

Abstract
The mitochondrial gene cytochrome‐c‐oxidase subunit 1 (COI) is useful in many taxa for phylogenetics, population genetics, metabarcoding, and rapid species identifications. However, the phylum Ctenophora (comb jellies) has historically been difficult to study due to divergent mitochondrial sequences and the corresponding inability to amplify COI with degenerate and standard COI “barcoding” primers. As a result, there are very few COI sequences available for ctenophores, despite over 200 described species in the phylum. Here, we designed new primers and amplified the COI fragment from members of all major groups of ctenophores, including many undescribed species. Phylogenetic analyses of the resulting COI sequences revealed high diversity within many groups that was not evident from more conserved 18S rDNA sequences, in particular among the Lobata (Ctenophora; Tentaculata; Lobata). The COI phylogenetic results also revealed unexpected community structure within the genus Bolinopsis, suggested new species within the genus Bathocyroe, and supported the ecological and morphological differences of some species such as Lampocteis cruentiventer and similar undescribed lobates (Lampocteis sp. “V” stratified by depth, and “A” differentiated by colour). The newly designed primers reported herein provide important tools to enable researchers to illuminate the diversity of ctenophores worldwide via quick molecular identifications, improve the ability to analyse environmental DNA by improving reference libraries and amplifications, and enable a new breadth of population genetic studies.
Keywords: barcoding, biodiversity, Ctenophora, cytochrome‐c oxidase, eDNA, metabarcoding, primers
1. INTRODUCTION
Ctenophora is a phylum of marine, gelatinous animals with ~200 named species (Mills, 2017) that are mostly planktonic. Unique genetic and morphological traits separate ctenophores from other gelatinous plankton and from cnidarians with which they are sometimes associated (Dunn et al., 2015). Ctenophores, also called comb jellies, are found throughout the ocean, from both poles to the equator and from the surface to the deep‐sea (Harbison et al., 1978), with the deepest ctenophore observed at over 7000 m depth (Lindsay & Miyake, 2007). Many species are common and well‐studied, particularly coastal species such as Mnemiopsis leidyi, or to an extent, Pleurobrachia pileus. However, most deep‐living ctenophores remain undescribed because specimens are delicate, difficult to access, and are often damaged during collection (Haddock, 2004). The use of remotely operated vehicles (ROVs) and specialized sampling equipment has expanded our ability to observe and collect ctenophores in the deep‐sea during the last 30 years (Haddock et al., 2017). Morphological investigations of specimens collected from the deep sea suggest that we have only begun to reveal the remarkable diversity within this phylum. However, there are few taxonomic experts on ctenophores, and morphological identifications often are stymied by damaged specimens, cryptic morphology, and poor preservation in all fixatives. Molecular data can provide relatively quick identifications, especially for taxa like ctenophores, although polymorphic loci and good reference libraries are critical to achieve this goal.
Sequence data from the nuclear 18S ribosomal gene provide a molecular phylogenetic framework for the broad relationships within ctenophores (Podar et al., 2001; Simion et al., 2015), and transcriptomes from a handful of species allow for more in‐depth studies of some representative diversity (Simion et al., 2017). However, the 18S rDNA gene fragment is highly conserved, and the phylogenies often do not effectively discriminate between many species and closely related genera, particularly in groups such as Lobata. For example, some genera, such as Bathocyroe, Eurhamphaea, Deiopea and Kiyohimea, have nearly identical 18S rDNA sequences, showing the limitations of the utility of the 18S fragment with respect to species delineation (Haddock et al., 2021). The “barcoding” mitochondrial cytochrome‐c‐oxidase subunit‐I (COI) sequence fragment is typically useful for species identification and delimitation because it is (usually) easily amplified with universal primers, variable between species, and easy to align between divergent taxa (DeSalle & Goldstein, 2019). Degenerate PCR primer sets are available to amplify COI from many taxa, spanning bacteria to humans (Folmer et al., 1994; Geller et al., 2013; Leray et al., 2013; Siddall et al., 2009). While the COI barcode locus can be successfully amplified for many taxa, it is often difficult for non‐model organisms (Vrijenhoek & Waples, 2012). For example, ctenophores have extremely high rates of mitochondrial evolution, are rich in adenine (A) and thymine (T) residues, and have variable gene order in the mitochondrial genome, even within a genus (Arafat et al., 2018; Kohn et al., 2012; Pett et al., 2011; Schultz et al., 2020). A consequence of high mitochondrial variability is that common primers are often poorly suited to amplify the COI fragment. Unsuccessful amplification of ctenophores by commonly used barcoding primers has a number of important ramifications including: a lack of quick and easy molecular identifications that results in difficulties for revealing diversity, few ctenophore sequences in public databases, and a deficiency of easily amplified phylogenetic and population‐genetic markers. The lack of COI primers and the resulting paucity of ctenophore sequences available in public repositories also hampers our understanding of the role of ctenophores in ocean ecology. Despite their seemingly low nutrient content, they are frequent prey for a large range of animals (Choy et al., 2017; Thiebot & McInnes, 2020; Yeh et al., 2020).
Metabarcoding and eDNA studies are powerful tools to assess community diversity, ecosystem monitoring, and function (Eble et al., 2020), but ctenophores are often excluded because they are either missing from reference libraries or are unable to be amplified because of divergent sequences. Some studies have noted the presence of ctenophores in zooplankton metabarcoding analyses based on 18S rDNA fragments (Günther et al., 2018; López‐Escardó et al., 2018; Preston et al., 2020; Schroeder et al., 2020; Yeh et al., 2020). However, since the fragment of 18S lacks resolution to discriminate between most species of ctenophores, often all members of the entire phylum are lumped together, obscuring diversity (Günther et al., 2018; López‐Escardó et al., 2018; Preston et al., 2020; Schroeder et al., 2020; Yeh et al., 2020). In studies that used multiple loci in a metabarcoding framework, a significant proportion of 18S sequences were from ctenophores, yet COI often failed to detect any (Djurhuus et al., 2018; Günther et al., 2018), or the ctenophore sequences had poor taxonomic assignments so the results were not informative (Lacoursière‐Roussel et al., 2018; Leray & Knowlton, 2015; Pitz et al., 2020).
For this study, we designed multiple primers to amplify COI from all major clades of ctenophores, including many deep‐living, undescribed species. We applied those primers across the phylum and tested taxonomic assignments for various groups. Finally, as a case study, we used our newly generated sequences as a library for an eDNA study along the eastern Pacific coast, and provided species‐level resolution for taxonomic assignments of ctenophores.
2. MATERIALS AND METHODS
2.1. Sample collection
Ctenophores were primarily collected by remotely operated vehicles (ROVs), blue‐water SCUBA diving, and midwater trawls, along with contributions from collaborators around the globe (Table S1). Tissue samples for genomic DNA or RNA were flash frozen in liquid nitrogen, then stored at –80°C. Genomic DNA was isolated from frozen samples using the DNeasy DNA Blood and Tissue kit (Qiagen) or the Monarch Genomic DNA Purification Kit (New England Biolabs) according to the manufacturers’ instructions. When possible, multiple individuals within a species from different localities and depths were separately amplified and sequenced.
2.2. Primer design
Initial ctenophore COI primers were designed based on COI sequences from our unpublished ctenophore transcriptome data, published mitochondrial sequences of Mnemiopsis leidyi (GenBank accession nos. NC016117, JF760210), Pleurobrachia pileus (GenBank accession no. JF760211), and from the ctenophores which we could successfully amplify with universal Folmer primers (Folmer et al., 1994; Figure 1). RNA extractions, transcriptome sequencing, and analyses were conducted as previously described (Francis et al., 2013). We designed primers for the mitochondrial COI gene with Primer3 (Untergasser et al., 2012) within Geneious Prime (v. 2020.2.3, www.geneious.com), using PrimerQuest (www.idtdna.com), or by eye based on nucleotide alignments. Primer design was an iterative process, with multiplexed PCR assays suggesting which variants and combinations of forward and reverse primers worked best. Primer positions were numbered relative to the sites as they occur in M. leidyi (JF760210; Pett et al., 2011), as indicated in Figure 1.
FIGURE 1.
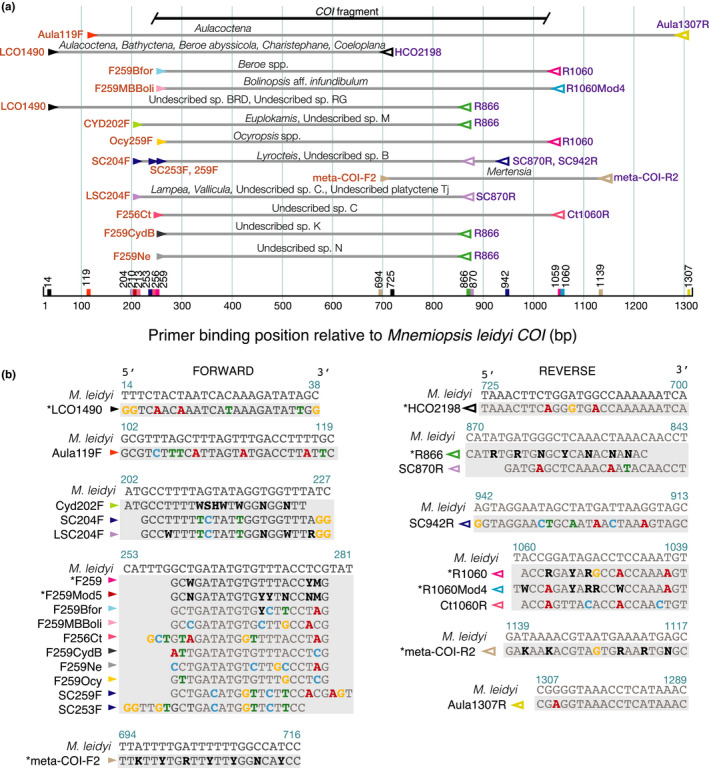
(a) Primer binding position, taxa amplified, and PCR reaction conditions for each primer set relative to published Mnemiopsis leidyi sequence, including the “Folmer” region and fragment sequences were trimmed to for phylogenetic analyses (b) primer sequences and position used to amplify and sequence all groups of ctenophores including LCO1490/HCO2198 (Folmer et al., 1994) and meta‐COI‐F2/meta‐COI‐R2 (Pett et al., 2011) (not used in this study). Differences from M. leidyi are indicated in bold and coloured according to base. Primers marked with * are more universal
2.3. Amplification and sequencing
Fragments of 18S rDNA were amplified using primers MitchA and MitchB (Medlin et al., 1988). Both 18S and COI fragments were amplified using Phusion High‐Fidelity PCR Master Mix with HF buffer (New England BioLabs) in a Veriti PCR thermal cycler (Life Technologies). PCR conditions were: 98°C for 30 s; 35 cycles of 98°C for 30 s, 50°C for 10 s, and 72°C for 10 s; 72°C for 5 min. Gene fragments were sequenced bidirectionally with PCR primers and the BigDyeTerminator v3.1 sequencing kit according to the manufacturer's protocol and analysed on a 3500xL Genetic Analyser (Life Technologies).
2.4. Statistical methods
Sequence fragments were assembled, edited, and aligned with MUSCLE (Edgar, 2004) within Geneious Prime. The COI alignment was translated with the Mold, Protozoan, and Coelenterate translation mitochondrial code table to ensure that the sequences were aligned in the correct reading frame based on annotated GenBank sequences and that no stop codons were present. Where available, we included published sequences for ctenophores for the COI fragment in phylogenetic analyses (Figure 2).
FIGURE 2.
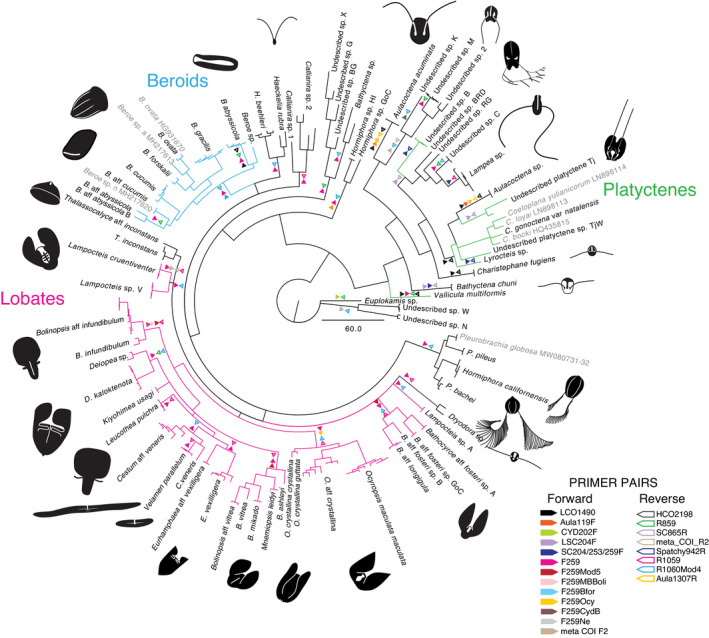
Bayesian phylogeny for 765 bp COI fragment for all ctenophores sequenced with successful primer pairs indicated by coloured triangles. Closed triangles indicate forward and open indicate reverse direction primers. Multiple triangles indicate multiple successful primer pairs. Branches coloured by group including; Lobates (pink), Beroids (blue), and Platyctenes (green)
We used jModelTest (Posada & Crandall, 1998) within Geneious to select the best evolutionary model. We estimated phylogenies with MrBayes (v3.2.7a, Ronquist et al., 2012) and IQ‐Tree 2 with 1000 bootstrap replicates (Minh et al., 2020) for all ctenophores available based on the most appropriate models selected by the AIC (Akaike, 1974). Bayesian phylogenies estimated with MrBayes included multiple runs of 5 × 106 generations with a 10% burnin, and six chains, that were sampled and printed every 1000 generations. Convergence was determined with TRACER (v1.7, Rambaut et al., 2018) and by comparing topologies of multiple runs.
Phylogenies were visualized with FigTree (v1.4.4, tree.bio.ed.ac.uk/software/figtree/). To illuminate within‐order diversity, alignments of lobate species for both COI and 18S fragments were analysed separately with the same parameters that had been used for the full alignment, and both Bayesian and maximum likelihood support values were reported on the phylogeny. The lobate‐specific phylogenies were rooted with the cydippid Pleurobrachia bachei as the outgroup.
To assess diversity within sequenced ctenophores for COI, we calculated percent general time reversible (GTR) distance within and between molecular operational taxonomic units (MOTUs). We considered a clade a MOTU when it was ≤4% GTR distance. Base composition was calculated with MegaX (v10.0.5, Kumar et al., 2018) and pairwise distances were calculated in Geneious Prime. We calculated the number of parsimony‐informative sites within the Lobata for both COI and 18S with DnaSP (v6.12.01, Rozas et al., 2017). We tested for saturation of observed proportions of transitions and transversions versus GTR distance among all MOTUs with the software DAMBE7 (v7, Xia, 2018). We also examined saturation, transitions, and transversions in five other loci from the published mitochondrial genomes of eight species of ctenophores with DAMBE7 to determine if the COI fragment was an outlier.
2.5. Environmental samples and metabarcoding
In 2012 on a transit from Monterey Bay, CA, USA to the mouth of the Gulf of California, Mexico, Pitz et al. (2020) sampled 15 offshore stations with 100 m‐depth vertical net tows which they sequenced for 18S and COI metabarcoding fragments. Primers mlCOIintF (Leray et al., 2013) and jgHCO2198 (Geller et al., 2013) were used to amplify the metabarcoding libraries for a ~300 bp fragment of COI. The authors found 9105 COI sequences from thirteen of the stations that were assigned only to the family level or higher (K. Pitz, personal communication). To illustrate how a more complete reference library can affect metabarcoding sequence assignments, we queried the COI sequences previously designated as ctenophores by the Banzai pipeline from Pitz et al. (2020) with our ctenophore‐specific library. Data for stations where no ctenophore sequences were recovered were not reported. We plotted original family and new species assignments based on the presence or absence of reads in each sample in Rstudio (Rstudio Team, 2015) with ggplot2 (Wickham, 2016).
3. RESULTS
In addition to 15 species with published data, we successfully amplified and sequenced 174 individuals of 72 distinct molecular operational taxonomic units (MOTUs). We included published species of Coeloplana, Pleurobrachia globosa, Beroe ovata, and Beroe sp. a and sp. n that we either did not have access to or were undescribed species (accession numbers in Figure 2). We defined a MOTU as a sequence or group of sequences that were ≤4% GTR divergent. Phylogenetic analyses of the resulting COI sequences revealed high diversity and a number of cryptic species within all groups, but especially within Lobata, which had not been evident from previous 18S rDNA sequence analyses. COI and 18S sequence fragments were deposited in GenBank with accession numbers (MW647008‐75, MW735695‐832, MW804236‐67, MZ391814) (Table S1). Taxon‐optimized primer sequences and positions are indicated in Figure 1.
3.1. Primer design and combinations
Successful primer pairs varied by taxon (Figure 1 and branches of Figure 2). Primer combinations amplified a range of fragment lengths (Figure 1), so we trimmed the ends of the alignments used to generate phylogenies, to exclude missing data. A few sets of primers such as F259/1060R worked well for many species, and reverse primer R866 was successful for many species of cydippids (Figure 2). However, amplification of many genera such as Euplokamis, Ocyropsis, Beroe, and Lampea required customized primers. Some primers designed for one species also worked for other closely related taxa, such as Bfor259F, which was specific to B. forskalii but also amplified most Beroids. For several species, multiple combinations of primer pairs successfully amplified the COI fragment (Figures 1 and 2).
3.2. Phylogenetics and species delimitation
Combined with published data, the COI alignment included 179 sequences representing 77 molecular operational taxonomic units (MOTUs). It was trimmed to a 765 base‐pair (bp) fragment (Figure 1) and we used a GTR+i+Γ model for phylogenetic analyses based on jModelTest results (Figure 2). Mitochondrial loci in ctenophores are generally rich in the bases adenine (A) and thymine (T) (Pett et al., 2011). Average base composition for all ctenophores sequenced were ~50% A, 21% T, 16% C, and 13% G, and the vast majority of changes were at the third codon position.
In order to examine saturation between and within taxa, we plotted the proportion of transitions and transversions versus GTR distance among all sequences (Figure 3a). For ctenophores sequenced, we found that the proportion of transversions was greater than that of transitions, which is in contrast to the general rule (for vertebrate and invertebrate mitochondrial DNA), where transitions are often two‐fold more common than transversions (Xia et al., 1996). Transitions were saturated for species that were more than ~30% distinct and transversions were saturated for species that were more than ~25% distinct. We saw the same pattern for the other mitochondrial loci that we could align without the presence of stop codons, including COII, COIII, CytB, ND4 and ND5 (Figure S1). In these genes as well, transversions were more common than transitions, and there were high levels of saturation.
FIGURE 3.
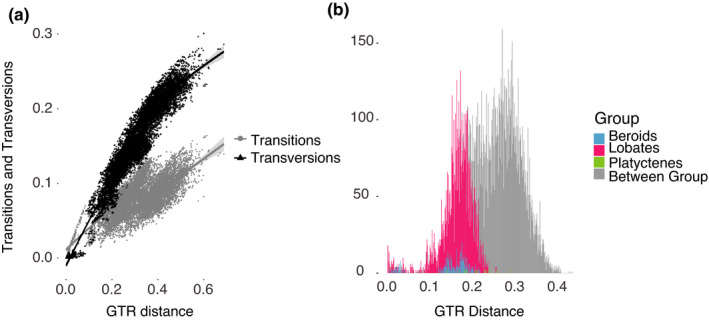
(a) Transitions and transversions versus GTR sequence divergence with best‐fit lines and confidence intervals and (b) Pairwise sequence distance (GTR) within and amongst all ctenophore species sequenced for the COI fragment
The high levels of saturation between distantly related species (Figure 3a) were reflected in the poor resolution of deeper level relationships amongst taxa in the Bayesian and maximum likelihood phylogeny trees. Phylogenetic relationships for COI among all the ctenophores were therefore portrayed in a tree without support values (Figure 2). In order to illustrate levels of diversity within the phylum, we also plotted within and between pairwise GTR distances for all individuals sequenced (Figure 3b). In comparison to other phyla, distances between specimen sequences were relatively large, ranging between from 0%–43%, and showed four peaks. The first peak represented within‐species variability, around 0%–4% GTR, although we had few within‐species samples for this estimate. The second small peak centred at 10% GTR distance, was represented by closely‐related species complexes. The two largest peaks were represented by the majority of our sequencing efforts and occurred at ~17% (within group; pink, blue, or green) and 30% GTR distance among different orders (grey) of ctenophores (Figure 3b).
The 18S alignment of the Lobata included 82 sequences for 29 MOTUs but included only 47 parsimony‐informative sites, was in sharp contrast to the high COI variability. We trimmed the fragment to 1780 bp and had a TrN+i+Γ model determined with jModel Test for phylogenetic analyses. Phylogenetic results showed little differentiation within and even between many genera (Figure 4a). Posterior probabilities and bootstrap values in the 18S tree were low for most nodes between species. Therefore, most phylogenetic relationships in the Lobata were unresolvable with 18S alone (Figure 4a). Distinguishing taxa using the COI fragment limited to the Lobata was more successful, since the marker is more variable than 18S. The COI fragment had 199 parsimony‐informative sites between 27 MOTUs (Figure 4b). Although deeper level relationships amongst more distantly related taxa were still not well supported and some saturation was evident (Figure 3a), analyses revealed high support for several cryptic species complexes and new MOTUs. Conversely, other species such as Ocyropsis maculata maculata and Beroe cucumis were revealed to be truly cosmopolitan with worldwide or at least ocean‐basin‐wide distributions (Figure 4b).
FIGURE 4.
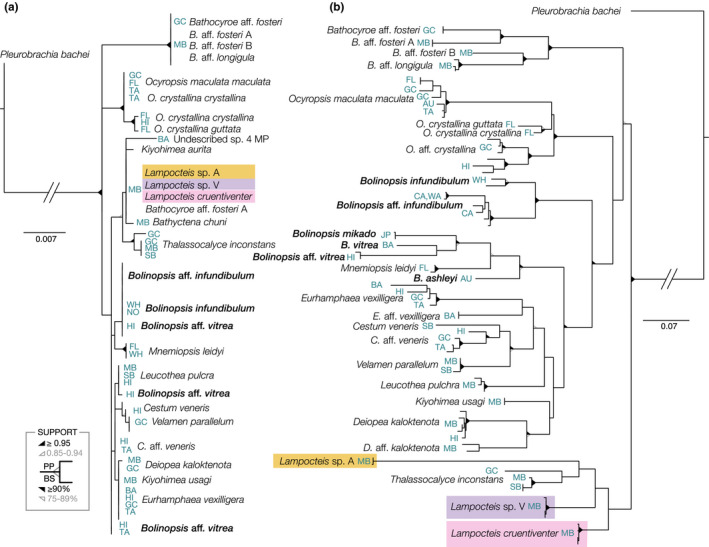
Bayesian and maximum likelihood estimated phylogenies for fragments of (a) 18S (1780 bp, TrN+i+Γ) and (b) COI (765 bp, GTR+i+Γ) limited to Lobate ctenophores. Bolinopsis species are bolded, showing the scattered distribution with 18S. Coloured backgrounds indicate the species of Lampocteis which are identical with 18S but form distinct clades with COI. Both likelihood and Bayesian support values are indicated by triangles on the nodes, with unlabelled nodes having low support (<75). Locations (Table S1) are indicated in the blue labels including: (AU) Australia, (BA) Bahamas, (CA) California, (FL) Florida, (GC) Gulf of California, (HI) Hawaii, (JP) Japan, (NO) Norway, (TA) Tahiti, (WA) Washington, (WH) Woods Hole, (SB) Santa Barbara and (MB) Monterey Bay
3.3. Metabarcoding ctenophores
Pitz et al. (2020) sampled a transect with 15 stations from California to Mexico with 100‐m vertical net tows in 2012. They amplified whole plankton samples for the COI metabarcoding fragment and found 9105 sequences that were identified as Ctenophores but were only able to assign taxonomy to the family level using the “Banzai pipeline”. In order to illustrate the utility and importance of a good reference library, we used our newly assembled ctenophore sequence library and assigned ctenophore sequences from the five nominal families that Pitz et al. (2020) found ten different species. We reduced species identifications to a presence/absence matrix (Figure 5).
FIGURE 5.
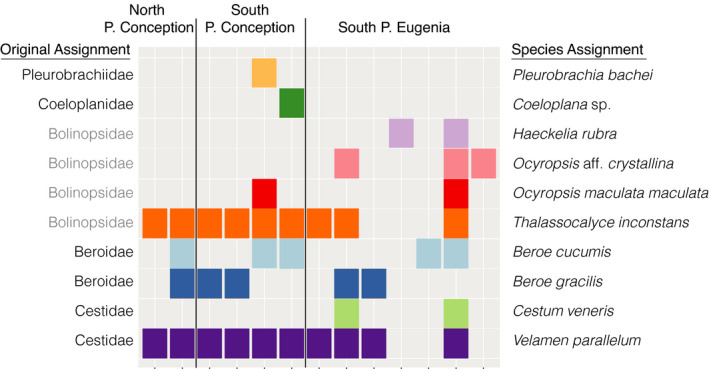
Presence and absence matrix of ctenophore family and species assignments for a 311 bp fragment of COI from 100 m depth net tows collected in 2012 along a transect that spanned from Central California, US, to Southern Baja California, Mexico from Pitz et al. (2020). Grey family names indicate sequences that were misassigned
4. DISCUSSION
4.1. Phylogenetic analyses
Newly‐designed primers combined with a few existing primer pairs enabled us to amplify ~700–1000 bp of the mitochondrial COI fragment for 77 MOTUs from all groups of ctenophores (Figure 2). The COI phylogeny illuminated a great deal of species‐level diversity that was not evident for the 18S fragment, within and between groups. However, the levels of divergence and saturation for mitochondrial genes among ctenophores are some of the highest in the Metazoa (Lavrov & Pett, 2016). Despite high levels of mitochondrial variation, within‐species diversity of ctenophores rarely exceeded 4% (Figure 3b). One example of high within‐species variation was B. infundibulum, which spanned a huge geographic range from the Northern Pacific to the Northern Atlantic Ocean with many geographic barriers within its range (Johnson et al., In preparation). Saturation within the phylum, especially at third codon positions, obscured many higher‐level relationships and resulted in basal polytomies or poorly supported deep nodes. However, here we were primarily interested in species delimitation rather than phylogenetic resolution. High levels of saturation and poor phylogenetic resolution were not surprising given the large amount of diversity we saw for the COI fragment in ctenophores. Phylogenetic analyses based on transcriptomic data provide better resolution within the phylum (S. Haddock, personal communication).
Curiously, we found a higher proportion of transversions than transitions for the COI fragment among ctenophores (Figure 3a). For protein‐coding loci, transitions are often more common than transversions because they usually result in synonymous mutations and involve the exchange of bases of similar shapes (Xia et al., 1996). It is plausible that the proportion of transitions within our data were overly saturated and additional mutations were obscured. The mitochondrial genomes of ctenophores are enriched with A/T residues, which also could contribute to a greater incidence of transversions (purines⇄pyrimidines) than transitions. Plots of other mitochondrial loci also revealed the same pattern, where transitions saturated quickly, often at ~20% GTR distance, and there were higher proportions of transversions (Figure S1). Despite high levels of saturation, closely related species and groups were well‐supported, especially within the Lobata (Figure 4b). Although saturation among divergent taxa resulted in poor phylogenetic resolution, and it was difficult to make strong conclusions based on the COI fragment alone, COI sequences were easily attributable to MOTUs, and proved useful for species delimitation.
4.2. The Lobata
When the COI phylogeny was limited to the lobate ctenophores, which form a monophyletic clade (including Thalassocalyce and Cestids; Podar et al., 2001), phylogenetic resolution was improved. The COI fragment revealed that many genera were polyphyletic although many relationships had low support so we were unable to draw strong conclusions (Figure 4b). Analysis of multiple isolates of Bolinopsis suggested that the genus is polyphyletic. Tropical species of Bolinopsis mikado (Japan), B. vitrea (Bahamas), B. aff. vitrea (Hawaii) and B. ashleyi (Australia) all formed a well‐supported clade with Mnemiopsis leidyi. The temperate species Bolinopsis infundibulum (Atlantic Ocean) and an undescribed Bolinopsis MOTU from the eastern Pacific were in a distinct and well‐supported clade (Figure 4b; and Johnson et al., In preparation). This was in contrast to the 18S fragment where species within Bolinopsis genus were undifferentiated, with the exception of a few individuals of B. aff. vitrea collected near Hawaii and Moorea (Figure 4a). Clearly, collections from more localities, along with transcriptomes and genomes, are critical in understanding phylogenetic relationships among ctenophores, but the COI gene provides a useful starting point.
4.3. Morphological and molecular concordance
Mitochondrial sequencing reinforced the designation of several undescribed species that returned one 18S MOTU, but differed morphologically or ecologically (Figure 4). Although we hesitate to make conclusions about deeper‐level phylogenetic relationships because of high degrees of saturation among more distantly related taxa, we found several MOTUs that confirmed morphological distinctions. For example, for species that were unresolved with the 18S fragment, we found support with the COI fragment for subtle morphological differences between undescribed species of Deiopea, Kiyohimea, and Eurhamphaea and within the genera Lampocteis and Bathocyroe. Specifically, L. cruentiventer (Figure 6a) and Lampocteis “sp. V” (Figure 6b), differed by ~10% GTR distance and were segregated by depth. A third suspected species within the genus was a colour morph called Lampocteis “sp. A,” (Figure 6c) which differed from the other two MOTUs by ~17% GTR distance, giving strong support for a distinct species.
FIGURE 6.
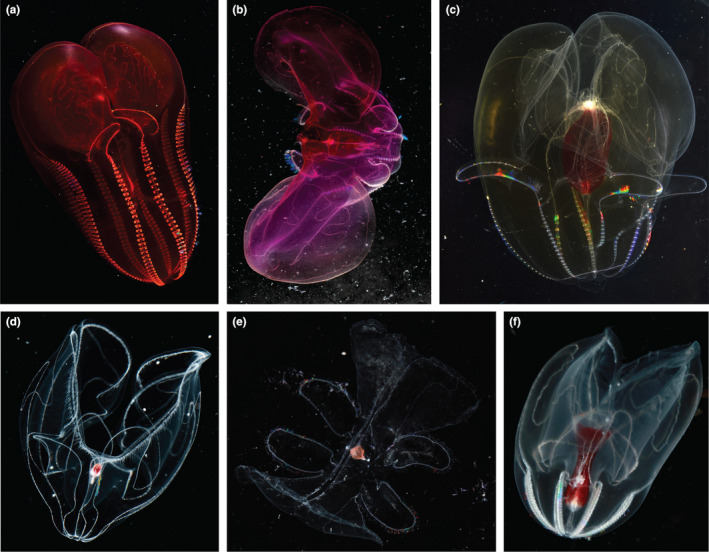
Images and known depths in meters of specimens of (a) Lampocteis cruentiventer (~250–1500 m) (b) Lampocteis sp. “V (~1500–3000 m), (c) Lampocteis sp. “A”, (~500–2800 m), (d) Bathocyroe aff. fosteri “A” (560–3572 m, small red gut), (e) Bathocyroe aff. fosteri “B” (>3000 m, peach gut), (f) Bathocyroe aff. longigula (1900–2858 m)
The COI fragment revealed three distinct Bathocyroe MOTUs collected from the central California coast, and one from the Gulf of California, Mexico, in contrast to only a single MOTU returned by the 18S fragment (Figures 4a,b and 6d–f). We could not definitively assign any Bathocyroe MOTUs to any of the three described species, B. fosteri (Madin & Harbison, 1978) from the Gulf of Mexico, B. paragaster (Ralph & Kaberry, 1950) from the South Western Pacific, and B. longigula (Horita et al., 2011) from shallow waters in Japan. We collected Bathocyroe aff. fosteri “A” (Figure 6d) from the central California coast, which matched the description of B. fosteri. Bathocyroe aff. longigula (Figure 6f), also from the central California coast, resembled B. longigula somewhat, in having an elongated stomodaeum, but it lacked any coloured spots along the meridional canals and the gut appeared wider than that of B. longigula. In addition, the B. aff. longigula was mainly found from ~2000 to 3000 m depth and B. longigula from Japanese waters was collected at the surface (although deep species are known to be occasionally upwelled to the surface in that region). Off the central California coast, B. aff. longigula (Figure 6f) and B. aff. fosteri “A” (Figure 6d) had overlapping depth ranges and distributions, but had small distinctions in gut shape and were the most divergent from one another molecularly (15% GTR). The third MOTU from the central California coast, Bathocyroe aff. fosteri “B” (Figure 6e), had a distinct gut shape and colour from all the other MOTUs, was relatively rare, and was only collected below 3000 m depth. Unfortunately, specimens from type localities of described species were unavailable for sequencing so it is unclear if any of the MOTUs we sequenced match those already described or new to science. However, with our new primers researchers worldwide can now amplify species of Bathocyroe and help to confirm species identifications.
Previous work posited that the large ctenophore Deiopea kaloktenota (Chun, 1880) might be a juvenile form of Kiyohimea usagi, in spite of the morphological differences between the two species (Matsumoto & Robison, 1992). By sequencing the COI fragment we resolved the relationships between Deiopea and Kiyohimea where rather than just one clade, we found three clades from central coast California, including two distinct Deiopea sp. and K. usagi. A single specimen collected offshore of the Hawaiian Islands also was the same MOTU as one of the Deiopea from central coast California, showing this species has high dispersal capabilities and a broad oceanic distribution (Figure 4b). Specimens of D. kaloktenota, which were described from the Mediterranean, and K. aurita (Komai & Tokioka, 1940) from Japan were unavailable for sequencing, so it is unclear again whether one or both species of Deiopea from Monterey Bay are undescribed.
4.4. Population subdivision of Ocyropsis
The high levels of within‐species diversity of ctenophores make the COI fragment, and possibly mitochondrial sequencing in general, a good marker for population genetics. Mitochondrial sequencing revealed many morphologically cryptic but genetically distinct species. Within Lobata, phylogenetic relationships were better supported and many cryptic species complexes were revealed for those with both sympatric distributions and allopatric isolation (Figure 4b).
The genus Ocyropsis was a good example. Currently, Ocyropsis contains four robustly described subspecies including O. crystallina crystallina and O. maculata maculata, which have sympatric distributions and were both described from the Atlantic Ocean (near Europe), but are thought to be distributed worldwide. The COI fragment for specimens of O. maculata maculata sequenced from Florida, Australia, and Tahiti were closely related and all represented one MOTU. In contrast, specimens of O. crystallina crystallina sequenced from Florida, the Gulf of California, and Hawaii, represented three distinct lineages. Each species of Ocyropsis has a subspecies based on coloration, originally described from the Gulf of Mexico. Of these, we sequenced O. crystallina guttata from Florida and confirmed that it was distinct from O. crystallina crystallina. Unfortunately, specimens from other regions were not available for sequencing. Regardless, the Ocyropsis genus represents just one interesting dichotomy where the subspecies O. crystallina crystallina may have many genetic and morphological distinctions that lead to many named subspecies, yet O. maculata maculata may have a nearly worldwide distribution (Johnson et al., In preparation).
4.5. eDNA and metabarcoding
In order to explore biodiversity, ecosystem function, and population genetics, a high‐resolution marker with a good reference library is critical. As of the writing of this manuscript there were COI sequences (including mitochondrial genomes) for 15 species of ctenophores on GenBank and BOLD databases. The paucity of mitochondrial sequencing data is mostly a result of the failure of commonly used barcoding primers to amplify the highly divergent phylum. In a study that took 100 m depth vertical net tows at stations from Monterey Bay, CA to the Gulf of California, Mexico, Pitz et al. (2020) successfully sequenced at least some ctenophores for COI and 18S, but was only able to assign taxonomy mostly to the family level, that were often incorrect. For example, all lobate ctenophores identified were mistakenly assigned to the family Bolinopsidae (Figure 5). When net tow data were queried against sequences generated by the new ctenophore primers, we were able to assign thousands of sequences to 10 distinct species of ctenophores, illuminating how diversity changed over two important biogeographic barriers, Point Conception and Punta Eugenia (Figure 5).
In conclusion, the primers designed herein will enable researchers worldwide to amplify and sequence ctenophores for the mitochondrial COI locus for quick identification, as a population genetic marker, and for metabarcoding studies. With the publication of our sequences amplified from our new primers, researchers can increase our understanding of marine biodiversity. As we continue to sequence a broader diversity of ctenophores, new sequence information will help with species identification and descriptions, and along with genomic data will also provide a better understanding of relationships within Ctenophora.
AUTHOR CONTRIBUTIONS
Lynne M. Christianson designed the study, primers, collected, amplified and sequenced most of the ctenophores, and helped draft the manuscript. Shannon B. Johnson helped to amplify and sequence some of the ctenophores, performed statistical analyses, and drafted the manuscript. Darrin T. Schultz provided valuable specimens, transcriptome data, mitochondrial sequences, and edited the manuscript. Steven H. D. Haddock helped conceive the study, provided specimens, funding, transcriptome data, performed statistical analyses and helped to draft the manuscript.
OPEN RESEARCH BADGES
This article has earned an Open Data badge for making publicly available the digitally‐shareable data necessary to reproduce the reported results. The data is available in GenBank with accession numbers (MW647008‐75, MW735695‐832, MW804236‐67, MZ391814).
Supporting information
Fig S1
Table S1
ACKNOWLEDGEMENTS
We gratefully acknowledge the crew and pilots of the R/V Western Flyer and ROV Doc Ricketts, R/V Rachel Carson and ROV Ventana for their help with collecting these delicate specimens and assistance with blue‐water diving. We would like to thank Warren Francis for generating many of our transcriptome data and for providing helpful comments on the manuscript. April Woods and Maile Prickett were instrumental in PCR and sequencing help, and we thank Kyra Schlining and Susan Von Thun for support at sea. We would also like to thank Jacob Winnikoff who created some of the silhouettes for Figure 2, helped collect specimens, and for his constructive comments on the manuscript. Numerous specimens were donated by Bruce Robison, Rob Sherlock, Kristine Walz, David Clague, Lonny Lundsten, William Browne, Stacy Kim, Keith Bayha, Larry Madin, Wyatt Patry (Monterey Bay Aquarium), California Academy of the Sciences, and Ocean Genome Legacy Centre to increase our sampling breadth. We also graciously acknowledge Katie Pitz, who provided ctenophore sequences from her metabarcoding library. We also benefitted from the constructive input of three anonymous reviewers who significantly improved our manuscript. This work was funded by the David and Lucile Packard Foundation, the National Science Foundation (NSF‐DEB 1542679 to SHDH) and the National Institutes of Health (R01‐GM087198).
Christianson, L. M. , Johnson, S. B. , Schultz, D. T. , & Haddock, S. H. D. (2022). Hidden diversity of Ctenophora revealed by new mitochondrial COI primers and sequences. Molecular Ecology Resources, 22, 283–294. 10.1111/1755-0998.13459
Contributor Information
Lynne M. Christianson, Email: lynne@mbari.org.
Shannon B. Johnson, Email: sjohnson@mbari.org.
Steven H. D. Haddock, Email: haddock@mbari.org.
DATA AVAILABILITY STATEMENT
Untrimmed fragments of COI and 18S sequence fragments have been deposited in GenBank with accession numbers (MW647008‐75, MW735695‐832, MW804236‐67, MZ391814) (Table S1). Primer sequences are indicated in Figure 1.
REFERENCES
- Akaike, H. (1974). A new look at the statistical model identification. IEEE Transactions on Automatic Control, 19, 716–723. 10.1109/TAC.1974.1100705 [DOI] [Google Scholar]
- Arafat, H. , Alamaru, A. , Gissi, C. , & Huchon, D. (2018). Extensive mitochondrial gene rearrangements in the Ctenophora: Insights from benthic Playtctenida. BMC Evolutionary Biology, 18, 65. 10.1186/s12862-018-1186-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy, C. A. , Haddock, S. H. D. , & Robison, B. H. (2017). Deep pelagic food web structure as revealed by in situ feeding observations. Proceedings of the Royal Society B: Biological Sciences, 284, 20172116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun, C. (1880). Die Ctenophoren des Golfes von Neapel und der angrenzenden Meeres‐Abschnitte. Flora und Fauna des Golfes von Neapel (pp. 1–311). Leipzig, W. Engelmann. [Google Scholar]
- DeSalle, R. , & Goldstein, P. (2019). Review and interpretation of trends in DNA barcoding. Frontiers in Ecology and Evolution, 7, 302. 10.3389/fevo.2019.00302 [DOI] [Google Scholar]
- Djurhuus, A. , Pitz, K. , Sawaya, N. A. , Rojas‐Márquez, J. , Michaud, B. , Montes, E. , Muller‐Karger, F. , & Breitbart, M. (2018). Evaluation of marine zooplankton community structure through environmental DNA metabarcoding. Limnology and Oceanography: Methods, 16, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn, C. W. , Leys, S. P. , & Haddock, S. H. D. (2015). The hidden biology of sponges and ctenophores. Trends in Ecology and Evolution, 30, 282–291. 10.1016/j.tree.2015.03.003 [DOI] [PubMed] [Google Scholar]
- Eble, J. A. , Daly‐Engel, T. S. , DiBattista, J. D. , Koziol, A. , & Gaither, M. R. (2020). Marine environmental DNA: Approaches, applications, and opportunities. In Sheppard C. (Ed.), Advances in marine biology (pp. 141–169). Elsevier Ltd. [DOI] [PubMed] [Google Scholar]
- Edgar, R. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32, 1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmer, O. , Black, M. B. , Hoeh, W. R. , Lutz, R. A. , & Vrijenhoek, R. C. (1994). DNA primers for amplification of mitochondrial cytochrome C oxidase subunit I from metazoan invertebrates. Molecular Marine Biology and Biotechnology, 3, 294–299. [PubMed] [Google Scholar]
- Francis, W. R. , Christianson, L. M. , Kiko, R. , Powers, M. L. , Shaner, N. C. , & D Haddock, S. H. (2013). A comparison across non‐model animals suggests an optimal sequencing depth for de novo transcriptome assembly. BMC Genomics, 14, 167. 10.1186/1471-2164-14-167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller, J. , Meyer, C. , Parker, M. , & Hawk, H. (2013). Redesign of PCR primers for mitochondrial cytochrome c subunit I for marine invertebrates and application in all‐taxa biotic surveys. Molecular Ecology Resources, 13, 851–861. 10.1111/1755-0998.12138 [DOI] [PubMed] [Google Scholar]
- Günther, B. , Knebelsberger, T. , Neumann, H. , Laakmann, S. , & Arbizu, P. M. (2018). Metabarcoding of marine environmental DNA based on mitochondrial and nuclear genes. Scientific Reports, 8, 14822. 10.1038/s41598-018-32917-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddock, S. H. D. (2004). A golden age of gelata: past and future research on planktonic ctenophores and cnidarians. Hydrobiologica, 530(531), 549–556. 10.1007/s10750-004-2653-9 [DOI] [Google Scholar]
- Haddock, S. , Christianson, L. , Francis, W. , Martini, S. , Powers, M. , Dunn, C. , Pugh, P. , Mills, C. , Osborn, K. , Seibel, B. , Choy, A. , Schnitzler, C. , Matsumoto, G. , Messié, M. , Schultz, D. , Winnikoff, J. , Gasca, R. , Browne, W. , Johnsen, S. , … Thuesen, E. (2017). Insights into the biodiversity, behavior, and bioluminescence of deep‐sea organisms using molecular and maritime technology. Oceanography, 30, 39–47. 10.5670/oceanog.2017.422 [DOI] [Google Scholar]
- Harbison, G. R. , Madin, L. P. , & Swanberg, N. R. (1978). On the natural history and distribution of oceanic ctenophores. Deep‐Sea Research, 25, 233–256. 10.1016/0146-6291(78)90590-8 [DOI] [Google Scholar]
- Horita, T. , Akiyama, H. , & Kubota, S. (2011). A new lobate ctenophore, Bathocyroe Longigula. Zoologische Mededelingen Leiden, 85, 877–886. [Google Scholar]
- Johnson, S. B. , Winnikoff, J. R. , Schultz, D. T. , Christianson, L. C. , Patry, W. L. , Mills, C. E. , & Haddock, S. H. D. (In preparation). Speciation in the midwater; invisible boundaries drive isolation of pelagic zooplankton in the eastern Pacific. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn, A. B. , Citarella, M. R. , Kocot, K. M. , Bobkova, Y. V. , Halanych, K. M. , & Moroz, L. L. (2012). Rapid evolution of the compact and unusual mitochondrial genome in the ctenophore, Pleurobrachia bachei . Molecular Phylogenetics and Evolution, 63, 203–207. 10.1016/j.ympev.2011.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komai, T. , & Tokioka, T. (1940). Kiyohimea aurita, n. gen., n. sp., type of a new family of lobate Ctenophora. Annotationes Zoologicae Japonenses, 19, 43–46. [Google Scholar]
- Kumar, S. , Stecher, G. , Li, M. , Knyaz, C. , & Tamura, K. (2018). MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Molecular Biology and Evolution, 35, 1547–1549. 10.1093/molbev/msy096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacoursière‐Roussel, A. , Howland, K. , Normandeau, E. , Grey, E. K. , Archambault, P. , Deiner, K. , Lodge, D. M. , Hernandez, C. , Leduc, N. , & Bernatchez, L. (2018). eDNA metabarcoding as a new surveillance approach for coastal Arctic biodiversity. Ecology and Evolution, 8, 7763–7777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavrov, D. V. , & Pett, W. (2016). Animal mitochondrial DNA as we do not know it: mt‐genome organization and evolution in nonbilaterian lineages. Genome Biology and Evolution, 8, 2896–2913. 10.1093/gbe/evw195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leray, M. , & Knowlton, N. (2015). DNA barcoding and metabarcoding of standardized samples reveal patterns of marine benthic diversity. Proceedings of the National Academy of Sciences, 112, 2076–2081. 10.1073/pnas.1424997112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leray, M. , Yang, J. Y. , Meyer, C. P. , Mills, S. C. , Agudelo, N. , Ranwez, V. , Boehm, J. T. , & Machida, R. J. (2013). A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: Application for characterizing coral reef fish gut contents. Frontiers in Zoology, 10, 34. 10.1186/1742-9994-10-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay, D. J. , & Miyake, H. (2007). A novel benthopelagic ctenophore from 7,217 m depth in the Ryukyu Trench, Japan, with notes on the taxonomy of deep‐sea cydippids. Plankton and Benthos Research, 2, 98–102. 10.3800/pbr.2.98 [DOI] [Google Scholar]
- López‐Escardó, D. , Paps, J. , de Vargas, C. , Massana, R. , Ruiz‐Trillo, I. , & del Campo, J. (2018). Metabarcoding analysis on European coastal samples reveals new molecular metazoan diversity. Scientific Reports, 8, 9106. 10.1038/s41598-018-27509-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madin, L. P. , & Harbison, G. R. (1978). Bathocyroe fosteri gen.nov., sp.nov.: A mesopelagic ctenophore observed and collected from a submersible. Journal of the Marine Biological Association of the United Kingdom, 58, 559–564. [Google Scholar]
- Matsumoto, G. I. , & Robison, B. H. (1992). Kiyohimea usagi, a new species of lobate ctenophore from the Monterey submarine canyon. Bulletin of Marine Science, 51, 19–29. [Google Scholar]
- Medlin, L. K. , Elwood, H. J. , Stickel, S. , & Sogin, M. L. (1988). The characterization of enzymatically amplified eukaryotic 16S‐like rRNA‐coding regions. Genetica, 71, 491–499. [DOI] [PubMed] [Google Scholar]
- Mills, C. E. (2017). Phylum ctenophora: list of all valid species names. https://faculty.washington.edu/cemills/Ctenolist.html [Google Scholar]
- Minh, B. Q. , Schmidt, H. A. , Chernomor, O. , Schrempf, D. , Woodhams, M. D. , von Haeseler, A. , & Lanfear, R. (2020). IQ‐TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Molecular Biology and Evolution, 37, 1530–1534. 10.1093/molbev/msaa015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pett, W. , Ryan, J. R. , Pang, K. , Mullikin, J. C. , Martindale, M. Q. , Baxevanis, A. D. , & Lavrov, D. V. (2011). Extreme mitochondrial evolution in the ctenophore Mnemiopsis leidyi: Insights from mtDNA and the nuclear genome. Mitochondrial DNA, 22, 130–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitz, K. , Guo, J. , Johnson, S. B. , Campbell, T. L. , Zhang, H. , Vrijenhoek, R. C. , Chavez, F. P. , & Geller, J. (2020). Zooplankton biogeographic boundaries in the California Current System as determined from metabarcoding. PLoS One, 15, e0231519–e0231520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podar, M. , Haddock, S. H. D. , Sogin, M. L. , & Harbison, G. R. (2001). A molecular phylogenetic framework for the phylum Ctenophora using 18S rRNA genes. Molecular Phylogenetics and Evolution, 21, 218–230. 10.1006/mpev.2001.1036 [DOI] [PubMed] [Google Scholar]
- Posada, D. , & Crandall, K. A. (1998). Modeltest: Testing the model of DNA substitution. Bioinformatics, 14, 817–818. 10.1093/bioinformatics/14.9.817 [DOI] [PubMed] [Google Scholar]
- Preston, C. M. , Durkin, C. A. , & Yamahara, K. M. (2020). DNA metabarcoding reveals organisms contributing to particulate matter flux to abyssal depths in the North East Pacific Ocean. Deep‐Sea Research Part II, 173, 104708. 10.1016/j.dsr2.2019.104708 [DOI] [Google Scholar]
- Rstudio Team . (2015). RStudio: Integrated development for R. Boston, MA: PBC. http://www.rstudio.com/ [Google Scholar]
- Ralph, P. M. , & Kaberry, C. (1950). New Zealand coelenterates. Ctenophores from cook straight (pp. 1–11). Zoology Publications from Victoria University College 3. [Google Scholar]
- Rambaut, A. , Drummond, A. , Xie, D. , Baele, G. , & Suchard, M. A. (2018). Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Systematic Biology, 67(5), 901–904. 10.1093/sysbio/syy032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist, F. , Teslenko, M. , van der Mark, P. , Ayres, D. L. , Darling, A. , Höhna, S. , Larget, B. , Liu, L. , Suchard, M. A. , & Huelsenbeck, J. P. (2012). MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology, 61, 539–542. 10.1093/sysbio/sys029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas, J. , Ferrer‐Mata, A. , Sánchez‐DelBarrio, J. C. , Guirao‐Rico, S. , Librado, P. , Ramos‐Onsins, S. E. , & Sánchez‐Gracia, A. (2017). DnaSP 6: DNA sequence polymorphism analysis of large datasets. Molecular Biology and Evolution, 34, 3299–3302. 10.1093/molbev/msx248 [DOI] [PubMed] [Google Scholar]
- Schroeder, A. , Stanković, D. , Pallavicini, A. , Gionechetti, F. , Pansera, M. , & Camatti, E. (2020). DNA metabarcoding and morphological analysis ‐ Assessment of zooplankton biodiversity in transitional waters. Marine Environmental Research, 160, 104946. 10.1016/j.marenvres.2020.104946 [DOI] [PubMed] [Google Scholar]
- Schultz, D. T. , Eizenga, J. M. , Corbett‐Detig, R. B. , Francis, W. R. , Christianson, L. M. , & Haddock, S. H. D. (2020). Conserved novel ORFs in the mitochondrial genome of the ctenophore Beroe forskalii . PeerJ, 8, e8356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddall, M. E. , Fontanella, F. M. , Watson, S. C. , Kvist, S. , & Erséus, C. (2009). Barcoding bamboozled by bacteria: convergence to metazoan mitochondrial primer targets by marine microbes. Systematic Biology, 4, 445–451. 10.1093/sysbio/syp033 [DOI] [PubMed] [Google Scholar]
- Simion, P. , Bekkouche, N. , Jager, M. , Quéinnec, E. , & Manuel, M. (2015). Exploring the potential of small RNA subunit and ITS sequences for resolving phylogenetic relationships within phylum Ctenophora. Zoology, 118, 102–114. 10.1016/j.zool.2014.06.004 [DOI] [PubMed] [Google Scholar]
- Simion, P. , Philippe, H. , Baurain, D. , Jager, M. , Richter, D. J. , Di Franco, A. , Roure, B. , Satoh, N. , Ereskovsky, A. , Lapébie, P. , Corre, E. , Delsuc, F. , King, N. , Wörheide, G. , & Manuel, M. (2017). A large and consistent phylogenomic dataset supports sponges as the sister group to all other animals. Current Biology, 27, 958–967. 10.1016/j.cub.2017.02.031 [DOI] [PubMed] [Google Scholar]
- Thiebot, J.‐B. , & McInnes, J. C. (2020). Why do marine endotherms eat gelatinous prey? ICES Journal of Marine Science, 77, 58–71. 10.1093/icesjms/fsz208 [DOI] [Google Scholar]
- Untergasser, A. , Cutcutache, I. , Koressaar, T. , Ye, J. , Faircloth, B. C. , Remm, M. , & Rozen, S. G. (2012). Primer3—New capabilities and interfaces. Nucleic Acids Research, 40, e115. 10.1093/nar/gks596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrijenhoek, R. C. , & Waples, R. S. (2012). Popular misconceptions. Molecular Ecology, 21, 4155–4156. 10.1111/j.1365-294X.2012.05658.x [DOI] [PubMed] [Google Scholar]
- Wickham, H. (2016). ggplot2: Elegant graphics for data analysis. New York: Springer‐Verlag. https://ggplot2.tidyverse.org [Google Scholar]
- Xia, X. (2018). DAMBE7: New and improved tools for data analysis in molecular biology and evolution. Molecular Biology and Evolution, 35, 1550–1552. 10.1093/molbev/msy073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, X. , Hafner, M. S. , & Sudman, P. D. (1996). On transition bias in mitochondrial genes of pocket gophers. Journal of Molecular Evolution, 43, 32–40. 10.1007/BF02352297 [DOI] [PubMed] [Google Scholar]
- Yeh, H. D. , Questel, J. M. , Maas, K. R. , & Bucklin, A. (2020). Metabarcoding analysis of regional variation in gut contents of the copepod Calanus finmarchicus in the North Atlantic Ocean. Deep Sea Research Part II: Topical Studies in Oceanography, 180, 104738. 10.1016/j.dsr2.2020.104738 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Table S1
Data Availability Statement
Untrimmed fragments of COI and 18S sequence fragments have been deposited in GenBank with accession numbers (MW647008‐75, MW735695‐832, MW804236‐67, MZ391814) (Table S1). Primer sequences are indicated in Figure 1.