

Abstract
Whole‐genome sequencing of non‐model organisms is now widely accessible and has allowed a range of questions in the field of molecular ecology to be investigated with greater power. However, some genomic regions that are of high biological interest remain problematic for assembly and data‐handling. Three such regions are the major histocompatibility complex (MHC), sex‐determining regions (SDRs) and the plant self‐incompatibility locus (S‐locus). Using these as examples, we illustrate the challenges of both assembling and resequencing these highly polymorphic regions and how bioinformatic and technological developments are enabling new approaches to their study. Mapping short‐read sequences against multiple alternative references improves genotyping comprehensiveness at the S‐locus thereby contributing to more accurate assessments of allelic frequencies. Long‐read sequencing, producing reads of several tens to hundreds of kilobase pairs in length, facilitates the assembly of such regions as single sequences can span the multiple duplicated gene copies of the MHC region, and sequence through repetitive stretches and translocations in SDRs and S‐locus haplotypes. These advances are adding value to short‐read genome resequencing approaches by allowing, for example, more accurate haplotype phasing across longer regions. Finally, we assessed further technical improvements, such as nanopore adaptive sequencing and bioinformatic tools using pangenomes, which have the potential to further expand our knowledge of a number of genomic regions that remain challenging to study with classical resequencing approaches.
Keywords: long‐read sequencing, major histocompatibility complex, self‐incompatibility locus, sex‐determining region, whole‐genome sequencing
1. INTRODUCTION
Whole‐genome sequencing has become an integral part of biological research, allowing a range of long‐standing ecological and evolutionary problems to be tackled (Bourgeois & Warren, 2021). Tremendous progress has, for instance, been made in associating ecologically‐relevant phenotypes to specific nucleotide variants using forward genetic approaches such as genome‐wide association studies (GWAS) in a growing number of non‐model species. In parallel, the possibility to describe the genetic diversity of natural populations at the whole genome level, rather than at a minute subsample of genetic markers, has provided the opportunity to pinpoint the targets of natural selection under diverse ecological conditions (Exposito‐Alonso et al., 2019; Feng et al., 2019; Wright et al., 2020).
In its simplest form, whole‐genome resequencing involves obtaining DNA from a set of individuals from one species, and constructing genomic libraries indexed to facilitate multiplexing and typically of short fragments, before proceeding to high‐throughput sequencing. These sequences are then mapped to a reference genome of the focal (or a related) species before variant calling is conducted. While this “classical” approach has demonstrated its power and has become routine, there are a number of situations where it will fail due to subtle or massive biases at various stages along the process. Sequence composition is for instance known to affect representation in the genomic libraries (Kozarewa et al., 2009). Repeated sequences found across the genome typically have a highly heterogeneous distribution, being enriched in centromeres, telomeres and more generally in regions with low recombination, and are highly problematic at the mapping stage, especially when the sequencing reads are shorter than the repeat length. Similar problems arise as paralogous sequences can be mistaken for alleles of a single gene, instead of separate recently diverged genes, or vice versa. Genomes also contain regions that are highly diverse across individuals, exhibiting gene copy number variation between individuals and high degrees of heterozygosity, preventing proper alignment to any single reference. Overall, while rapid progress is being made to improve sequencing methods and to generate unbiased markers for phylogenomics (Allen et al., 2017; Zhang et al., 2019), some genomic regions still present persistent problems in assembly or data handling and interpretation. Yet, these problematic regions can be of considerable ecological and evolutionary interest, often associated with the control of very important fitness‐related traits, for example mating preferences, immune responses or other major adaptive phenotypes. These regions commonly exhibit low levels of recombination which leads to the accumulation of repeats (Figure 1). They may show long stretches of sequences in linkage disequilibrium, and also include many genomic regions or genetic systems that are evolving under strong balancing selection.
FIGURE 1.
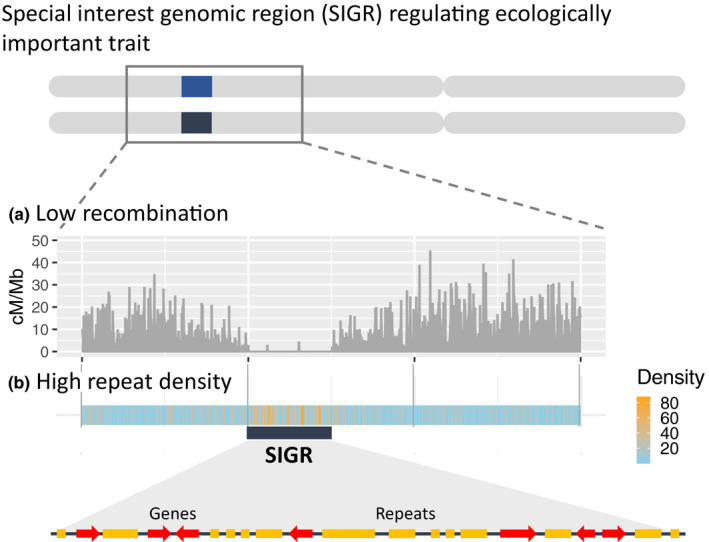
Special interest genomic regions (SIGRs) regulate ecologically important traits, but are difficult to analyse using classical genomic methods since suppressed recombination leads to high repeat densities. Haplotype divergence, represented as distinct blue and black SIGR boxes, further hampers the use of single reference genome assemblies. (a) For illustration purposes, example recombination frequencies in centimorgan (cM) per megabase pair (Mb) are shown along a chromosomal region including a SIGR. (b) The repeat density (in %) is shown for the same genomic region. Blue and orange colours indicate low and high densities of repetitive DNA sequences, respectively. In the close‐up of the SIGR, genes are shown as red arrows and repeats as orange boxes
Here, we refer to these regions collectively as “special interest genomic regions” (SIGRs). We define SIGRs as medium to large genomic blocks (>10 kb), of greater complexity than a single gene, with a particular biological function that is the target of study, and commonly with a distinctive mode of evolution. Examples of SIGRs are: major histocompatibility complex gene clusters (Yamaguchi & Dijkstra, 2019); plant heterostyly supergene (Huu et al., 2020); butterfly mimicry supergene (Jay et al., 2018); ant supergene of social organization (Yan et al., 2020); fungal mating‐type regions (Branco et al., 2017); plant multiallelic self‐incompatibility locus (Castric & Vekemans, 2004); sex‐determining regions, including recently evolved (Geraldes et al., 2015) or old sex chromosomes (Xu et al., 2019). However, some of the challenges we describe also apply to a broader set of genomic regions such as nucleolar organizing regions containing tandemly repeated sequences of ribosomal RNA genes (Handa et al., 2018; McStay, 2016), telomeres (Baird, 2018; Peska & Garcia, 2020), centromeres (Han et al., 2020; Mandakova et al., 2020) and large haplotype blocks (Todesco et al., 2020).
Special interest genomic regions present a set of characteristic challenges in simple resequencing studies. However, as we outline in greater detail below, accurate assessment of intrapopulation polymorphisms in these regions is crucial for addressing questions about the impact of important demographic events (e.g., population bottlenecks, long‐range dispersal, sudden increase in habitat fragmentation), of speciation processes, or of genomic events (e.g., whole‐genome duplication), on the biological functions associated with a given SIGR. Patterns of sequence divergence and sequence diversity in these SIGRs, when correctly evaluated, may be used to infer past evolutionary history of the organisms as they may carry unique molecular signatures of drastic changes in, for example, the mating system, or they may host genetic barriers responsible for recent speciation events. Also, comparative genomic analyses of these regions, among closely or distantly related species, may help identify the functional sequences in what are sometimes still poorly explored biological systems. Finally, accurate characterization of these regions is essential for understanding how processes of molecular evolution, such as transposable element dynamics, or accumulation of deleterious mutations, are triggered in genomic regions of reduced recombination.
In this review, we first highlight how recent technical advances are promising to substantially alleviate methodological barriers associated with the study of SIGRs, including (i) the use of long‐read sequencing technologies to improve genotyping and de novo assembly accuracy, and (ii) variation graph or pangenome approaches to better integrate haplotype diversity and haplotype divergence in bioinformatic pipelines. We then illustrate a series of approaches that have been developed to tackle such regions using examples from the major histocompatibility complex (MHC), using amplicon and long‐read sequencing (Box 1), sex‐determining regions (SDRs), using long‐read sequencing to improve de novo assembly (Box 2) and the plant self‐incompatibility locus (S‐locus), using multiple reference mapping of short‐read resequencing data (Box 3). Finally, we discuss some of the questions raised above, using results from the literature, and highlighting areas where further progress is likely to be made.
BOX 1. Assessing multiallelic diversity and copy number variation in the major histocompatibility complex (MHC) using amplicon and long‐read sequencing.
In a majority of vertebrate clades, antigen presentation by classical MHC class I and class II molecules is the first step in T‐cell dependent adaptive immune responses. T‐cells then recognize antigens as either “self” or “foreign”, and when the antigen is “foreign”, an appropriate adaptive immune response is triggered. Each MHC molecule can present a limited number of antigens and there is therefore selection for expressing several different MHC molecules to gain the ability to eliminate a wider range of pathogens (Murphy & Weaver, 2017). Classical MHC genes are among the most polymorphic genes known in vertebrates. In humans, for example, there have been 7,967 different alleles reported for the HLA‐B gene, as reported on the HLA alleles website (http://hla.alleles.org/nomenclature/stats.html; accessed May 2021). The MHC genes are also polygenic, meaning that there are several gene copies (paralogues) with different degrees of similarity. The high MHC polymorphism is mainly maintained by selection from a wide range of pathogens, the selective mechanisms being negative‐frequency dependent selection, fluctuating selection and heterozygote advantage (Doherty & Zinkernagel, 1975; Hedrick, 2002; Takahata & Nei, 1990). However, a considerable number of studies have also shown that an MHC‐based mate choice can play a significant role in maintaining high MHC polymorphism. This can for example be in the form of mate choice for partners with specific MHC haplotypes, or mate choice to maximise MHC‐diversity (number of MHC alleles per individual including several paralogues) in the offspring (Kamiya et al., 2014).
Human MHC genes are found spread over a 7 Mb (megabase pair) genomic region on chromosome 6, where the MHC genes are linked but interspersed with non‐MHC genes (Chin et al., 2020; Jain et al., 2018). Human MHC (human leucocyte antigen [HLA]) class I alleles in open reading frame are found at six different loci (HLA‐A, ‐B, ‐C, ‐E, ‐F, ‐G) and the majority of these have orthologous loci in other great apes (Hominidae), lesser apes (Hylobatidae), and other primate clades (monkeys) (Shiina & Blancher, 2019). Interestingly, several of the MHC class I genes have been tandemly duplicated in monkeys as characterized in the cynomolgus macaque, Macaca fascicularis (Shiina & Blancher, 2019). Cynomolgus macaques have several MHC class I loci with a large number of paralogues, whereas humans only have a single MHC class I gene copy per locus (Figure 3). The structural genomic organization of MHC is relatively conserved among mammals but strikingly different in other vertebrate groups such as fish and birds (Balakrishnan et al., 2010; Chen et al., 2015; Shiina et al., 2007; Yamaguchi & Dijkstra, 2019). There has been a particular interest in studying MHC in wild birds, probably because early on it was hypothesized that MHC could influence traits that affect mate choice (Zelano & Edwards, 2002). Songbirds have highly duplicated MHC class I and IIB genes, many paralogues, and although the number of MHC alleles per individual, over a large number of paralogues, can be measured using high‐throughput amplicon sequencing, the genomic organization of the MHC genes is still to a large extent unknown (Balakrishnan et al., 2010; Biedrzycka et al., 2017; O'Connor et al., 2016; Sutton et al., 2018).
FIGURE 3.
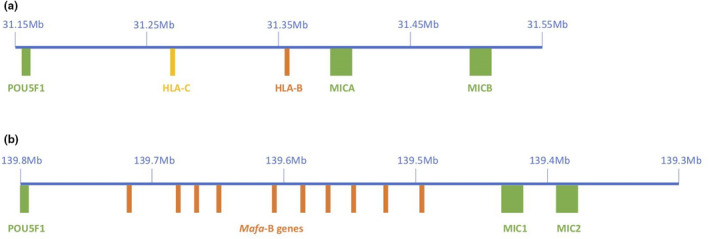
The core MHC region in humans (Chromosome 6) holds six MHC‐I genes in open reading frame (HLA‐A to ‐C and HLA‐E to ‐G), and the core MHC region in cynomolgus macaque Macaca fascicularis (Chromosome 4) holds five orthologous MHC‐I loci (note: no MHC‐C locus). A subset (400 kb in human and 500 kb in cynomolgus macaque) of these homologous genomic MHC regions is shown here, the flanking genes POU5F1 and MIC genes (green boxes), and the MHC‐I genes (orange and yellow boxes) being indicated. (a) MHC class I region in human (HLA) with single HLA‐C and HLA‐B gene copies (locations from ensembl GRCh38.p13 assembly chromosome 6) and (b) MHC class I region in cynomolgus macaque with tandemly duplicated MHC‐B gene copies (Mafa, MHC‐B genes (Watanabe et al., 2007), POU5F1 and MIC locations from ensembl Macaca_fascicularis_5.0 assembly chromosome 4). Note: MHC class I gene(s) at the B locus is called HLA‐B in humans and Mafa‐B in cynomolgus macaque
In summary, classical MHC genes; (i) have high polymorphism, (ii) have high sequence divergence among alleles within species in the exons that encode the antigen binding region but low sequence divergence in the exons that encode the structural parts, (iii) are often found in a synteny of tandemly duplicated paralogues within loci, and (iv) are found in genomic regions with accumulation of repetitive sequences (TEs).
Amplicon‐based technologies to characterize MHC diversity have been used successfully in a wide range of non‐model organisms (Minias et al., 2019; O'Connor et al., 2018), whereas short‐read de novo assembly approaches are challenging due to gene copy number variation between individuals within species. Added to this is the high polymorphism and low allelic divergence in both newly duplicated gene copies and genes subjected to gene conversion. Due to the repetitive nature of the MHC region it is difficult to assemble, as individual sequence reads from short‐read technology do not span gene copies and hence several MHC gene copies or clusters are often collapsed into a single MHC gene or cluster. This results in an underestimation of the true number of MHC genes. However, due to the high degree of heterozygosity the true number of MHC gene copies can also be overestimated by mistakenly counting MHC alleles instead of MHC genes. Long‐read technologies are helping to address these issues. For example, a human genome assembled from ONT data assembled all class I HLA genes on a single 3 Mb contig (Jain et al., 2018). Similarly, a de novo genome assembled for the water buffalo using PacBio reads resulted in characterizing all MHC class II genes on a single 218 kb contig, whereas this region previously had 26 gaps when assembled using short reads (Low et al., 2019).
A further advantage of long‐read sequences is the ability to phase assemblies and haplotype contigs in order to recover information on allelic variation. In de novo human genomes the accurate recovery of known HLA alleles has become a benchmark to assess the success of various long‐read sequencing and assembly strategies. However, it should be noted that the HLA genes are single‐copy, whereas in cynomolgus macaques, for example, they are tandemly duplicated with short intergenic distances (see, e.g., the studies of Chin et al., 2020; Jain et al., 2018; Nurk et al., 2020).
Long‐read sequencing is providing us with increasingly contiguous reference assemblies, although population‐level assessment of MHC diversity within these genomes is still challenging. Long‐amplicon sequencing of full‐length HLA genes can circumvent some of the allele‐calling issues with short‐read sequencing. Another promising technique on the horizon is ONT’s adaptive sequencing, where selective sequencing of parts of the genome is possible without specific wet‐laboratory preparation, as discussed for example in Dilthey (2021) and Payne et al. (2021).
BOX 2. Characterizing sex‐determining regions (SDRs) in plants using long‐read sequencing technologies and de novo assembly.
The problems and potential of studying SDRs are broadly the same in both animals and plants (Charlesworth & Mank, 2010); however, here we will concentrate on plant systems as they are considered to be particularly labile (Käfer et al., 2017). Land plants (Embryophyta) have “alternation of generations” and alternate between diploid and haploid stages of the life cycle, with the haploid stage producing sperm and eggs. In plants such as mosses, in which the haploid stage is dominant, separate sexes (when present) are determined genetically by a UV system: plants either inherit a U chromosome (female) or a V chromosome (male). The diploid stages are sexless having both U and V chromosomes. Seed plants, in contrast, have a dominant diploid stage in which sex is genetically determined by an XY or ZW system, while the haploid stage acquires its sex epigenetically from the diploid parent. In fact, most land plants are cosexual, having both male and female reproductive structures on the same individual. However, unisexuality (dioecy) has evolved frequently, and instances are scattered across the flowering plant phylogeny (Renner, 2014). Nevertheless, 43% of all dioecious angiosperms are found in just 34 entirely dioecious clades (Renner & Müller, 2021).
The evolution of separate sexes has considerable ecological consequences (Lloyd, 1982), in plants no less than in animals, as males and females may face different selection pressures, particularly if their costs of reproduction differ (Queenborough et al., 2007). There are a number of studies showing that males and females can occupy different ecological niches (Freeman et al., 1976). Dioecy is a major mechanism promoting outbreeding, along with the self‐incompatibility (SI) systems discussed below. It has been suggested that dioecy may evolve more easily than SI systems (Thomson & Barrett, 1981), and through different alternative evolutionary pathways (Dufay et al., 2014). This supposed easy evolvability might explain the frequency of dioecy on certain islands, where it has apparently recently evolved from self‐compatible immigrants (Baker & Cox, 1984; Thomson & Barrett, 1981). The dioecious mating system also interacts strongly with hybrid zone dynamics (Pickup et al., 2019), and provides a uniparentally inherited marker of considerable use in phylogeography (Jobling & Tyler‐Smith, 2017). Comparative whole‐genome sequencing studies, at the population or interspecific level, are particularly promising for studying contrasting evolutionary histories of male and female lineages. For instance, paternal versus maternal haplotypes may show different directionalities of introgression across hybrid zones, may have different geographical origin, and may have different times to the most recent common ancestor. Such differences can point to important undiscovered aspects of species biology.
A further question concerns the molecular pathways involved in the evolutionary transitions between different patterns of sexual development, for example, between hermaphrodite flowers and unisexual flowers (monoecy and dioecy) in which male or female floral organs have been deleted or reduced. It is probable that many different molecular mechanisms underlie the evolution of unisexual flower development (Diggle et al., 2011). Parallel evolution (i.e., the reuse of the same underlying molecular developmental mechanisms) could also play a role. To answer this question, again it is crucial to identify the sex‐determining genes or sequences in diverse dioecious plant species.
With the advent of long‐read sequencing, entirely new possibilities are available to the molecular ecology community. High quality draft genomes can be assembled with unprecedented levels of contiguity. Examples of the use of long‐read sequencing for the elucidation of sex determination are provided by studies in asparagus and poplar (Harkess et al., 2017, 2020; Müller et al., 2020). In both genera, hemizygous sequences in the male‐specific region of the Y chromosome (MSY) are essential for sex determination. In poplar, a strong candidate gene for sex determination, the response regulator ARR17, was proposed before (Geraldes et al., 2015). However, only with the use of long ONT and PacBio reads, partial duplicates of the ARR17 gene were revealed in the MSY of different poplar species (Müller et al., 2020) (Figure 4). With short reads from the Illumina sequencing platform, these duplicated sequences collapse into a single genomic region. This is a common problem with duplicated sequences that still have high similarity to each other. Long‐read sequencing helps to overcome this difficulty.
FIGURE 4.
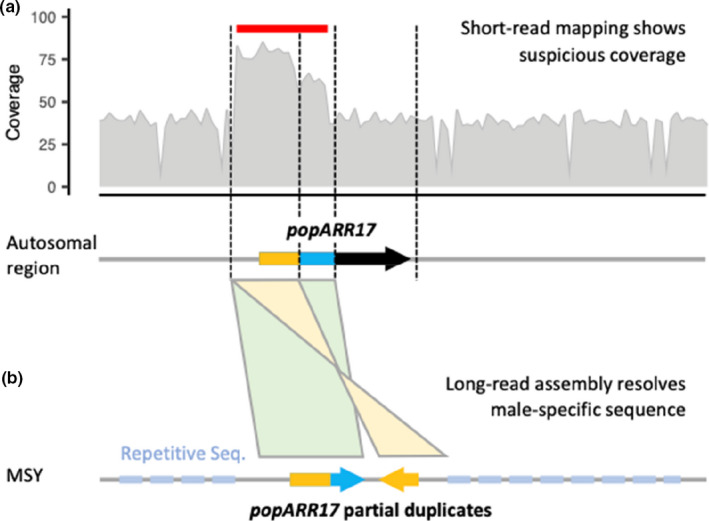
Long‐read sequencing allows assembly of complex genomic regions. (a) Coverage plot of a short‐read mapping of a male Populus individual against the (female) reference genome, showing that the genome region with the popARR17 gene exhibits suspicious sequencing coverage. This region, indicated by a red vertical bar, exhibits 1.5‐ to 2‐fold higher coverage than expected. (b) Long‐read assembly of the male‐specific sequence of the Y chromosome (MSY) reveals partial popARR17 duplicates nested within repetitive sequences, readily explaining inconsistencies in short‐read mappings. In this case the regional repeat architecture was resolved largely using ONT sequencing (Müller et al., 2020)
BOX 3. Analysing balanced polymorphisms at the plant self‐incompatibility locus using multiple reference mapping of short‐read resequencing data.
About 40% of hermaphrodite flowering plant species possess a self‐incompatibility (SI) system that enforces outcrossing by recognition and rejection of self‐pollen (Igic et al., 2008). In many cases, a single genomic region (called the S‐locus) contains the SI genes, which typically present extreme variability because of a combination of high allelic diversity and high divergence caused by the maintenance of haplotypes at these genes over extended periods of time. This is driven by a form of balancing selection, specifically negative frequency‐dependent selection (Castric & Vekemans, 2004). SI systems are an example of a field studied by two distinct scientific communities in parallel. First, molecular physiologists have worked out in exquisite detail the mechanisms by which self‐pollen is recognized and rejected in different plant families. This has highlighted the existence of two categories of SI systems: self‐recognition systems and nonself‐recognition systems. The self‐recognition systems involve only two cognate genes, one expressed in the pollen and the other in the pistil. Conversely, the nonself‐recognition systems involve a single pistil gene but up to twenty pollen‐expressed genes located at the S‐locus that collaboratively determine the paternal SI phenotype (Iwano & Takayama, 2012). Second, in parallel, population geneticists have developed detailed predictions for how variation at these genes should be influenced by natural selection (Charlesworth et al., 2005). At the population level, the frequency at which S‐alleles segregate is predicted to be strongly affected by negative frequency‐dependent selection, in the simplest cases causing alleles to be found at frequencies that are more homogeneous than expected for genes evolving under selective neutrality. Furthermore, negative frequency‐dependent selection promotes higher effective gene flow among populations (Schierup et al., 2000), leading again to greater homogeneity than expected from neutrality. Whole‐genome resequencing surveys in families that evolved independent SI systems could be highly valuable to test theoretical predictions about the distribution of S‐alleles within and among populations, in relation to their demographic history, as part of a search for convergent evolutionary properties. However, population‐level variation has remained poorly documented in the empirical literature on SI systems as the S‐locus combines several features that make it technically challenging to analyse. These are: (i) very high allelic diversity, (ii) high sequence divergence among alleles, (iii) the occurrence of multiple genes at the S‐locus functioning collaboratively to produce the male self‐incompatible phenotype in nonself recognition systems (e.g., S‐RNase system of Solanaceae), (iv) strong accumulation of repetitive sequences (TEs) associated with the absence of recombination at the S‐locus (Goubet et al., 2012), and (iv) the existence of many paralogues for the SI genes including co‐evolving paralogues that modulate the SI reaction, for example, the SLG paralogue in Brassica (Takasaki et al., 2000). Together, these features have strongly hindered the large‐scale use of both amplicon‐based technologies and short‐read de novo assembly approaches in the survey of S‐locus diversity in population studies (reviewed in Bachmann et al., 2018 and Genete et al., 2020).
Recently, however, short‐read resequencing data were shown to be of high value when used in a mapping approach that attempts to map sequence reads sequentially against each of the previously obtained reference S‐locus sequences, instead of against a single genomic reference (Figure 5). Such an approach has been used successfully to obtain exhaustive genotypic characterization of the S‐locus in cultivars of the apple tree, Malus domestica (De Franceschi et al., 2018), and in populations of Arabidopsis halleri (Genete et al., 2020) and A. lyrata (Mable et al., 2018; Takou et al., 2021), two species where molecular polymorphism of the SRK gene had been the focus of several previous studies (though it remained incompletely described). A dedicated bioinformatic pipeline implementing this approach has been developed (Genete et al., 2020) and is available at https://github.com/mathieu‐genete/NGSgenotyp. It computes a series of mapping statistics to help identify the matching reference S‐alleles (Figure 5c) and also uses a de novo assembly approach to detect new S‐allele sequences that can in turn enrich the reference database (Figure 5b). The power of this approach was demonstrated by obtaining complete genotyping tables from the analysis of short‐read resequencing data from 56 and 46 individuals of A. halleri (Genete et al., 2020) and A. lyrata (Takou et al., 2021), respectively. These data allowed the detection of nine and 12 putative new S‐alleles for these two species, bringing the overall species‐wide number of S‐alleles currently detected in A. halleri and A. lyrata to 63 and 58, respectively, with about 50 alleles shared between species (X. Vekemans, unpublished data). This pipeline could, in principle, be applied to other highly polymorphic loci such as the S‐RNase self‐incompatibility system of Solanaceae, Plantaginaceae or Rosaceae, the MHC (only applicable to MHC genes for which paralogous sequences can be excluded), the sex‐determining gene in honeybee (CSD gene), or multiallelic mating‐type loci in fungi.
FIGURE 5.
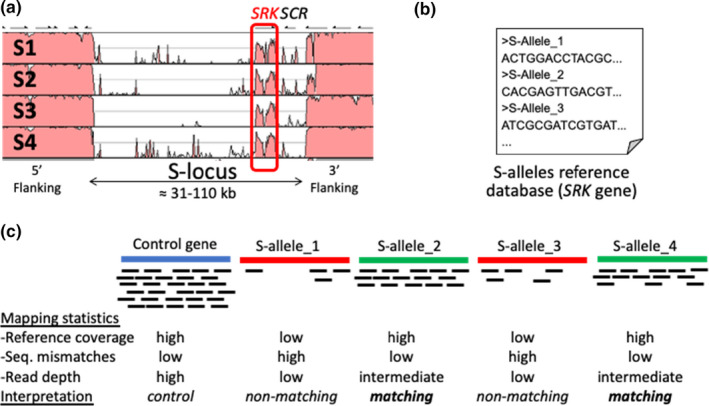
Strategy to infer S‐locus genotypes from individual short‐read resequencing data. (a) VISTA plot of haplotypes at the S‐locus of Arabidopsis halleri showing extremely low sequence conservation, except for the pistil‐expressed gene SRK (adapted from Goubet et al., 2012). (b) Schematic representation of a database of fasta sequences from previously known alleles at the SRK gene, which are used as references for sequential mapping of short‐reads from individual resequencing data. (c) Schematic representation of the results of sequential mapping against control genes (i.e., single‐copy genes with low polymorphism) or against nonmatching versus matching S‐alleles. The focal individual is heterozygous for S‐allele_2 and S‐allele_4, as mapping statistics against these two references are reporting high coverage of the reference sequence (proportion of positions with at least 1 read aligned), low sequence mismatches (between the reads consensus and the reference sequence), and intermediate (about half) read depth as compared to that for control genes (because of heterozygosity at the S‐locus)
However, some further difficulties persist in some of these systems. For instance, in S‐RNase SI systems of Maloideae (e.g., apple or pear trees), the pistil expressed gene, S‐RNase, has only two short exons separated by an intron highly variable in size and rich in repeat elements, for example, Dreesen et al. (2010). In such cases, the mapping approach that involves sequential mapping against each of the known reference S‐allele sequences (Figure 5) may be less successful than in SI systems that have a single large exon covering the pollen‐pistil recognition domains. Hence, long‐read sequencing technologies may be necessary to allow successful genotyping in those systems. Other developments of the approach could include using a previously established reference database of S‐allele sequences to generate multiple targets for gene capture experiments, followed by multiplexed Illumina sequencing, thus allowing the production of a powerful and affordable S‐locus genotyping platform for large population surveys. Alternatively, such a reference database could be used, in combination with the new real‐time Oxford Nanopore Technology, to perform selective sequencing of the S‐locus alleles.
2. TECHNICAL ADVANCES AND SOLUTIONS
2.1. Third generation sequencing: long reads, single molecules
Long‐read sequencing, also called third generation sequencing, became widely available in 2011, when Pacific Biosciences (PacBio) commercialized the “single‐molecule real‐time” (SMRT) methodology (Amarasinghe et al., 2020; Eid et al., 2009). In 2014, Oxford Nanopore Technologies (ONT) followed this by releasing the MinION device, enabling Nanopore sequencing in almost any laboratory. Currently, both technologies generate long reads with N50 values of approximately 30 kb (that is: half of the sequencing data consists of reads ≥30 kb). In addition, the ONT platform allows for ultra‐long read sequencing with N50s exceeding 100 kb. However, the DNA preparation for generating ultra‐long reads can be challenging, especially when working with difficult organisms, with tough cell walls and problematic secondary chemistry, such as xerophytic plants (Schalamun et al., 2019).
The PacBio and ONT sequencing technologies follow very different principles. The PacBio SMRT sequencers measure fluorescence signals from labelled bases incorporated by a polymerase that synthesizes the complementary strand of a single circular DNA molecule. By sequencing the same molecule repeatedly, sequencing errors can be corrected. On the ONT platform, single DNA strands are passed through biological nanopores with an enzyme attached, measuring the changes of the electrical current. Different bases have different resistances, therefore allowing the sequence to be inferred from the current changes. The base calling algorithm is crucial in defining the raw read accuracy. Both methods suffered initially from comparatively high error rates of more than 10%, but recent developments have led to dramatic improvements, notably PacBio's highly accurate HiFi sequencing giving per base accuracies of >99.9%. This opens completely new opportunities for genome assembly (Nurk et al., 2020).
For the assembly of complex genomic regions, PacBio HiFi and ONT ultra‐long read sequencing are at present complementary approaches and the optimum technology to use depends on the specific characteristic of the genome of interest. In the case of near‐identical large repeats, the longer read length of the ONT platform provides an advantage, while highly repetitive segmental duplications can be better resolved using HiFi data (Nurk et al., 2020). The two methods, taken together, enable genome assemblies of impressive quality, and can generate single contigs representing chromosomes from telomere to telomere, even spanning complex centromeres (Belser et al., 2021; Jain et al., 2018; Miga et al., 2020; Phillippy, 2020). Special interest genomic regions like the MHC‐locus (Box 1) and the plant S‐locus (Box 3) may be used as a benchmark of the quality of an assembly. Indeed, previous attempts using short‐read technology have proven to be ineffective for proper assembly of the S‐locus region that is strongly enriched in transposable elements (TEs), even when sequenced from bacterial artificial chromosome (BAC) genomic libraries (Figure 2a) (Goubet et al., 2012). Improvements of the approach involving BAC clones was provided by using long‐read PacBio sequencing technology, which produced an exhaustive assembly of the S‐locus region in several haplotypes (Figure 2b), although it still necessitates the construction and screening of BAC genomic libraries (Bachmann et al., 2018). However, it has been shown recently that long‐reads obtained by ONT allow reconstruction of the entire S‐locus region in Brassica rapa and B. oleracea using direct individual whole‐genome de novo assembly (Belser et al., 2018). In addition, for an individual of Arabidopsis halleri heterozygous at the S‐locus, it has proved possible to obtain sequences of the full S‐locus region, for both S‐haplotypes, using the latter technology (Figure 2c; V. Castric, unpublished data).
FIGURE 2.
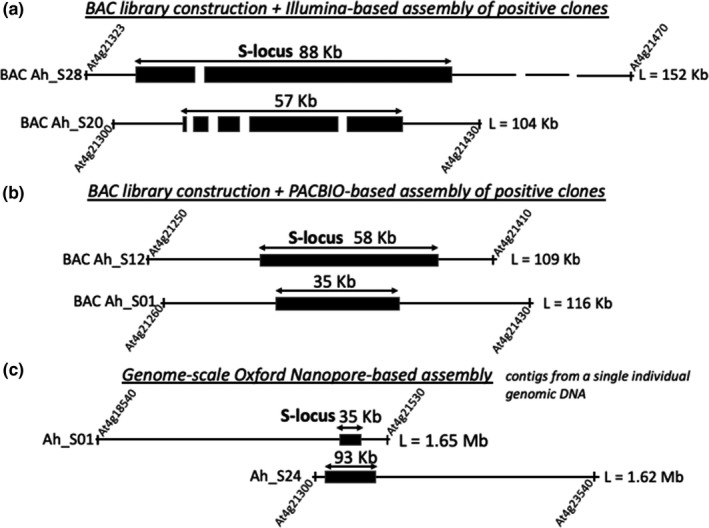
Influence of sequencing technology on level of assembly of individual haplotypes at the self‐incompatibility locus (S‐locus) in Arabidopsis halleri. (a) BAC clones sequenced using short‐read Illumina technology, showing fragmented assembly of the S‐locus despite starting from BAC clones (Goubet et al., 2012). (b) BAC clones sequenced using long‐read PacBio technology, showing complete assembly of the S‐locus after construction of BAC libraries and screening for the S‐locus region (V. Castric, unpublished data). (c) Whole‐genome sequencing using Oxford Nanopore technology, showing large contig sizes encompassing the full S‐locus for the two S‐haplotypes of a heterozygous individual (V. Castric, unpublished data). Limits of the contigs are annotated using A. thaliana gene IDs
Badouin et al. (2021) recently proposed a new generic method using third generation sequencing technology to identify genetic systems evolving under strong balancing selection, such as plant self‐incompatibility systems, in unexplored lineages. The strategy involves a pooled transcriptomic approach based on long‐reads obtained from tissues of interest (e.g., pollen and pistil for self‐incompatibility). The full transcripts are then mapped on a reference genome. By focusing on candidate loci that exhibit simultaneously high haplotype diversity and high sequence divergence among haplotypes, the method avoids potential pitfalls associated with hidden paralogy.
2.2. Variation graph and pangenome approaches
The extent of progress made on the quality of reference genome assemblies is impressive. However, it is now possible to go beyond this and determine the extent of structural variation (i.e., genetic polymorphisms in which sections of a genome differ in structure between individuals of the same species). Structural variation is now being revealed with unprecedented accuracy, a recent example being the discovery of polymorphism of large haplotype blocks in the genome of sunflower (Todesco et al., 2020). The majority of structural variants (SVs) are associated with the activity of transposable elements, and a substantial fraction of structural variation overlaps genes or regulatory elements. Their contribution to phenotypic diversity (and their long‐term evolutionary impact) is becoming more apparent, with SVs responsible for changes in gene expression or dosage (Alonge et al., 2020; Baduel et al., 2021). Recent bioinformatic methods have been developed to take full advantage of this better appreciation of genomic diversity, in the form of variant graphs (Garrison et al., 2018; Hickey et al., 2020). In these approaches, the genome is not simplified as a single linear stretch of nucleotides, but is rather represented as an enhanced data structure (a graph) that integrates the full collection of single nucleotide or structural variants (the “pangenome”). Sequencing reads can then be aligned and compared, and variants called on this pangenome rather than on just a single reference genome. This technical difference is particularly important in the context of highly heterozygous diploid genomes, where the strategy of aligning sequencing reads on a haploid reference may be especially problematic. Similar concerns also arise in cases where samples from closely related species are aligned to the reference genome of just one (model) species, introducing potentially important biases in the mapping properties among samples. Overall, taking into account the structure of pangenomes is likely to result in more accurate mapping for short‐read data, for example, Llamas et al. (2019).
Structural variation or high divergence among haplotypes are inherent to many SIGRs, and there are several approaches to take full advantage of the properties of pangenomes. For example, as the MHC genes are highly polymorphic it can be problematic to assign amplicon sequences or RNA‐seq reads to a specific locus in the reference genome (Box 1). Indeed, mapping of reads using a reference that included alternative haplotypes led to more accurate estimates of allele‐specific expression in a study with RNA‐seq data from human HLA genes (Lee et al., 2018), and mapping and SNP‐calling were also improved in a genome‐wide resequencing analysis using a pig pangenome (Tian et al., 2020). Along the same lines, Tsuchimatsu et al. (2017), followed by Genete et al. (2020), used sequential alignment to a series of S‐locus references for efficient and high throughput genotyping of large collections of individuals from natural populations in Arabidopsis thaliana and A. halleri, something that had remained a major technical challenge in the field (Box 3).
3. DISCUSSION
3.1. Finding functional determinants in SIGRs
The approaches described above have the potential to help characterize the functional determinants of the phenotypic traits encoded within these regions, such as the choice of mating partners or the spectrum of immune response. When characterizing the immune response, knowledge about the synteny of MHC genes is important as it can help unravel function, for example single‐copy MHC genes and tandemly duplicated MHC genes might encode MHC molecules with different functions. Moreover, the different gene copies among tandemly duplicated genes may, for example, have evolved neofunctionalizations or different degrees of expression (Greene et al., 2011; Shiina & Blancher, 2019). We do not yet know how to interpret the limited information available about synteny in the MHC genomic regions in non‐model organisms, and it is probably too early to draw conclusions about synteny differences between species outside mammals. However, with better knowledge about the organization of MHC genomic regions from a larger number of jawed vertebrates, it will be possible to interpret synteny and to measure the degree of heterozygosity per MHC gene instead of across all MHC gene copies. This information will help to clarify to what extent MHC genes encode MHC molecules with similar function in the immune system and hence can be placed in a single category, and to what extent they encode MHC molecules with possibly different functions and hence preferably should be placed in several different categories, in terms of ecological and evolutionary processes. Both human and chicken have well assembled MHC genomic regions and the synteny of the MHC genes can therefore be reflected upon (Shiina et al., 2007, 2009). Humans have single copy MHC‐I genes, with different functions: the MHC genes HLA‐A to ‐C encode classical highly polymorphic MHC molecules with antigen presenting ability, whereas the MHC genes HLA‐E to ‐G encode nonclassical MHC molecules that are more monomorphic and with less clear tasks in the immune system and in self/nonself recognition. A different synteny and genomic organization is seen in domestic chicken (Kaufman et al., 1999; Shiina et al., 2007). Here, the single copy MHC‐I genes are classical (i.e., acting via T cells) whereas the tandemly duplicated MHC‐I genes are nonclassical (i.e., acting via natural killer, NK, cells; e.g., MHC‐Y). This is just a conceptual comparison (the classical and nonclassical MHC‐I genes in humans and chicken are not orthologous) to point out the drastically different MHC organizations we expect to be found in accurate assemblies of the MHC genomic regions in future long‐read genomes.
In plant self‐incompatibility, the availability of high‐quality assemblies of several highly divergent haplotypes at the S‐locus in A. halleri (Goubet et al., 2012) allowed identification of a dozen of small RNA‐producing loci that control the dominance relationships among self‐incompatibility alleles, acting as Fisherian dominance modifiers (Durand et al., 2014, 2020). In Petunia (Solanaceae), the S‐locus is also highly polymorphic and extends over about 15 to 20 Mb (Kubo et al., 2015), making it very difficult to assemble. Extensive analyses using amplicon techniques and BAC library screening identified up to 20 pollen‐expressed genes within a single haplotype at the S‐locus of Petunia, which are collaboratively contributing to recognition and detoxification of the nonself pistil‐expressed proteins (S‐RNases) produced by mating partners (Kubo et al., 2015; Wu et al., 2020). In order to fully understand this collaborative functional process, and its evolution, studies using more efficient sequencing and assembly approaches are necessary to obtain full S‐locus sequences from many haplotypes.
In sex chromosome studies in non‐model organisms, the first step consists of identifying the SDR according to different strategies (Palmer et al., 2019). The first approaches relate to the absolute numbers of resequencing reads mapping to scaffolds. Scaffolds with low coverage are suggestive of Y or W linkage. Alternatively, the ratio of male to female reads that map can be used. However, these approaches assume that there are correctly assembled scaffolds covering the SDR. Misassembly of the SDR renders these methods uninformative at best, highly misleading at worst. In such cases an assembly‐free k‐mer strategy is preferred. Genomes from males and females are broken into k‐mers (i.e., all possible subsequences of length k), then k‐mers that are autosomal, versus those which are sex‐linked, can be determined by read count (Akagi et al., 2014; Carvalho & Clark, 2013; Neves et al., 2020; Torres et al., 2018). Another approach involves whole genome re‐sequencing of a population of males and females followed by a genome‐wide association study (GWAS) to find sex‐associated SNPs (Geraldes et al., 2015). GWAS may be used in wild populations but requires a relatively high number of individuals in the resequencing population to eliminate false positives. The combination of resequencing results from closely related species can greatly assist the elimination of false positives (Geraldes et al., 2015). An alternative to GWAS is to study segregation directly in a cross under a probabilistic framework, as implemented by the SEX‐DETector model (Muyle et al., 2016). SNP data from parents and progeny are used to probabilistically assign SNPs to three segregation types: autosomal, X/Y‐linked pairs and hemizygous. Examples of the use of the SEX‐DETector model are grapevine (Badouin et al., 2020) and Cannabis (Prentout et al., 2020). An important point to be borne in mind is the necessity of properly accounting for linkage disequilibrium (LD). Some studies exclude loci that exhibit high LD, whereas it can be a signal of reduced recombination around features of interest including sex‐determining regions (McKinney et al., 2020).
3.2. Inferring demographic or life history changes from SIGR polymorphism data
When a high‐quality assembly of a SIGR is available, carefully deployed short reads from a resequencing experiment using genome reduction techniques can be used to elucidate the evolutionary and ecological ramifications, such as sex‐specific demographic changes due to life‐history evolutionary changes or due to cultural changes, to mention just a few examples. In humans, as a result of Y‐genotyping, patrilineal demographic events can be precisely dated. One extraordinary finding has been that if the effective population size of males and females is modelled back in time using matrilineal (mitochondrial) and patrilineal (Y‐chromosome) data, the male line (but not the female) shows a marked bottleneck around 7–5 kya (Karmin et al., 2015). This observation has been attributed to the sociocultural phenomenon of increased formation of, and competition between, patrilineal kin groups (Zeng et al., 2018). Such inferences, and many like them, would have been impossible without two decades of intense study of Y haplogroups. Long‐read whole‐genome resequencing will be of considerable importance going forward, particularly to reveal Y‐sequences in individuals that are not present in the reference genomes. Population genomic analyses of SDRs can also reveal sex‐linkage of species isolating factors. In docks (Rumex) (Beaudry et al., 2020), a large demographic study has been carried out on R. hastatulus to test the importance of sex chromosomes on reproductive isolation (using genome reduction methods rather than whole‐genome resequencing). In this case the species is polymorphic for two noninterbreeding sex‐systems (XY and XYY) and the formation of the XYY cytotype (estimated at ~200 kya) was apparently soon followed by genetic isolation, consistent with the hypothesis that the origin of the XYY contributed to reproductive isolation. This particular example lends itself to genome reduction methods, but in cases of very small SDRs whole‐genome resequencing approaches are not only advantageous but necessary.
Application of novel approaches to exploit whole‐genome resequencing data generated for other purposes, allows to extract accurate polymorphism data from SIGRs such as the plant S‐locus (Box 3). These should in particular be valuable for studying the effect of major demographic or genomic (e.g., polyploidy) events on S‐locus diversity and evolution. For instance, based on short‐read resequencing data from two populations of A. lyrata, one of which having experienced a strong genetic bottleneck about 70,000 generations ago, no difference in S‐allele diversity was observed between the two populations, suggesting strong resilience of the S‐locus to demographic events thanks to strong balancing selection (Takou et al., 2021). Analyses of population data could also reveal possible shifts in mating systems in peripheral populations, as for instance a shift to a selfing regime, which is considered the most common evolutionary trend in flowering plant reproduction (Stebbins, 1974). Such shift would lead to a signature of drastic reduction in allelic diversity at the S‐locus (Novikova et al., 2017; Shimizu et al., 2004; Vekemans et al., 2014) besides other consequences affecting genome‐wide patterns of molecular evolution (Wright et al., 2008). Even very ancient shifts in mating systems could be detected by comparing phylogenetic patterns of S‐alleles among species, as suggested by Leducq et al. (2014) who observed two phylogenetic clusters of S‐alleles at the S‐locus of Biscutella neustriaca which they interpret as signatures of a transitory loss of ancestral SI, followed by re‐activation of functionality and allelic rediversification from two ancestral S‐allele lineages.
Similarly, maintenance of functional diversity after strong bottlenecks has been observed at MHC in the critically endangered Raso lark, Alauda razae (Stervander et al., 2020). Indeed, despite low homozygosity at most MHC loci, diversity was maintained through retention of a high number of gene copies, aided by cosegregation of multiple haplotypes comprising 2–8 linked MHC‐I loci. This highlights the importance of assessing not only single locus polymorphism, but also copy number variation at the MHC. In contrast, very low allelic diversity, and only three MHC loci were observed in the Chinese alligator that went almost extinct in the 1970s and is the subject of active conservation management (Zhai et al., 2017).
3.3. Studying patterns of molecular evolution within SIGRs
The availability of entire sequences of SIGRs in different haplotypes would allow to better understand patterns of molecular evolution in these highly heterozygous and nonrecombining regions. In particular, such regions are expected to accumulate a “sheltered” load of deleterious variants whose removal by purifying selection is rendered less efficient by linkage to the balanced polymorphism (Jay et al., 2021; Uyenoyama, 2005). In self‐incompatible plants, revealing this sheltered load requires carefully designed controlled crosses, and so far only two empirical studies have documented this effect (Llaurens et al., 2009; Stone, 2004). First, there is a clear need to extend this kind of analysis to more study systems to determine how general this phenomenon actually is. Second, a major puzzle is that the S‐locus region typically contains very few genes, being even limited in Arabidopsis to only the genes directly involved in the self‐recognition phenotype to the exclusion of any other protein‐coding gene. Hence, the deleterious load can only be caused by variants of the genes that are flanking the S‐locus region. In these conditions, obtaining reliably reconstructed haplotypes not only of the S‐locus region itself, but also of the linked region will be crucial to determine the extent of the genomic tract upon which the strong balancing selection acting on the S‐locus negatively interferes with the removal of linked deleterious variants by purifying selection. Similarly, accurate assembly, using 10x linked‐reads technology, of divergent haplotypes generated by large inversion polymorphisms associated with different wing colour patterns in mimetic butterflies, has been instrumental in highlighting evidence for accumulation of deleterious mutations sheltered in heterozygous genotypes (Jay et al., 2021). Once this load of deleterious mutations has accumulated in different haplotypes, it has been demonstrated to play an important role in maintaining high levels of heterozygosity and hence in maintaining the balanced polymorphism. A similar enrichment of deleterious variants in regions linked to balanced polymorphisms has been documented in the human MHC region (Lenz et al., 2016).
4. CONCLUSION
Overall, our goal with this review is to summarize recent progress in a variety of study systems that control essential biological functions but have proven challenging to study because of technical limitations in the sequencing methods. We show that by adapting molecular and bioinformatic methods to particular systems, each with their own set of peculiarities, it is possible to obtain reliable information on large samples of natural populations, sometimes even from short‐read data alone. Our hope is that in the next few years, many of these technical challenges will be lifted by further progress in sequencing methods, allowing many of these regions to finally be properly represented in population‐scale de novo assemblies of non‐model organisms. How fast this will happen remains to be determined, but meanwhile we hope that our review will inspire the study of these fascinating genetic systems in a broader range of study species, as well as encourage the development of further technical improvements extending to other difficult study systems.
AUTHOR CONTRIBUTIONS
All authors contributed to conceptualization of ideas, and to writing and editing the manuscript.
5. CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGEMENTS
We wish to thank Andrew Foote, Evelyn Jensen, Rebecca Taylor and David Coltman, editors of the special issue "The use of whole‐genome sequences in molecular ecology" for inviting us to write this review. We thank three anonymous reviewers for their helpful comments which greatly improved the manuscript. The work of X.V. and V.C. is supported by the Agence Nationale de la Recherche (TE‐MoMa project, grant ANR‐18‐CE02‐0020‐01) and the European Research Council (NOVEL project, grant number 648321); N.A.M. received funding from the German Research Foundation (DFG MU 4357/1‐1); HW received funding from the European Research Council (Optimal‐Immunity project, grant number 679799); QC acknowledges support from the Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grants Program (grant no. RGPIN‐2014‐05820).
Vekemans, X. , Castric, V. , Hipperson, H. , Müller, N. A. , Westerdahl, H. , & Cronk, Q. (2021). Whole‐genome sequencing and genome regions of special interest: Lessons from major histocompatibility complex, sex determination, and plant self‐incompatibility. Molecular Ecology, 30, 6072–6086. 10.1111/mec.16020
DATA AVAILABILITY STATEMENT
Data sharing not applicable – no new data generated.
REFERENCES
- Akagi, T. , Henry, I. M. , Tao, R. , & Comai, L. (2014). A Y‐chromosome–encoded small RNA acts as a sex determinant in persimmons. Science, 346(6209), 646–650. 10.1126/science.1257225 [DOI] [PubMed] [Google Scholar]
- Allen, J. M. , Boyd, B. , Nguyen, N.‐P. , Vachaspati, P. , Warnow, T. , Huang, D. I. , Grady, P. G. S. , Bell, K. C. , Cronk, Q. C. B. , Mugisha, L. , Pittendrigh, B. R. , Soledad Leonardi, M. , Reed, D. L. , & Johnson, K. P. (2017). Phylogenomics from whole genome sequences using aTRAM. Systematic Biology, 66(5), 786–798. 10.1093/sysbio/syw105 [DOI] [PubMed] [Google Scholar]
- Alonge, M. , Wang, X. , Benoit, M. , Soyk, S. , Pereira, L. , Zhang, L. , Suresh, H. , Ramakrishnan, S. , Maumus, F. , Ciren, D. , Levy, Y. , Harel, T. H. , Shalev‐Schlosser, G. , Amsellem, Z. , Razifard, H. , Caicedo, A. L. , Tieman, D. M. , Klee, H. , Kirsche, M. , … Lippman, Z. B. (2020). Major impacts of widespread structural variation on gene expression and crop improvement in tomato. Cell, 182(1), 145–161. 10.1016/j.cell.2020.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amarasinghe, S. L. , Su, S. , Dong, X. Y. , Zappia, L. , Ritchie, M. E. , & Gouil, Q. (2020). Opportunities and challenges in long‐read sequencing data analysis. Genome Biology, 21(1), 10.1186/s13059-020-1935-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann, J. A. , Tedder, A. , Laenen, B. , Steige, K. A. , & Slotte, T. (2018). Targeted long‐read sequencing of a locus under long‐term balancing selection in Capsella . G3 Genes|genomes|genetics, 8(4), 1327–1333. 10.1534/g3.117.300467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badouin, H. , Boniface, M.‐C. , Pouilly, N. , Fuchs, A.‐L. , Vear, F. , Langlade, N. B. Gouzy, J. & Muños, S. (2021). Pooled Single‐Molecule transcriptomics identifies a giant gene under balancing selection in sunflower. bioRxiv. 10.1101/2021.03.17.435796 [DOI] [Google Scholar]
- Badouin, H. , Velt, A. , Gindraud, F. , Flutre, T. , Dumas, V. , Vautrin, S. , Marande, W. , Corbi, J. , Sallet, E. , Ganofsky, J. , Santoni, S. , Guyot, D. , Ricciardelli, E. , Jepsen, K. , Käfer, J. , Berges, H. , Duchêne, E. , Picard, F. , Hugueney, P. , … Marais, G. A. B. (2020). The wild grape genome sequence provides insights into the transition from dioecy to hermaphroditism during grape domestication. Genome Biology, 21(1), 223. 10.1186/s13059-020-02131-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baduel, P. , Leduque, B. , Ignace, A. , Gy, I. , Gil, J. , Loudet, O. , Colot, V. , & Quadrana, L. (2021). Genetic and environmental modulation of transposition shapes the evolutionary potential of Arabidopsis thaliana . Genome Biology, 22(1), 138. 10.1186/s13059-021-02348-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird, D. M. (2018). Telomeres and genomic evolution. Philosophical Transactions of the Royal Society B‐Biological Sciences, 373(1741), 10.1098/rstb.2016.0437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, H. G. , & Cox, P. A. (1984). Further thoughts on dioecism and islands. Annals of the Missouri Botanical Garden, 71, 244–253. 10.2307/2399068 [DOI] [Google Scholar]
- Balakrishnan, C. N. , Ekblom, R. , Völker, M. , Westerdahl, H. , Godinez, R. , Kotkiewicz, H. , Burt, D. W. , Graves, T. , Griffin, D. K. , Warren, W. C. , & Edwards, S. V. (2010). Gene duplication and fragmentation in the zebra finch major histocompatibility complex. Bmc Biology, 8, 29. 10.1186/1741-7007-8-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudry, F. E. G. , Barrett, S. C. H. , & Wright, S. I. (2020). Ancestral and neo‐sex chromosomes contribute to population divergence in a dioecious plant. Evolution, 74(2), 256–269. 10.1111/evo.13892 [DOI] [PubMed] [Google Scholar]
- Belser, C. , Baurens, F.‐C. , Noel, B. , Martin, G. , Cruaud, C. , Istace, B. , Yahiaoui, N. , Labadie, K. , Hribova, E. , Doležel, J. , Lemainque, A. , Wincker, P. , D'Hont, A. , & Aury, J.‐M. (2021). Telomere‐to‐telomere gapless chromosomes of banana using nanopore sequencing. bioRxiv. 10.1101/2021.04.16.440017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belser, C. , Istace, B. , Denis, E. , Dubarry, M. , Baurens, F.‐C. , Falentin, C. , Genete, M. , Berrabah, W. , Chèvre, A.‐M. , Delourme, R. , Deniot, G. , Denoeud, F. , Duffé, P. , Engelen, S. , Lemainque, A. , Manzanares‐Dauleux, M. , Martin, G. , Morice, J. , Noel, B. , … Aury, J.‐M. (2018). Chromosome‐scale assemblies of plant genomes using nanopore long reads and optical maps. Nature Plants, 4(11), 879–887. 10.1038/s41477-018-0289-4 [DOI] [PubMed] [Google Scholar]
- Biedrzycka, A. , O’Connor, E. , Sebastian, A. , Migalska, M. , Radwan, J. , Zając, T. , Bielański, W. , Solarz, W. , Ćmiel, A. , & Westerdahl, H. (2017). Extreme MHC class I diversity in the sedge warbler (Acrocephalus schoenobaenus); selection patterns and allelic divergence suggest that different genes have different functions. Bmc Evolutionary Biology, 17, 159. 10.1186/s12862-017-0997-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeois, Y. X. C. , & Warren, B. H. (2021). An overview of current population genomics methods for the analysis of whole‐genome resequencing data in eukaryotes. Molecular Ecology, 10.1111/mec.15989 [DOI] [PubMed] [Google Scholar]
- Branco, S. , Badouin, H. , Rodríguez de la Vega, R. C. , Gouzy, J. , Carpentier, F. , Aguileta, G. , Siguenza, S. , Brandenburg, J. T. , Coelho, M. A. , & Hood, M. E. & Giraud, T. (2017). Evolutionary strata on mating‐type chromosomes. Proceedings of the National Academy of Sciences of the United States of America, 114(27), 7067–7072. 10.1073/pnas.1701658114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho, A. B. , & Clark, A. G. (2013). Efficient identification of Y chromosome sequences in the human and Drosophila genomes. Genome Research, 23(11), 1894–1907. 10.1101/gr.156034.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castric, V. , & Vekemans, X. (2004). Plant self‐incompatibility in natural populations: A critical assessment of recent theoretical and empirical advances. Molecular Ecology, 13(10), 2873–2889. 10.1111/j.1365-294X.2004.02267.x [DOI] [PubMed] [Google Scholar]
- Charlesworth, D. , & Mank, J. E. (2010). The birds and the bees and the flowers and the trees: Lessons from genetic mapping of sex determination in plants and animals. Genetics, 186(1), 9–31. 10.1534/genetics.110.117697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, D. , Vekemans, X. , Castric, V. , & Glemin, S. (2005). Plant self‐incompatibility systems: a molecular evolutionary perspective. New Phytologist, 168(1), 61–69. 10.1111/j.1469-8137.2005.01443.x [DOI] [PubMed] [Google Scholar]
- Chen, L. C. , Lan, H. , Sun, L. , Deng, Y. L. , Tang, K. Y. , & Wan, Q. H. (2015). Genomic organization of the crested ibis MHC provides new insight into ancestral avian MHC structure. Scientific Reports, 5, 7963. 10.1038/srep07963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin, C.‐S. , Wagner, J. , Zeng, Q. , Garrison, E. , Garg, S. , Fungtammasan, A. , Rautiainen, M. , Aganezov, S. , Kirsche, M. , Zarate, S. , Schatz, M. C. , Xiao, C. , Rowell, W. J. , Markello, C. , Farek, J. , Sedlazeck, F. J. , Bansal, V. , Yoo, B. , Miller, N. , … Zook, J. M. (2020). A diploid assembly‐based benchmark for variants in the major histocompatibility complex. Nature Communications, 11(1), 4794. 10.1038/s41467-020-18564-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Franceschi, P. , Bianco, L. , Cestaro, A. , Dondini, L. , & Velasco, R. (2018). Characterization of 25 full‐length S‐RNase alleles, including flanking regions, from a pool of resequenced apple cultivars. Plant Molecular Biology, 97, 279–296. 10.1007/s11103-018-0741-x [DOI] [PubMed] [Google Scholar]
- Diggle, P. K. , Di Stilio, V. S. , Gschwend, A. R. , Golenberg, E. M. , Moore, R. C. , Russell, J. R. W. , & Sinclair, J. P. (2011). Multiple developmental processes underlie sex differentiation in angiosperms. Trends in Genetics, 27(9), 368–376. 10.1016/j.tig.2011.05.003 [DOI] [PubMed] [Google Scholar]
- Dilthey, A. T. (2021). State‐of‐the‐art genome inference in the human MHC. The International Journal of Biochemistry & Cell Biology, 131, 105882. 10.1016/j.biocel.2020.105882 [DOI] [PubMed] [Google Scholar]
- Doherty, P. C. , & Zinkernagel, R. M. (1975). Enhanced immunological surveillance in mice heterozygous at the H‐2 gene complex. Nature, 256(5512), 50–52. 10.1038/256050a0 [DOI] [PubMed] [Google Scholar]
- Dreesen, R. S. G. , Vanholme, B. T. M. , Luyten, K. , Van Wynsberghe, L. , Fazio, G. , Roldan‐Ruiz, I. , & Keulemans, J. (2010). Analysis of Malus S‐RNase gene diversity based on a comparative study of old and modern apple cultivars and European wild apple. Molecular Breeding, 26(4), 693–709. 10.1007/s11032-010-9405-5 [DOI] [Google Scholar]
- Dufay, M. , Champelovier, P. , Käfer, J. , Henry, J. P. , Mousset, S. , & Marais, G. A. B. (2014). An angiosperm‐wide analysis of the gynodioecy–dioecy pathway. Annals of Botany, 114, 539–548. 10.1093/aob/mcu134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand, E. , Chantreau, M. , Le Veve, A. , Stetsenko, R. , Dubin, M. , Genete, M. , Llaurens, V. , Poux, C. , Roux, C. , Billiard, S. , Vekemans, X. , & Castric, V. (2020). Evolution of self‐incompatibility in the Brassicaceae: Lessons from a textbook example of natural selection. Evolutionary Applications, 13(6), 1279–1297. 10.1111/eva.12933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand, E. , Méheust, R. , Soucaze, M. , Goubet, P. M. , Gallina, S. , Poux, C. , Fobis‐Loisy, I. , Guillon, E. , Gaude, T. , Sarazin, A. , Figeac, M. , Prat, E. , Marande, W. , Bergès, H. , Vekemans, X. , Billiard, S. , & Castric, V. (2014). Dominance hierarchy arising from the evolution of a complex small RNA regulatory network. Science, 346(6214), 1200–1205. 10.1126/science.1259442 [DOI] [PubMed] [Google Scholar]
- Eid, J. , Fehr, A. , Gray, J. , Luong, K. , Lyle, J. , Otto, G. , Peluso, P. , Rank, D. , Baybayan, P. , Bettman, B. , Bibillo, A. , Bjornson, K. , Chaudhuri, B. , Christians, F. , Cicero, R. , Clark, S. , Dalal, R. , deWinter, A. , Dixon, J. , … Turner, S. (2009). Real‐time DNA sequencing from single polymerase molecules. Science, 323(5910), 133–138. 10.1126/science.1162986 [DOI] [PubMed] [Google Scholar]
- Exposito‐Alonso, M. , 500 Genomes Field Experiment Team , Burbano, H. A. , Bossdorf, O. , Nielsen, R. , & Weigel, D. (2019). Natural selection on the Arabidopsis thaliana genome in present and future climates. Nature, 573(7772), 126–129. 10.1038/s41586-019-1520-9 [DOI] [PubMed] [Google Scholar]
- Feng, S. , Fang, Q. I. , Barnett, R. , Li, C. , Han, S. , Kuhlwilm, M. , Zhou, L. , Pan, H. , Deng, Y. , Chen, G. , Gamauf, A. , Woog, F. , Prys‐Jones, R. , Marques‐Bonet, T. , Gilbert, M. T. P. , & Zhang, G. (2019). The genomic footprints of the fall and recovery of the Crested Ibis. Current Biology, 29(2), 340–349. 10.1016/j.cub.2018.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman, D. C. , Klikoff, L. G. , & Harper, K. T. (1976). Differential resource utilization by sexes of dioecious plants. Science, 193(4253), 597–599. 10.1126/science.193.4253.597 [DOI] [PubMed] [Google Scholar]
- Garrison, E. , Sirén, J. , Novak, A. M. , Hickey, G. , Eizenga, J. M. , Dawson, E. T. , Jones, W. , Garg, S. , Markello, C. , Lin, M. F. , Paten, B. , & Durbin, R. (2018). Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nature Biotechnology, 36(9), 875–879. 10.1038/nbt.4227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genete, M. , Castric, V. , & Vekemans, X. (2020). Genotyping and de novo discovery of allelic variants at the Brassicaceae self‐incompatibility locus from short‐read sequencing data. Molecular Biology and Evolution, 37(4), 1193–1201. 10.1093/molbev/msz258 [DOI] [PubMed] [Google Scholar]
- Geraldes, A. , Hefer, C. A. , Capron, A. , Kolosova, N. , Martinez‐Nunez, F. , Soolanayakanahally, R. Y. , Stanton, B. , Guy, R. D. , Mansfield, S. D. , Douglas, C. J. & Cronk, Q. C. B. (2015). Recent Y chromosome divergence despite ancient origin of dioecy in poplars (Populus). Molecular Ecology, 24(13), 3243–3256. 10.1111/mec.13126 [DOI] [PubMed] [Google Scholar]
- Goubet, P. M. , Bergès, H. , Bellec, A. , Prat, E. , Helmstetter, N. , Mangenot, S. , Gallina, S. , Holl, A.‐C. , Fobis‐Loisy, I. , Vekemans, X. , & Castric, V. (2012). Contrasted patterns of molecular evolution in dominant and recessive self‐incompatibility haplotypes in Arabidopsis . Plos Genetics, 8(3), e1002495. 10.1371/journal.pgen.1002495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene, J. M. , Wiseman, R. W. , Lank, S. M. , Bimber, B. N. , Karl, J. A. , Burwitz, B. J. , Lhost, J. J. , Hawkins, O. E. , Kunstman, K. J. , Broman, K. W. , Wolinsky, S. M. , Hildebrand, W. H. , & O'Connor, D. H. (2011). Differential MHC class I expression in distinct leukocyte subsets. Bmc Immunology, 12, 39. 10.1186/1471-2172-12-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, M. , Yang, Y. , Zhang, M. , & Wang, K. (2020). Considerations regarding centromere assembly in plant whole‐genome sequencing. Methods, 187, 54–56. 10.1016/j.ymeth.2020.09.006 [DOI] [PubMed] [Google Scholar]
- Handa, H. , Kanamori, H. , Tanaka, T. , Murata, K. , Kobayashi, F. , Robinson, S. J. , Koh, C. S. , Pozniak, C. J. , Sharpe, A. G. , Paux, E. , International Wheat Genome Sequencing Consortium , & Nasuda, S. (2018). Structural features of two major nucleolar organizer regions (NORs), Nor‐B1 and Nor‐B2, and chromosome‐specific rRNA gene expression in wheat. Plant Journal, 96(6), 1148–1159. 10.1111/tpj.14094 [DOI] [PubMed] [Google Scholar]
- Harkess, A. , Huang, K. , van der Hulst, R. , Tissen, B. , Caplan, J. L. , Koppula, A. , Batish, M. , Meyers, B. C. , & Leebens‐Mack, J. (2020). Sex determination by two Y‐linked genes in garden asparagus. The Plant Cell, 32(6), 1790–1796. 10.1105/tpc.19.00859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkess, A. , Zhou, J. , Xu, C. , Bowers, J. E. , Van der Hulst, R. , Ayyampalayam, S. , Mercati, F. , Riccardi, P. , McKain, M. R. , Kakrana, A. , Tang, H. , Ray, J. , Groenendijk, J. , Arikit, S. , Mathioni, S. M. , Nakano, M. , Shan, H. , Telgmann‐Rauber, A. , Kanno, A. , … Chen, G. (2017). The asparagus genome sheds light on the origin and evolution of a young Y chromosome. Nature Communications, 8, 1279. 10.1038/s41467-017-01064-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick, P. W. (2002). Pathogen resistance and genetic variation at mhc loci. Evolution, 56(10), 1902–1908. 10.1111/j.0014-3820.2002.tb00116.x [DOI] [PubMed] [Google Scholar]
- Hickey, G. , Heller, D. , Monlong, J. , Sibbesen, J. A. , Sirén, J. , Eizenga, J. , Dawson, E. T. , Garrison, E. , Novak, A. M. , & Paten, B. (2020). Genotyping structural variants in pangenome graphs using the vg toolkit. Genome Biology, 21(1), 35. 10.1186/s13059-020-1941-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huu, C. N. , Keller, B. , Conti, E. , Kappela, C. , & Lenhard, M. (2020). Supergene evolution via stepwise duplications and neofunctionalization of a floral‐organ identity gene. Proceedings of the National Academy of Sciences of the United States of America, 117, 23148–23157. 10.1073/pnas.2006296117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igic, B. , Lande, R. , & Kohn, J. R. (2008). Loss of self‐incompatibility and its evolutionary consequences. International Journal of Plant Sciences, 169(1), 93–104. 10.1086/523362 [DOI] [Google Scholar]
- Iwano, M. , & Takayama, S. (2012). Self/non‐self discrimination in angiosperm self‐incompatibility. Current Opinion in Plant Biology, 15(1), 78–83. 10.1016/j.pbi.2011.09.003 [DOI] [PubMed] [Google Scholar]
- Jain, M. , Olsen, H. E. , Turner, D. J. , Stoddart, D. , Bulazel, K. V. , Paten, B. , Haussler, D. , Willard, H. F. , Akeson, M. , & Miga, K. H. (2018). Linear assembly of a human centromere on the Y chromosome. Nature Biotechnology, 36(4), 321–323. 10.1038/nbt.4109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay, P. , Tezenas, E. , & Giraud, T. (2021). A deleterious mutation‐sheltering theory for the evolution of sex chromosomes and supergenes. bioRxiv. 10.1101/2021.05.17.444504 [DOI] [Google Scholar]
- Jay, P. , Whibley, A. , Frézal, L. , Rodríguez de Cara, M. Á. , Nowell, R. W. , Mallet, J. , Dasmahapatra, K. K. , & Joron, M. (2018). Supergene evolution triggered by the introgression of a chromosomal inversion. Current Biology, 28(11), 1839–1945. 10.1016/j.cub.2018.04.072 [DOI] [PubMed] [Google Scholar]
- Jobling, M. A. , & Tyler‐Smith, C. (2017). Human Y‐chromosome variation in the genome‐sequencing era. Nature Reviews Genetics, 18(8), 485–497. 10.1038/nrg.2017.36 [DOI] [PubMed] [Google Scholar]
- Käfer, J. , Marais, G. A. , & Pannell, J. R. (2017). On the rarity of dioecy in flowering plants. Molecular Ecology, 26(5), 1225–1241. 10.1111/mec.14020 [DOI] [PubMed] [Google Scholar]
- Kamiya, T. , O’Dwyer, K. , Westerdahl, H. , Senior, A. , & Nakagawa, S. (2014). A quantitative review of MHC‐based mating preference: The role of diversity and dissimilarity. Molecular Ecology, 23(21), 5151–5163. 10.1111/mec.12934 [DOI] [PubMed] [Google Scholar]
- Karmin, M. , Saag, L. , Vicente, M. , Sayres, M. A. W. , Järve, M. , Talas, U. G. , Rootsi, S. , Ilumäe, A.‐M. , Mägi, R. , Mitt, M. , Pagani, L. , Puurand, T. , Faltyskova, Z. , Clemente, F. , Cardona, A. , Metspalu, E. , Sahakyan, H. , Yunusbayev, B. , Hudjashov, G. , … Kivisild, T. (2015). A recent bottleneck of Y chromosome diversity coincides with a global change in culture. Genome Research, 25(4), 459–466. 10.1101/gr.186684.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman, J. , Milne, S. , Göbel, T. W. F. , Walker, B. A. , Jacob, J. P. , Auffray, C. , Zoorob, R. , & Beck, S. (1999). The chicken B locus is a minimal essential major histocompatibility complex. ‐, 401(6756), 923–925. 10.1038/44856 [DOI] [PubMed] [Google Scholar]
- Kozarewa, I. , Ning, Z. , Quail, M. A. , Sanders, M. J. , Berriman, M. , & Turner, D. J. (2009). Amplification‐free Illumina sequencing‐library preparation facilitates improved mapping and assembly of (G+C)‐biased genomes. Nature Methods, 6(4), 291–295. 10.1038/nmeth.1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo, K.‐I. , Paape, T. , Hatakeyama, M. , Entani, T. , Takara, A. , Kajihara, K. , Tsukahara, M. , Shimizu‐Inatsugi, R. , Shimizu, K. K. , & Takayama, S. (2015). Gene duplication and genetic exchange drive the evolution of S‐RNase‐based self‐incompatibility in Petunia . Nature Plants, 1(1), 14005. 10.1038/nplants.2014.5 [DOI] [PubMed] [Google Scholar]
- Leducq, J. B. , Gosset, C. C. , Gries, R. , Calin, K. , Schmitt, E. , Castric, V. , & Vekemans, X. (2014). Self‐incompatibility in Brassicaceae: Identification and characterization of SRK‐like sequences linked to the S‐locus in the tribe Biscutelleae. G3 Genes|genomes|genetics, 4(6), 983–992. 10.1534/g3.114.010843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, W. , Plant, K. , Humburg, P. , & Knight, J. C. (2018). AltHapAlignR: Improved accuracy of RNA‐seq analyses through the use of alternative haplotypes. Bioinformatics, 34(14), 2401–2408. 10.1093/bioinformatics/bty125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz, T. L. , Spirin, V. , Jordan, D. M. , & Sunyaev, S. R. (2016). Excess of deleterious mutations around HLA genes reveals evolutionary cost of balancing selection. Molecular Biology and Evolution, 33(10), 2555–2564. 10.1093/molbev/msw127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llamas, B. , Narzisi, G. , Schneider, V. , Audano, P. A. , Biederstedt, E. , Blauvelt, L. , Bradbury, P. , Chang, X. , Chin, C.‐S. , Fungtammasan, A. , Clarke, W. E. , Cleary, A. , Ebler, J. , Eizenga, J. , Sibbesen, J. A. , Markello, C. J. , Garrison, E. , Garg, S. , Hickey, G. , … Busby, B. (2019). A strategy for building and using a human reference pangenome. F1000Research, 8, 1751. 10.12688/f1000research.19630.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llaurens, V. , Billiard, S. , Castric, V. , & Vekemans, X. (2009). Evolution of dominance in sporophytic self‐incompatibility systems: I. Genetic load and coevolution of levels of dominance in pollen and pistil. Evolution, 63(9), 2427–2437. 10.1111/j.1558-5646.2009.00709.x [DOI] [PubMed] [Google Scholar]
- Lloyd, D. G. (1982). Selection of combined versus separate sexes in seed plants. American Naturalist, 120(5), 571–585. 10.1086/284014 [DOI] [Google Scholar]
- Low, W. Y. , Tearle, R. , Bickhart, D. M. , Rosen, B. D. , Kingan, S. B. , Swale, T. , Thibaud‐Nissen, F. , Murphy, T. D. , Young, R. , Lefevre, L. , Hume, D. A. , Collins, A. , Ajmone‐Marsan, P. , Smith, T. P. L. , & Williams, J. L. (2019). Chromosome‐level assembly of the water buffalo genome surpasses human and goat genomes in sequence contiguity. Nature Communications, 10, 260. 10.1038/s41467-018-08260-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mable, B. K. , Brysting, A. K. , Jørgensen, M. H. , Carbonell, A. K. Z. , Kiefer, C. , Ruiz‐Duarte, P. , Lagesen, K. , & Koch, M. A. (2018). Adding complexity to complexity: Gene family evolution in polyploids. Frontiers in Ecology and Evolution, 6, 114. 10.3389/fevo.2018.00114 [DOI] [Google Scholar]
- Mandakova, T. , Hlouskova, P. , Koch, M. A. , & Lysak, M. A. (2020). Genome evolution in Arabideae was marked by frequent centromere repositioning. The Plant Cell, 32(3), 650–665. 10.1105/tpc.19.00557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney, G. , McPhee, M. V. , Pascal, C. , Seeb, J. E. , & Seeb, L. W. (2020). Network analysis of linkage disequilibrium reveals genome architecture in chum salmon. G3 Genes|genomes|genetics, 10(5), 1553–1561. 10.1534/g3.119.400972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McStay, B. (2016). Nucleolar organizer regions: Genomic ‘dark matter’ requiring illumination. Genes & Development, 30(14), 1598–1610. 10.1101/gad.283838.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miga, K. H. , Koren, S. , Rhie, A. , Vollger, M. R. , Gershman, A. , Bzikadze, A. , Brooks, S. , Howe, E. , Porubsky, D. , Logsdon, G. A. , Schneider, V. A. , Potapova, T. , Wood, J. , Chow, W. , Armstrong, J. , Fredrickson, J. , Pak, E. , Tigyi, K. , Kremitzki, M. , … Phillippy, A. M. (2020). Telomere‐to‐telomere assembly of a complete human X chromosome. Nature, 585(7823), 79–84. 10.1038/s41586-020-2547-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minias, P. , Pikus, E. , Whittingham, L. A. , & Dunn, P. O. (2019). Evolution of copy number at the MHC varies across the avian tree of life. Genome Biology and Evolution, 11(1), 17–28. 10.1093/gbe/evy253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, N. A. , Kersten, B. , Leite Montalvão, A. P. , Mähler, N. , Bernhardsson, C. , Bräutigam, K. , Carracedo Lorenzo, Z. , Hoenicka, H. , Kumar, V. , Mader, M. , Pakull, B. , Robinson, K. M. , Sabatti, M. , Vettori, C. , Ingvarsson, P. K. , Cronk, Q. , Street, N. R. , & Fladung, M. (2020). A single gene underlies the dynamic evolution of poplar sex determination. Nature Plants, 6(6), 630–637. 10.1038/s41477-020-0672-9 [DOI] [PubMed] [Google Scholar]
- Murphy, K. , & Weaver, C. (2017). Janeway’s immunobiology. New York: Garland Science. [Google Scholar]
- Muyle, A. , Kafer, J. , Zemp, N. , Mousset, S. , Picard, F. , & Marais, G. A. B. (2016). SEX‐DETector: A probabilistic approach to study sex chromosomes in non‐model organisms. Genome Biology and Evolution, 8(8), 2530–2543. 10.1093/gbe/evw172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves, C. J. , Matzrafi, M. , Thiele, M. , Lorant, A. , Mesgaran, M. B. , & Stetter, M. G. (2020). Male linked genomic region determines sex in dioecious Amaranthus palmeri . Journal of Heredity, esaa04. 10.1093/jhered/esaa047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novikova, P. Y. , Tsuchimatsu, T. , Simon, S. , Nizhynska, V. , Voronin, V. , Burns, R. , Fedorenko, O. M. , Holm, S. , Säll, T. , Prat, E. , Marande, W. , Castric, V. , & Nordborg, M. (2017). Genome sequencing reveals the origin of the allotetraploid Arabidopsis suecica . Molecular Biology and Evolution, 34(4), 957–968. 10.1093/molbev/msw299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk, S. , Walenz, B. P. , Rhie, A. , Vollger, M. R. , Logsdon, G. A. , Grothe, R. , Miga, K. H. , Eichler, E. E. , Phillippy, A. M. , & Koren, S. (2020). HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high‐fidelity long reads. Genome Research, 30(9), 1291–1305. 10.1101/gr.263566.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor, E. A. , Cornwallis, C. K. , Hasselquist, D. , Nilsson, J.‐Å. , & Westerdahl, H. (2018). The evolution of immunity in relation to colonization and migration. Nature Ecology & Evolution, 2(5), 841–849. 10.1038/s41559-018-0509-3 [DOI] [PubMed] [Google Scholar]
- O'Connor, E. A. , Strandh, M. , Hasselquist, D. , Nilsson, J. A. , & Westerdahl, H. (2016). The evolution of highly variable immunity genes across a passerine bird radiation. Molecular Ecology, 25(4), 977–989. 10.1111/mec.13530 [DOI] [PubMed] [Google Scholar]
- Palmer, D. H. , Rogers, T. F. , Dean, R. , & Wright, A. E. (2019). How to identify sex chromosomes and their turnover. Molecular Ecology, 28(21), 4709–4724. 10.1111/mec.15245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne, A. , Holmes, N. , Clarke, T. , Munro, R. , Debebe, B. , & Loose, M. (2021). Readfish enables targeted nanopore sequencing of gigabase‐sized genomes. Nature Biotechnology, 39, 442–450. 10.1038/s41587-020-00746-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peska, V. , & Garcia, S. (2020). Origin, diversity, and evolution of telomere sequences in plants. Frontiers in Plant Science, 11, 117. 10.3389/fpls.2020.00117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillippy, A. M. (2020). The (near) complete sequence of a human genome. Retrieved from https://genomeinformatics.github.io/CHM13v1/ [Google Scholar]
- Pickup, M. , Brandvain, Y. , Fraisse, C. , Yakimowski, S. , Barton, N. H. , Dixit, T. , Lexer, C. , Cereghetti, E. , & Field, D. L. (2019). Mating system variation in hybrid zones: Facilitation, barriers and asymmetries to gene flow. New Phytologist, 224(3), 1035–1047. 10.5061/dryad.dm7cs86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentout, D. , Razumova, O. , Rhone, B. , Badouin, H. , Henri, H. , Feng, C. , Käfer, J. , & Karlov, G. , & Marais, G. A. B. (2020). An efficient RNA‐seq‐based segregation analysis identifies the sex chromosomes of Cannabis sativa . Genome Research, 30(2), 164–172. 10.1101/gr.251207.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queenborough, S. A. , Burslem, D. , Garwood, N. C. , & Valencia, R. (2007). Determinants of biased sex ratios and inter‐sex costs of reproduction in dioecious tropical forest trees. American Journal of Botany, 94(1), 67–78. 10.3732/ajb.94.1.67 [DOI] [PubMed] [Google Scholar]
- Renner, S. S. (2014). The relative and absolute frequencies of angiosperm sexual systems: Dioecy, monoecy, gynodioecy, and an updated online database. American Journal of Botany, 101(10), 1588–1596. 10.3732/ajb.1400196 [DOI] [PubMed] [Google Scholar]
- Renner, S. S. , & Müller, N. A. (2021). Plant sex chromosomes defy evolutionary models of expanding recombination suppression and genetic degeneration. Nature Plants, 7, 392–402. 10.1038/s41477-021-00884-3 [DOI] [PubMed] [Google Scholar]
- Schalamun, M. , Nagar, R. , Kainer, D. , Beavan, E. , Eccles, D. , Rathjen, J. P. , & Schwessinger, B. (2019). Harnessing the MinION: An example of how to establish long‐read sequencing in a laboratory using challenging plant tissue from Eucalyptus pauciflora . Molecular Ecology Resources, 19(1), 77–89. 10.1111/1755-0998.12938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierup, M. H. , Charlesworth, D. , & Vekemans, X. (2000). The effect of hitch‐hiking on genes linked to a balanced polymorphism in a subdivided population. Genetical Research, 76(1), 63–73. 10.1017/s0016672300004547 [DOI] [PubMed] [Google Scholar]
- Shiina, T. , & Blancher, A. (2019). The Cynomolgus macaque MHC polymorphism in experimental medicine. Cells, 8(9), 978. 10.3390/cells8090978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiina, T. , Briles, W. E. , Goto, R. M. , Hosomichi, K. , Yanagiya, K. , Shimizu, S. , Inoko, M. & Miller, M. M. (2007). Extended gene map reveals tripartite motif, C‐type lectin, and Ig superfamily type genes within a subregion of the chicken MHC‐B affecting infectious disease. Journal of Immunology, 178(11), 7162–7172. 10.4049/jimmunol.178.11.7162 [DOI] [PubMed] [Google Scholar]
- Shiina, T. , Hosomichi, K. , Inoko, H. , & Kulski, J. K. (2009). The HLA genomic loci map: Expression, interaction, diversity and disease. Journal of Human Genetics, 54(1), 15–39. 10.1038/jhg.2008.5 [DOI] [PubMed] [Google Scholar]
- Shimizu, K. K. , Cork, J. M. , Caicedo, A. L. , Mays, C. A. , Moore, R. C. , Olsen, K. M. , & Purugganan, M. D. (2004). Darwinian selection on a selfing locus. Science, 306(5704), 2081–2084. 10.1126/science.1103776 [DOI] [PubMed] [Google Scholar]
- Stebbins, G. L. (1974). Flowering plants: Evolution above the species level. Belknap Press. [Google Scholar]
- Stervander, M. , Dierickx, E. G. , Thorley, J. , Brooke, M. d. L. , & Westerdahl, H. (2020). High MHC gene copy number maintains diversity despite homozygosity in a Critically Endangered single‐island endemic bird, but no evidence of MHC‐based mate choice. [‐ 2020/10/01]. Molecular Ecology, 29, 3578–3592. 10.1111/mec.15471 [DOI] [PubMed] [Google Scholar]
- Stone, J. L. (2004). Sheltered load associated with S‐alleles in Solanum carolinense . Heredity, 92(4), 335–342. 10.1038/sj.hdy.6800425 [DOI] [PubMed] [Google Scholar]
- Sutton, J. T. , Helmkampf, M. , Steiner, C. C. , Bellinger, M. R. , Korlach, J. , Hall, R. , Baybayan, P. , Muehling, J. , Gu, J. , Kingan, S. , Masuda, B. M. , & Ryder, O. A. (2018). A high‐quality, long‐read de novo genome assembly to aid conservation of Hawaii's last remaining crow species. Genes, 9(8), 393. 10.3390/genes9080393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata, N. , & Nei, M. (1990). Allelic genealogy under overdominant and frequency‐dependent selection and polymorphism of major histocompatibility complex loci. Genetics, 124(4), 967–978. 10.1093/genetics/124.4.967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasaki, T. , Hatakeyama, K. , Suzuki, G. , Watanabe, M. , Isogai, A. , & Hinata, K. (2000). The S receptor kinase determines self‐incompatibility in Brassica stigma. Nature, 403(6772), 913–916. 10.1038/35002628 [DOI] [PubMed] [Google Scholar]
- Takou, M. , Hämälä, T. , Koch, E. , Steige, K. A. , Dittberner, H. , Yant, L. , Genete, M. , Sunyaev, S. , Castric, V. , Vekemans, X. , Savolainen, O. , & De Meaux, J. (2021). Maintenance of adaptive dynamics in a bottlenecked range‐edge population that retained outcrossing. Molecular Biology and Evolution, 38, 1820–1836. 10.1093/molbev/msaa322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson, J. D. , & Barrett, S. C. H. (1981). Selection for outcrossing, sexual selection, and the evolution of dioecy in plants. American Naturalist, 118(3), 443–449. 10.1086/283837 [DOI] [Google Scholar]
- Tian, X. , Li, R. , Fu, W. , Li, Y. , Wang, X. , Li, M. , Du, D. , Tang, Q. , Cai, Y. , Long, Y. , Zhao, Y. , Li, M. , & Jiang, Y. U. (2020). Building a sequence map of the pig pan‐genome from multiple de novo assemblies and Hi‐C data. Science China‐Life Sciences, 63(5), 750–763. 10.1007/s11427-019-9551-7 [DOI] [PubMed] [Google Scholar]
- Todesco, M. , Owens, G. L. , Bercovich, N. , Légaré, J.‐S. , Soudi, S. , Burge, D. O. , Huang, K. , Ostevik, K. L. , Drummond, E. B. M. , Imerovski, I. , Lande, K. , Pascual‐Robles, M. A. , Nanavati, M. , Jahani, M. , Cheung, W. , Staton, S. E. , Muños, S. , Nielsen, R. , Donovan, L. A. , … Rieseberg, L. H. (2020). Massive haplotypes underlie ecotypic differentiation in sunflowers. Nature, 584(7822), 602–607. 10.1038/s41586-020-2467-6 [DOI] [PubMed] [Google Scholar]
- Torres, M. F. , Mathew, L. S. , Ahmed, I. , Al‐Azwani, I. K. , Krueger, R. , Rivera‐Nuñez, D. , Mohamoud, Y. A. , Clark, A. G. , Suhre, K. , & Malek, J. A. (2018). Genus‐wide sequencing supports a two‐locus model for sex‐determination in Phoenix . Nature Communications, 9, 3969. 10.1038/s41467-018-06375-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchimatsu, T. , Goubet, P. M. , Gallina, S. , Holl, A.‐C. , Fobis‐Loisy, I. , Bergès, H. , Marande, W. , Prat, E. , Meng, D. , Long, Q. , Platzer, A. , Nordborg, M. , Vekemans, X. , & Castric, V. (2017). Patterns of Polymorphism at the self‐incompatibility locus in 1,083 Arabidopsis thaliana genomes. Molecular Biology and Evolution, 34(8), 1878–1889. 10.1093/molbev/msx122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyenoyama, M. K. (2005). Evolution under tight linkage to mating type. New Phytologist, 165(1), 63–70. 10.1111/j.1469-8137.2004.01246.x [DOI] [PubMed] [Google Scholar]
- Vekemans, X. , Poux, C. , Goubet, P. M. , & Castric, V. (2014). The evolution of selfing from outcrossing ancestors in Brassicaceae: What have we learned from variation at the S‐locus? Journal of Evolutionary Biology, 27(7), 1372–1385. 10.1111/jeb.12372 [DOI] [PubMed] [Google Scholar]
- Watanabe, A. , Shiina, T. , Shimizu, S. , Hosomichi, K. , Yanagiya, K. , Kita, Y.F. , Kimura, T. , Soeda, E. , Torii, R. , Ogasawara, K. , Kulski, J.K. , & Inoko, H. (2007). A BAC‐based contig map of the cynomolgus macaque (Macaca fascicularis) major histocompatibility complex genomic region. Genomics, 89(3), 402–412. 10.1016/j.ygeno.2006.11.002 [DOI] [PubMed] [Google Scholar]
- Wright, B. R. , Farquharson, K. A. , McLennan, E. A. , Belov, K. , Hogg, C. J. , & Grueber, C. E. (2020). A demonstration of conservation genomics for threatened species management. Molecular Ecology Resources, 20(6), 1526–1541. 10.1111/1755-0998.13211 [DOI] [PubMed] [Google Scholar]
- Wright, S. I. , Ness, R. W. , Foxe, J. P. , & Barrett, S. C. (2008). Genomic consequences of outcrossing and selfing in plants. International Journal of Plant Sciences, 169(1), 105–118. 10.1086/523366 [DOI] [Google Scholar]
- Wu, L. H. , Williams, J. S. , Sun, L. H. , & Kao, T. H. (2020). Sequence analysis of the Petunia inflata S‐locus region containing 17 S‐Locus F‐Box genes and the S‐RNase gene involved in self‐incompatibility. Plant Journal, 104(5), 1348–1368. 10.1111/tpj.15005 [DOI] [PubMed] [Google Scholar]
- Xu, L. , Auer, G. , Peona, V. , Suh, A. , Deng, Y. , Feng, S. , Zhang, G. , Blom, M. P. K. , Christidis, L. , Prost, S. , Irestedt, M. , & Zhou, Q. I. (2019). Dynamic evolutionary history and gene content of sex chromosomes across diverse songbirds. Nature Ecology & Evolution, 3, 834–844. 10.1038/s41559-019-0850-1 [DOI] [PubMed] [Google Scholar]
- Yamaguchi, T. , & Dijkstra, J. M. (2019). Major Histocompatibility Complex (MHC) genes and disease resistance in fish. Cells, 8(4), 378. 10.3390/cells8040378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, Z. , Martin, S. H. , Gotzek, D. , Arsenault, S. V. , Duchen, P. , Helleu, Q. , Riba‐Grognuz, O. , Hunt, B. G. , Salamin, N. , Shoemaker, D. W. , Ross, K. G. , & Keller, L. (2020). Evolution of a supergene that regulates a trans‐species social polymorphism. Nature Ecology & Evolution, 4(2), 240–249. 10.1038/s41559-019-1081-1 [DOI] [PubMed] [Google Scholar]
- Zelano, B. , & Edwards, S. V. (2002). An MHC component to kin recognition and mate choice in birds: Predictions, progress, and prospects. American Naturalist, 160, S225–S237. 10.1086/342897 [DOI] [PubMed] [Google Scholar]
- Zeng, T. C. , Aw, A. J. , & Feldman, M. W. (2018). Cultural hitchhiking and competition between patrilineal kin groups explain the post‐Neolithic Y‐chromosome bottleneck. Nature Communications, 9, 10.1038/s41467-018-04375-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai, T. , Yang, H. Q. , Zhang, R. C. , Fang, L. M. , Zhong, G. H. , & Fang, S. G. (2017). Effects of population bottleneck and balancing selection on the Chinese Alligator. Scientific Reports, 7(1), 5549. 10.1038/s41598-017-05640-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, F. , Ding, Y. H. , Zhu, C. D. , Zhou, X. , Orr, M. C. , Scheu, S. , & Luan, Y. X. (2019). Phylogenomics from low‐coverage whole‐genome sequencing. Methods in Ecology and Evolution, 10(4), 507–517. 10.1111/2041-210x.13145 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable – no new data generated.