

Abstract
Aims
To evaluate the safety, pharmacokinetics and pharmacodynamics of single‐ and multiple‐rising doses (MRDs) of BI 705564 and establish proof of mechanism.
Methods
BI 705564 was studied in 2 placebo‐controlled, Phase I clinical trials testing single‐rising doses (1–160 mg) and MRDs (1–80 mg) of BI 705564 over 14 days in healthy male volunteers. Blood samples were analysed for BI 705564 plasma concentration, Bruton's tyrosine kinase (BTK) target occupancy (TO) and CD69 expression in B cells stimulated ex vivo. A substudy was conducted in allergic, otherwise healthy, MRD participants. Safety was assessed in both studies.
Results
All doses of BI 705564 were well tolerated. Geometric mean BI 705564 plasma terminal half‐life ranged from 10.1 to 16.9 hours across tested doses, with no relevant accumulation after multiple dosing. Doses ≥20 mg resulted in ≥85% average TO that was maintained for ≥48 hours after single‐dose administration. Functional effects of BTK signalling were demonstrated by dose‐dependent inhibition of CD69 expression. In allergic participants, BI 705564 treatment showed a trend in wheal size reduction in a skin prick test and complete inhibition of basophil activation. Mild bleeding‐related adverse events were observed with BI 705564; bleeding time increased in 1/12 participants (8.3%) who received placebo vs 26/48 (54.2%) treated with BI 705564.
Conclusion
BI 705564 showed efficient target engagement through durable TO and inhibition of ex vivo B‐cell activation, and proof of mechanism through effects on allergic skin responses. Mild bleeding‐related adverse events were probably related to inhibition of platelet aggregation by BTK inhibition.
Keywords: Bruton's tyrosine kinase, BTK inhibitor, nephritis, systemic lupus erythematosus
What is already known about this subject
Despite therapeutic progress in recent years, there is still a need for more effective and safe treatments for autoimmune diseases including rheumatoid arthritis, systemic lupus erythematosus and lupus nephritis.
Bruton's tyrosine kinase (BTK), a member of the Tec family of immune kinases, plays an important role in diverse, disease‐relevant signal transduction cascades and is a potential target for therapeutic intervention.
Several BTK inhibitors have demonstrated efficacy in models of autoimmune disease and subsequent clinical studies, but have been associated with an increased risk of bleeding‐related adverse events in patients treated for haematological malignancies.
What this study adds
In 2 placebo‐controlled, Phase I clinical trials, the BTK inhibitor BI 705564 showed dose‐dependent inhibition of CD69 expression and, in allergic participants, resulted in reduction of wheal size in a skin prick test and complete inhibition of basophil activation.
BI 705564 was well tolerated but was associated with mild bleeding‐related adverse events.
The occurrence of bleeding‐related effects despite high BTK selectivity indicate that the effect on platelets is most likely to be mediated via BTK and not via Tec inhibition.
1. INTRODUCTION
Despite therapeutic progress in recent years, there is a need for more effective and safe treatments for autoimmune diseases, including rheumatoid arthritis (RA), systemic lupus erythematosus (SLE) and lupus nephritis (LN). 1 , 2 , 3 , 4 In RA and SLE/LN, pathogenic mechanisms involve B‐cell activation and activation of innate immune and stromal cells, leading to the production of autoantibodies and subsequent complement fixation and antigen presentation to T cells in RA 5 and SLE, 6 , 7 , 8 and deposition of immunoglobulin (Ig)G autoantibodies/immune complexes within the renal glomerulus in LN. 8 , 9 , 10
Bruton's tyrosine kinase (BTK), a member of the Tec family of immune kinases, plays an important role in signal transduction cascades downstream from key cell surface receptors in lymphoid and myeloid cell types. 11 , 12 , 13 The defective B‐cell maturation and decreased antibody levels observed in individuals with X‐linked agammaglobulinaemia harbouring loss‐of‐function Btk mutations, exemplifies the essential role of BTK in B cells. 11 BTK is needed for Fc‐gamma receptor signalling in monocytes/macrophages, 14 Fc‐epsilon receptor (FcεR) signalling in basophils and mast cells 15 , 16 and glycoprotein VI (GPVI) receptor signalling in platelets. 17 , 18 The function of BTK across diverse, disease‐relevant signalling pathways provided the stimulus for identifying and developing selective BTK inhibitors for preclinical testing in models of autoimmune disease and subsequent clinical studies. 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 Conflicting data on potential redundancy with the related Tec kinase for platelet function, combined with the observed risk of bleeding events with the approved BTK inhibitors ibrutinib and acalabrutinib, have made the risk of bleeding‐related events for BTK inhibitors difficult to predict prior to clinical trials. 17 , 32 , 33 , 34
BI 705564 is an irreversible, covalent, small‐molecule inhibitor of BTK, developed as a candidate for the treatment of SLE/LN and RA. In a kinetic binding assay, BI 705564 inhibited BTK enzyme binding, with a mean half maximal inhibitory concentration of 0.28 nM (n = 6). BI 705564 profiling against a panel of 282 kinases revealed significant selectivity for BTK. Only 3 other kinases (BMX, TXK and Tec) exhibited >80% inhibition at 3 μM, corresponding to half maximal inhibitory concentration values >30‐fold higher than that for BTK (unpublished data). A structurally related analogue of BI 705564, BI‐BTK‐1, was assessed in the SLE/LN‐relevant nephrotoxic serum nephritis in vivo mouse model. 19 , 35 Given the irreversible binding of BI‐BTK‐1 to BTK, determination of the ex vivo percentage target occupancy (TO) in this model allowed the relationship between target engagement and efficacy to be ascertained. Treatment with BI‐BTK‐1 at doses that provided ≥85% BTK TO resulted in prevention of proteinuria and almost complete amelioration of renal disease. 19 , 35 Treatment with BI‐BTK‐1 resulted in dose‐responsive protection of renal function, with daily doses of 10 and 30 mg/kg achieving 87 and 89% inhibition of proteinuria, respectively; considerable kidney protection, as measured by renal glomerular and tubule histology analyses, was also observed with 97 and 91% inhibition of a summed kidney histology score, respectively. In addition, testing of BI‐BTK‐1 in the chronic NZB/W F1 model of LN demonstrated dose‐responsive inhibition of proteinuria, circulating anti‐double‐stranded DNA IgG levels and renal protection as measured by histopathology. 19 , 35
Here we report the findings of 2 Phase I trials testing single‐ and multiple‐rising doses (SRDs/MRDs) of oral BI 705564 in healthy male volunteers. The primary objectives were to investigate the safety and tolerability of BI 705564; secondary objectives included pharmacokinetics (PK) and pharmacodynamics (PD). As it has been suggested that BTK inhibition could decrease reactivity to allergens by preventing IgE‐dependent basophil and mast cell activation, 16 , 36 , 37 the effect of BI 705564 on a skin prick test in participants with allergies was assessed as a proof of mechanism.
2. METHODS
2.1. Study designs and participants
The SRD study was a single‐blind, partially randomised, placebo‐controlled trial carried out at the Human Pharmacology Centre, Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach an der Riss, Germany, between May 2017 and February 2018 (www.ClinicalTrials.gov: NCT03123185). The MRD study was a double‐blind, randomised, placebo‐controlled trial carried out at the Clinical Research Services Mannheim GmbH, Mannheim, Germany, between November 2017 and November 2018 (www.ClinicalTrials.gov: NCT03325712). Both trials were conducted in accordance with the Declaration of Helsinki, the ICH GCP guidelines and local laws. All local ethics committees were informed and approved the trial and written informed consent was obtained from all participants prior to admission into the trial.
Participants in both studies were healthy men aged 18–50 years with a body mass index of 18.5–29.9 kg/m2 (inclusive). Main exclusion criteria were clinically relevant pre‐existing diseases or abnormalities in the screening examination, including infectious serology (hepatitis, human immunodeficiency virus), marked baseline prolongation of QT/QTc interval (such as QTc intervals repeatedly >450 ms), any other relevant electrocardiogram (ECG) finding or a history of additional risk factors for torsades de pointes (e.g. heart failure, hypokalaemia or family history of long QT syndrome).
In both studies, all doses of study medication were administered orally under the supervision of the principal investigator or an authorised designee. In the SRD study, 47 participants received treatment under fasted conditions in 6 consecutive groups, and 32 participants received treatment under fed conditions (with a high‐fat meal) in 4 consecutive groups. Each group included 7 or 8 participants: 5 or 6 receiving BI 705564 and 2 receiving placebo. BI 705564 was given in single doses from 1 mg rising to 80 mg in the fasted groups, and from 20 mg rising to 160 mg in the fed groups. The decision to proceed to the next dose group was based upon the safety, tolerability and PK data of the preceding dose group. A further food effect analysis was performed in an open‐label, 2‐way crossover, including 12 participants randomly assigned to receive 10 mg BI 705564 in a fed state followed by a fasted state, or vice versa, with a 10‐day washout between treatments. Participants were kept under close medical surveillance at the study site for at least 24 hours after drug administration. Venous blood samples were taken at regular intervals up to 24 hours post administration and then daily through Day 10.
In the MRD study, 60 participants received treatment in 5 sequential groups. Each group included 10 participants: 8 receiving BI 705564 and 2 receiving placebo. The BI 705564 dose groups were 10, 20, 40, 60 or 80 mg daily for 14 days, administered after consumption of a standard breakfast. Blood samples were taken daily from Day −1 to Day 17. After completion of the dose‐escalation phase, an additional double‐blind, randomised, placebo‐controlled cohort was added (via a protocol amendment) to test 40 mg BI 705564 daily for 4 weeks in participants with a known allergy to house dust mite (HDM) or cat allergen. The purpose of this additional cohort was to test safety for a longer duration and evaluate the effect of BI 705564 on an allergic reaction in a skin prick test as a preliminary proof of clinical concept. Inclusion criteria for the participants in this skin prick test group (SPTG) were the same as those for the previous MRD cohorts, with the additional requirement of a history of at least 1 year of IgE‐mediated perennial allergies as documented by a positive skin prick test (wheal at screening at least 5 mm in diameter at longest measurement).
2.2. Safety assessments
Safety and tolerability were assessed in both studies by recording treatment‐emergent adverse events (TEAEs), vital signs, ECG and clinical laboratory assessments. TEAEs reported between study drug administration and the last study visit were considered on‐treatment.
As other BTK inhibitors tested in individuals diagnosed with haematological disorders have been associated with increased bleeding rates, 32 , 33 measurement of bleeding time was included in all treatment groups in the MRD study, including the SPTG. Bleeding times were assessed at baseline and at the post‐treatment examination for all treatment groups, and additionally on Days 7 and 14 for the MRD cohort and Days 8, 15, 22 and 28 for the SPTG. Bleeding time was measured according to a modified Ivy method (Surgicutt). The upper limit of normal (ULN) was 480 seconds.
In the SPTG, platelet function (adhesion and aggregation) was assessed using the INNOVANCE Platelet Function Analyser (PFA)‐200 System with Dade PFA collagen/epinephrine and collagen/ADP. This test was performed at the Gerinnungszentrum Mannheim, Mannheim, Germany.
The safety analysis in the MRD study pooled the participants who received placebo in the MRD cohort and those who received placebo in the SPTG cohort (the treatment duration was 4 weeks in the SPTG compared with 2 weeks in the MRD cohort).
2.3. PK and PD assessments
Plasma concentrations of BI 705564 were measured using a validated liquid chromatography–tandem mass spectrometry method with a calibration range of 0.100–100 nmol/L and a lower limit of quantification of 0.1 nmol/L. The assay accuracy ranged from 99.4 to 100.4% and the precision was between 2.9 and 6.1% coefficient of variation. The analyses were performed by NUVISAN GmbH (Neu‐Ulm, Germany).
For determination of TO, peripheral blood mononuclear cells were isolated from whole blood samples, followed by preparation of cell lysates. The amount of free BTK (not occupied by BI 705564) in the lysate was determined by adding a biotin‐conjugated probe that binds to the same site as BI 705564. The biotin‐labelled BTK was then detected by streptavidin in a meso‐scale discovery‐based ligand binding immunoassay. The concentration of total BTK (i.e. free BTK and BTK bound by BI 705564) was determined by 2 different anti‐BTK antibodies for capture and detection in a meso‐scale discovery‐based ligand binding immunoassay. Free BTK levels were normalised to total BTK, and percent changes relative to baseline were calculated. Free BTK and total BTK assays were validated at NUVISAN GmbH and clinical samples were analysed in accordance with the applicable principles of Good Clinical Practice.
To evaluate the effect of BI 705564 treatment on B‐cell activation, whole blood samples were stimulated ex vivo with anti‐IgD, and the percentage of CD69 + B cells among all CD19 + B cells was measured by flow cytometry, as previously described, 38 with modifications to adapt to the study design. Percent inhibition relative to baseline was calculated.
For analysis of basophil activation in the MRD study, whole blood samples were stimulated ex vivo with anti‐IgE, followed by measurement of the percentage of CD63 + basophils by flow cytometry, as previously described, 37 with modifications to adapt to the study design. Percent inhibition relative to baseline was calculated.
2.4. Skin prick tests
Commercially available skin prick test solutions (Bencard Allergie GmbH, Munich, Germany), including those for HDM (Dermatophagoides farinae and Dermatophagoides pteronyssinus), cat fur, dog hair, histamine solution (positive control) and normal saline/solvent (negative control), were administered as a single drop to the volar aspect of the lower arm, followed by piercing of the skin through the drop with a needle or lancet.
During screening, the skin prick test was performed with multiple allergens, and only positive allergens (>5 mm wheal diameter) were retested on Day −1, Day 8 and Day 28. On Day −1, only 4 subjects had wheal diameter assessments, thus, measurements from all subjects at screening were taken as baseline.
The appearance of a wheal surrounded by a red flare (erythema) was considered a positive reaction. The wheal size, i.e. the largest diameter in millimetres, was measured directly on the skin using a ruler, with the final results recorded after 15 minutes. Wheal area (A) was calculated based on the wheal diameter (d): .
2.5. Statistical analysis
All analyses were reported using descriptive statistics for safety, PK and PD. In each trial, the safety population included all participants who had received at least 1 dose of study drug. The PK and PD populations included all participants who had received the study drug and provided evaluable data for PK and PD analysis without relevant violations. PK parameters were calculated using noncompartmental methods with Phoenix WinNonlin 6.3 software. Dose proportionality was assessed using a power model (as detailed in Appendix A). In the SPTG, the percent change of wheal area from baseline was analysed by a mixed effects model (using R version 3.6.0), where treatment, visit and treatment by visit interaction were considered fixed effects and participants and types of allergen were considered random effects.
2.6. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY.
3. RESULTS
3.1. Participants
Demographics and baseline characteristics were similar across treatment groups in the SRD and MRD studies, with minor differences between groups for age, height and body mass index (Tables 1 and 2). All participants treated in the SRD study completed treatment, whereas 2 participants treated with 40 mg BI 705564 in the MRD study discontinued: 1 due to urinary tract infection and haematuria, and 1 refused to comply with the protocol.
TABLE 1.
Demographics and baseline characteristics of participants in the single‐rising dose study
| Placebo fasted + fed (n = 20) | BI 705564 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fasted | Fed | Food effect | ||||||||||
| 1 mg (n = 6) | 3 mg (n = 5) | 10 mg (n = 6) | 20 mg (n = 6) | 40 mg (n = 6) | 80 mg (n = 6) | 20 mg (n = 6) | 40 mg (n = 6) | 80 mg (n = 6) | 160 mg (n = 6) | 10 mg a (n = 12) | ||
| Age (y), median (range) | 39 (19–48) | 35 (31–43) | 29 (20–43) | 34 (22–46) | 32 (24–48) | 37 (26–49) | 35 (26–50) | 36 (29–49) | 30 (21–39) | 33 (25–48) | 39 (29–48) | 30 (19–50) |
| Height (cm), mean (SD) | 180 (5) | 180 (7) | 177 (4) | 180 (5) | 181 (8) | 175 (9) | 176 (5) | 180 (6) | 179 (9) | 184 (6) | 176 (5) | 178 (5) |
| BMI (kg/m2), mean (SD) | 25 (2) | 24 (3) | 23 (2) | 24 (3) | 24 (3) | 25 (3) | 25 (2) | 26 (3) | 23 (2) | 25 (3) | 27 (2) | 24 (3) |
Two participants in the 10 mg BI 705564 food effect group were also included in other groups (1 in the placebo group and 1 in the 3 mg BI 705564 group).
BMI, body mass index; SD, standard deviation.
TABLE 2.
Demographics and baseline characteristics of participants in the multiple‐rising dose study
| Placebo (n = 12) a | BI 705564 | ||||||
|---|---|---|---|---|---|---|---|
| 10 mg (n = 8) | 20 mg (n = 8) | 40 mg (n = 8) | 60 mg (n = 8) | 80 mg (n = 8) | 40 mg SPTG (n = 8) | ||
| Age (y), median (range) | 38 (27–50) | 34 (23–48) | 46 (35–51) | 32 (21–47) | 38 (28–48) | 46 (26–51) | 25 (18–49) |
| Height (cm), mean (SD) | 181 (10) | 176 (4) | 182 (4) | 178 (4) | 177 (5) | 178 (7) | 183 (8) |
| BMI (kg/m2), mean (SD) | 26 (3) | 25 (3) | 25 (2) | 23 (3) | 26 (2) | 26 (3) | 26 (3) |
Includes all participants who received placebo in the trial.
BMI, body mass index; SD, standard deviation; SPTG, skin prick test group.
3.2. Safety
In both studies, all doses of BI 705564 were well tolerated (Tables 3 and 4). TEAEs were mostly of mild intensity and not dose limiting; there were no serious TEAEs, deaths or adverse events (AEs) of severe intensity reported for either study. The overall percentage of participants who experienced a TEAE was slightly lower in the combined BI 705564 groups compared with placebo in the SRD (26/71 [37%] vs 9/20 [45%]) and MRD (23/48 [48%] vs 7/12 [58%]) studies, respectively. Headache was the most frequently reported TEAE in both the SRD (BI 705564 combined groups 8/71 [11%], placebo 4/20 [20%]) and MRD (BI 705564 combined groups 7/48 [15%], placebo 1/12 [8%]) studies. In the MRD study, gastrointestinal disorders (diarrhoea, abdominal pain, toothache and nausea) were also common and were reported by a greater proportion of participants in the combined BI 705564 groups (9/48 [19%]) than the placebo group (1/12 [8%]). In the SRD study, minimal differences were observed between BI 705564 treatment groups when stratified by their fasting status.
TABLE 3.
Summary of treatment‐emergent adverse events in the single‐rising dose study
| n (%) | Placebo fasted + fed (n = 20) | BI 705564 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fasted | Fed | Food effect | Total (N = 71) | ||||||||||||
| 1 mg (n = 6) | 3 mg (n = 5) | 10 mg (n = 6) | 20 mg (n = 6) | 40 mg (n = 6) | 80 mg (n = 6) | 20 mg (n = 6) | 40 mg (n = 6) | 80 mg (n = 6) | 160 mg (n = 6) | Fasted 10 mg a (n = 12)b | Fed 10 mg a (n = 11)b | Total 10 mg a (n = 12)b | |||
| TEAEs | 9 (45) | 3 (50) | 1 (20) | 2 (33) | 1 (17) | 3 (50) | 2 (33) | 3 (50) | 4 (67) | 0 | 3 (50) | 2 (17) | 2 (18) | 4 (33) | 26 (37) |
| Drug‐related TEAEs | 4 (20) | 1 (17) | 0 | 0 | 0 | 1 (17) | 1 (17) | 0 | 4 (67) | 0 | 0 | 0 | 1 (9) | 1 (8) | 8 (11) |
| TEAEs in ≥2 participants overall, system organ class/preferred term | |||||||||||||||
| Nervous system disorders | 4 (20) | 2 (33) | 0 | 1 (17) | 0 | 1 (17) | 1 (17) | 1 (17) | 1 (17) | 0 | 1 (17) | 0 | 0 | 0 | 8 (11 |
| Headache | 4 (20) | 2 (33) | 0 | 1 (17) | 0 | 1 (17) | 1 (17) | 1 (17) | 1 (17) | 0 | 1 (17) | 0 | 0 | 0 | 8 (11) |
| Gastrointestinal disorders | 2 (10) | 0 | 0 | 0 | 0 | 1 (17) | 0 | 1 (17) | 2 (33) | 0 | 0 | 0 | 1 (9) | 1 (8) | 5 (7) |
| Nausea | 1 (5) | 0 | 0 | 0 | 0 | 1 (17) | 0 | 0 | 1 (17) | 0 | 0 | 0 | 1 (9) | 1 (8) | 3 (4) |
| Diarrhoea | 1 (5) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (17) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1) |
| Vomiting | 1 (5) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (17) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1) |
| Vascular disorders | 2 (10) | 0 | 0 | 1 (17) | 1 (17) | 1 (17) | 0 | 0 | 0 | 0 | 1 (17) | 0 | 0 | 0 | 4 (6) |
| Haematoma | 1 (5) | 0 | 0 | 1 (17) | 1 (17) | 0 | 0 | 0 | 0 | 0 | 1 (17) | 0 | 0 | 0 | 3 (4) |
| Infections and infestations | 1 (5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (33) | 0 | 1 (17) | 0 | 0 | 0 | 3 (4) |
| Nasopharyngitis | 1 (5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (17) | 0 | 0 | 0 | 1 (1) |
| Oral herpes | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (17) | 0 | 0 | 0 | 0 | 0 | 1 (1) |
| Rhinitis | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (17) | 0 | 0 | 0 | 0 | 0 | 1 (1) |
| Injury, poisoning and procedural complications | 1 (5) | 0 | 1 (20) | 0 | 0 | 1 (17) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (9) | 1 (8) | 3 (4) |
| Respiratory, thoracic and mediastinal disorders | 0 | 0 | 0 | 1 (17) | 0 | 0 | 0 | 1 (17) | 0 | 0 | 0 | 1 (8) | 0 | 1 (8) | 3 (4) |
| Oropharyngeal pain | 0 | 0 | 0 | 1 (17) | 0 | 0 | 0 | 1 (17) | 0 | 0 | 0 | 1 (8) | 0 | 1 (8) | 3 (4) |
| Skin and subcutaneous disorders | 1 (5) | 1 (17) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (9) | 1 (8) | 2 (3) |
| Erythema | 1 (5) | 1 (17) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1) |
| General disorders and admin site conditions | 0 | 0 | 0 | 0 | 0 | 0 | 1 (17) | 0 | 0 | 0 | 0 | 1 (8) | 1 (9) | 2 (17) | 3 (4) |
| Chest pain | 0 | 0 | 0 | 0 | 0 | 0 | 1 (17) | 0 | 0 | 0 | 0 | 0 | 1 (9) | 1 (8) | 2 (3) |
Food effect analysis (two‐way crossover); bTwo participants in the food effect group were also included in other groups (one in the placebo group and 1 in the 3 mg BI 705564 group).
TEAE, treatment‐emergent adverse event.
TABLE 4.
Summary of treatment‐emergent adverse events in the multiple‐rising dose study
| n (%) | Placebo (n = 12) a | BI 705564 | ||||||
|---|---|---|---|---|---|---|---|---|
| 10 mg (n = 8) | 20 mg (n = 8) | 40 mg (n = 8) | 60 mg (n = 8) | 80 mg (n = 8) | 40 mg SPTG (n = 8) | Total (N = 48) | ||
| TEAEs | 7 (58) | 3 (38) | 2 (25) | 4 (50) | 5 (63) | 2 (25) | 7 (88) | 23 (48) |
| Drug‐related TEAEs | 3 (25) | 1 (13) | 0 | 4 (50) | 4 (50) | 1 (13) | 6 (75) | 16 (33) |
| TEAEs in ≥2 participants overall, system organ class/preferred term | ||||||||
| Nervous system disorders | 3 (25) | 2 (25) | 0 | 1 (13) | 1 (13) | 0 | 5 (63) | 9 (19) |
| Headache | 1 (8) | 2 (25) | 0 | 1 (13) | 0 | 0 | 4 (50) | 7 (15) |
| Dizziness | 0 | 0 | 0 | 0 | 1 (13) | 0 | 2 (25) | 3 (6) |
| Orthostatic intolerance | 1 (8) | 0 | 0 | 1 (13) | 0 | 0 | 0 | 1 (2) |
| Gastrointestinal disorders | 1 (8) | 1 (13) | 1 (13) | 3 (38) | 1 (13) | 0 | 3 (38) | 9 (19) |
| Diarrhoea | 0 | 0 | 0 | 1 (13) | 0 | 0 | 3 (38) | 4 (8) |
| Abdominal pain, upper | 0 | 0 | 0 | 0 | 1 (13) | 0 | 1 (13) | 2 (4) |
| Toothache | 0 | 0 | 1 (13) | 1 (13) | 0 | 0 | 0 | 2 (4) |
| Nausea | 1 (8) | 0 | 0 | 0 | 0 | 0 | 1 (13) | 1 (2) |
| Musculoskeletal and connective tissue disorders | 2 (17) | 0 | 0 | 1 (13) | 1 (13) | 0 | 2 (25) | 4 (8) |
| Myalgia | 0 | 0 | 0 | 0 | 0 | 0 | 2 (25) | 2 (4) |
| Back pain | 1 (8) | 0 | 0 | 1 (13) | 0 | 0 | 0 | 1 (2) |
| Arthralgia | 2 (17) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Vascular disorders | 1 (8) | 0 | 0 | 0 | 0 | 0 | 1 (13) | 1 (2) |
| Haematoma | 1 (8) | 0 | 0 | 0 | 0 | 0 | 1 (13) | 1 (2) |
| Infections and infestations | 2 (17) | 0 | 1 (13) | 1 (13) | 0 | 0 | 2 (25) | 4 (8) |
| Nasopharyngitis | 2 (17) | 0 | 0 | 0 | 0 | 0 | 2 (25) | 2 (4) |
| Herpes simplex | 0 | 0 | 1 (13) | 0 | 0 | 0 | 0 | 1 (2) |
| Urinary tract infection | 0 | 0 | 0 | 1 (13) | 0 | 0 | 0 | 1(2) |
| Injury, poisoning and procedural complications | 0 | 0 | 0 | 0 | 1 (13) | 1 (13) | 0 | 2 (4) |
| Respiratory, thoracic and mediastinal disorders | 0 | 0 | 0 | 0 | 0 | 0 | 3 (38) | 3 (6) |
| Oropharyngeal pain | 0 | 0 | 0 | 0 | 0 | 0 | 3 (38) | 3 (6) |
| Epistaxis | 0 | 0 | 0 | 0 | 0 | 0 | 2 (25) | 2 (4) |
| Skin and subcutaneous disorders | 0 | 0 | 0 | 1 (13) | 2 (25) | 0 | 2 (25) | 5 (10) |
| Petechiae | 0 | 0 | 0 | 0 | 2 (25) | 0 | 2 (25) | 4 (8) |
| General disorders and admin site conditions | 2 (17) | 0 | 0 | 0 | 0 | 1 (13) | 3 (38) | 4 (8) |
| Fatigue | 2 (17) | 0 | 0 | 0 | 0 | 1 (13) | 2 (25) | 3 (6) |
Includes all participants who received placebo in the trial; the participants who received placebo in the MRD cohort and those who received placebo in the SPTG were pooled (the treatment duration was 4 weeks in the SPTG compared with 2 weeks in the MRD cohort).
MRD, multiple rising dose; SPTG, skin prick test group; TEAE, treatment‐emergent adverse event.
Infections occurred in 1/20 participants (5%) treated with placebo (nasopharyngitis) and 3/71 participants (4%) treated with BI 705564 (nasopharyngitis, oral herpes, rhinitis) in the SRD study, and in 2/12 participants (17%) treated with placebo (nasopharyngitis) and 4/48 participants (8%) treated with BI 705564 (nasopharyngitis [n = 2], herpes simplex, urinary tract infection) in the MRD study.
In the SRD study, haematoma occurred in 1/20 participants (5%) in the placebo group and 3/71 (4%) participants in the combined BI 705564 groups. In the MRD study, bleeding‐related TEAEs were only reported in participants treated with BI 705564 and included petechiae and vessel puncture site haematoma (n = 1), petechiae (n = 3), epistaxis (n = 2), haematoma (n = 1) and haematuria (n = 1). The frequency of bleeding‐associated TEAEs appeared to be higher in the 40‐mg SPTG compared with the other treatment groups (Table 4), which may be related to the longer duration of treatment. All bleeding‐associated TEAEs were mild in intensity and resolved without sequelae.
TEAEs were considered drug related by the investigator in 9/71 participants (13%) treated with BI 705564 vs 4/20 (20%) who received placebo in the SRD study. In the MRD study, the frequencies of drug‐related TEAEs in the BI 705564 treatment groups ranged from 0% in the 20‐mg group to 50% (4/8) in the 40‐ and 60‐mg groups and 75% (6/8) in the 40‐mg SPTG compared with 3/12 (25%) who received placebo. All bleeding‐related TEAEs were considered drug related.
In the MRD study (including the SPTG), all participants had a bleeding time below the ULN at baseline (Figure 1). In the placebo groups, bleeding time remained within the normal range for all participants, except 1. In the BI 705564 MRD groups, prolongation of bleeding time was detected during treatment in a total of 18 participants, with a maximum bleeding time of 2 × ULN observed in 2 participants in the 60‐mg group, and returned to normal in all cases during follow‐up. In addition, 1 participant in the BI 705564 40‐mg group had a transient prolongation of bleeding time after the end of treatment. In the SPTG, bleeding time was prolonged in 7/8 participants during treatment with BI 705564 40 mg; bleeding time decreased again during follow‐up in all cases, but only returned to normal in 4/7 participants. A maximum bleeding time of 3.9 × ULN was observed in 1 participant at Day 28 and this decreased to 1.7 × ULN during follow‐up.
FIGURE 1.
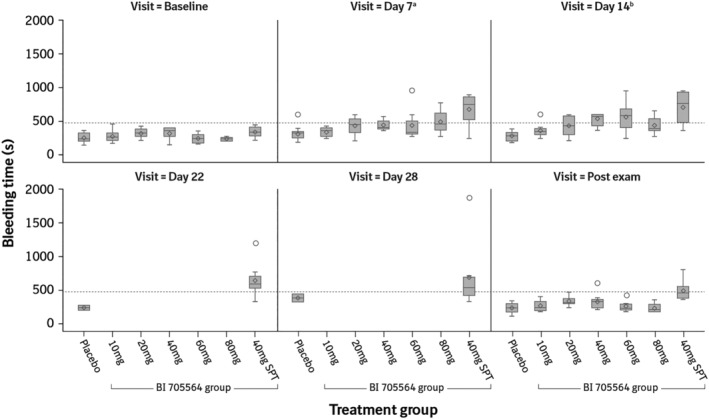
Bleeding time at different timepoints in the multiple‐rising dose study by treatment group and visit. Data are presented as median (horizontal line), interquartile range (boxes), and minimum and maximum (vertical lines). The open circles represent outliers (values less than the 25% quartile or greater than the 75% quartile by >1.5× the interquartile range). The dashed line represents the upper limit of normal (480 s). Maximum bleeding time was truncated at 600 s for the BI 705564 10, 20 and 40 mg dose groups, thus the values represented in the figure could be underestimated for these groups. aDay 8 for the SPT group; bDay 15 for the SPT group. SPT, skin prick test
In the SPTG, collagen/epinephrine‐dependent PFA‐200 was prolonged in all 8 participants treated with BI 705564 and in 1 participant treated with placebo (although this individual had elevated PFA‐200 at baseline); the other participants treated with placebo remained in the normal range. In contrast, collagen/ADP‐dependent PFA‐200 was prolonged in only 1 participant at a single timepoint and closure times appeared similar between participants treated with BI 705564 and those who received placebo.
Across all treatment groups in the SRD and MRD studies, there were no clinically relevant mean or median changes from baseline in any of the clinical laboratory parameters assessed (haematology, differentials, coagulation, electrolytes, enzymes, substrates, plasma proteins, hormones or inflammatory parameters). Notably, there were no relevant changes in thrombocyte counts or coagulation parameters (PTT, aPTT, INR or fibrinogen), nor were there any clinically significant changes in neutrophil or lymphocyte counts (mean and median values remained stable).
In the MRD study, 1 participant in the 40‐mg treatment group had ca. 250 erythrocytes/μL in the urine analysis and TEAEs of mild urinary tract infection and mild haematuria. An additional 3 participants had erythrocytes detected in the urine analysis but these were not assessed as TEAS by the investigator (ca. 5–10 erythrocytes/μL in 1 participant in the 60‐mg group; ca. 50 erythrocytes/μL in 1 participant in the 20‐mg treatment group and 1 participant in the 40‐mg group); these all resolved by the end of treatment examination at the latest. The presence of microhaematuria did not show a clear relationship with a prolonged bleeding time and no participant had faecal occult blood. There were no relevant changes in the urine analysis in the SRD study and there were no relevant drug‐related changes in vital signs or ECGs in either study.
3.3. PK
After single‐ or multiple‐dose administration, BI 705564 plasma concentrations peaked at 1–4 hours, before declining in a biphasic manner (Figure 2). In the SRD study, the geometric mean apparent terminal half‐life for BI 705564 ranged from 10.1 to 16.9 hours under both fasted and fed conditions. Food increased BI 705564 exposure (Figure 2). The single‐dose PK profile of BI 705564 was predictive of its multiple‐dose PK profile. Following multiple dosing, there was no relevant accumulation of BI 705564; in most dose groups, steady state was reached by approximately Day 2.
FIGURE 2.

Plasma concentration–time profiles of BI 705564 in the single‐rising dose study. Data are presented as geometric mean on a semi‐log scale. Blood samples were analysed for BI 705564 plasma concentrations using liquid chromatography tandem mass spectrometry, with a lower limit of quantification of 0.1 nmol/L
3.4. BTK target occupancy
After single‐dose administration of BI 705564, dose‐dependent increases in BTK TO were observed (Figure 3A). Single doses of 40 and 80 mg BI 705564 under fasted conditions, and doses of ≥20 mg under fed conditions, resulted in ≥85% average TO at 24 hours and 48 hours postdose. The time profile indicated a rapid increase in TO within 2 hours of dosing, which was maintained up to 72 hours, followed by a slow decline over the following 6 days (Figure 3A). This extended PD effect is consistent with the mechanism of a covalent BTK inhibitor. Similarly, in the MRD study, TO increased dose dependently, with doses of 20 mg and above resulting in ≥85% average TO at steady state (Figure 3B). Differences in TO above 85% may not be detectable due to variability and the limited sensitivity of the assay.
FIGURE 3.
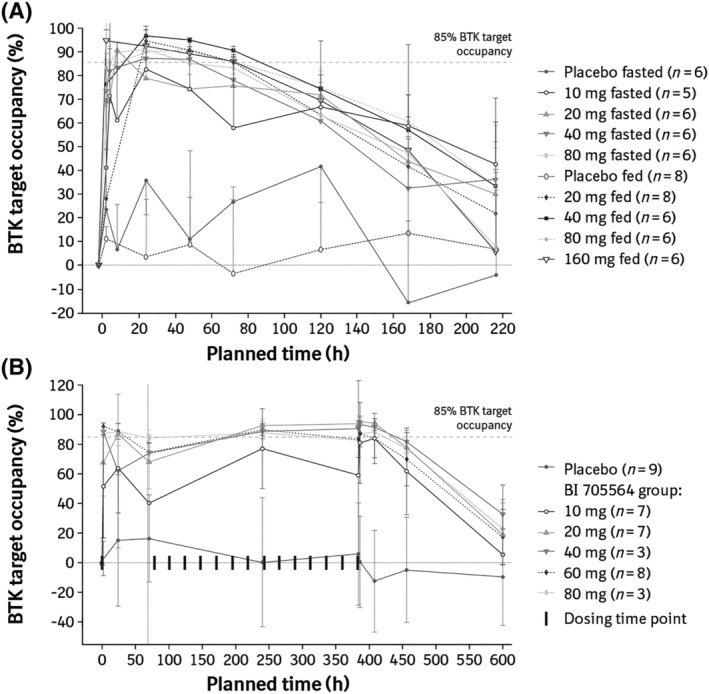
BTK target occupancy in (A) the single‐rising dose studya and (B) the multiple‐rising dose study. (A) Mean BTK TO (+SD) after single‐dose administration of BI 705564 under fasted and fed (high fat) conditions. (B) Mean BTK TO (±SD) after single‐ and multiple‐dose administration of BI 705564 under fed (standard breakfast) conditions. Data are shown for those participants who had sufficient valid data available. The most common reason for exclusion was baseline TO levels being too low for inhibition to be measured. aData for the 1‐ and 3‐mg BI 705564 groups are not presented owing to missing samples and nonvalid data. BTK, Bruton's tyrosine kinase; SD, standard deviation; TO, target occupancy
3.5. CD69 expression on ex vivo stimulated B cells
CD69 expression on ex vivo stimulated B cells was analysed after single‐dose administration of BI 705564 under fasted and fed conditions. Dose‐dependent inhibition of CD69 + B cells, with clear separation from placebo, was observed with BI 705564 doses of 20 mg and above (Figure 4). Doses between 20 and 160 mg showed maximal inhibition of 70–100% at 24 hours, followed by a decline to baseline levels on Day 6 (approximately 120 hours). The inhibition of CD697 + B‐cell frequency correlates well with TO, although inhibition appeared to decrease more rapidly than TO.
FIGURE 4.
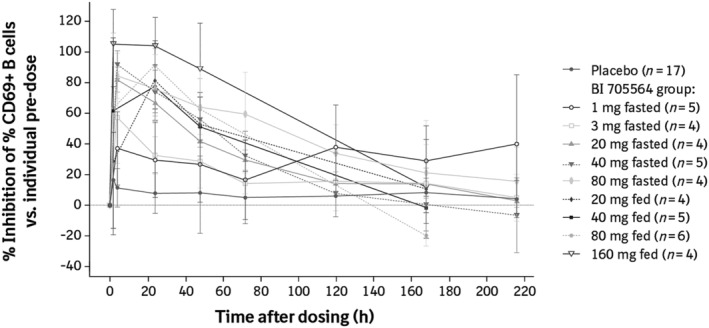
Inhibition of CD69 + B cells after administration of BI 705564 in the single‐rising dose studya. Data are presented as mean ± standard deviation. To evaluate the effect of BI 705564 on B‐cell activation, whole blood samples were stimulated with anti‐IgD, and CD69 + B cells were subsequently measured as a percentage of all CD19 + B cells using flow cytometry. aData for the 10‐mg BI 705564 groups are not presented owing to an insufficient number of participants with a predose stimulation response of >10% CD69 + B cells. IgD, immunoglobulin D
3.6. Skin prick test and biomarker analysis in allergic participants
In the SPTG of the MRD study, the median skin prick test wheal area at screening was 50 mm2 and decreased by 28 and 36% at Day 8 and Day 28, respectively, after treatment with BI 705564 (Figure 5 and Appendix B). The 2 participants who received placebo showed variable effects; however, there was no consistent reduction of the wheal area.
FIGURE 5.
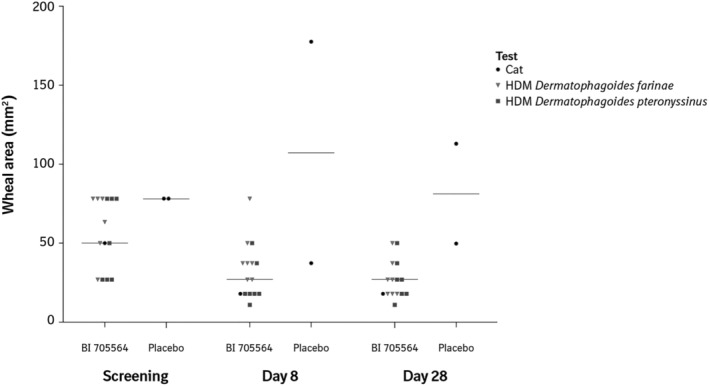
Wheal area at different visits and by treatment group (skin prick test group). Wheal area is calculated based on wheal diameter. Symbols represent individual measures by allergen: Cat, HDM Dermatophagoides farinae and HDM Dermatophagoides pteronyssinus. Horizontal bars represent medians. BI 705564 (n = 7), placebo (n = 2). HDM, house dust mite
In parallel to the skin prick test, effects on peripheral biomarkers, basophil CD63 and B‐cell CD69 expression upon ex vivo stimulation were analysed at baseline, and on Days 8 and 28 (Figure 6). The frequency of CD63 + basophils (corrected by unstimulated values) was in the range of 18–69% and was completely inhibited at Day 8 and Day 28. One placebo‐treated participant showed 18% CD63 + basophils pretreatment, which was inhibited by 32% at Day 8 and by 44% at Day 28 after treatment. Placebo responses were unexpectedly high compared with findings from other studies and prestudy assay validation work. Two participants (1 who received placebo and 1 BI 705564‐treated) showed insufficient baseline induction of CD63 expression (<10% CD63 + basophils) and were therefore excluded from the analysis. The frequency of B cells positive for CD69 expression (corrected by unstimulated values) was in the range of 11–56% and was potently inhibited at Day 8 and Day 28 by a mean of 77 and 70%, respectively. One BI 705564‐treated participant showed insufficient induction of CD69 expression at baseline (<10% CD69 + B cells) and was therefore excluded from the analysis.
FIGURE 6.
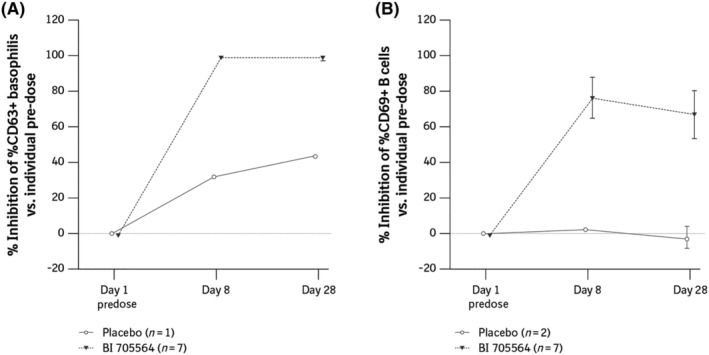
Inhibition of CD63 + basophils (A) and inhibition of CD69 + B cells (B) after administration of BI 705564 in the skin prick test group. Data are presented as mean ± standard deviation. (A) To evaluate the effect of BI 705564 on basophil activation, whole blood samples were stimulated with anti‐IgE and CD63 + basophils were analysed by flow cytometry. Percent inhibition relative to predose CD63 was calculated for Day 8 and Day 28. (B) To evaluate the effect of BI 705564 on B‐cell activation, whole blood samples were stimulated with anti‐IgD, and CD69 + B cells were subsequently measured as a percentage of all CD19 + B cells using flow cytometry. Percent inhibition relative to predose CD69 was calculated for Day 8 and Day 28. Ig, immunoglobulin
4. DISCUSSION
In the MRD study reported here, the frequency of BI 705564‐related TEAEs, in particular treatment‐related bleeding‐associated TEAEs, appeared to be dependent on dose and treatment duration; the highest frequency was seen in participants in the SPTG who received 40 mg of BI 705564 for 4 weeks, and the lowest frequency in participants who received 20 mg for 2 weeks. All bleeding‐associated TEAEs were mild in intensity and resolved without sequelae; however, the occurrence of bleeding‐related TEAEs and the increase in bleeding time indicate that there is potential risk of bleeding with BI 705564, thus there is a need for caution, particularly if studies were to be conducted in patients with a risk of decreased thrombocyte count, such as those with SLE.
The occurrence of petechiae and other mild bleeding‐associated AEs is in line with the inhibition of platelet activation that has also been described for other BTK inhibitors, such as ibrutinib. 39 The inhibition of platelet aggregation with collagen/epinephrine (but not with collagen/ADP) seen in the SPTG of our study is similar to results seen with ibrutinib; 40 since only epinephrine‐induced aggregation requires BTK action for GPVI signalling, this suggests a mechanism‐based inhibition of platelet aggregation. The selectivity of BI 705564 for BTK over Tec suggests that the inhibition of platelet aggregation is likely to be mediated by BTK. In this case, potentially all BTK inhibitors could be affected by some increased risk of bleeding‐associated TEAEs. Second‐generation selective BTK inhibitors, such as acalabrutinib, 41 , 42 , 43 tirabrutinib (ONO/GS‐4059), 44 , 45 zanubrutinib (BGB‐3111), 46 evobrutinib 47 and fenebrutinib (GDC‐0853) 48 are associated with fewer bleeding‐associated AEs than ibrutinib. 17 , 34 The role of BTK inhibition as the primary driver for inhibition of platelet aggregation is supported by results from an in vitro study comparing the inhibitory effects of ibrutinib and acalabrutinib, which found that neither inhibitor was more selective for BTK over Tec, but that the potency of suppression of platelet aggregation correlated with the potency of on‐target BTK inhibition. 17 Other studies in healthy volunteers and patients with chronic lymphoid leukaemia suggest that differences in the risk of bleeding events between ibrutinib and second‐generation BTK inhibitors may be related to off‐target inhibition of Src family kinases. 34 , 49 , 50 At high concentrations, ibrutinib irreversibly inhibits Tec and Src kinases, in addition to BTK, thus completely blocking GPVI signalling; lower doses could be sufficient to block GPVI‐mediated platelet activation without impacting platelet response to stimuli of physiological haemostasis. 34 , 49 , 50 Notably, fenebrutinib, a noncovalent, reversible inhibitor that is highly selective for BTK, was well tolerated in Phase I studies in healthy volunteers, with no reported bleeding events; 51 however, this compound failed to meet its primary efficacy endpoint in a Phase II study in patients with moderate‐to‐severe SLE. 52
Gastrointestinal AEs are also common with ibrutinib 33 , 53 and have been reported with several other BTK inhibitors. 29 , 54 , 55 In the MRD study, gastrointestinal AEs were more frequent in BI 705564 groups than the placebo group, and 3/8 participants in the SPTG reported diarrhoea. Evobrutinib and BMS‐986142 have also been associated with increases in alanine aminotransferase, aspartate aminotransferase and lipase. 55 , 56 There was no increase in alanine aminotransferase and aspartate aminotransferase observed during treatment with BI 705564.
While grade ≥3 lymphocytosis and neutropenia have been reported in patients treated with approved BTK inhibitors, 57 , 58 there were no clinically relevant changes in lymphocyte or neutrophil counts in any of the subjects treated in the studies reported here.
In the SPTG, a reduction in allergen reactivity with BI 705564 was shown by decreased wheal area and inhibition of basophil CD63 and B‐cell CD69 expression. A reduction in allergen reactivity has previously been shown following ibrutinib treatment in 6 participants with peanut or tree nut allergies 59 and in 2 patients with chronic lymphocytic leukaemia and allergies to cats and ragweed. 37 The effects of ibrutinib on wheal size were somewhat stronger than those of BI 705564 in our study; however, both studies had limited sample sizes and there were several differences between the 2 studies: Dispenza et al. 59 used food allergens for the skin prick test; ibrutinib was administered at a dose >10‐fold higher (420 mg) than the dose of BI 705564 in the current study; and the ibrutinib study did not include a placebo control. The ibrutinib study showed a trend towards smaller baseline skin prick test reactions being more likely to become completely negative during treatment. 59 The fact that our study did not include small baseline reactions (e.g. wheal areas <20 mm2) may in part explain why we did not observe conversion from positive to negative skin prick test reactions. While the reduction in wheal size with BI 705564 appeared modest, full inhibition of IgE‐stimulated CD63 + basophils was seen, similar to the effects shown with ibrutinib. 37 , 59 The basophil biomarker assay detects the activation of peripheral circulating basophils and only serves as an indicator for Fcε‐mediated target engagement, whereas the skin prick involves resident mast cells in the skin. BI 705564 is a P‐glycoprotein (P‐gp) substrate, whereas ibrutinib is a P‐gp inhibitor, 33 thus, in mast cells, while P‐gp‐mediated efflux could reduce the local concentration of BI 705564, ibrutinib could potentially reach higher concentrations. While basophils also express P‐gp, it is possible that in vivo, better enrichment may be reached in the peripheral circulation than the skin.
Inhibition of CD69 expression by BI 705564 correlated well with TO; however, inhibition decreased more rapidly over time relative to the decrease in TO. The different kinetics in the PD effects could be related to the amplification in the downstream signalling leading to CD69 expression. Consequently, inhibition of CD69 expression may require a high level of TO and after falling below this threshold the inhibition drops off quickly. Differences between TO and effects on CD69 have also been observed for other BTK inhibitors such as acalabrutinib. 31
In conclusion, BI 705564 was well tolerated in both SRD and MRD studies. It showed efficient target engagement through durable TO and inhibition of ex vivo B‐cell activation, and proof of mechanism by effects on allergic skin responses. However, the mild bleeding‐related AEs, which were probably related to inhibition of platelet aggregation by BTK inhibition, could be a concern for the clinical development of BTK inhibitors for therapeutic use in autoimmune conditions that are associated with a decreased thrombocyte count.
CONTRIBUTORS
T.L., J.S., F.M., A.G., M.F. and A.H. were involved the conception and design of the studies included in this report; F.M. and A.S. were responsible for the acquisition of the data; T.L., J.S., E.B., F.M., E.K., M.R., C.H., J.W., S.W., X.L., M.F., S.P., S.V., A.H. and J.H. were all involved in the analysis and interpretation of the results of the studies. All authors collaborated on the writing of the manuscript and made the decision to submit the manuscript for publication. We thank the volunteers who participated in the clinical studies described here.
COMPETING INTERESTS
Tobias Litzenburger, Jürgen Steffgen, Ewald Benediktus, Fabian Müller, Elliott Klein, Meera Ramanujam, Christian Harcken, Alpana Gupta, Jing Wu, Sabrina Wiebe, Xiujiang Li, Mary Flack, Steven J. Padula, Sudha Visvanathan, Andreas Hünnemeyer and Jianan Hui are employees of Boehringer Ingelheim. Armin Schultz is an employee of CRS Clinical Research Services Mannheim GmbH, contracted by Boehringer Ingelheim.
ACKNOWLEDGEMENTS
This study was funded by Boehringer Ingelheim. Editorial assistance in the preparation of this manuscript was provided by Esther Race, PhD and Leigh Church, PhD of OPEN Health Medical Communications (Choice, London, UK) and funded by Boehringer Ingelheim. Agreements between Boehringer Ingelheim and the authors included the confidentiality of the study data.
APPENDIX A. ASSESSMENT OF DOSE PROPORTIONALITY
Dose proportionality was assessed for the secondary PK endpoints. The basic model for the investigation of dose proportionality is a power model that describes the functional relationship between the dose and PK endpoints:
The model consists of a regression model applied to log‐transformed data. The corresponding ANCOVA model includes the logarithm of the dose as a covariate. Together with α′ = exp(α) and , taking natural logarithms converts this model to a linear form as follows:
Where:
Y ij : logarithm of the PK endpoint for participant j at dose level i; where i = 1, 2, …, 5, j = 1, 2, …, 8;
α: intercept parameter;
β: slope parameter;
X i : logarithm of dose I;
ε ij : random error associated with participant j at dose level i (assumed to be independent and identically normally distributed).
This equation can be fitted as a linear regression model. Perfect dose proportionality would correspond to a slope of 1. The assumption of a linear relationship between the log‐transformed PK endpoint and the log‐transformed dose was checked.
APPENDIX B. SCREENING AND DAY 28 WHEAL AREA FOR INDIVIDUAL PARTICIPANTS TREATED WITH BI 705564 40 mg AND PLACEBO IN THE SPTG
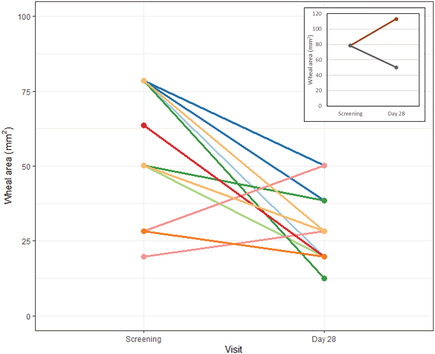
Wheal area is calculated based on wheal diameter. In the main figure, each line represents an individual participant treated with BI 705564 40 mg and tested with a single allergen (cat, HDM Dermatophagoides farinae or HDM Dermatophagoides pteronyssinus). Lines of the same colour represent the same participant tested with different allergens; the inlay represents 2 participants treated with placebo and tested with a single allergen.
Litzenburger T, Steffgen J, Benediktus E, et al. Safety, pharmacokinetics and pharmacodynamics of BI 705564, a highly selective, covalent inhibitor of Bruton's tyrosine kinase, in Phase I clinical trials in healthy volunteers. Br J Clin Pharmacol. 2021;87:1824–1838. 10.1111/bcp.14571
The authors confirm that the principal investigators for this paper are Fabian Müller and Armin Schultz and that they had direct clinical responsibility for the participants enrolled into these studies.
DATA AVAILABILITY STATEMENT
To ensure independent interpretation of clinical study results, Boehringer Ingelheim grants all external authors access to all relevant material, including participant‐level clinical study data, and relevant material as needed by them to fulfill their role and obligations as authors under the ICMJE criteria.
Furthermore, clinical study documents (e.g. study report, study protocol, statistical analysis plan) and participant clinical study data are available to be shared after publication of the primary manuscript in a peer‐reviewed journal and if regulatory activities are complete and other criteria met per the BI Policy on Transparency and Publication of Clinical Study Data: https://trials.boehringer-ingelheim.com/transparency_policy.html
Prior to providing access, documents will be examined and, if necessary, redacted and the data will be de‐identified, to protect the personal data of study participants and personnel, and to respect the boundaries of the informed consent of the study participants.
Clinical Study Reports and Related Clinical Documents can be requested via this link: https://trials.boehringer-ingelheim.com/trial_results/clinical_submission_documents.html
All such requests will be governed by a Document Sharing Agreement.
Bona fide, qualified scientific and medical researchers may request access to de‐identified, analysable participant clinical study data with corresponding documentation describing the structure and content of the datasets. Upon approval, and governed by a Data Sharing Agreement, data are shared in a secured data‐access system for a limited period of 1 year, which may be extended upon request.
Researchers should use https://clinicalstudydatarequest.com to request access to study data.
REFERENCES
- 1. Menez SP, El Essawy B, Atta MG. Lupus nephritis: current treatment paradigm and unmet needs. Rev Recent Clin Trials. 2018;13(2):105‐113. [DOI] [PubMed] [Google Scholar]
- 2. Olsen IC, Lie E, Vasilescu R, Wallenstein G, Strengholt S, Kvien TK. Assessments of the unmet need in the management of patients with rheumatoid arthritis: analyses from the NOR‐DMARD registry. Rheumatology (Oxford). 2019;58(3):481‐491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Radawski C, Genovese MC, Hauber B, et al. Patient perceptions of unmet medical need in rheumatoid arthritis: a cross‐sectional survey in the USA. Rheumatol Ther. 2019;6(3):461‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bakshi J, Segura BT, Wincup C, Rahman A. Unmet needs in the pathogenesis and treatment of systemic lupus erythematosus. Clin Rev Allergy Immunol. 2018;55(3):352‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen SJ, Lin GJ, Chen JW, et al. Immunopathogenic mechanisms and novel immune‐modulated therapies in rheumatoid arthritis. Int J Mol Sci. 2019;20:pii: E1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak‐Rothstein A. Chromatin‐IgG complexes activate B cells by dual engagement of IgM and toll‐like receptors. Nature. 2002;416(6881):603‐607. [DOI] [PubMed] [Google Scholar]
- 7. Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365(22):2110‐2121. [DOI] [PubMed] [Google Scholar]
- 8. Schwartz N, Goilav B, Putterman C. The pathogenesis, diagnosis and treatment of lupus nephritis. Curr Opin Rheumatol. 2014;26(5):502‐509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Almaani S, Meara A, Rovin BH. Update on lupus nephritis. Clin J am Soc Nephrol. 2017;12(5):825‐835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hahn BH, McMahon MA, Wilkinson A, et al. American College of Rheumatology guidelines for screening, treatment, and management of lupus nephritis. Arthritis Care Res (Hoboken). 2012;64(6):797‐808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Conley ME, Dobbs AK, Farmer DM, et al. Primary B cell immunodeficiencies: comparisons and contrasts. Annu Rev Immunol. 2009;27:199‐227. [DOI] [PubMed] [Google Scholar]
- 12. Weber ANR, Bittner Z, Liu X, Dang TM, Radsak MP, Brunner C. Bruton's tyrosine kinase: an emerging key player in innate immunity. Front Immunol. 2017;8:1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rawlings DJ, Saffran DC, Tsukada S, et al. Mutation of unique region of Bruton's tyrosine kinase in immunodeficient XID mice. Science. 1993;261(5119):358‐361. [DOI] [PubMed] [Google Scholar]
- 14. Jongstra‐Bilen J, Puig Cano A, Hasija M, Xiao H, Smith CI, Cybulsky MI. Dual functions of Bruton's tyrosine kinase and Tec kinase during Fcgamma receptor‐induced signaling and phagocytosis. J Immunol. 2008;181:288‐298. [DOI] [PubMed] [Google Scholar]
- 15. Lusková P, Draber P. Modulation of the Fcepsilon receptor I signaling by tyrosine kinase inhibitors: search for therapeutic targets of inflammatory and allergy diseases. Curr Pharm Des. 2004;10(15):1727‐1737. [DOI] [PubMed] [Google Scholar]
- 16. Smiljkovic D, Blatt K, Stefanzl G, et al. BTK inhibition is a potent approach to block IgE‐mediated histamine release in human basophils. Allergy. 2017;72(11):1666‐1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen J, Kinoshita T, Gururaja T, et al. The effect of Bruton's tyrosine kinase (BTK) inhibitors on collagen‐induced platelet aggregation, BTK, and tyrosine kinase expressed in hepatocellular carcinoma (TEC). Eur J Haematol. 2018;101:604‐612. [DOI] [PubMed] [Google Scholar]
- 18. Quek LS, Bolen J. Watson SP. a role for Bruton's tyrosine kinase (Btk) in platelet activation by collagen. Curr Biol. 1998;8(20):1137‐1140. [DOI] [PubMed] [Google Scholar]
- 19. Chalmers SA, Doerner J, Bosanac T, et al. Therapeutic blockade of immune complex‐mediated glomerulonephritis by highly selective inhibition of Bruton's tyrosine kinase. Sci Rep. 2016;6(1):26164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chalmers SA, Wen J, Doerner J, et al. Highly selective inhibition of Bruton's tyrosine kinase attenuates skin and brain disease in murine lupus. Arthritis Res Ther. 2018;20(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Evans EK, Tester R, Aslanian S, et al. Inhibition of Btk with CC‐292 provides early pharmacodynamic assessment of activity in mice and humans. J Pharmacol Exp Ther. 2013;346(2):219‐228. [DOI] [PubMed] [Google Scholar]
- 22. Gillooly KM, Pulicicchio C, Pattoli MA, et al. Bruton's tyrosine kinase inhibitor BMS‐986142 in experimental models of rheumatoid arthritis enhances efficacy of agents representing clinical standard‐of‐care. PLoS ONE. 2017;12(7):e0181782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Honigberg LA, Smith AM, Sirisawad M, et al. The Bruton tyrosine kinase inhibitor PCI‐32765 blocks B‐cell activation and is efficacious in models of autoimmune disease and B‐cell malignancy. Proc Natl Acad Sci U S a. 2010;107(29):13075‐13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim YY, Park KT, Jang SY, et al. HM71224, a selective Bruton's tyrosine kinase inhibitor, attenuates the development of murine lupus. Arthritis Res Ther. 2017;19(1):211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mina‐Osorio P, LaStant J, Keirstead N, et al. Suppression of glomerulonephritis in lupus‐prone NZB x NZW mice by RN486, a selective inhibitor of Bruton's tyrosine kinase. Arthritis Rheum. 2013;65(9):2380‐2391. [DOI] [PubMed] [Google Scholar]
- 26. Norman P. Investigational Bruton's tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Expert Opin Investig Drugs. 2016;25(8):891‐899. [DOI] [PubMed] [Google Scholar]
- 27. Pan Z, Scheerens H, Li SJ, et al. Discovery of selective irreversible inhibitors for Bruton's tyrosine kinase. ChemMedChem. 2007;2(1):58‐61. [DOI] [PubMed] [Google Scholar]
- 28. Rankin AL, Seth N, Keegan S, et al. Selective inhibition of BTK prevents murine lupus and antibody‐mediated glomerulonephritis. J Immunol. 2013;191(9):4540‐4550. [DOI] [PubMed] [Google Scholar]
- 29. Smith PF, Krishnarajah J, Nunn PA, et al. A phase I trial of PRN1008, a novel reversible covalent inhibitor of Bruton's tyrosine kinase, in healthy volunteers. Br J Clin Pharmacol. 2017;83(11):2367‐2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu H, Huang Q, Qi Z, et al. Irreversible inhibition of BTK kinase by a novel highly selective inhibitor CHMFL‐BTK‐11 suppresses inflammatory response in rheumatoid arthritis model. Sci Rep. 2017;7(1):466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barf T, Covey T, Izumi R, et al. Acalabrutinib (ACP‐196): a covalent Bruton tyrosine kinase inhibitor with a differentiated selectivity and in vivo potency profile. J Pharmacol Exp Ther. 2017;363(2):240‐252. [DOI] [PubMed] [Google Scholar]
- 32. AstraZeneca . CALQUENCE® (acalabrutinib) Prescribing Information. October 2017.
- 33. Pharmacyclics LLC . IMBRUVICA® (ibrutinib) Prescribing Information. July 2019.
- 34. Denzinger V, Busygina K, Jamasbi J, et al. Optimizing platelet GPVI inhibition versus haemostatic impairment by the Btk inhibitors ibrutinib, acalabrutinib, ONO/GS‐4059, BGB‐3111 and evobrutinib. Thromb Haemost. 2019;119(3):397‐406. [DOI] [PubMed] [Google Scholar]
- 35. Chalmers SA, Glynn E, Garcia SJ, et al. BTK inhibition ameliorates kidney disease in spontaneous lupus nephritis. Clin Immunol. 2018;197:205‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dispenza MC, Regan JA, Bochner BS. Potential applications of Bruton's tyrosine kinase inhibitors for the prevention of allergic reactions. Expert Rev Clin Immunol. 2017;13(10):921‐923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Regan JA, Cao Y, Dispenza MC, et al. Ibrutinib, a Bruton's tyrosine kinase inhibitor used for treatment of lymphoproliferative disorders, eliminates both aeroallergen skin test and basophil activation test reactivity. J Allergy Clin Immunol. 2017;140(3):875‐79.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lamb DJ, Wollin SL, Schnapp A, et al. BI 1002494, a novel potent and selective Oral spleen tyrosine kinase inhibitor, displays differential potency in human basophils and B cells. J Pharmacol Exp Ther. 2016;357(3):554‐561. [DOI] [PubMed] [Google Scholar]
- 39. Tran PN, O'Brien S. The safety of Bruton's tyrosine kinase inhibitors for the treatment of chronic lymphocytic leukemia. Expert Opin Drug Saf. 2017;16(9):1079‐1088. [DOI] [PubMed] [Google Scholar]
- 40. Kamel S, Horton L, Ysebaert L, et al. Ibrutinib inhibits collagen‐mediated but not ADP‐mediated platelet aggregation. Leukemia. 2015;29(4):783‐787. [DOI] [PubMed] [Google Scholar]
- 41. Bye AP, Unsworth AJ, Desborough MJ, et al. Severe platelet dysfunction in NHL patients receiving ibrutinib is absent in patients receiving acalabrutinib. Blood Adv. 2017;1(26):2610‐2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Byrd JC, Harrington B, O'Brien S, et al. Acalabrutinib (ACP‐196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):323‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Patel V, Balakrishnan K, Bibikova E, et al. Comparison of Acalabrutinib, a selective Bruton tyrosine kinase inhibitor, with Ibrutinib in chronic lymphocytic leukemia cells. Clin Cancer Res. 2017;23(14):3734‐3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Walter HS, Rule SA, Dyer MJ, et al. A phase 1 clinical trial of the selective BTK inhibitor ONO/GS‐4059 in relapsed and refractory mature B‐cell malignancies. Blood. 2016;127(4):411‐419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Munakata W, Ando K, Hatake K, et al. Phase I study of tirabrutinib (ONO‐4059/GS‐4059) in patients with relapsed or refractory B‐cell malignancies in Japan. Cancer Sci. 2019;110(5):1686‐1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guo Y, Liu Y, Hu N, et al. Discovery of zanubrutinib (BGB‐3111), a novel, potent, and selective covalent inhibitor of Bruton's tyrosine kinase. J Med Chem. 2019;62(17):7923‐7940. [DOI] [PubMed] [Google Scholar]
- 47. Becker A, Martin EC, Mitchell DY, et al. Safety, tolerability, pharmacokinetics, target occupancy, and concentration‐QT analysis of the novel BTK inhibitor evobrutinib in healthy volunteers. Clin Transl Sci. 2020;13(2):325‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Crawford JJ, Johnson AR, Misner DL, et al. Discovery of GDC‐0853: a potent, selective, and noncovalent Bruton's tyrosine kinase inhibitor in early clinical development. J Med Chem. 2018;61(6):2227‐2245. [DOI] [PubMed] [Google Scholar]
- 49. Nicolson PLR, Hughes CE, Watson S, et al. Inhibition of Btk by Btk‐specific concentrations of ibrutinib and acalabrutinib delays but does not block platelet aggregation mediated by glycoprotein VI. Haematologica. 2018;103(12):2097‐2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Series J, Garcia C, Levade M, et al. Differences and similarities in ibrutinib and acalabrutinib effects on platelet functions. Haematologica. 2019;104(11):2292‐2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Herman AE, Chinn LW, Kotwal SG, et al. Safety, pharmacokinetics, and pharmacodynamics in healthy volunteers treated with GDC‐0853, a selective reversible Bruton's tyrosine kinase inhibitor. Clin Pharmacol Ther. 2018;103(6):1020‐1028. [DOI] [PubMed] [Google Scholar]
- 52. Isenberg D, Furie E, Jones N, et al. Efficacy, safety, and pharmacodynamic effects of the Bruton's tyrosine kinase inhibitor, fenebrutinib (GDC‐0853), in moderate to severe systemic lupus erythematosus: results of a Phase 2 randomized controlled trial [abstract]. Arthritis Rheumatol 2109; 71. https://acrabstracts.org/abstract/efficacy‐safety‐and‐pharmacodynamic‐effects‐of‐the‐brutons‐tyrosine‐kinase‐inhibitor‐fenebrutinib‐gdc‐0853‐in‐moderate‐to‐severe‐systemic‐lupus‐erythematosus‐results‐of‐a‐phase‐2‐rando/. Accessed November 14, 2019. [DOI] [PubMed]
- 53. de Weerdt I, Koopmans SM, Kater AP, van Gelder M. Incidence and management of toxicity associated with ibrutinib and idelalisib: a practical approach. Haematologica. 2017;102(10):1629‐1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Byrd JC, Smith S, Wagner‐Johnston N, et al. First‐in‐human phase 1 study of the BTK inhibitor GDC‐0853 in relapsed or refractory B‐cell NHL and CLL. Oncotarget. 2018;9(16):13023‐13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lee SK, Xing J, Catlett IM, et al. Safety, pharmacokinetics, and pharmacodynamics of BMS‐986142, a novel reversible BTK inhibitor, in healthy participants. Eur J Clin Pharmacol. 2017;73(6):689‐698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Montalban X, Arnold DL, Weber MS, et al. Placebo‐controlled trial of an Oral BTK inhibitor in multiple sclerosis. N Engl J Med. 2019;380(25):2406‐2417. [DOI] [PubMed] [Google Scholar]
- 57. Herman SE, Niemann CU, Farooqui M, et al. Ibrutinib‐induced lymphocytosis in patients with chronic lymphocytic leukemia: correlative analyses from a phase II study. Leukemia. 2014;28(11):2188‐2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Parmar S, Patel K, Pinilla‐Ibarz J. Ibrutinib (imbruvica): a novel targeted therapy for chronic lymphocytic leukemia. P T. 2014;39(7):483‐519. [PMC free article] [PubMed] [Google Scholar]
- 59. Dispenza MC, Pongracic JA, Singh AM, Bochner BS. Short‐term ibrutinib therapy suppresses skin test responses and eliminates IgE‐mediated basophil activation in adults with peanut or tree nut allergy. J Allergy Clin Immunol. 2018;141:1914‐16.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
To ensure independent interpretation of clinical study results, Boehringer Ingelheim grants all external authors access to all relevant material, including participant‐level clinical study data, and relevant material as needed by them to fulfill their role and obligations as authors under the ICMJE criteria.
Furthermore, clinical study documents (e.g. study report, study protocol, statistical analysis plan) and participant clinical study data are available to be shared after publication of the primary manuscript in a peer‐reviewed journal and if regulatory activities are complete and other criteria met per the BI Policy on Transparency and Publication of Clinical Study Data: https://trials.boehringer-ingelheim.com/transparency_policy.html
Prior to providing access, documents will be examined and, if necessary, redacted and the data will be de‐identified, to protect the personal data of study participants and personnel, and to respect the boundaries of the informed consent of the study participants.
Clinical Study Reports and Related Clinical Documents can be requested via this link: https://trials.boehringer-ingelheim.com/trial_results/clinical_submission_documents.html
All such requests will be governed by a Document Sharing Agreement.
Bona fide, qualified scientific and medical researchers may request access to de‐identified, analysable participant clinical study data with corresponding documentation describing the structure and content of the datasets. Upon approval, and governed by a Data Sharing Agreement, data are shared in a secured data‐access system for a limited period of 1 year, which may be extended upon request.
Researchers should use https://clinicalstudydatarequest.com to request access to study data.