

Abstract
Autosomal dominant polycystic kidney disease (ADPKD), caused by mutations of PKD1 or PKD2 genes, is characterized by development and growth of cysts causing progressive kidney enlargement. Reduced resting cytosolic calcium and increased cAMP levels associated with the tonic action of vasopressin are two central biochemical defects in ADPKD. Here we show that co‐targeting two GPCRs, the vasopressin V2 receptor (V2R) and the calcium sensing receptor, using the novel V2R antagonist lixivaptan in combination with the calcimimetic R‐568, reduced cyst progression in two animal models of human PKD. Lixivaptan is expected to have a safer liver profile compared to tolvaptan, the only drug approved to delay PKD progression, based on computational model results and initial clinical evidence. PCK rat and Pkd1 RC/RC mouse littermates were fed without or with lixivaptan (0.5%) and R‐568 (0.025% for rats and 0.04% for mice), alone or in combination, for 7 (rats) or 13 (mice) weeks. In PCK rats, the combined treatment strongly decreased kidney weight, cyst and fibrosis volumes by 20%, 49%, and 73%, respectively, compared to untreated animals. In Pkd1 RC/RC mice, the same parameters were reduced by 20%, 56%, and 69%, respectively. In both cases the combined treatment appeared nominally more effective than the individual drugs used alone. These data point to an intriguing new application for two existing drugs in PKD treatment. The potential for synergy between these two compounds suggested in these animal studies, if confirmed in appropriate clinical investigations, would represent a welcome advancement in the treatment of ADPKD.
Keywords: calcimimetics, calcium‐sensing receptor, GPCRs, polycystic kidney disease, vaptans, vasopressin V2 receptor
Abbreviations
- ADPKD
autosomal dominant polycystic kidney disease
- AMPK
AMP‐activated protein kinase
- ARPKD
autosomal recessive polycystic kidney disease
- CaSR
calcium sensing receptor
- CFTR
cystic fibrosis transmembrane conductance regulator
- ciPTEC
conditionally immortalized proximal tubular epithelial cells
- CKD
chronic kidney disease
- FDA
Food and Drug Administration
- GPCRs
G protein coupled receptors
- LXV
lixivaptan
- mTOR
mechanistic target of rapamycin
- PC1
polycystin‐1
- PC2
polycystin‐2
- PTH
parathyroid hormone
- REMS
risk evaluation and mitigation strategy
- V2R
vasopressin V2 receptor
1. INTRODUCTION
Autosomal dominant polycystic kidney disease (ADPKD) is a dominant inherited disease characterized by progressive and bilateral cyst development and growth in the kidneys, but also in the liver and other organs. 1 By the age of 60 years, most patients develop kidney failure and 50% of them require renal replacement therapy. 2 ADPKD is caused primarily by loss‐of‐function mutations in the PKD1 or PKD2 genes, encoding polycystin‐1 (PC1) and polycystin‐2 (PC2), respectively. Loss‐of‐function of PC1 or PC2 is associated with decreased cytosolic calcium content and increased intracellular cAMP levels, with consequent disruption of molecular pathways controlling tubulogenesis, cellular proliferation, and fluid secretion, eventually causing the development of fluid‐filled cysts. 1 , 3
Currently, the only drug that is specifically approved for the treatment of patients with rapidly progressive ADPKD in Europe, Japan, Canada and the United States is tolvaptan, a vasopressin V2 receptor (V2R) antagonist. Two phase 3 clinical studies in ADPKD patients demonstrated its efficacy in reducing total kidney volume growth and slowing eGFR decline. 4 , 5 However, tolvaptan was also found to cause liver toxicity in ADPKD patients. 6 On this basis, the US Food and Drug Administration recommended using tolvaptan only under a risk evaluation and mitigation strategy, which is aimed at preventing serious and potentially life‐threatening liver injury. 7 , 8 Therefore, although tolvaptan is effective in slowing ADPKD progression in patients, is important to develop interventions that are both effective and safe especially in lifelong treatments.
Recently, a novel V2R antagonist, lixivaptan, which had been initially developed for the treatment of hyponatremia, 9 was shown to reduce kidney cystic burden and cAMP levels in PCK rats, an orthologous model of human PKD. 10 In cortical collecting duct cells, lixivaptan prevented the increase in intracellular cAMP, aquaporin‐2 trafficking and water reabsorption in response to vasopressin, providing a mechanistic explanation for the aquaretic effect observed in vivo. 11
In addition, lixivaptan and its three major metabolites were represented in DILIsym, a quantitative systems toxicology platform model of drug‐induced liver injury, in order to predict lixivaptan's potential risk of hepatotoxicity at doses intended for the treatment of ADPKD. 12 The results of the DILIsym simulation for lixivaptan, compared to the previously published simulation for tolvaptan, 13 suggest that lixivaptan is less likely to induce hepatotoxicity than tolvaptan at their respective therapeutic doses for ADPKD treatment. 12 Subsequently, lixivaptan was tested in a phase 2 clinical trial in ADPKD patients with chronic kidney disease (NCT03487913). Preliminary results of the study confirmed that lixivaptan is a potent V2R antagonist in ADPKD patients. The study also defined the dose range for an open‐label clinical trial designed to assess the liver safety of lixivaptan in subjects who previously experienced liver chemistry test abnormalities while treated with tolvaptan (NCT04152837), and for an upcoming Phase 3, double‐blind, placebo‐controlled, randomized trial to demonstrate the efficacy and safety of lixivaptan in subjects with ADPKD (NCT04064346).
Two of the most crucial molecular dysregulations in ADPKD which are considered pro‐proliferative events, i.e., decreased cytosolic calcium content and increased intracellular cAMP levels, 14 are both modulated in the kidney by the activation of the extracellular calcium‐sensing receptor (CaSR). 15 CaSR is a G‐protein‐coupled receptor which senses changes in extracellular calcium concentrations and regulates parathyroid hormone secretion and renal tubular calcium reabsorption to maintain serum calcium levels within the normal range. 15 Recently, we have shown that conditionally immortalized human proximal tubular epithelial cells (ciPTEC) isolated from urine of a healthy subject or of a PKD1 patient, or upon stable down‐regulation of Pkd1, express a functional CaSR. 16 , 17 Interestingly, specific activation of CaSR elicited by its allosteric modulator, the calcimimetic R568, induced an increase of cytosolic Ca2+, a decrease in intracellular cAMP level, and a reduction of mechanistic target of rapamycin activity. 17 In addition, R568 rescued the cell energy status by increasing intracellular ATP levels and mitochondrial calcium content. 18 In animal models of PKD (Cy/+ Han:SPRD rats and pcy mice), calcimimetics inhibited late‐stage cyst growth by increasing intracellular calcium. 19 , 20 Conversely, in other animal models (PCK rats and Pkd2 WS25/− mice), no significant effect of calcimimetics was reported—except for reduced fibrosis—possibly because the potential positive effect of the calcimimetics was offset by marked hypocalcemia. 21 It is currently unknown whether reduced doses of calcimimetic could retain efficacy in the treatment of ADPKD without eliciting the potential detrimental effect of hypocalcemia.
In the present study, we investigated the effect of combination treatment with the calcimimetic R568 and lixivaptan in two animal models of human PKD, PCK rats and Pkd1 RC/RC mice, using a lower dose of R568, to examine the relative role of their two GPCR targets. Specifically, we evaluated the endpoint manifestations of disease such as kidney weight, kidney cysts, kidney fibrosis volume and renal cAMP.
2. MATERIALS AND METHODS
2.1. Experimental animals and study design
All experiments were performed in PCK rats and Pkd1 RC/RC mice maintained in the animal facilities of the Department of Veterinary Medicine of the Mayo Clinic (Rochester, MN, USA). All animal procedures were approved by the Institutional Animal Care and Utilization Committee.
The PCK rat, a Sprague‐Dawley strain, is a model orthologous to ARPKD. It is characterized by an aberrant structure and function of the fibrocystin protein, which is caused by a splicing mutation that leads to a frameshift in Pkhd1 gene, and which in turn results in progressive PKD. 22 , 23 The Pkd1 RC/RC mouse model, inbred into the C57BL/6 background, is homozygous for the hypomorphic Pkd1 p.R3277C allele, orthologous to ADPKD and exhibits slowly progressive PKD. 24
The animals were fed ground rodent chow ad libitum (Labdiet 5053, Purina Mills, Richmond, IN, USA). At the 4th week of age, they were divided into four groups on a control diet or a diet containing lixivaptan and/or R568. Rats (n = 80, 10 animals per group and gender) received ground rodent chow containing 0.5% lixivaptan, 0.025% R568, 0.5% lixivaptan and 0.025% R568 together, or rodent chow without drugs (control group) for 7 weeks. Mice (n = 80, 10 animals per treatment group and gender) were fed with ground rodent chow containing 0.5% lixivaptan, 0.04% R568, 0.5% lixivaptan and 0.04% R568 together, or rodent chow without drugs (control group) for 13 weeks. One week before the scheduled sacrifice, animals were housed in metabolic cages to collect 24‐h urine outputs. At 10 weeks (rats) or 16 weeks (mice) of age, animals were weighed and anesthetized with ketamine (90 mg/kg) and xylazine (10 mg/kg) by intraperitoneal injection. Blood was obtained by cardiac puncture and was used for plasma calcium, creatinine and urea levels determination. The right kidney was placed into pre‐weighed vials containing 10% formaldehyde/phosphate buffer saline (pH 7.4). Tissues were embedded in paraffin for histological and histomorphometric analysis. The left kidneys were immediately frozen in liquid nitrogen for cAMP or PKA activity measurements.
2.2. cAMP content and PKA activity of whole kidneys
For cAMP analysis, whole kidneys were ground to fine powder under liquid nitrogen and homogenized in 0.1 M HCl at room temperature. Total protein content was determined with the BCA Protein Assay Kit (Pierce, IL, USA). Samples were centrifuged at 10 000 rpm for 10 min and the supernatant was used to measure cAMP levels with Direct cAMP ELISA kit, according to the manufacturer's instructions (Enzo Life Sciences, Inc, Farmingdale, NY, USA).
PKA activity determination was assessed by PKA Colorimetric Activity Kit (ThermoFisher Scientific Inc, Monza, Italy). Whole kidney slices were homogenized in Lysis Buffer supplied by the manufacturer. After total protein concentration measurement, samples were centrifuged at 10 000 rpm for 10 min at 4°C and the supernatant was used for PKA activity analysis, performed following manufacturer's instructions.
2.3. Gel electrophoresis and immunoblotting
Proteins were separated on 10% polyacrylamide gels (Bio‐Rad Laboratories, Inc, Hercules, CA, USA) under reducing conditions. Protein bands were electrophoretically transferred onto Immobilon‐P membranes (Merck Millipore, Darmstadt, Germany) and incubated with primary antibodies overnight. Anti‐Phospho‐AMPK (Thr172 of its α subunit) and anti‐Tot AMPK antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Anti‐ERK 1/2 and anti‐Phospho‐ERK 1/2 (Thr185/Tyr187) were from Merck Millipore. Bands were normalized to total protein using stain‐free technology. Densitometry analysis was performed using ImageLab (Bio‐Rad Laboratories, Inc, Hercules, CA, USA) and analyzed using GraphPad Prism (GraphPad Software, San Diego, CA, USA).
2.4. Histomorphometric and histological analysis
Tissue cross‐sections (5 μm) of whole kidney were stained with hematoxylin‐eosin, for cyst volume measurement, and picrosirius red for collagen, to analyze fibrosis. 25 Stained sections were visualized under a Nikon microscope and digital images were acquired using a high‐resolution color digital camera (Nikon DS‐Ri1). Mice cyst and fibrosis volumes were calculated using three cross‐sections per kidney. For renal fibrosis, sequential images were collected using a 10× objective for rats and a 20× objective for mice sections, moving from one end of the available cortex to the other, without overlapping, excluding renal medulla. Analysis of light microscopy images was performed using the NIS‐Elements AR software system (Nikon, Elgin, IL, USA), applying a colored threshold to separate the objects from their background. Renal cyst volume was calculated from the percentage cystic area of the sections adjusted to kidney weight. Fibrosis volume was calculated by measuring the percentage fibrotic area of 8 cortical images adjusted to kidney weight.
2.5. Calcium and phosphorus determinations
Plasma calcium and phosphorus were measured using a Hitachi Modular System.
2.6. Statistical analysis
One‐way ANOVA followed by Tukey's multiple comparisons test was used for the statistical analysis. All values are expressed as means ± SEM. A difference of p < .05 was considered statistically significant.
3. RESULTS
3.1. Effect of combined lixivaptan and R568 treatment on cAMP content in whole kidneys
Increased renal cAMP levels are one of the principal biochemical hallmarks of ADPKD. In PCK rats, both lixivaptan or R568 monotherapy displayed a trend in reducing total renal cAMP (33.3% reduction vs. control in lixivaptan treated animals and 20.5% reduction vs. control in R568 treated animals); however, only the combined treatment induced a statistically significant reduction in cAMP levels (38.7%, p = .033) compared to animals fed with standard diet (Figure 1A).
FIGURE 1.
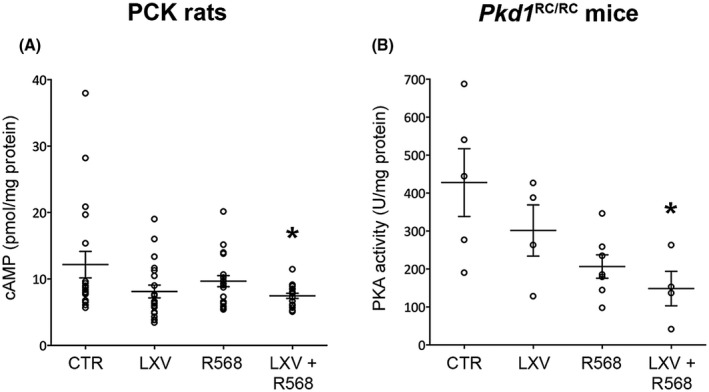
Effect of lixivaptan and R568 treatment on renal cAMP levels or PKA activity. (A) Renal cAMP in PCK rats, expressed as pmol cAMP on mg of total proteins. (B) PKA activity in kidney homogenates in Pkd1 RC/RC mice, measured as units (U) on mg of total proteins. Data are expressed as means ± SEM, *p < .05 versus CTR
In Pkd1 RC/RC mice, renal cAMP levels were indirectly determined by measuring PKA activity (Figure 1B). Once again, whereas both lixivaptan and R568 monotherapy showed a trend toward reduction of PKA activity, only the combined lixivaptan and R568 treatment statistically significantly reduced PKA activity (65.3% vs. CTR, p = .024; Figure 1B). Interestingly, in this animal model of PKD, R568 appeared to have a more potent effect (51.7% reduction) than lixivaptan (29.5% reduction) relative to control animals, although the difference did not reach statistical significance.
3.2. Effect of combined lixivaptan and R568 treatment on AMPK and ERK1/2 expression
AMP‐activated protein kinase (AMPK) is a key regulator of the defective aerobic glycolysis and proliferative processes in PKD. 26 Reduced AMPK signaling has been observed in PKD and, consistent with this observation, AMPK activation has been proved to significantly reduce cystic growth in both in vitro and ex vivo models of renal cystogenesis. 27 , 28 The effect of the combined treatment on active AMPK was evaluated by measuring the levels of AMPK phosphorylated at residue Thr172 of its α subunit. Interestingly, in PCK rats, AMPK activity was significantly increased under combined treatment (CTR = 1.0 ± 0.1, LXV + R568 = 1.7 ± 0.2, p < .01 vs. CTR; LXV = 1.1 ± 0.1, R568 = 1.3 ± 0.1, n.s. vs. CTR; Figure 2A,B). In Pkd1 RC/RC mice, the two drugs in combination induced a five‐fold increase in pAMPK levels (CTR = 1.0 ± 0.1, LXV + R568 = 5.1 ± 0.7, p < .0001; Figure 2C,D). Both lixivaptan and R568 monotherapy increased pAMPK, although only treatment with calcimimetic resulted in a statistically significant increase compared to CTR (LXV = 2.2 ± 0.3, n.s. vs. CTR; R568 = 3.3 ± 0.4, p < .01 vs. CTR; Figure 2D). Combined treatment with the two drugs had a clear significant effect in increasing pAMPK expression with respect to both lixivaptan and R568 monotherapy (p < .001 vs. LXV, p < .05 vs. R568).
FIGURE 2.
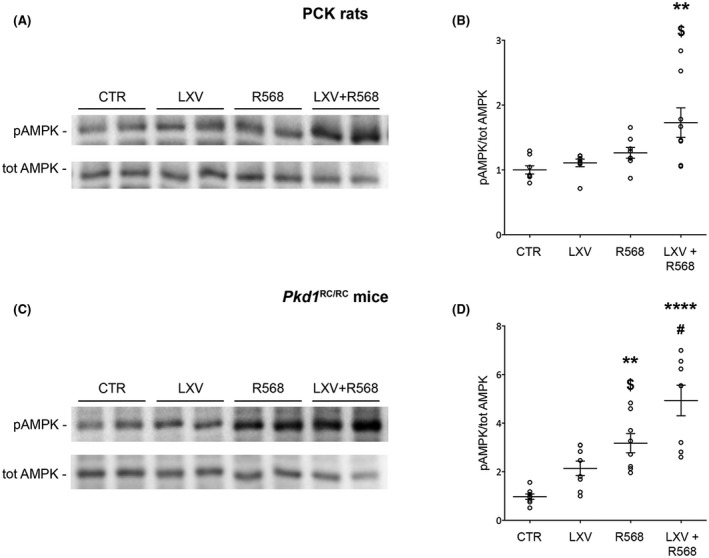
AMPK phosphorylation levels in PCK rats and Pkd1 RC/RC mice. (A–C) Representative blots for total AMPK or for pT172‐AMPK (pAMPK). (B) Densitometric analysis and statistical studies (means ± SEM) in PCK rats (n = 8, 4 animals per group and gender), **p < .01 versus CTR, $ p < .05 versus LXV. (D) Densitometric analysis and statistical studies (means ± SEM) in Pkd1 RC/RC mice (n = 8, 4 animals per group and gender), ****p < .0001 versus CTR, # p < .001 versus LXV, **p < .01 versus CTR, $ p < .05 versus LXV + R568
Various pro‐proliferative pathways are upregulated in ADPKD. Several studies demonstrated the activation of the MAPK/ERK pathway in cellular and animal PKD models. 29 , 30 Evaluation of pERK1/2 levels revealed that cotreatment with lixivaptan and R568 in both animal models resulted in a significant reduction in pERK1/2 levels (PCK rats: CTR = 1.0 ± 0.04, LXV + R568 = 0.7 ± 0.05, p < .01 vs. CTR; LXV = 0.95 ± 0.09, R568 = 0.8 ± 0.05, n.s. vs. CTR, Figure 3A,B; Pkd1 RC/RC mice: CTR = 1.0 ± 0.06, LXV + R568 = 0.7 ± 0.05, p < .01 vs. CTR and LXV; LXV = 1.02 ± 0.05 n.s. vs. CTR, R568 = 0.8 ± 0.08 n.s. vs. CTR, p < .01 vs. LXV, Figure 3C,D).
FIGURE 3.
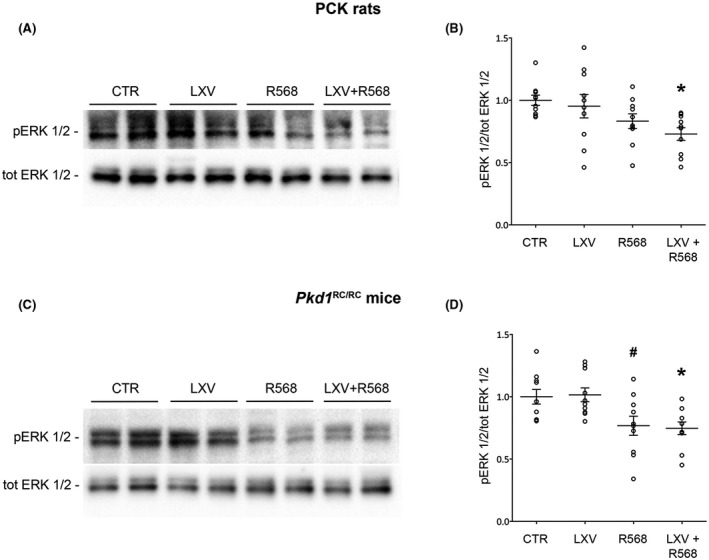
ERK phosphorylation levels in PCK rats and Pkd1 RC/RC mice. (A–C) Representative blots for total ERK1/2 or ERK1/2 phosphorylated at Thr185/Tyr187 (pERK 1/2). (B) Densitometric analysis and statistical studies (means ± SEM) in PCK rats (n = 10, 5 animals per group and gender), *p < .01 versus CTR. (D) Densitometric analysis and statistical studies (means ± SEM) in Pkd1 RC/RC mice (n = 10, 5 animals per group and gender), *p < .01 versus CTR and LXV, # p < .01 versus LXV
3.3. Effect of combined lixivaptan and R568 treatment on renal cyst burden and fibrosis
The effect of the treatments on the ADPKD phenotype was evaluated by analyzing kidney weight (expressed as percentage of body weight), cyst volume and fibrosis volume. In both animal models, untreated animals displayed a typical PKD phenotype characterized by enlarged kidneys and relevant renal cysts development and fibrosis, as expected.
In PCK rats, treatment with either lixivaptan or R568 statistically significantly reduced kidney weight (normalized to body weight, Figure 4A). Interestingly, the reduction in kidney weight in rats treated with lixivaptan combined with R568 versus untreated animals reached a higher level of statistical significance compared to the monotherapy groups (LXV 19.7% reduction, p = .0000074 vs. CTR; R568 9.2% reduction, p < .05 vs. CTR; combined treatment 20.4% reduction, p = .0000047 vs. CTR) (Figure 4A).
FIGURE 4.
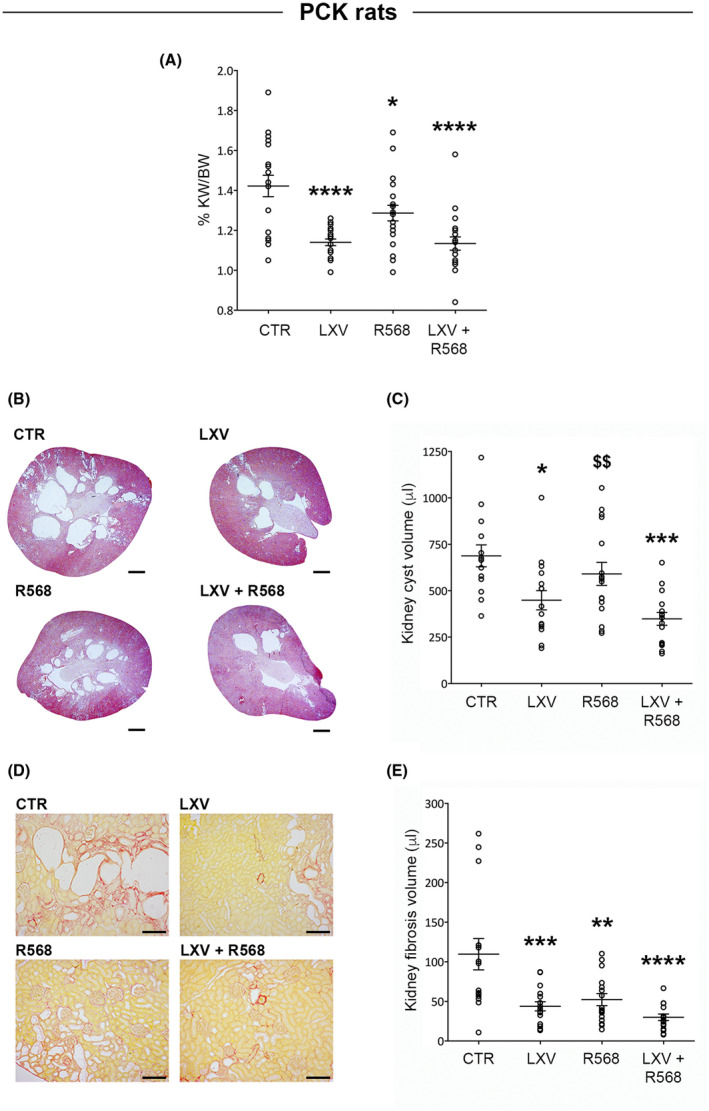
Effect of lixivaptan and R568 treatment on renal morphology in PCK rats. (A) Evaluation of the effect of the drugs on kidney weight expressed as percentage of body weight. Data are expressed as means ± SEM, ****p < .0001 versus CTR, *p < .05 versus CTR or LXV or LXV + R568. (B) Representative histological images of kidney cross‐sections (scale bars: 2 mm) stained with hematoxylin‐eosin for cyst volume analysis, showed in the histogram (C). Data are expressed as means ± SEM, *p < .05, ***p < .001 versus CTR, $$ p < .01 versus LXV + R568. (D) Representative histological images of transverse kidney sections (scale bars: 200 mm) stained with picrosirius red for fibrosis measurement, quantified in the histogram (E). Data are expressed as means ± SEM, ***p < .001, **p < .01, ****p < .0001 versus CTR
Figure 4B shows representative tissue cross‐sections of whole kidney from the four experimental conditions after staining with hematoxylin‐eosin. As expected, kidney sections from control animals exhibit the presence of large cysts mainly restricted to the medullary region. Cyst burden appeared qualitatively reduced in all treated groups. Quantitative analysis of kidney cyst volume (Figure 4C) revealed that lixivaptan alone was more effective (34.8% reduction, p < .05 vs. CTR) compared to R568 (14.1% reduction, n.s. vs. CTR) in reducing cyst burden. Of note, PCK rats treated with lixivaptan and R568 together showed the highest degree of cyst volume reduction (49.4% reduction, p < .001 vs. CTR).
Besides cyst burden, a major pathological feature of PKD is the development of interstitial inflammation and fibrosis, which is associated with accumulation of inflammatory cells. 31 , 32 , 33 Interestingly, qualitative (Figure 4D) and quantitative (Figure 4E) evaluation of kidney fibrosis by picrosirius red staining for collagen demonstrated that both lixivaptan and R568 monotherapy had a strong, statistically significant effect in reducing renal fibrosis volume (LXV 60% reduction, p < .001 vs. CTR; R568 52.3% reduction, p < .01 vs. CTR) (Figure 4E). Of interest, the combined treatment had the strongest effect in reducing cyst fibrosis (72.7%, p < .0001 vs. CTR) (Figure 4E).
The effect of the combined lixivaptan and R568 treatment on renal cysts was also tested in Pkd1 RC/RC mice, a PKD1 model characterized by gradually increasing cyst burden, which makes it optimal for therapeutic testing. 24 In Pkd1 RC/RC mice, only lixivaptan alone or in combination with R568 significantly reduced kidney weight; however, the combined treatment induced a nominally higher reduction, and the reduction reached a higher level of statistical significance, compared to animals treated with lixivaptan alone (Figure 5A). Specifically, kidney weight decreased by 15.5% in animals treated with lixivaptan alone (p < .01 vs. CTR), and by 20.1% in animals fed with lixivaptan and R568 diet (p < .0001 vs. CTR). R568 alone induced a non‐significant decrease (8.1%) compared to control animals. Histochemical qualitative (Figure 5B) and quantitative (Figure 5C) analysis of kidney cyst volume from tissue cross‐sections of whole kidneys demonstrated that both lixivaptan and R568 monotherapy were effective in reducing kidney cyst burden in Pkd1 RC/RC mice; however, in animals treated with the two drugs in combination, once again a nominally stronger effect was obtained and the comparison with control animals reached a higher level of statistical significance (LXV 38.9%, p < .05 vs. CTR; R568 42.7%, p < .01 vs. CTR; combined treatment 55.5%, p < .001 vs. CTR, Figure 5C). An analogous result was observed upon analysis of renal fibrosis volume after picrosirius red staining (Figure 5D). Lixivaptan and R568 monotherapy had a similar significantly strong effect in reducing renal fibrosis volume compared to animals fed with regular diet (LXV 64.4% reduction, p < .001 vs. CTR; R568 64.9% reduction, p < .001 vs. CTR). Importantly, once again the combined treatment showed a nominally greater effect, and the comparison with control animals reached a higher level of statistical significance, in reducing renal fibrosis (69.1% reduction, p < .0001 vs. CTR, Figure 5E).
FIGURE 5.
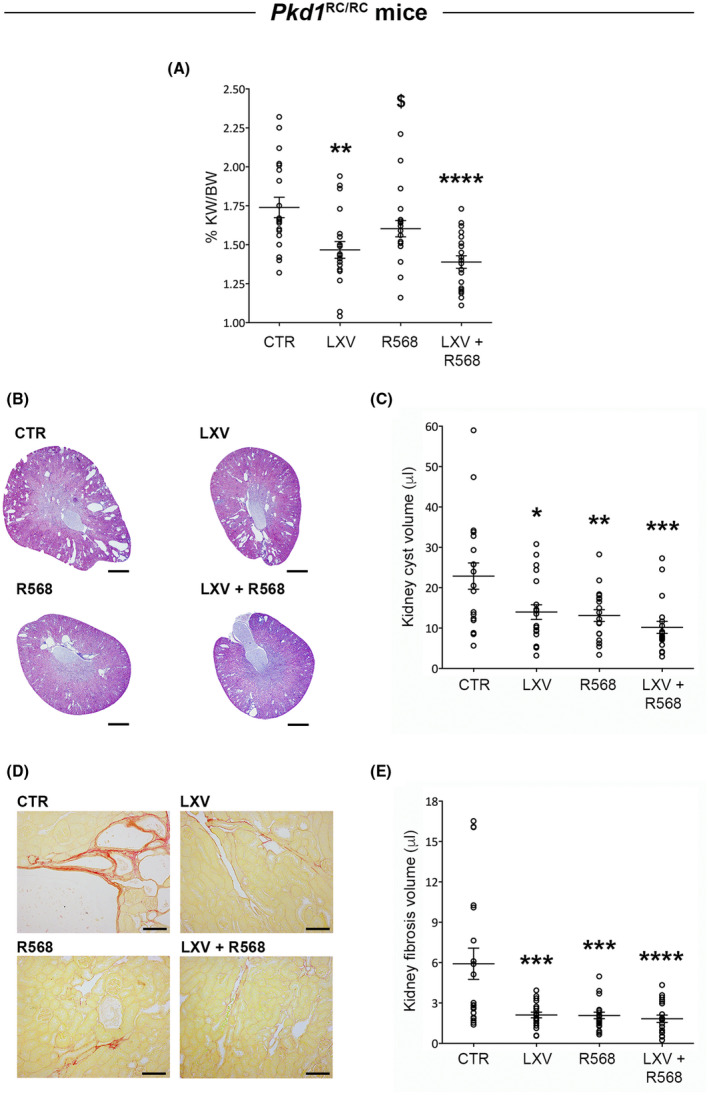
Lixivaptan and R568 treatment effect on renal morphology in Pkd1 RC/RC mice. (A) Evaluation of the effect of the drugs on kidney weight expressed as percentage of body weight. Data are expressed as means ± SEM, **p < .01, ****p < .0001 versus CTR, $ p < .05 versus LXV + R568. (B) Representative histological images of transverse kidney sections (scale bars: 1 mm) stained with hematoxylin‐eosin for cyst volume analysis, showed in the histogram (C). Data are expressed as means ± SEM, *p < .05, **p < .01, ***p < .001 versus CTR. (D) Representative histological images of kidney cross‐sections (scale bars: 100 mm) stained with picrosirius red for fibrosis measurement, quantified in the histogram (E). Data are expressed as means ± SEM, ***p < .001, ****p < .0001 versus CTR
Taken together, these data, summarized in Tables 1 and 2, demonstrate that combination treatment with lixivaptan and R568 has similar effects in two different PKD animal models and may be more effective in reducing disease manifestations than the single drugs used as monotherapies.
TABLE 1.
Effect of lixivaptan and R568 on parameters involved in PKD development in PCK rats
| Parameters | Values ± SEM | p values | |||
|---|---|---|---|---|---|
| CTR (n = 20) | LXV (n = 20) | R568 (n = 20) | LXV + R568 (n = 20) | ||
| Kidney weight/BW (%) | |||||
| CTR | 1.42 ± 0.05 | – | <.0001* | .0627 | <.0001* |
| LXV | 1.14 ± 0.02 | <.0001* | – | .0345* | .9995 |
| R568 | 1.29 ± 0.04 | .0627 | .0345* | – | .0256* |
| LXV + R568 | 1.13 ± 0.03 | <.0001* | .9995 | .0256* | – |
| cAMP (pmol/mg protein) | |||||
| CTR | 12.17 ± 1.99 | – | .0819 | .4498 | .0341* |
| LXV | 8.12 ± 0.96 | .0819 | – | .7807 | .9792 |
| R568 | 9.68 ± 0.83 | .4498 | .7807 | – | .5493 |
| LXV + R568 | 7.46 ± 0.38 | .0341* | .9792 | .5493 | – |
| Cystic volume (μl) | |||||
| CTR | 687.9 ± 59.08 | – | .0134* | .5787 | .0002* |
| LXV | 448.8 ± 51.83 | .0134* | – | .2249 | .5229 |
| R568 | 590.7 ± 62.27 | .5787 | .2249 | – | .0086* |
| LXV + R568 | 348.3 ± 34.41 | .0002* | .5229 | .0086* | – |
| Fibrotic volume (μl) | |||||
| CTR | 109.6 ± 19.79 | – | .0004* | .0024* | <.0001* |
| LXV | 43.85 ± 5.78 | .0004* | – | .9442 | .795 |
| R568 | 52.29 ± 7.62 | .0024* | .9442 | – | .4584 |
| LXV + R568 | 29.93 ± 4.14 | <.0001* | .795 | .4584 | – |
| Plasma phosphorus (mg/dl) | |||||
| CTR | 8.61 ± 0.31 | – | .9854 | .0295* | .0048* |
| LXV | 8.45 ± 0.34 | .9854 | – | .0103* | .0013* |
| R568 | 9.96 ± 0.35 | .0295* | .0103* | – | .9464 |
| LXV + R568 | 10.22 ± 0.31 | .0048* | .0013* | .9464 | – |
| 24 h urine volume (ml) | |||||
| CTR | 14.72 ± 0.7 | – | <.0001* | .3414 | <.0001* |
| LXV | 48.37 ± 2.56 | <.0001* | – | <.0001* | .4656 |
| R568 | 10.65 ± 0.72 | .3414 | <.0001* | – | <.0001* |
| LXV + R568 | 44.88 ± 1.97 | <.0001* | .4656 | <.0001* | – |
| Body weight (g) | |||||
| CTR | 325.3 ± 19.41 | – | .9499 | .9997 | .8991 |
| LXV | 311.3 ± 19.37 | .9499 | – | .9703 | .9985 |
| R568 | 322.8 ± 17.67 | .9997 | .9703 | – | .9301 |
| LXV + R568 | 307.2 ± 16.63 | .8991 | .9985 | .9301 | – |
Data are expressed as means ± SEM.
p < .05.
TABLE 2.
Effect of lixivaptan and R568 on parameters involved in PKD development in Pkd1 RC/RC mice
| Parameters | Values ± SEM | p values | |||
|---|---|---|---|---|---|
| CTR (n = 20) | LXV (n = 20) | R568 (n = 20) | LXV + R568 (n = 20) | ||
| Kidney weight/BW (%) | |||||
| CTR | 1.74 ± 0.07 | – | .0033* | .2835 | <.0001* |
| LXV | 1.47 ± 0.05 | .0033* | – | .2866 | .7348 |
| R568 | 1.6 ± 0.05 | .2835 | .2866 | – | .0309* |
| LXV + R568 | 1.39 ± 0.04 | <.0001* | .7348 | .0309* | – |
| cAMP (U/mg prot.) a | |||||
| CTR | 427.8 ± 89.34 | – | .5025 | .05* | .0282* |
| LXV | 301.6 ± 67.44 | .5025 | – | .6668 | .3855 |
| R568 | 206.6 ± 30.81 | .05* | .6668 | – | .8945 |
| LXV + R568 | 148.5 ± 45.42 | .0282* | .3855 | .8945 | – |
| Cystic volume (μl) | |||||
| CTR | 22.88 ± 3.26 | – | .0202* | .0099* | .0004* |
| LXV | 13.97 ± 1.82 | .0202* | – | .9914 | .5874 |
| R568 | 13.1 ± 1.45 | .0099* | .9914 | – | .771 |
| LXV + R568 | 10.18 ± 1.49 | .0004* | .5874 | .771 | – |
| Fibrotic volume (μl) | |||||
| CTR | 5.92 ± 1.16 | – | .0002* | .0002* | <.0001* |
| LXV | 2.11 ± 0.21 | .0002* | – | >.9999 | .9884 |
| R568 | 2.08 ± 0.24 | .0002* | >.9999 | – | .9918 |
| LXV + R568 | 1.83 ± 0.27 | <.0001* | .9884 | .9918 | – |
| Plasma calcium (mg/dl) | |||||
| CTR | 7.79 ± 0.19 | – | .9993 | .9792 | .9075 |
| LXV | 7.76 ± 0.28 | .9993 | – | .9507 | .8452 |
| R568 | 7.9 ± 0.11 | .9792 | .9507 | – | .9918 |
| LXV + R568 | 7.97 ± 0.12 | .9075 | .8452 | .9918 | – |
| Plasma phosphorus (mg/dl) | |||||
| CTR | 7.72 ± 0.55 | – | .9307 | .9995 | .7357 |
| LXV | 7.31 ± 0.37 | .9307 | – | .9607 | .9747 |
| R568 | 7.64 ± 0.65 | .9995 | .9607 | – | .7981 |
| LXV + R568 | 7.02 ± 0.23 | .7357 | .9747 | .7981 | – |
| 24 h urine volume (ml) | |||||
| CTR | 1.27 ± 0.14 | – | <.0001* | .9976 | <.0001* |
| LXV | 3.95 ± 0.44 | <.0001* | – | <.0001* | .7153 |
| R568 | 1.19 ± 0.15 | .9976 | <.0001* | – | <.0001* |
| LXV + R568 | 3.54 ± 0.27 | <.0001* | .7153 | <.0001* | – |
| Body weight (g) | |||||
| CTR | 24.5 ± 0.65 | – | .8962 | .9988 | .9996 |
| LXV | 23.79 ± 0.74 | .8962 | – | .8304 | .8517 |
| R568 | 24.65 ± 0.81 | .9988 | .8304 | – | >.9999 |
| LXV + R568 | 24.6 ± 0.66 | .9996 | .8517 | >.9999 | – |
Data are expressed as means ± SEM.
Indirectly measured as PKA activity.
p < .05.
To evaluate whether the combined treatment had different effects in males and females, the data were also analyzed considering the sex. For most outcomes (cAMP, KW/BW, cyst and fibrosis volumes), the significance is randomly retained in males or females. In the cases where the significance is lost, due to the small sample size, there is a clear trend in the same direction and the effects of treatment are confirmed (Figures S1–S3).
3.4. Effect of combined lixivaptan and R586 treatment on parameters related to renal function
Tables 1 and 2 report the effect of combined lixivaptan and R586 treatment on parameters related to renal function in PCK rats and Pkd1 RC/RC mice, respectively. No effect on total body weight was observed in any of the treatment groups compared to controls (untreated PKD animal models).
In both PKD models, PCK rats and Pkd1 RC/RC mice, lixivaptan treatment caused a 3‐fold increase in 24‐h urine output compared to untreated control animals, confirming the strong aquaretic effect of the drug. 10 Conversely, R568 alone used at 0.025% in rats and 0.04% in mice had no effect on the 24‐h diuresis both in PCK rats and Pkd1 RC/RC mice. This result is not in agreement with what was observed in a previous study in PCK rats treated with R568 0.05% or 0.1% 21 and is likely due to the reduced dose employed in the present study. Finally, in both animal models, combination therapy with lixivaptan and R568 did not increase urine output compared to lixivaptan monotherapy, confirming that the low doses of calcimimetic in this study do not exacerbate the aquaretic effect of lixivaptan.
In both animal models, there was no effect on plasma urea and creatinine with any treatment, either alone or combined, although a trend toward a decrease in plasma creatinine in PCK rats and in plasma urea in Pkd1 RC/RC mice was observed with the combined treatment with the two drugs (data not shown). Plasma phosphorus was significantly increased in PCK rats, but not in Pkd1 RC/RC mice, with R568 treatment either alone or combined with lixivaptan. Importantly, plasma calcium measured in Pkd1 RC/RC mice was not affected by the calcimimetic (Table 2), confirming that the results of this study with lower doses of R568 are not confounded by hypocalcemia. The calcium levels for PCK rats were not available as samples were compromised and not eligible for measurement.
4. DISCUSSION
In this work, we investigated, with positive outcome, whether dual targeting of V2R and CaSR could have additive effects in attenuating PKD progression in two well‐established PKD animal models, the PCK rat and Pkd1 RC/RC mouse. Specifically, the main results of this work can be summarized as follows: (i) in PCK rats, the combined treatment strongly decreased kidney weight, cyst volume and fibrosis volume by 20%, 49%, and 73%, respectively, compared to untreated animals; (ii) in Pkd1 RC/RC mice, the same parameters were reduced by 20%, 56%, and 69%, respectively; (iii) in both animal models, combination therapy appeared nominally more effective than the individual drugs used alone.
In recent years, it has become clear that disruption of vasopressin signaling has a crucial pathophysiological role in the progression of PKD and that vasopressin directly regulates cyst growth. 34 Accordingly, treatment with a vasopressin V2‐receptor antagonist was proven to delay PKD progression in humans. 4 , 5 Thus, the potentiation of the efficacy of a combination of V2R antagonist and calcimimetic compared to vaptan monotherapy can be explained by several lines of evidence showing that CaSR signaling counteracts the known vasopressin alterations 15 , 35 , 36 , 37 in PKD. Moreover, besides collecting duct, CaSR is expressed across the entire length of the nephron, including the proximal tubule, where it is expressed apically, and the basolateral membrane of the TAL, where it has the highest expression. 15 This broad expression can represent an advantage for the proposed therapeutic strategy.
The strategy to simultaneously target CaSR and V2R is strictly based on the connection between calcium signals and the onset of PKD. 3 Another intriguing observation in support of this strategy is that external Ca2+ modulates whole cell conductance of wild type LLC‐PK1 renal epithelial cells by regulating PC2‐associated currents, consistent with the stimulation of CaSR. 38 Intracellular calcium homeostasis is impaired in ADPKD and is believed to promote cAMP accumulation and abnormal cell proliferative response to cAMP. 3 However, calcium has effects beyond its impact on cAMP levels. The cellular response to cAMP is proliferative when intracellular calcium is low 39 and anti‐proliferative when intracellular calcium is increased. 40 Therefore, there is a strong rationale in favor of targeting two GPCRs acting on each second messenger in a way that is expected to improve ADPKD dysregulations.
The PCK rat is a model orthologous to ARPKD, characterized by progressive PKD with a renal phenotype which resembles that of ADPKD. 22 , 23 The Pkd1 RC/RC mouse model closely mimics human ADPKD with slowly progressive PKD. 24 The lixivaptan dose used in this work was established as a fully effective dose based on previous work by the authors which showed that 0.5% lixivaptan was effective in reducing cyst growth in PCK rats. 10 Conversely, other previous evidence demonstrated that treatment with 0.05% and 0.1% of R568 induced profound hypocalcemia, which might have hindered a potential positive effect of the drug. 21 Therefore, in this work we used the calcimimetic R568 at lower concentration (0.04% for mice and 0.025% for rats) to minimize hypocalcemia. Of note, we found that plasma calcium levels in Pkd1 RC/RC mice treated with the combined treatment were not altered with respect to non‐treated animals. Interestingly we wish to underline that the R‐568 alone (0.04%) did not produce any effects on serum calcium levels and induced significant positive effects in reducing cAMP, cystic and fibrotic volumes, in contrast to the findings obtained by Wang et al. 21 at higher concentration. Moreover, we report here that the 24 h urine volume was slightly but significantly reduced whereas Wang et al. reported a significant increase, supporting the positive effects of R‐568 concentration used in the present contribution. On the other hand, the dual therapy did not result in an apparent improvement in plasma levels of BUN and creatinine. This is probably due to the fact that the animals were sacrificed at an age at which the worsening of BUN and creatinine levels cannot be appreciated 24 , 41 and therefore the treatment on these parameters cannot be assessed.
Several in vivo studies demonstrated the pivotal role played by cAMP in PKD development, for example by promoting cyst growth through a proliferative stimulus and the secretion of fluids into the cyst lumen mediated by the cystic fibrosis transmembrane conductance regulator chloride channel. 42 , 43 , 44 , 45 Both lixivaptan and R568 alone showed a clear trend in reducing cAMP levels, measured directly or by the evaluation of PKA activity, but had an additive and significant effect if used together. In addition, in both animal models, the combined treatment showed a significant increase in the expression of active AMPK, whose activity is downregulated in PKD and involved in cystogenesis. 27 , 28
Animal model differences may explain the higher efficacy of lixivaptan monotherapy in PCK rats and R568 monotherapy in Pkd1 RC/RC mice; however, the differences did not reach statistical significance. Previous work has shown that another vaptan, tolvaptan, has a dose dependent effect on kidney levels of cAMP and on PKD manifestations. 46 In one study, 41 monotherapy with tolvaptan did not induce a statistical reduction of cAMP levels in Pkd1 RC/RC mice compared to control animals, whereas tolvaptan plus pasireotide combination treatment did. The rationale in this approach is that a combination of drugs targeting simultaneously the complex signaling in ADPKD, may have a potential synergism and may provide clinicians the opportunity to reduce drug doses and in turn side effects.
In this work, we show that the combined treatment with lixivaptan and R568 is indeed more effective compared to each monotherapy also in morphological disease manifestations in the kidney. In PCK rats, kidney weight reduction was statistically comparable in animals treated with lixivaptan alone or combined with R568; however, combination therapy elicited a reduction of kidney cyst volume and fibrosis volume that reached a higher level of statistical significance compared to the effect of each monotherapy. A similar finding was observed in Pkd1 RC/RC mice with respect to kidney weight, cyst volume and fibrosis volume. Indeed, besides cystogenesis, fibrotic deposition is another major cause of renal function loss in ADPKD. Recently, V2R activation has been proven to increase renal interstitial myofibroblast population and ECM deposition in cystic epithelial cells. 47 In vitro studies in human ADPKD cystic epithelial cells showed that V2R increased fibrosis by an ERK1/2‐dependent mechanism, 47 which is also regulated by CaSR signaling. Of note, in ciPTEC stably silenced for PC1, CaSR activation elicited by the calcimimetic R568 inhibited ERK1/2 pathway. 17 The present study corroborates these results, demonstrating how V2R antagonism and CaSR activation may potentiate each other leading to a statistically significant fibrosis reduction only in animals treated with combination therapy, consistent with ERK1/2 activity reduction.
Despite practical considerations imposing a limit to the sample size that can be studied in an animal experiment, we are encouraged by the fact that, in both animal models, combination therapy showed a nominally greater effect for every disease‐related endpoint measured in this study compared to both monotherapies and that, for every such endpoint, the treatment effect of combination therapy consistently reached a higher level of statistical significance compared to the effect of the two monotherapies. Thus, we believe that this may represent a true, important effect of combination therapy, especially when considering that the doses of lixivaptan in this study were considered fully effective doses. 10
The potential additional beneficial effect of the use of calcimimetics in the treatment of PKD is strengthened by a recent elegant study demonstrating that tubular crystal deposition of calcium oxalate causes tubule dilation, activation of PKD‐associated signaling pathways, and increased cystogenesis. 48 Interestingly, the authors observed that, in a cohort of ADPKD patients, lower levels of urinary excretion of citrate, an endogenous inhibitor of calcium crystal formation, were correlated with increased disease severity. These findings suggest that supplementation with citrate as a chelator of calcium to reduce renal crystal formation may slow progression in PKD. Remarkably, in the kidney, CaSR promotes citrate excretion, protecting the kidney against the risk of crystal precipitates in urine. 49 This suggests that combination therapy with R568 and lixivaptan may delay disease progression in PKD not only because CaSR counteracts vasopressin signaling (by reducing cAMP concentrations and increasing intracellular calcium), but also because it simultaneously reduces the risk of crystal deposition.
In conclusion, we demonstrated that combined treatment with lixivaptan and R568 displayed an additive effect in reducing three of the most serious hallmarks of ADPKD, specifically renal intracellular cAMP, kidney cyst burden and kidney fibrosis. These data suggest an intriguing new clinical application for two classes of existing drugs. Vaptans and calcimimetics could be used in combination to achieve greater efficacy in the treatment of ADPKD or, in patients who cannot tolerate the aquaretic effects of a full dose vaptan treatment regimen, to reduce the vaptan dose without sacrificing therapeutic efficacy. These findings are suggestive enough to justify further investigation and possibly an exploratory clinical trial.
DISCLOSURES
LP is an employee and owns stock in Palladio Biosciences, Inc, the sponsor of lixivaptan. R568 was supplied by Amgen Inc.
AUTHOR CONTRIBUTIONS
Annarita Di Mise, Vicente E. Torres and Giovanna Valenti designed research and supervised the project; Annarita Di Mise, Xiaofang Wang and Hong Ye performed research and analyzed data; Annarita Di Mise, Vicente E. Torres and Giovanna Valenti designed and interpreted experiments; Annarita Di Mise, Vicente E. Torres and Giovanna Valenti wrote and revised the paper; Lorenzo Pellegrini revised the paper; all authors commented on the manuscript.
Supporting information
Fig S1‐S3
ACKNOWLEDGMENTS
We thank Amgen, Inc for providing the calcimimetic R‐568. This research was funded by Regional project POR Puglia Innonetwork (grant number H6GG787 to GV), by the National Institute of Diabetes and Digestive and Kidney Diseases (grant numbers DK‐44863 and DK‐90728 to VET) and by the Mayo Clinic Robert M. and Billie Kelley Pirnie Translational PKD Research Center. Annarita Di Mise is supported by “Attrazione e Mobilità dei Ricercatori, PON “R&I” 2014–2020, Azione I.2” (code AIM1893457‐3, linea 1). Open Access Funding provided by Universita degli Studi di Bari Aldo Moro within the CRUI‐CARE Agreement. [Correction added on May 31, 2022, after first online publication: CRUI‐CARE Funding statement has been added.]
Di Mise A, Wang X, Ye H, Pellegrini L, Torres VE, Valenti G. Pre‐clinical evaluation of dual targeting of the GPCRs CaSR and V2R as therapeutic strategy for autosomal dominant polycystic kidney disease. FASEB J. 2021;35:e21874. 10.1096/fj.202100774R
Vicente E. Torres and Giovanna Valenti contributed equally to this work.
Contributor Information
Annarita Di Mise, Email: annarita.dimise@uniba.it.
Giovanna Valenti, Email: giovanna.valenti@uniba.it.
REFERENCES
- 1. Chebib FT, Torres VE. Autosomal dominant polycystic kidney disease: core curriculum 2016. Am J Kidney Dis. 2016;67(5):792‐810. doi: 10.1053/j.ajkd.2015.07.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Spithoven EM, Kramer A, Meijer E, et al. Renal replacement therapy for autosomal dominant polycystic kidney disease (ADPKD) in Europe: prevalence and survival—an analysis of data from the ERA‐EDTA registry. Nephrol Dial Transplant. 2014;29(suppl 4):iv15‐iv25. doi: 10.1093/ndt/gfu017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sussman CR, Wang X, Chebib FT, Torres VE. Modulation of polycystic kidney disease by G‐protein coupled receptors and cyclic AMP signaling. Cell Signal. 2020;72:109649. doi: 10.1016/j.cellsig.2020.109649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367(25):2407‐2418. doi: 10.1056/NEJMoa1205511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in later‐stage autosomal dominant polycystic kidney disease. N Engl J Med. 2017;377(20):1930‐1942. doi: 10.1056/NEJMoa1710030 [DOI] [PubMed] [Google Scholar]
- 6. Watkins PB, Lewis JH, Kaplowitz N, et al. Clinical pattern of tolvaptan‐associated liver injury in subjects with autosomal dominant polycystic kidney disease: analysis of clinical trials database. Drug Saf. 2015;38(11):1103‐1113. doi: 10.1007/s40264-015-0327-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chebib FT, Perrone RD, Chapman AB, et al. A practical guide for treatment of rapidly progressive ADPKD with tolvaptan. J Am Soc Nephrol. 2018;29(10):2458‐2470. doi: 10.1681/ASN.2018060590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Torres VE, Chapman AB, Devuyst O, et al. Multicenter, open‐label, extension trial to evaluate the long‐term efficacy and safety of early versus delayed treatment with tolvaptan in autosomal dominant polycystic kidney disease: the TEMPO 4:4 Trial. Nephrol Dial Transplant. 2018:33(3):477‐489. doi: 10.1093/ndt/gfx043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bowman BT, Rosner MH. Lixivaptan ‐ an evidence‐based review of its clinical potential in the treatment of hyponatremia. Core Evid. 2013;8:47‐56. doi: 10.2147/CE.S36744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang X, Constans MM, Chebib FT, Torres VE, Pellegrini L. Effect of a vasopressin V2 receptor antagonist on polycystic kidney disease development in a rat model. Am J Nephrol. 2019;49(6):487‐493. doi: 10.1159/000500667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Di Mise A, Venneri M, Ranieri M, et al. Lixivaptan, a new generation diuretic, counteracts vasopressin‐induced aquaporin‐2 trafficking and function in renal collecting duct cells. Int J Mol Sci. 2019;21(1):183. doi: 10.3390/ijms21010183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Woodhead JL, Pellegrini L, Shoda LKM, Howell BA. Comparison of the hepatotoxic potential of two treatments for autosomal‐dominant polycystic kidney diseaseusing quantitative systems toxicology modeling. Pharm Res. 2020;37(2):24. doi: 10.1007/s11095-019-2726-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Woodhead JL, Brock WJ, Roth SE, et al. Application of a mechanistic model to evaluate putative mechanisms of tolvaptan drug‐induced liver injury and identify patient susceptibility factors. Toxicol Sci. 2017;155(1):61‐74. doi: 10.1093/toxsci/kfw193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Torres VE, Harris PC. Mechanisms of disease: autosomal dominant and recessive polycystic kidney diseases. Nat Clin Pract Nephrol. 2006;2(1):40‐55; quiz 55. doi: 10.1038/ncpneph0070 [DOI] [PubMed] [Google Scholar]
- 15. Riccardi D, Valenti G. Localization and function of the renal calcium‐sensing receptor. Nat Rev Nephrol. 2016;12(7):414‐425. doi: 10.1038/nrneph.2016.59 [DOI] [PubMed] [Google Scholar]
- 16. Di Mise A, Tamma G, Ranieri M, et al. Conditionally immortalized human proximal tubular epithelial cells isolated from the urine of a healthy subject express functional calcium‐sensing receptor. Am J Physiol Renal Physiol. 2015;308(11):F1200‐F1206. doi: 10.1152/ajprenal.00352.2014 [DOI] [PubMed] [Google Scholar]
- 17. Di Mise A, Tamma G, Ranieri M, et al. Activation of calcium‐sensing receptor increases intracellular calcium and decreases cAMP and mTOR in PKD1 deficient cells. Sci Rep. 2018;8(1) 5704. doi: 10.1038/s41598-018-23732-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Di Mise A, Ranieri M, Centrone M, et al. Activation of the calcium‐sensing receptor corrects the impaired mitochondrial energy status observed in renal polycystin‐1 knockdown cells modeling autosomal dominant polycystic kidney disease. Front Mol Biosci. 2018;5:77. doi: 10.3389/fmolb.2018.00077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gattone VH, Chen NX, Sinders RM, et al. Calcimimetic inhibits late‐stage cyst growth in ADPKD. J Am Soc Nephrol. 2009;20(7):1527‐1532. doi: 10.1681/ASN.2008090927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen NX, Moe SM, Eggleston‐Gulyas T, et al. Calcimimetics inhibit renal pathology in rodent nephronophthisis. Kidney Int. 2011;80(6):612‐619. doi: 10.1038/ki.2011.139 [DOI] [PubMed] [Google Scholar]
- 21. Wang X, Harris PC, Somlo S, Batlle D, Torres VE. Effect of calcium‐sensing receptor activation in models of autosomal recessive or dominant polycystic kidney disease. Nephrol Dial Transplant. 2009;24(2):526‐534. doi: 10.1093/ndt/gfn527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Katsuyama M, Masuyama T, Komura I, Hibino T, Takahashi H. Characterization of a novel polycystic kidney rat model with accompanying polycystic liver. Exp Anim. 2000;49(1):51‐55. doi: 10.1538/expanim.49.51 [DOI] [PubMed] [Google Scholar]
- 23. Lager DJ, Qian Q, Bengal RJ, Ishibashi M, Torres VE. The pck rat: a new model that resembles human autosomal dominant polycystic kidney and liver disease. Kidney Int. 2001;59(1):126‐136. doi: 10.1046/j.1523-1755.2001.00473.x [DOI] [PubMed] [Google Scholar]
- 24. Hopp K, Ward CJ, Hommerding CJ, et al. Functional polycystin‐1 dosage governs autosomal dominant polycystic kidney disease severity. J Clin Invest. 2012;122(11):4257‐4273. doi: 10.1172/JCI64313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang X, Gattone V 2nd, Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC‐31260 and OPC‐41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol. 2005;16(4):846‐851. doi: 10.1681/ASN.2004121090 [DOI] [PubMed] [Google Scholar]
- 26. Rowe I, Chiaravalli M, Mannella V, et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med. 2013;19(4):488‐493. doi: 10.1038/nm.3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Takiar V, Nishio S, Seo‐Mayer P, et al. Activating AMP‐activated protein kinase (AMPK) slows renal cystogenesis. Proc Natl Acad Sci U S A. 2011;108(6):2462‐2467. doi: 10.1073/pnas.1011498108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pathomthongtaweechai N, Soodvilai S, Chatsudthipong V, Muanprasat C. Pranlukast inhibits renal epithelial cyst progression via activation of AMP‐activated protein kinase. Eur J Pharmacol. 2014;724:67‐76. doi: 10.1016/j.ejphar.2013.12.013 [DOI] [PubMed] [Google Scholar]
- 29. Yamaguchi T, Nagao S, Wallace DP, et al. Cyclic AMP activates B‐Raf and ERK in cyst epithelial cells from autosomal‐dominant polycystic kidneys. Kidney Int. 2003;63(6):1983‐1994. doi: 10.1046/j.1523-1755.2003.00023.x [DOI] [PubMed] [Google Scholar]
- 30. Omori S, Hida M, Fujita H, et al. Extracellular signal‐regulated kinase inhibition slows disease progression in mice with polycystic kidney disease. J Am Soc Nephrol. 2006;17(6):1604‐1614. doi: 10.1681/asn.2004090800 [DOI] [PubMed] [Google Scholar]
- 31. Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med. 2008;359(14):1477‐1485. doi: 10.1056/NEJMcp0804458 [DOI] [PubMed] [Google Scholar]
- 32. Zeier M, Fehrenbach P, Geberth S, Mohring K, Waldherr R, Ritz E. Renal histology in polycystic kidney disease with incipient and advanced renal failure. Kidney Int. 1992;42(5):1259‐1265. doi: 10.1038/ki.1992.413 [DOI] [PubMed] [Google Scholar]
- 33. Ibrahim S. Increased apoptosis and proliferative capacity are early events in cyst formation in autosomal‐dominant, polycystic kidney disease. ScientificWorldJournal. 2007;7:1757‐1767. doi: 10.1100/tsw.2007.274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. van Gastel MDA, Torres VE. polycystic kidney disease and the vasopressin pathway. Ann Nutr Metab. 2017;70(Suppl 1):43‐50. doi: 10.1159/000463063 [DOI] [PubMed] [Google Scholar]
- 35. Ranieri M, Di Mise A, Tamma G, Valenti G. Vasopressin‐aquaporin‐2 pathway: recent advances in understanding water balance disorders. F1000Res. 2019;8:149. doi: 10.12688/f1000research.16654.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ranieri M, Di Mise A, Tamma G, Valenti G. Calcium sensing receptor exerts a negative regulatory action toward vasopressin‐induced aquaporin‐2 expression and trafficking in renal collecting duct. Vitam Horm. 2020;112:289‐310. doi: 10.1016/bs.vh.2019.08.008 [DOI] [PubMed] [Google Scholar]
- 37. Ranieri M, Zahedi K, Tamma G, et al. CaSR signaling down‐regulates AQP2 expression via a novel microRNA pathway in pendrin and NaCl cotransporter knockout mice. FASEB J. 2018;32(4):2148‐2159. doi: 10.1096/fj.201700412RR [DOI] [PubMed] [Google Scholar]
- 38. Dai XQ, Perez PL, Soria G, et al. External Ca2+ regulates polycystin‐2 (TRPP2) cation currents in LLC‐PK1 renal epithelial cells. Exp Cell Res. 2017;350(1):50‐61. doi: 10.1016/j.yexcr.2016.11.004 [DOI] [PubMed] [Google Scholar]
- 39. Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B‐Raf/ERK pathway, switching cells to a cAMP‐dependent growth‐stimulated phenotype. J Biol Chem. 2004;279(39):40419‐40430. doi: 10.1074/jbc.M405079200 [DOI] [PubMed] [Google Scholar]
- 40. Yamaguchi T, Hempson SJ, Reif GA, Hedge AM, Wallace DP. Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J Am Soc Nephrol. 2006;17(1):178‐187. doi: 10.1681/asn.2005060645 [DOI] [PubMed] [Google Scholar]
- 41. Hopp K, Hommerding CJ, Wang X, Ye H, Harris PC, Torres VE. Tolvaptan plus pasireotide shows enhanced efficacy in a PKD1 model. J Am Soc Nephrol. 2015;26(1):39‐47. doi: 10.1681/asn.2013121312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gattone VH 2nd, Maser RL, Tian C, Rosenberg JM, Branden MG. Developmental expression of urine concentration‐associated genes and their altered expression in murine infantile‐type polycystic kidney disease. Dev Genet. 1999;24(3‐4):309‐318. doi: [DOI] [PubMed] [Google Scholar]
- 43. Gattone VH 2nd, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med. 2003;9(10):1323‐1326. doi: 10.1038/nm935 [DOI] [PubMed] [Google Scholar]
- 44. Torres VE, Wang X, Qian Q, Somlo S, Harris PC, Gattone VH 2nd. Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat Med. 2004;10(4):363‐364. doi: 10.1038/nm1004 [DOI] [PubMed] [Google Scholar]
- 45. Testa F, Magistroni R. ADPKD current management and ongoing trials. J Nephrol. 2020;33(2):223‐237. doi: 10.1007/s40620-019-00679-y [DOI] [PubMed] [Google Scholar]
- 46. Aihara M, Fujiki H, Mizuguchi H, et al. Tolvaptan delays the onset of end‐stage renal disease in a polycystic kidney disease model by suppressing increases in kidney volume and renal injury. J Pharmacol Exp Ther. 2014;349(2):258‐267. doi: 10.1124/jpet.114.213256 [DOI] [PubMed] [Google Scholar]
- 47. Dwivedi N, Tao S, Jamadar A, et al. Epithelial vasopressin type‐2 receptors regulate myofibroblasts by a YAP‐CCN2‐dependent mechanism in polycystic kidney disease. J Am Soc Nephrol. 2020;31(8):1697‐1710. doi: 10.1681/asn.2020020190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Torres JA, Rezaei M, Broderick C, et al. Crystal deposition triggers tubule dilation that accelerates cystogenesis in polycystic kidney disease. J Clin Invest. 2019;129(10):4506‐4522. doi: 10.1172/JCI128503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vezzoli G, Macrina L, Magni G, Arcidiacono T. Calcium‐sensing receptor: evidence and hypothesis for its role in nephrolithiasis. Urolithiasis. 2019;47(1):23‐33. doi: 10.1007/s00240-018-1096-0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S3