dC
5-aza-2′-deoxycytidine
- 5-mC
5-Methylcytosine
- B2B
BEAS-2B cells
- BSA
bovine serum albumin
- BSP
bisulfate-sequencing PCR
- ChIP
chromatin immunoprecipitation
- Cr
chromium
- Cr(VI)
Hexavalent chromium
- CTA
cell transformation assays
- CXCL5
C-X-C Motif Chemokine Ligand 5
- DNase
deoxyribonuclease
- EMT
epithelial to mesenchyme transition
- FITC
fluorescence isothiocyanate
- GI
genome instability
- HAT
histone acetyltransferase
- HDACs
histone deacetylases
- HDAC1-DNMT1
histone deacetylase 1-DNA methyltransferase 1
- HFL1
human fetal lung fibroblast 1
- hMLHl
DNA of human mutant L homolog 1
- HUVEC
human umbilical vein endothelial cells
- IHC
staining immunohistochemical staining
- KO
knockout
- MeDIP
methylated DNA immunoprecipitation
- MSP
methylation-specific PCR
- PBMCs
peripheral blood monocytes
- pRL-TK
renilla luciferase-thymidine kinase
- qRT-PCR
quantitative real-time PCR
- TRITC
tetramethylrhodamine isothiocyanate
- TSA
Trichostatin A
- TSS
transcription start site
- TWA
8 h-time-weighted-average
- ZEB1
zinc finger E-box binding homeobox 1
- ZnCrO44Zn(OH)2
zinc chromate
Footnotes
CRediT authorship contribution statement
Xin Ge: Methodology, Formal data analysis, Investigation, Visualization, Writing-original draft. Jun He: Methodology, Formal dada analysis, Investigation, Validation. Lin Wang: Methodology, Investigation. Lei Zhao: Methodology; Investigation. Yifang Wang: Methodology, Validation. Gang Wu: Methodology, Resources. Weitao Liu: Formal analysis. Wenjing Liu: Investigation. Yongqian Shu: Methodology. Wei Gong: Methodology, Data curation. Xin-Liang Ma: data analysis. Yajing Wang: Methodology. Bing-Hua Jiang: Conceptualization, Supervision, Methodology, funding acquisition. Ling-Zhi Liu: Conceptualization, Supervision, Methodology, Writing-review & editing, funding acquisition.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.scitotenv.2022.155713.
References
- Amente S, Lania L, Majello B, 2011. Epigenetic reprogramming of Myc target genes. Am. J. Cancer Res. 1, 413–418. [PMC free article] [PubMed] [Google Scholar]
- Arenberg DA, Keane MP, DiGiovine B, Kunkel SL, Morris SB, Xue YY, et al. , 1998. Epithelial-neutrophil activating peptide (ENA-78) is an important angiogenic factor in non-small cell lung cancer. J. Clin. Invest. 102, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley LA, Kasina S, Mehra R, Adsule S, Admon AJ, Lonigro RJ, et al. , 2008. CXCL5 promotes prostate cancer progression. Neoplasia 10, 244–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedar H, Bergman Y, 2009. Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet 10, 295–304. [DOI] [PubMed] [Google Scholar]
- Chen C, Xu ZQ, Zong YP, Ou BG, Shen XH, Feng H, et al. , 2019. CXCL5 induces tumor angiogenesis via enhancing the expression of FOXD1 mediated by the AKT/NF-kappaB pathway in colorectal cancer. Cell Death Dis. 10, 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho MH, Park JH, Choi HJ, Park MK, Won HY, Park YJ, et al. , 2015. DOT1L cooperates with the c-Myc-p300 complex to epigenetically derepress CDH1 transcription factors in breast cancer progression. Nat. Commun. 6, 7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV, 2012. MYC on the path to cancer. Cell 149, 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV, O’Donnell KA, Zeller KL, Nguyen T, Osthus RC, Li F, 2006. The c-Myc target gene network. Semin. Cancer Biol. 16, 253–264. [DOI] [PubMed] [Google Scholar]
- De Flora S, Camoirano A, Bagnasco M, Bennicelli C, Corbett GE, Kerger BD, 1997. Estimates of the chromium(VI) reducing capacity in human body compartments as a mechanism for attenuating its potential toxicity and carcinogenicity. Carcinogenesis 18,531–537. [DOI] [PubMed] [Google Scholar]
- Ding SZ, Yang YX, Li XL, Michelli-Rivera A, Han SY, Wang L, et al. , 2013. Epithelial-mesenchymal transition during oncogenic transformation induced by hexavalent chromium involves reactive oxygen species-dependent mechanism in lung epithelial cells. Toxicol. Appl. Pharmacol. 269, 61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disteldorf EM, Krebs CF, Paust HJ, Turner JE, Nouailles G, Tittel A, et al. , 2015. CXCL5 drives neutrophil recruitment in TH17-mediated GN. J. Am. Soc. Nephrol. 26, 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X, Pan MH, Wang L, Li W, Jiang C, He J, et al. , 2018. Hypoxia-mediated mitochondria apoptosis inhibition induces temozolomide treatment resistance through miR-26a/Bad/Bax axis. Cell Death Dis. 9, 1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman RD, Khuon S, Chou YH, Opal P, Steinert PM, 1996. The function of intermediate filaments in cell shape and cytoskeletal integrity. J. Cell Biol. 134, 971–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haensel D, Sun P, MacLean AL, Ma X, Zhou Y, Stemmier MP, et al. , 2019. An Ovol2-Zebl transcriptional circuit regulates epithelial directional migration and proliferation. EMBO Rep. 20, e46273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Zhang X, Wang D, Baccarelli A, 2012. Environmental chemical exposures and human epigenetics. Int. J. Epidemiol. 41, 79–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YL, Hou MF, Kuo PL, Huang YF, Tsai EM, 2013. Breast tumor-associated osteoblast-derived CXCL5 increases cancer progression by ERK/MSKl/Elk-l/snail signal ing pathway. Oncogene 32, 4436–4447. [DOI] [PubMed] [Google Scholar]
- Hu G, Li P, Cui X, Li Y, Zhang J, Zhai X, et al. , 2018. Cr(VI)-induced methylation and down-regulation of DNA repair genes and its association with markers of genetic damage in workers and 16HBE cells. Environ. Pollut. 238, 833–843. [DOI] [PubMed] [Google Scholar]
- Kakunaga T, 1973. A quantitative system for assay of malignant transformation by chemical carcinogens using a clone derived from BALB-3T3. Int. J. Cancer 12, 463–473. [DOI] [PubMed] [Google Scholar]
- Kawamura M, Toiyama Y, Tanaka K, Saigusa S, Okugawa Y, Hiro J, et al. , 2012. CXCL5, a promoter of cell proliferation, migration and invasion, is a novel serum prognostic marker in patients with colorectal cancer. Eur. J. Cancer 48, 2244–2251. [DOI] [PubMed] [Google Scholar]
- Lamouille S, Xu J, Derynck R, 2014. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen JE, Nathan V, Osborne JK, Farrow RK, Deb D, Sullivan JP, et al. , 2016. ZEB1 drives epithelial-to-mesenchymal transition in lung cancer. J. Clin. Invest. 126, 3219–3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, King J, Moro A, Sugi MD, Dawson DW, Kaplan J, et al. , 2011. Overexpression of CXCL5 is associated with poor survival in patients with pancreatic cancer. Am. J. Pathol. 178, 1340–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou J, Wang Y, Yao C, Jin L, Wang X, Xiao Y, et al. , 2013. Role of DNA methylation in cell cycle arrest induced by Cr (VI) in two cell lines. PLoS One 8, e71031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascolo MG, Perdichizzi S, Rotondo F, Morandi E, Guerrini A, Silingardi P, et al. , 2010. BALB/c 3T3 cell transformation assay for the prediction of carcinogenic potential of chemicals and environmental mixtures. Toxicol. in Vitro 24, 1292–1300. [DOI] [PubMed] [Google Scholar]
- Matthews EJ, Spalding JW, Tennant RW, 1993. Transformation of BALB/C-3T3 cells: V. Transformation responses of 168 chemicals compared with mutagenicity in Salmonella and carcinogenicity in rodent bioassays. Environ. Health Perspect. 101 (Suppl. 2), 347–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehra R, Juneja M, 2005. Fingernails as biological indices of metal exposure. J. Biosci. 30, 253–257. [DOI] [PubMed] [Google Scholar]
- Miyazaki H, Patel V, Wang H, Edmunds RK, Gutkind JS, Yeudall WA, 2006. Downregulation of CXCL5 inhibits squamous carcinogenesis. Cancer Res. 66, 4279–4284. [DOI] [PubMed] [Google Scholar]
- Nakajima S, Doi R, Toyoda E, Tsuji S, Wada M, Koizumi M, et al. , 2004. N-cadherin expression and epithelial-mesenchymal transition in pancreatic carcinoma. Clin. Cancer Res. 10, 4125–33. [DOI] [PubMed] [Google Scholar]
- Nickens KP, Patierno SR, Ceryak S, 2010. Chromium genotoxicity: a double-edged sword. Chem. Biol. Interact 188, 276–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien KM, White AJ, Jackson BP, Karagas MR, Sandler DP, Weinberg CR, 2019. Toenail-based metal concentrations and young-onset breast cancer. Am. J. Epidemiol. 188, 646–655. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Park JY, Park KH, Bang S, Kim MH, Lee JE, Gang J, et al. , 2007. CXCL5 overexpression is associated with late stage gastric cancer. J. Cancer Res. Clin. Oncol. 133, 835–840. [DOI] [PubMed] [Google Scholar]
- Pratheeshkumar P, Son YO, Divya SP, Turcios L, Roy RV, Hitron JA, et al. , 2016. Hexavalent chromium induces malignant transformation of human lung bronchial epithelial cells via ROS-dependent activation of miR-21-PDCD4 signaling. Oncotarget 7, 51193–51210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor DM, Suh M, Mittal L, Hirsch S, Valdes Salgado R, Bartlett C, et al. , 2016. Inhalation cancer risk assessment of hexavalent chromium based on updated mortality for Painesville chromate production workers. J. Expo. Sci. Environ. Epidemiol. 26, 224–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Moreno R, Curtis KJ, Coughlin TR, Miranda-Vergara MC, Dutta S, Natarajan A, et al. , 2019. The CXCL5/CXCR2 axis is sufficient to promote breast cancer colonization during bone metastasis. Nat. Commun. 10, 4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saintigny P, Massarelli E, Lin S, Ahn YH, Chen Y, Goswami S, et al. , 2013. CXCR2 expression in tumor cells is a poor prognostic factor and promotes invasion and metastasis in lung adenocarcinoma. Cancer Res. 73, 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnekenburger M, Talaska G, Puga A, 2007. Chromium cross links histone deacetylase 1-DNA methyltransferase 1 complexes to chromatin, inhibiting histone-remodeling marks critical for transcriptional activation. Mol. Cell. Biol. 27, 7089–7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedman RM, Beaumont J, McDonald TA, Reynolds S, Krowech G, Howd R, 2006. Review of the evidence regarding the carcinogenicity of hexavalent chromium in drinking water. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 24, 155–182. [DOI] [PubMed] [Google Scholar]
- Shaul YD, Yuan B, Thiru P, Nutter-Upham A, McCallum S, Lanzkron C, et al. , 2016. MERAV: a tool for comparing gene expression across human tissues and cell types. Nucleic Acids Res. 44, D560–D566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stillie R, Farooq SM, Gordon JR, Stadnyk AW, 2009. The functional significance behind expressing two IL-8 receptor types on PMN. J. Leukoc. Biol. 86, 529–543. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Kondo K, Hirose T, Nakagawa H, Tsuyuguchi M, Hashimoto M, et al. , 2005. Microsatellite instability and protein expression of the DNA mismatch repair gene, hMLHl, of lung cancer in chromate-exposed workers. Mol. Carcinog. 42, 150–158. [DOI] [PubMed] [Google Scholar]
- Tikhanovich I, Zhao J, Bridges B, Kumer S, Roberts B, Weinman SA, 2017. Arginine methylation regulates c-Myc-dependent transcription by altering promoter recruitment of the acetyltransferase p300. J. Biol. Chem. 292, 13333–13344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervoorts J, Luscher-Firzlaff JM, Rottmann S, Lilischkis R, Walsemann G, Dohmann K, et al. , 2003. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep. 4, 484–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YM, Zhou P, Wang LY, Li ZH, Zhang YN, Zhang YX, 2012. Correlation between DNase I hypersensitive site distribution and gene expression in HeLa S3 cells. PLoS One 7, e42414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Shi L, Gu J, Zhan C, Xi J, Ding J, et al. , 2018. CXCL5 regulation of proliferation and migration in human non-small cell lung cancer cells. J. Physiol. Biochem. 74, 313–324. [DOI] [PubMed] [Google Scholar]
- Wang L, Bayanbold K, Zhao L, Wang Y, Adamcakova-Dodd A, Thome PS, et al. , 2022. Redox sensitive miR-27a/b/Nrf2 signaling in Cr(VI)-induced carcinogenesis. Sci. Total Environ. 809, 151118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm M, Hafner D, Lombeck L, Ohnesorge FK, 1991. Monitoring of cadmium, copper, lead and zinc status in young children using toenails: comparison with scalp hair. Sci. Total Environ. 103, 199–207. [DOI] [PubMed] [Google Scholar]
- Wu K, Yu S, Liu Q, Bai X, Zheng X, Wu K, 2017. The clinical significance of CXCL5 in non-small cell lung cancer. Onco Targets Ther. 10, 5561–5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Holmgren S, Andrews DM, Wolfe MS, 2017. Evaluating the impact of the U.S. National Toxicology Program: a case study on hexavalent chromium. Environ. Health Perspect. 125, 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Jiang Y, Yin Y, Li Q, He J, Jing Y, et al. , 2013. A regulatory circuit of miR-148a/152 and DNMT1 in modulating cell transformation and tumor angiogenesis through IGF-IR and IRS1. J. Mol. Cell Biol. 5, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Ou B, Han D, Wang P, Zong Y, Zhu C, et al. , 2017. Tumor-derived CXCL5 promotes human colorectal cancer metastasis through activation of the ERK/Elk-1/Snail and AKT/GSK3beta/beta-catenin pathways. Mol. Cancer 16, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou SL, Dai Z, Zhou ZJ, Wang XY, Yang GH, Wang Z, et al. , 2012. Overexpression of CXCL5 mediates neutrophil infiltration and indicates poor prognosis for hepatocellular carcinoma. Hepatology 56, 2242–2254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.