

Abstract
This analysis aimed to describe the pharmacokinetics (PK) of acalabrutinib and its active metabolite, ACP‐5862. A total of 8935 acalabrutinib samples from 712 subjects and 2394 ACP‐5862 samples from 304 subjects from 12 clinical studies in patients with B‐cell malignancies and healthy subjects were analysed by nonlinear mixed‐effects modelling. Acalabrutinib PK was characterized by a 2‐compartment model with first‐order elimination. The large variability in absorption was adequately described by transit compartment chain and first‐order absorption, with between‐occasion variability on the mean transit time and relative bioavailability. The PK of ACP‐5862 was characterized by a 2‐compartment model with first‐order elimination, and the formation rate was defined as the acalabrutinib clearance multiplied by the fraction metabolized. Health status, Eastern Cooperative Oncology Group performance status, and coadministration of proton‐pump inhibitors were significant covariates. However, none of the investigated covariates led to clinically meaningful changes in exposure, supporting a flat dosing of acalabrutinib.
Keywords: acalabrutinib, ACP‐5862, B‐cell malignancies, between‐occasion variability, pH‐dependent solubility, pharmacokinetics, population pharmacokinetics
What is already known about this subject
Acalabrutinib is a highly selective, potent, covalent Bruton tyrosine kinase (BTK) inhibitor approved for the treatment of previously treated mantle cell lymphoma as well as previously untreated and relapsed/refractory chronic lymphocytic leukaemia/small lymphocytic lymphoma.
A previous population pharmacokinetic (PK) model for acalabrutinib and its active metabolite, ACP‐5862, developed using data from 285 healthy subjects and 292 patients, concluded that no adjustments from acalabrutinib 100 mg twice‐daily dosing were needed based on covariate analyses; however, ACP‐5862 concentration data were only available in 48 patients.
What this study adds
A significant amount of acalabrutinib and ACP‐5862 concentration data in patients have become available since the last population PK evaluation and are included in the current population PK model.
The improved PK model for acalabrutinib and ACP‐5862 better characterizes the variable absorption of acalabrutinib and PK of ACP‐5862 and was considered adequate for deriving exposure estimates to be used in subsequent PK/pharmacodynamic analyses relating exposure of acalabrutinib and ACP‐5862 to clinical outcomes.
In the current analysis, health status, Eastern Cooperative Oncology Group performance status and proton‐pump inhibitor use were identified as significant covariates (vs. dose and body weight in the previous analysis); however, none of the covariate–PK parameter relationships revealed clinically meaningful changes in acalabrutinib or ACP‐5862 exposure.
See Original Article here .
1. INTRODUCTION
Acalabrutinib is a potent, highly selective, covalent Bruton tyrosine kinase (BTK) inhibitor. 1 , 2 ACP‐5862 has been identified as the major pharmacologically active metabolite of acalabrutinib in plasma. A population pharmacokinetic (PK) analysis of acalabrutinib and ACP‐5862 has been reported 3 ; subsequently, a substantial amount of data for both acalabrutinib and ACP‐5862 has been generated in patients with B‐cell malignancies. The solubility of acalabrutinib decreases with increasing pH; consequently, the absorption of acalabrutinib is highly variable and affected by changes in gastric pH and gastric emptying time. 4 For example, concomitant proton‐pump inhibitor (PPI) use or coadministration with food causes a delay in peak concentration and, in the case of PPIs, a reduction in area under the curve (AUC). These variables were allowed but not recorded in detail in the clinical studies. A population PK model can help distinguish the between‐occasion variability (BOV) in absorption within a subject and variability between subjects due to these and other factors.
The mean exposure of ACP‐5862 is approximately 2‐fold higher than that of acalabrutinib. 5 ACP‐5862 is approximately half as potent as acalabrutinib for BTK inhibition and has a similar kinase selectivity profile. 6 Taken together, these results indicate that ACP‐5862 contributes to the efficacy and safety of acalabrutinib. Therefore, quantifying total exposure to acalabrutinib and ACP‐5862 may facilitate a better understanding of acalabrutinib safety and efficacy outcomes.
The objectives of the current analysis were to update the previously developed model 3 with additional acalabrutinib and ACP‐5862 concentration data, revise the model to better characterize the variability in absorption, and investigate the impact of covariates on exposure to acalabrutinib and ACP‐5862 in a larger patient population.
2. METHODS
2.1. Study population and plasma samples
Data were obtained from eight phase 1–3 trials in adults with B‐cell malignancies and 4 phase 1 trials in healthy subjects. The details of included studies are provided in Table S1.
2.2. PK model development
One‐, 2‐ and 3‐compartment models with various different absorption models were tested to describe acalabrutinib PK. Similar to the previous model, the formation rate of ACP‐5862 was defined as the apparent clearance (CL/F) of acalabrutinib multiplied by the fraction metabolized, 3 which was fixed to 0.4 based on results of metabolite profiling in a human absorption, distribution, metabolism and excretion study of acalabrutinib. All ACP‐5862 and acalabrutinib model parameters were simultaneously estimated. Between‐subject variability (BSV) and BOV for PK parameters were evaluated using exponential models. BSV was evaluated on all PK parameters. BOV was not investigated for metabolite parameters as it was deemed necessary to describe acalabrutinib absorption. Residual error was described with an exponential model (additive on log‐transformed data).
2.2.1. Covariate model
A full covariate modelling procedure was conducted. 7 A list of candidate covariate–parameter relationships was identified based on exploratory graphical analyses, scientific interest, clinical judgement, mechanistic plausibility and/or prior knowledge. Covariates were selected to avoid correlation or collinearity in predictors. The covariate effect was not considered relevant if the change of the structural PK parameter value and its associated 95% confidence interval (CI) was within a 0.8–1.25 interval (i.e., <20% change). Covariates for which the final estimate and associated 95% CI were found outside the 0.8–1.25 interval or those for which the 95% CI did not cross the no‐effect point estimate of 1 were kept in the reduced covariate model.
The impact and clinical relevance of covariates were evaluated for their effect on acalabrutinib and ACP‐5862 exposure at steady‐state (AUC24h,ss and maximum plasma concentration [Cmax,ss]) through stochastic simulations. Additional information regarding missing data handling, data cleaning, covariate model details, model evaluation methods, and hardware and software details are provided in Supplemental Information.
2.3. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20. 8
3. RESULTS
3.1. Study population and acalabrutinib and ACP‐5862 concentration–time data
A total of 138 healthy subjects and 575 patients with B‐cell malignancies were included (subject characteristics in Tables S2 and S3). The analysis dataset contained 8935 (7.1% below lower limit of quantification [BLQ]) acalabrutinib samples from 712 subjects and 2394 (4.3% BLQ) ACP‐5862 samples from 304 subjects. One subject had metabolite samples only; hence, the total number of subjects was 713. The median (range) number of samples per subject was 12 (1–28) for acalabrutinib and 7 (3–28) for ACP‐5862 (Table S4). BLQ data were observed in both the absorption and elimination phases (Figure S1). BLQ data were included and modelled as a probability of being BLQ. Most subjects had ≥1 rich sampling profile, defined as ≥3 samples per occasion, and sparse samples collected on ≥2 different dosing occasions. Overall, 20.1% (n = 1793) of acalabrutinib and 10.8% (n = 258) of ACP‐5862 observations were collected as sparse data. For modelling purposes, sparse samples collected on different dosing days were lumped into 1 unique sparse occasion for identifiability of occasional‐level random effects (Figure S2).
3.2. Acalabrutinib and ACP‐5862 PK model
The acalabrutinib concentration–time data were adequately described with a 2‐compartment model with a 5‐transit compartment chain and first‐order absorption. The data supported inclusion of BOV on mean transit time (MTT; change in objective function value [∆OFV] = −2410) and relative bioavailability (F1; ∆OFV = −214), which also significantly improved goodness of fit and individual fits.
ACP‐5862 data were adequately described with a 2‐compartment model with a first‐order formation rate of 0.4*CL/F, similar to the previous analysis. 3
Residual error models were exponential models (additive on the log‐scale) with separate terms for parent and metabolite. Separate residual error parameters were included for samples obtained on occasions for which rich profiles vs. sparse samples had been collected. The rationale for the 2 different error terms was: (i) sparse samples had a much higher proportion of imputed dose times vs. occasions with rich sampling (~50% vs. < 1% [acalabrutinib]; 98% vs. < 1% [ACP‐5862]); and (ii) there was lumping of sparse samples into 1 occasion for BOV models.
The following covariates were added to the model: Eastern Cooperative Oncology Group performance status (ECOG PS), race, body weight, health status, PPI use, H2 receptor antagonist use, estimated glomerular filtration rate and hepatic impairment. CD19+ count was excluded from covariate evaluation due to the high proportion of subjects with missing values (n = 233, 33%). Age and sex were excluded from covariate evaluation due to the high correlation with body weight and health status and were instead graphically assessed to rule out their impact on acalabrutinib and ACP‐5862 exposure. Relationships with an estimate and 95% CI outside the 0.8–1.25 interval included health status on CL/F and apparent peripheral volume of distribution (Vp/F) and concomitant PPI use on F1 (Figure S3). Further, although the ECOG PS of ≥2 on CL/F point estimate was within the 0.8–1.25 interval, its 95% CI did not cross 1; therefore, the relationship was deemed significant and was retained in the model. Race (i.e., Black/African American) on apparent central volume of distribution (Vc/F) was outside the 0.8–1.25 interval but could not be well estimated (relative standard errors [RSE] > 110%) and was therefore removed.
All final structural parameters were estimated with an RSE ≤ 20%, whereas covariate and random effects were estimated with an RSE ≤ 40% (Table 1). As expected, the residual error was higher for sparse samples vs. samples from rich profiles. Because metabolite data were available only in some of the studies, overall shrinkage in BSV for metabolite parameters was high. 9
TABLE 1.
Parameter estimates for acalabrutinib and ACP‐5862 base and final model
| Final model OFV: 8345, condition no.: 24.7 | |||
|---|---|---|---|
| Parameter | Estimate (%RSE) | Bootstrap a | Shrinkage b |
| Fixed effects | |||
| CL/F (L/h) | 134 (1.97) | 132 [126, 139] | |
| Vc/F (L) | 31.0 (15.4) | 30.3 [25.3, 35.7] | |
| Q/F (L/h) | 20.9 (3.94) | 20.8 [18.6, 23.0] | |
| Vp/F (L) | 110 (3.45) | 108 [97.6, 120] | |
| Ka (h−1) | 1.48 (2.63) | 1.48 [1.41, 1.55] | |
| MTT (h) | 0.459 (4.16) | 0.460 [0.426, 0.492] | |
| CLM/F (L/h) | 21.8 (1.98) | 21.6 [20.2, 22.6] | |
| VcM/F (L) | 22.7 (6.66) | 22.7 [19.5, 25.8] | |
| QM/F (L/h) | 26.7 (8.65) | 26.4 [22.9, 29.9] | |
| VpM/F (L) | 89.2 (4.58) | 88.1 [77.6, 97] | |
| Covariate effects c | |||
| Healthy subject—CL/F | 0.467 (19.3) | 0.467 [0.367, 0.569] | |
| Healthy subject—Vp/F | −0.556 (5.59) | −0.554 [−0.6, −0.496] | |
| ECOG 2—CL/F | −0.171 (36.2) | −0.158 [−0.274, −0.032] | |
| PPI—F1 | −0.358 (7.01) | −0.368 [−0.49, −0.228] | |
| Random effects | |||
| BSV CL/F (CV%) | 23.8 (5.86) | 24.1 [20.4, 28] | 36.1 |
| BSV Vc/F (CV%) | 270 (6.51) | 284 [225, 367] | 25.9 |
| BSV Vp/F (CV%) | 33.7 (6.96) | 33.9 [25, 42.4] | 49.6 |
| BSV CLM/F (CV%) | 11.8 (21.1) | 13.1 [9.22, 20.2] | 70.2 |
| BSV VcM/F (CV%) | 47.2 (12.1) | 53.3 [36.9, 71.1] | 66.5 |
| BSV QM/F (CV%) | 40.7 (14.4) | 40 [23.8, 59.2] | 72.0 |
| BSV VpM/F (CV%) | 19.2 (30.5) | 21.2 [15.3, 41.2] | 79.6 |
| BOV MTT (CV%) | 118 (2.39) | 119 [104, 137] | 45.4 |
| BOV F1 (CV%) | 56.1 (1.80) | 55.3 [50.4, 61.1] | 42.4 |
| Residual error (SD) | 0.586 (0.558) | 0.583 [0.555, 0.612] | 13.0 |
| Residual error sparse (SD) | 0.856 (1.16) | 0.843 [0.762, 0.917] | 7.92 |
| Residual error metabolite (SD) | 0.334 (1.48) | 0.332 [0.304, 0.357] | 14.6 |
| Residual error metabolite sparse (SD) | 0.234 (4.25) | 0.225 [0, 0.325] | 37.4 |
Abbreviations: BOV, between‐occasion variability; BSV, between‐subject variability; CL/F, apparent clearance; CLM/F, apparent clearance metabolite; CV, coefficient of variation; ECOG, Eastern Cooperative Oncology Group; F1, relative bioavailability; Ka, first‐order absorption rate constant; MTT, mean transit time; OFV, objective function value; PPI, proton‐pump inhibitor; Q/F, apparent intercompartment clearance; QM/F, apparent intercompartment clearance metabolite; RSE, relative standard error; SD, standard deviation; Vc/F, apparent central volume of distribution; VcM/F, apparent central volume of distribution metabolite; Vp/F, apparent peripheral volume of distribution; VpM/F, apparent peripheral volume of distribution metabolite.
50th [2.5th, 97.5th percentile].
Shrinkage for BOV is provided as the mean of 4 occasions.
Relative change (1 + estimate).
Goodness‐of‐fit plots did not reveal any general model misspecifications, except a slight bias at low concentrations close to the lower limit of quantification, which was more pronounced in healthy subjects (Figures S4–7). Prediction‐corrected visual predictive check (pcVPC) indicated an adequate model performance for the pooled data (Figure 1), although variability still appeared overpredicted for healthy subjects for both acalabrutinib and ACP‐5862 (Figure S8).
FIGURE 1.
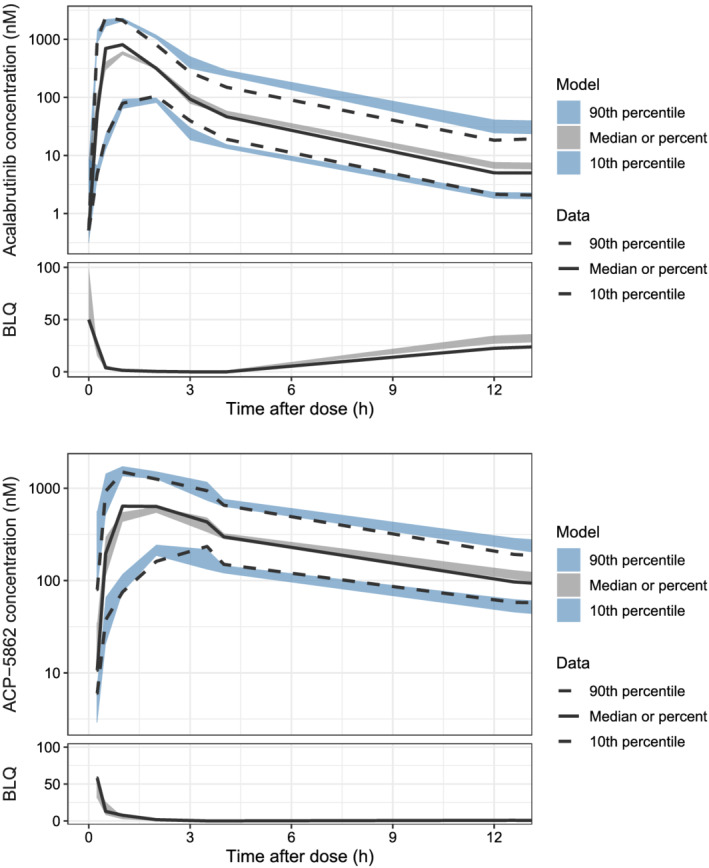
Prediction‐corrected visual predictive check for the final model (12‐hour profile). The top panels show acalabrutinib data and the bottom show ACP‐5862 data on log‐scale. The observed data have been omitted to better visualize the percentiles. The solid and dashed lines are the median and the 10th and 90th percentiles of the observations. The shaded areas are the 95% confidence intervals of the median and the 10th and 90th percentiles predicted by the model. Note: prediction correction could result in values originally above lower limit of quantification to appear to be below (BLQ) after the correction (2.1 nM and 10 nM for acalabrutinib and ACP‐5862, respectively)
The posterior predictive check (Figure 2) showed that the observed median AUC0‐last and Cmax fell within the distribution of simulated statistic values for most scenarios (across doses and studies), confirming good predictive performance of the model.
FIGURE 2.
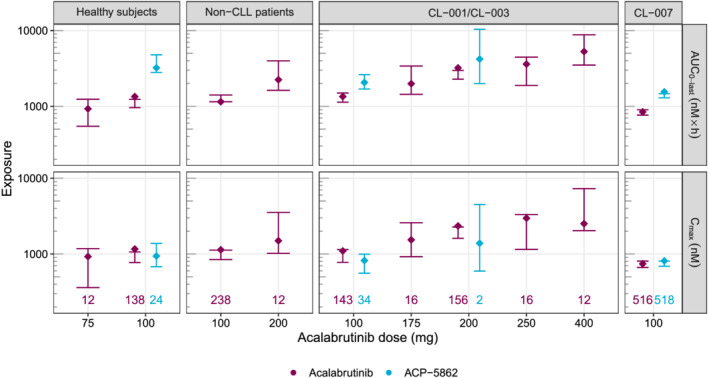
Posterior predictive check of final model for acalabrutinib and ACP‐5862 AUC0‐last (top row) and Cmax (bottom row) by dose group. Studies with a similar sampling schedule have been grouped (columns). Diamonds represent the computed median AUC0‐last or Cmax on the observed data (across subjects and rich occasions). The coloured numbers at the bottom of each column represent the number of rich occasions for each scenario. Error bars represent the 95% confidence intervals based on 300 simulations. AUC0‐last, area under the concentration–time curve from time 0 to last observation; CLL, chronic lymphocytic leukaemia; Cmax, maximum plasma concentration
3.3. Acalabrutinib and ACP‐5862 exposures
The model‐predicted medians (90% prediction interval) for acalabrutinib AUC24h,ss and Cmax,ss for the reference population at 100 mg BID (patients with B‐cell malignancy, ECOG PS ≤ 1 and without concomitant use of PPIs) were 1668 (1094–2536) ng·h/mL and 461 (199.8–783.3) ng/mL, respectively. The model‐predicted ACP‐5862 median (90% prediction interval) AUC24h,ss and Cmax,ss for the reference population at 100 mg BID were 4175 (3256–5430) ng·h/mL and 461.3 (263.7–678.4) ng/mL, respectively.
Relative to a reference patient, acalabrutinib and ACP‐5862 exposures were 36% lower with concomitant PPI use (Figure S9). Body weight, estimated glomerular filtration rate, hepatic impairment and race were not identified to impact acalabrutinib or ACP‐5862 exposure. Graphics assessing predicted individual exposures at 100 mg BID vs. covariates align with these results (data not shown) and did not reveal any differences with age or between different categories of ethnicity, line of therapy, indication, or sex.
4. DISCUSSION
In this analysis, a parent–metabolite population PK model was constructed for acalabrutinib and ACP‐5862. The model included a 2‐compartment model with a transit chain, first‐order absorption and linear elimination for describing acalabrutinib, and a 2‐compartment model with linear elimination for describing ACP‐5862 concentration–time profiles. Covariate analysis identified baseline ECOG PS on CL/F, health status on CL/F and Vp/F, and administration of PPI on F1 as significant covariates. None of the bootstrapped 95% CIs for covariate effect parameters included zero, confirming the statistical significance of included covariates. The final model (Figure S10) was numerically stable, estimated parameters with adequate precision and had adequate predictive performance.
The solubility of acalabrutinib decreases with increasing gut pH. Therefore, it is currently recommended that coadministration of acalabrutinib with acid‐reducing agents either use a staggered dosing approach (antacids and H2‐receptor antagonists) or be avoided (PPIs). 2 However, these recommendations were not in place at the time of PK sampling in some of the patient studies. Similarly, studies exploring food effects on acalabrutinib exposure identified a delayed and decreased Cmax but similar mean AUC when acalabrutinib is administered with a high‐calorie meal vs. fasting conditions; therefore, acalabrutinib can be given with or without food. 2 Food status was not recorded in the patient studies and could not be used as a covariate. Together, this may explain part of the large unexplained variability in the absorption phase observed in this analysis.
In clinical drug–drug interaction studies in healthy subjects, coadministration with omeprazole (PPI) decreased acalabrutinib AUC by 43% (data on file). In this population PK study, despite having less detailed patient data, coadministration of PPIs was observed to reduce acalabrutinib and ACP‐5862 AUC24h,ss by 36%, consistent with the results of dedicated drug–drug interaction studies. Remaining variability in absorption (not related to PPIs) was described by BOV on F1 and MTT, estimated to be 56% and 118%, respectively. The current strategy to describe absorption proved to be more robust, successfully separated the sources of variability, improved model descriptive performance and allowed for more reliable interpretation of parameter estimates vs. the previous analysis. 3
pcVPCs split by health status indicated that the variability appeared to be overpredicted in healthy subjects, which is expected due to the more controlled settings, particularly with respect to food and acid‐reducing agents, in studies of healthy volunteers; thus, any population simulations of healthy subjects may suffer from overdispersion. This model was only used to predict PK for subjects with B‐cell malignancies, for which the variability was well characterized. In addition, posterior predictive checks confirmed the model's predictive performance for generating AUC and Cmax. Hence, the model was deemed fit for purpose and considered adequate for deriving exposure estimates for use in subsequent PK/PD analyses (see also Edlund et al.10).
Relative to a reference cancer population, acalabrutinib exposures were lower in healthy subjects (32 and 24% for AUC24h,ss and Cmax,ss, respectively) and higher in subjects with ECOG PS ≥ 2 (21% and 14%, respectively). Acalabrutinib and ACP‐5862 exposures were 36% lower with concomitant PPI use. Considering the magnitude of change for each covariate (maximum 36% decrease or 21% increase), which was comparable with the overall variability in exposures, none of the included covariates were deemed to have a clinically relevant impact on acalabrutinib/ACP‐5862 exposure.
Therefore, the updated population PK analysis supports the use of the approved dose and schedule of acalabrutinib in various patient populations and demonstrates no clinically relevant changes in acalabrutinib or ACP‐5862 exposure based on patients' characteristics (age, sex, weight, race, ethnicity, alanine aminotransferase/aspartate transaminase, renal function, hepatic function, health status, indication, line of therapy or ECOG PS), confirming the results of the previous analysis. 3
COMPETING INTERESTS
Núria Buil‐Bruna, Helena Edlund, Huan Liu and Helen Tomkinson are employees of AstraZeneca. Karthick Vishwanathan is a current employee and stock shareholder of AstraZeneca. Shringi Sharma is an employee of Acerta Pharma. Francesco Bellanti is an employee of Certara. Joseph Ware is a former employee of Acerta Pharma and retains AstraZeneca stock. The study was funded by Acerta Pharma, South San Francisco, CA, a member of the AstraZeneca Group.
CONTRIBUTORS
H.E. contributed to the analysis and interpretation of the data, and to writing, reviewing and approving the manuscript.
F.B. contributed to the analysis and interpretation of the data, and to reviewing and approving the manuscript.
H.L. contributed to the preparation of the data, and to reviewing and approving the manuscript.
H.T. contributed to the analysis and interpretation of the data, and to writing, reviewing and approving the manuscript.
J.A.W. contributed to the analysis strategy, clinical insights and interpretation of the data, and to writing, reviewing and approving the manuscript.
S.S. contributed to the analysis and interpretation of the data, and to writing, reviewing and approving the manuscript.
N.B‐B. contributed to the analysis and interpretation of the data, and to writing, reviewing and approving the manuscript.
K.V. contributed to the study design and to reviewing and approving the manuscript.
Supporting information
Table S1. Details of studies and included data
Table S2. Demographics and subject characteristics (categorical variables)
Table S3. Demographics and subject characteristics (continuous variables)
Table S4. Tabulation of pharmacokinetic samples included
Figure S1. Distribution of acalabrutinib (left) and ACP‐5862 (right) pharmacokinetic sample collection times. Note: only the first BLQ of the elimination phase is included for each subject. X‐axes are cut at 32 hours, removing 2 acalabrutinib samples. BLQ, below the lower limit of quantification.
Figure S2. Acalabrutinib and ACP‐5862 concentrations vs. time after dose at different sampling times (rows). Occasions 1–3 include dense sampling occasions and sparse data include all observations collected on sparse sampling days. Studies with a similar sampling schedules have been grouped (columns). Lines connect data from the same dosing occasion within a subject. Dashed horizontal lines depict lower limit of quantification (2.1 and 10.4 nM for acalabrutinib and ACP‐5862, respectively). Data below the LLOQ are shown at LLOQ/2. CL, chronic lymphocytic leukaemia; LLOQ, lower limit of quantification.
Figure S3. Impact of covariates on typical parameter estimates in the full covariate model. The dots represent the final estimate and the bars indicate a 95% CI based on parameter precision. The vertical dashed line (value = 1) represents the no‐effect point estimate, which suggests no impact of a covariate on the typical value estimate. The shaded area represents the no‐effect range and corresponds to an approximate change of 20% with respect to the typical value estimate. BW, body weight; CI, confidence interval; CL/F, apparent clearance; CLM/F, apparent clearance metabolite; ECOG, Eastern Cooperative Oncology Group; eGFR, estimated glomerular filtration rate; F, bioavailability; HV, healthy volunteers; H2RA, H2‐receptor antagonist; Ka, absorption rate; MTT, mean transit time; PPI, proton‐pump inhibitor; Vc/F, central apparent volume of distribution; VcM/F, apparent central volume of distribution metabolite; Vp/F, apparent peripheral volume of distribution.
Figure S4. Observed versus predicted concentrations for acalabrutinib (top) and ACP‐5862 (bottom). The red line is the line of unity. The blue line is the non‐parametric smoother, and the shaded area is the 95% confidence interval. The dashed horizontal line is the lower limit of quantification (LLOQ, 2.1 nM and 10.4 nM for acalabrutinib and ACP‐5862, respectively). Observations below the LLOQ are not displayed. Axes for acalabrutinib data are cut at 0.4 nM, removing 22 acalabrutinib population predictions.
Figure S5. NPDE vs. time after dose. Non‐BLQ samples are plotted as circles and BLQ samples are plotted as asterisks. The red dashed line indicates zero. The blue line is the nonparametric smoother, and the shaded area is the 95% confidence interval. The x‐axis is cut at 32 hours (removing 2 acalabrutinib samples) to better illustrate earlier time points. BLQ, below the lower limit of quantification; NPDE, normalized prediction distribution errors.
Figure S6. NPDE vs. time after dose for healthy subjects (top) and patients (bottom). Non‐BLQ samples are plotted as circles and BLQ samples are plotted as asterisks. The red dashed line indicates zero. The blue line is the nonparametric smoother, and the shaded area is the 95% confidence interval. The x‐axis is cut at 32 hours (removing 2 acalabrutinib samples) to better illustrate earlier time points. BLQ, below the lower limit of quantification; NPDE, normalized prediction distribution errors.
Figure S7. NPDE vs. population predictions. The blue line is the nonparametric smoother, and the shaded area is the 95% confidence interval. The vertical horizontal line is the lower limit of quantification. Observations BLQ are not displayed in the figure. Axes for acalabrutinib data are cut at 0.4 nM, removing 22 acalabrutinib population predictions. BLQ, below the limit of quantification; NPDE, normalized prediction distribution errors.
Figure S8. Prediction‐corrected visual predictive check stratified by health status. The top panels show acalabrutinib data and the bottom show ACP‐5862 data. The observed data have been omitted to better visualize the percentiles. The solid and dashed lines are the median and the 10th and 90th percentiles of the observations, respectively. The shaded areas are the 95% confidence intervals of the median and the 10th and 90th percentiles predicted by the model. Data were modelled on the log scale. Prediction correction could result in values originally above LLOQ to appear to be BLQ after the correction (2.1 nM and 10 nM for acalabrutinib and ACP‐5862, respectively). BLQ, below LLOQ; LLOQ, lower limit of quantification.
Figure S9. Effect of covariates on acalabrutinib (top) and ACP‐5862 (bottom) AUC24h,ss (left) and Cmax,ss (right). The diamonds are the point estimate and the bars indicate a 95% prediction interval based on between‐subject and between‐occasion variability relative to the reference. The reference is defined as the most prevalent scenario in the population: a patient with ECOG performance status of 0 or 1 and no concomitant PPI. The dashed line indicates 1 (i.e., the reference value). AUC24h,ss, area under the plasma concentration–time curve for 24 hours at steady‐state; Cmax,ss, maximum plasma concentration at steady‐state; ECOG, Eastern Cooperative Oncology Group; PPI, proton‐pump inhibitor.
Figure S10. Acalabrutinib and ACP‐5862 final model. CL/F, apparent clearance; CLM/F, apparent clearance of the metabolite; ECOG, Eastern Cooperative Oncology Group; F1, relative bioavailability; Fm, fraction metabolized; Ka, first‐ order absorption rate constant; Ktr, transit absorption rate constant [MTT (mean transit time) = (Ntr + 1)/Ktr]; PPI, proton‐pump inhibitor; Q/F, apparent intercompartment clearance; QM/F, apparent intercompartment clearance of the metabolite.
ACKNOWLEDGEMENTS
The authors thank Magnus Åstrand, Ulrika Wählby Hamrén, Jaap Mandema and Rik de Greef for critical review and input to the model development.
The authors acknowledge the Acerta Pharma study teams. They also thank the patients and healthy volunteers who participated in these studies as well as their friends and family who supported them. The underlying clinical studies that informed this analysis were funded by Acerta Pharma, South San Francisco, CA, a member of the AstraZeneca Group. Medical writing assistance, funded by Acerta Pharma, was provided by Allison Green, PhD, and Cindy Gobbel, PhD, of Peloton Advantage, LLC, an OPEN Health company.
Edlund H, Bellanti F, Liu H, et al. Improved characterization of the pharmacokinetics of acalabrutinib and its pharmacologically active metabolite, ACP‐5862, in patients with B‐cell malignancies and in healthy subjects using a population pharmacokinetic approach. Br J Clin Pharmacol. 2022;88(2):846-852. 10.1111/bcp.14988
A principal investigator was not included in the author byline because the reported data are from a population PK model developed using the data from 12 different studies. The principle investigators of the original studies were not involved in performing these analyses or interpreting the data; therefore, they did not qualify for authorship according to ICMJE criteria.
Funding information Acerta Pharma, South San Francisco, CA, a member of the AstraZeneca Group
DATA AVAILABILITY STATEMENT
Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca's data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
REFERENCES
- 1. Barf T, Covey T, Izumi R, et al. Acalabrutinib (ACP‐196): A covalent Bruton tyrosine kinase inhibitor with a differentiated selectivity and in vivo potency profile. J Pharmacol Exp Ther. 2017;363(2):240‐252. [DOI] [PubMed] [Google Scholar]
- 2. Calquence [package insert] . Wilmington, DE: AstraZeneca Pharmaceuticals; 2019.
- 3. Edlund H, Lee SK, Andrew MA, Slatter JG, Aksenov S, Al‐Huniti N. Population pharmacokinetics of the BTK inhibitor acalabrutinib and its active metabolite in healthy volunteers and patients with B‐Cell malignancies. Clin Pharmacokinet. 2019;58(5):659‐672. [DOI] [PubMed] [Google Scholar]
- 4. Pepin XJH, Moir AJ, Mann JC, et al. Bridging in vitro dissolution and in vivo exposure for acalabrutinib. Part II. A mechanistic PBPK model for IR formulation comparison, proton pump inhibitor drug interactions, and administration with acidic juices. Eur J Pharm Biopharm. 2019;142:435‐448. [DOI] [PubMed] [Google Scholar]
- 5. Calquence [summary of product characteristics] . Bedfordshire, United Kingdom: AstraZeneca UK Limited; 2020.
- 6. Kaptein A, Podoll T, de Bruin G, et al. Preclinical pharmacological profiling of ACP‐5862, the major metabolite of the covalent BTK inhibitor acalabrutinib, displays intrinsic BTK inhibitory activity [abstract]. Cancer Res. 2019;79(13 suppl):2194. [Google Scholar]
- 7. Ravva P, Gastonguay MR, Tensfeldt TG, Faessel HM. Population pharmacokinetic analysis of varenicline in adult smokers. Br J Clin Pharmacol. 2009;68(5):669‐681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Alexander SPH, Kelly E, Mathie A, et al. The concise guide to PHARMACOLOGY 2019/20: introduction and other protein targets. Br J Pharmacol. 2019;176(Suppl 1):S1‐s20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Savic RM, Karlsson MO. Importance of shrinkage in empirical Bayes estimates for diagnostics: problems and solutions. AAPS J. 2009;11(3):558‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Edlund et al. Exposure–response analysis of acalabrutinib and its active metabolite, ACP‐5862, in patients with B‐Cell malignancies.. Br J Clin Pharmacol. 2021. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Details of studies and included data
Table S2. Demographics and subject characteristics (categorical variables)
Table S3. Demographics and subject characteristics (continuous variables)
Table S4. Tabulation of pharmacokinetic samples included
Figure S1. Distribution of acalabrutinib (left) and ACP‐5862 (right) pharmacokinetic sample collection times. Note: only the first BLQ of the elimination phase is included for each subject. X‐axes are cut at 32 hours, removing 2 acalabrutinib samples. BLQ, below the lower limit of quantification.
Figure S2. Acalabrutinib and ACP‐5862 concentrations vs. time after dose at different sampling times (rows). Occasions 1–3 include dense sampling occasions and sparse data include all observations collected on sparse sampling days. Studies with a similar sampling schedules have been grouped (columns). Lines connect data from the same dosing occasion within a subject. Dashed horizontal lines depict lower limit of quantification (2.1 and 10.4 nM for acalabrutinib and ACP‐5862, respectively). Data below the LLOQ are shown at LLOQ/2. CL, chronic lymphocytic leukaemia; LLOQ, lower limit of quantification.
Figure S3. Impact of covariates on typical parameter estimates in the full covariate model. The dots represent the final estimate and the bars indicate a 95% CI based on parameter precision. The vertical dashed line (value = 1) represents the no‐effect point estimate, which suggests no impact of a covariate on the typical value estimate. The shaded area represents the no‐effect range and corresponds to an approximate change of 20% with respect to the typical value estimate. BW, body weight; CI, confidence interval; CL/F, apparent clearance; CLM/F, apparent clearance metabolite; ECOG, Eastern Cooperative Oncology Group; eGFR, estimated glomerular filtration rate; F, bioavailability; HV, healthy volunteers; H2RA, H2‐receptor antagonist; Ka, absorption rate; MTT, mean transit time; PPI, proton‐pump inhibitor; Vc/F, central apparent volume of distribution; VcM/F, apparent central volume of distribution metabolite; Vp/F, apparent peripheral volume of distribution.
Figure S4. Observed versus predicted concentrations for acalabrutinib (top) and ACP‐5862 (bottom). The red line is the line of unity. The blue line is the non‐parametric smoother, and the shaded area is the 95% confidence interval. The dashed horizontal line is the lower limit of quantification (LLOQ, 2.1 nM and 10.4 nM for acalabrutinib and ACP‐5862, respectively). Observations below the LLOQ are not displayed. Axes for acalabrutinib data are cut at 0.4 nM, removing 22 acalabrutinib population predictions.
Figure S5. NPDE vs. time after dose. Non‐BLQ samples are plotted as circles and BLQ samples are plotted as asterisks. The red dashed line indicates zero. The blue line is the nonparametric smoother, and the shaded area is the 95% confidence interval. The x‐axis is cut at 32 hours (removing 2 acalabrutinib samples) to better illustrate earlier time points. BLQ, below the lower limit of quantification; NPDE, normalized prediction distribution errors.
Figure S6. NPDE vs. time after dose for healthy subjects (top) and patients (bottom). Non‐BLQ samples are plotted as circles and BLQ samples are plotted as asterisks. The red dashed line indicates zero. The blue line is the nonparametric smoother, and the shaded area is the 95% confidence interval. The x‐axis is cut at 32 hours (removing 2 acalabrutinib samples) to better illustrate earlier time points. BLQ, below the lower limit of quantification; NPDE, normalized prediction distribution errors.
Figure S7. NPDE vs. population predictions. The blue line is the nonparametric smoother, and the shaded area is the 95% confidence interval. The vertical horizontal line is the lower limit of quantification. Observations BLQ are not displayed in the figure. Axes for acalabrutinib data are cut at 0.4 nM, removing 22 acalabrutinib population predictions. BLQ, below the limit of quantification; NPDE, normalized prediction distribution errors.
Figure S8. Prediction‐corrected visual predictive check stratified by health status. The top panels show acalabrutinib data and the bottom show ACP‐5862 data. The observed data have been omitted to better visualize the percentiles. The solid and dashed lines are the median and the 10th and 90th percentiles of the observations, respectively. The shaded areas are the 95% confidence intervals of the median and the 10th and 90th percentiles predicted by the model. Data were modelled on the log scale. Prediction correction could result in values originally above LLOQ to appear to be BLQ after the correction (2.1 nM and 10 nM for acalabrutinib and ACP‐5862, respectively). BLQ, below LLOQ; LLOQ, lower limit of quantification.
Figure S9. Effect of covariates on acalabrutinib (top) and ACP‐5862 (bottom) AUC24h,ss (left) and Cmax,ss (right). The diamonds are the point estimate and the bars indicate a 95% prediction interval based on between‐subject and between‐occasion variability relative to the reference. The reference is defined as the most prevalent scenario in the population: a patient with ECOG performance status of 0 or 1 and no concomitant PPI. The dashed line indicates 1 (i.e., the reference value). AUC24h,ss, area under the plasma concentration–time curve for 24 hours at steady‐state; Cmax,ss, maximum plasma concentration at steady‐state; ECOG, Eastern Cooperative Oncology Group; PPI, proton‐pump inhibitor.
Figure S10. Acalabrutinib and ACP‐5862 final model. CL/F, apparent clearance; CLM/F, apparent clearance of the metabolite; ECOG, Eastern Cooperative Oncology Group; F1, relative bioavailability; Fm, fraction metabolized; Ka, first‐ order absorption rate constant; Ktr, transit absorption rate constant [MTT (mean transit time) = (Ntr + 1)/Ktr]; PPI, proton‐pump inhibitor; Q/F, apparent intercompartment clearance; QM/F, apparent intercompartment clearance of the metabolite.
Data Availability Statement
Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca's data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.