

Summary
Background
Bile acids are important endocrine modulators of intestinal and hepatic signalling cascades orchestrating critical pathophysiological processes in various liver diseases. Increasing knowledge on bile acid signalling has stimulated the development of synthetic ligands of nuclear bile acid receptors and other bile acid analogues.
Aim
This review summarises important aspects of bile acid‐mediated crosstalk between the gut and the liver (“gut‐liver axis”) as well as recent findings from experimental and clinical studies.
Methods
We performed a literature review on bile acid signalling, and therapeutic applications in chronic liver disease.
Results
Intestinal and hepatic bile acid signalling pathways maintain bile acid homeostasis. Perturbations of bile acid‐mediated gut‐liver crosstalk dysregulate transcriptional networks involved in inflammation, fibrosis and endothelial dysfunction. Bile acids induce enterohepatic feedback signalling by the release of intestinal hormones, and regulate enterohepatic circulation. Importantly, bile acid signalling plays a central role in maintaining intestinal barrier integrity and antibacterial defense, which is particularly relevant in cirrhosis, where bacterial translocation has a profound impact on disease progression. The nuclear bile acid farnesoid X receptor (FXR) is a central intersection in bile acid signalling and has emerged as a relevant therapeutic target.
Conclusions
Experimental evidence suggests that bile acid signalling improves the intestinal barrier and protects against bacterial translocation in cirrhosis. FXR agonists have displayed efficacy for the treatment of cholestatic and metabolic liver disease in randomised controlled clinical trials. However, similar effects remain to be shown in advanced liver disease, particularly in patients with decompensated cirrhosis.
Bile acid signalling in the gut‐liver axis modulates numerous signalling cascades in the intestine and the liver that are vital for the maintenance of bile acid homeostasis, enterohepatic circulation, metabolism and intestinal barrier integrity.
1. INTRODUCTION
Bile acids (BAs) modulate numerous signalling cascades in the intestine and the liver that are vital for the maintenance of BA homeostasis, enterohepatic circulation, metabolism and intestinal barrier integrity. Dysregulation of BA signalling becomes particularly relevant in chronic liver diseases, as perturbations of BA‐mediated gut‐liver crosstalk may affect transcriptional networks involved in inflammation, fibrosis, endothelial dysfunction and ultimately affect disease progression. The physiological interactions between the gut and liver include (a) the uptake and metabolism of nutrients and BAs, (b) endocrine signalling between gut and the liver, and (c) the hepatic immune response to gut‐derived (inflammatory) signals and pathogens and are commonly subsumed by the term “gut‐liver axis.” In this review, we summarise how BA signalling in the gut‐liver axis affects BA synthesis and enterohepatic circulation, intestinal barrier integrity and bacterial translocation, the relevance of BA signalling in (advanced) chronic liver disease and discuss emerging therapeutic strategies.
2. REVIEW CRITERIA
Literature research was conducted on PubMed, considering relevant papers on BA signalling without publication date restrictions. For well‐established concepts and general knowledge on BA physiology, review articles from peer‐reviewed high‐impact journals were cited. Regarding experimental studies, we focused on publications investigating the molecular mechanisms of BA signalling, as well as therapeutic interventions targeting the gut‐liver axis, specifically considering animal liver disease models. Regarding clinical studies, we prioritised randomised clinical trials (RCTs) and interventional studies using bile acid analogues or synthetic ligands of nuclear BA receptors registered at “ClinicalTrials.gov.” Peer‐reviewed publications in English and recently published abstracts from RCTs were taken into consideration. This review is not designed as a systematic review.
3. BILE ACIDS AND TARGET RECEPTORS
3.1. Bile acid species, chemical features and synthesis
BAs are produced from cholesterol in the liver and mediate the resorption of dietary lipids and fat‐soluble vitamins. 1 BAs are classified by certain biochemical characteristics that determine their physiological activity. First, BAs are categorised as primary BAs (BAs synthesised by the liver) or secondary BAs (primary BAs chemically modified by gut bacteria). 2 The primary BA pool in humans essentially consists of cholic acid (CA) and chenodeoxycholic acid (CDCA), accounting for approximately 70%–80% of the total BA pool, while the secondary BA pool comprises lithocholic acid (LCA), deoxycholic acid (DCA) and ursodeoxycholic acid (UDCA). 2 Second, BAs are differentiated as unconjugated and conjugated BA (bound to taurine or glycine). 1 , 3 Third, BA subtypes exhibit different degrees of hydrophobicity, which is determined by hydroxyl groups and the stereochemistry of BAs. 4 The BA pool displays considerable plasticity which may impact digestive function, nuclear receptor binding, bile fluid solubility, and BA‐associated toxicity 4 and significantly shifts between healthy individuals and patients with liver diseases. 5 , 6
De‐novo BA synthesis by hepatocytes follows either the classical (“neutral”) or the alternative (“acidic”) biosynthetic pathway, summarised in Figure 1. In the classical pathway, cholesterol is modified by the cholesterol 7α‐hydroxylase (encoded by the CYP7A1 gene). 7 Further metabolisation by CYP8B1 leads to the formation of CA or, alternatively, modification by CYP27A1 produces CDCA. 7 , 8 The expression of CYP7A1 is considered the rate‐limiting enzymatic step for overall BA synthesis, 9 while the expression of CYP8B1 defines the BA pool as the synthesis of CA is contingent on CYP8B1. 10 The paradigm that the alternative pathway exclusively results in the synthesis of CDCA 7 was recently challenged since the knockout of CYP7A1 in mice resulted in a shift towards the alternative pathway while CA was still synthesised, 11 potentially explained by a subsequent study reporting that human CYP8B1 (expressed by cultured yeast cells) is able to convert CDCA to CA. 12
FIGURE 1.
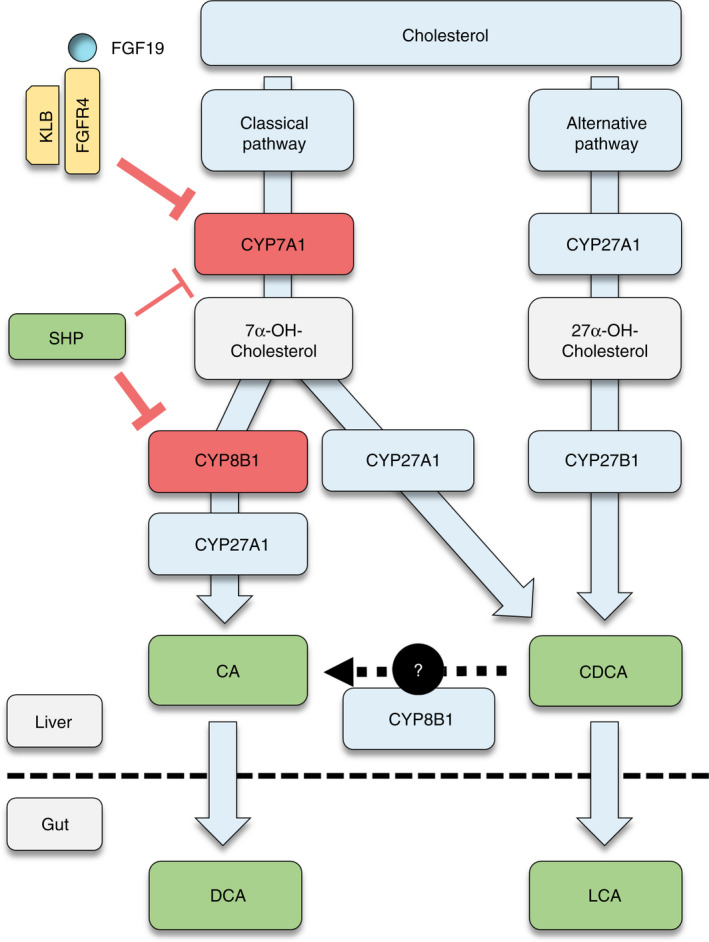
Regulatory pathways of bile acid synthesis. Primary bile acids (BA) are synthesised from cholesterol in hepatocytes, following either the “classical” or the “alternative” pathway. In the classical pathway, cholesterol is first modified by CYP7A1 and subsequently by CYP8B1 to produce cholic acid (CA) or, alternatively by CYP27A1 to produce chenodeoxycholic acid (CDCA). In the alternative pathway, CYP27A1 initiates cholesterol modification, followed by CYP7B1‐dependent biotransformation, resulting in formation of CDCA. Expression of CYP7A1 is the rate‐limiting enzymatic step for BA synthesis, while CYP8B1 defines the BA pool as it is critical for production of CA. Metabolisation of BAs by gut bacteria leads to the formation of the secondary BAs deoxycholic acid (DCA) and lithocholic acid (LCA). BA synthesis is mainly regulated by two mechanisms: First, intrahepatic farnesoid X receptor (FXR) activation induces the expression of small heterodimer partner (SHP), which suppresses the expression of CYP8B1 and (to a lesser extent) CYP7A1. Second, activation of FXR in the intestines induces the release of fibroblast growth factor‐19 (FGF19) into the portal venous system. FGF19 binds to the FGF receptor 4 (FGFR4) and its co‐receptor beta‐Klotho (KLB) on hepatocytes, thereby strongly suppressing CYP7A1 expression. Abbreviations: BA, bile acid; CA, cholic acid; CDCA, chenodeoxycholic acid; DCA, deoxycholic acid; FGF19, fibroblast growth factor‐19; FGFR4, fibroblast growth factor receptor 4; FXR, farnesoid X receptor; KLB, beta‐Klotho; LCA, lithocholic acid; SHP, small heterodimer partner
3.2. Bile acid transporters and enterohepatic circulation
The majority of BAs undergoes efficient “recycling” by enterohepatic circulation, which is characterised by the intestinal re‐uptake of BAs (predominantly in the ileum), delivery to the liver via the portal venous system, uptake in the liver and, again, secretion into the bile canaliculi (Figure 2). Hence, only about 5% of circulating BAs is lost in the faeces. 13 Efficacious transport mechanisms are important for enterohepatic circulation, since only a few (primarily unconjugated) BA are absorbed passively in the gut and the liver, whereas intestinal absorption and hepatic uptake of most BA relies on active transport across cell membranes. 14 More specifically, enterohepatic circulation of BA in the gut‐liver axis is mainly contingent on four transporters with specific expression patterns and location at the cellular membranes of intestinal epithelial cells and/or hepatocytes (summarised in Figure 3): The apical sodium‐dependent bile acid transporter (ASBT, SLC10A2), organic solute transporter‐α and ‐β (OST‐α, SLC51A; OST‐β, SLC51B), Na+‐taurocholate cotransporting polypeptide (NTCP, SLC10A1) and the bile salt export pump (BSEP; ABCB11). 15
FIGURE 2.
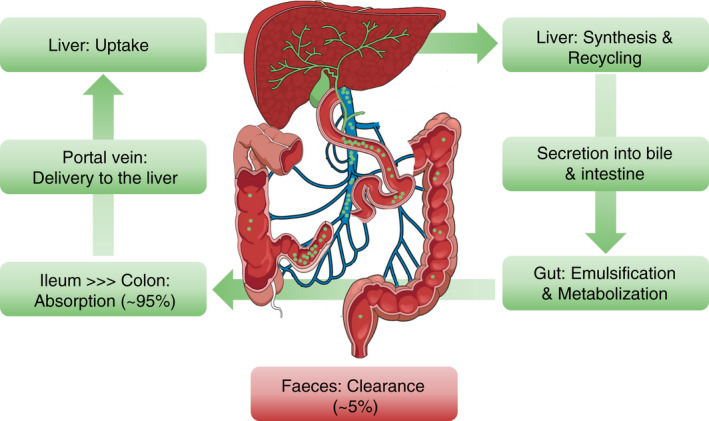
Enterohepatic circulation of bile acids: anatomical overview. Primary bile acids (BA; indicated as green dots) are released into bile canaliculi and secreted into the duodenum, where they mediate absorption of nutrients, for example emulsification of lipids and fat‐soluble vitamins, and regulate signalling pathways in the gut‐liver axis. Metabolisation of BA by gut bacteria leads to the formation of secondary BAs. Most BAs undergo enterohepatic circulation: the highest proportion of BAs is reabsorbed in the ileum, transported into the portal circulation, and delivered back to the liver via the portal vein. After reuptake in hepatocytes, BAs are recycled and secreted into the bile, and again reach the gut, thus completing the enterohepatic cycle. Abbreviation: BA, bile acid
FIGURE 3.
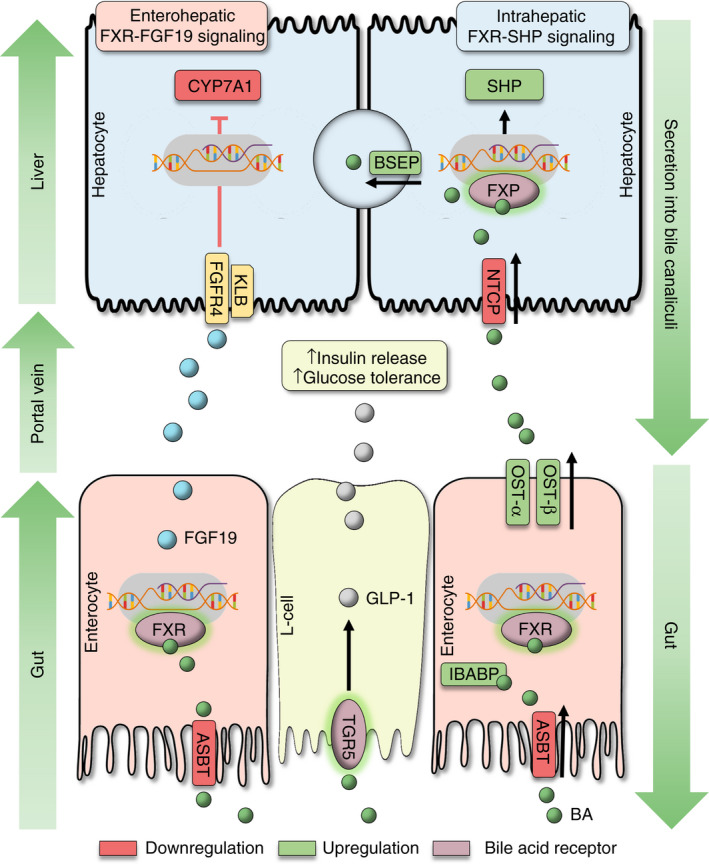
Bile acid gut‐liver axis signalling modulates the enterohepatic circulation, bile acid synthesis and glucose homeostasis. (i) Enterohepatic circulation of bile acids (BA) is contingent on four important transporters in the gut‐liver axis: ASBT, OST‐α/‐β, NTCP and BSEP. ASBT mediates BA uptake from the intestinal lumen (primarily ileum), whereas OST‐α/‐β releases BAs into the portal circulation. Ileal bile acid binding protein (IBABP) binds intracellular BAs and protects enterocytes from BA toxicity. NTCP facilitates BA uptake from sinusoidal blood into hepatocytes, and BSEP mediates BA secretion into bile canaliculi. Activation of FXR downregulates ASBT and NTCP and upregulates OST‐α/‐β, BSEP and IBABP (indicated in red and green color). (ii) Bile acid synthesis is regulated by the enterohepatic FXR‐FGF19 pathway, and the intrahepatic FXR‐SHP pathway. Activation of FXR in the intestines (primarily in the ileum) leads to release of FGF19 into the portal circulation, which represses BA synthesis (CYP7A1 gene) by binding to FGFR4/KLB (‘enterohepatic’ feedback signalling). Activation of FXR in hepatocytes induces expression of SHP, which acts as a transrepressor of genes for BA synthesis (“intrahepatic” feedback signalling). (iii) Activation of TGR5 on intestinal L‐cells induces release of GLP‐1, which induces insulin secretion and enhances glucose tolerance. Abbreviations: (ASBT) apical sodium dependent bile acid transporter; (BA) bile acids; (BSEP) bile salt export pump; (FXR) farnesoid X receptor; (FGF19) fibroblast growth factor 19; (FGFR4) FGF receptor 4; (GLP‐1) glucagon‐like peptide‐1; (IBABP) ileal bile acid binding protein; (KLB) beta‐klotho; (NTCP) sodium/taurocholate cotransporting polypeptide; (OST‐α/‐β) organic solute transporter alpha and beta; (SHP) small heterodimer partner; (TGR5) transmembrane G protein‐coupled receptor‐5
3.3. Farnesoid X receptor
Farnesoid X receptor (FXR) belongs to the family of nuclear hormone receptors (NHR; ie, receptors exerting transcription factor activity upon modulation by ligands) and is a nuclear receptor for BAs. 16 , 17 Upon activation by ligands, FXR binds to target promoter regions in heterodimerisation with retinoid X receptor‐alpha; 18 , 19 however, FXR can also modulate gene transcription in monomeric conformation. 20 FXR signalling regulates numerous downstream signalling cascades, for example, transcriptional networks involving peroxisome proliferator‐activated receptors (PPAR). 21 Expression mapping of nuclear receptors in mice revealed that FXR is predominantly expressed in the liver, intestines, and kidney. 22 Along the different sections of the gastrointestinal tract of mice, FXR displays the highest expression in the ileum. 22 , 23 In the mouse liver, FXR is most abundantly expressed by hepatocytes, and to considerably lower extent in liver sinusoidal endothelial cells (LSEC) and Kupffer cells, 24 whereas hepatic stellate cells (HSC) reportedly do not express FXR. 25 Several synthetic FXR agonists were developed in recent years. These compounds may be differentiated as steroidal vs non‐steroidal FXR agonists, the latter being characterised by improved pharmacokinetics and side effect profile. Detailed biochemical and pharmacological features of FXR agonists were recently reviewed. 26 , 27
3.4. Takeda G protein‐coupled receptor‐5
Takeda G protein‐coupled receptor‐5 (TGR5; encoded by GPBAR1) is an epithelial receptor for BAs, which marks an important difference to nuclear BA receptors. 28 , 29 Similar to NHRs, TGR5 modulates downstream transcriptional activity, for example, via the intracellular protein kinase A, 29 , 30 protein kinase B, 31 extracellular signal‐related kinase 1/2, 32 or Rho kinase 33 signalling pathways. Expression of TGR5 varies across different anatomical compartments and displays high expression in gallbladder epithelium 34 , 35 and the intestine (particularly ileum and colon), while exhibiting lower expression in the liver. 28 , 36 TGR5 is also expressed in other organs such as skeletal muscle and brown adipose tissue. 29 , 30 , 34 , 37 Again, these expression profiles are mostly deduced from animal tissues. In the liver, TGR5 is expressed by cholangiocytes, 32 , 35 Kupffer cells, 38 LSEC 39 and, against earlier assumptions, also in hepatocytes. 40 , 41
3.5. Other nuclear bile acid receptors
BAs also interact with the vitamin D receptor (VDR; NR1I1), pregnane X receptor (PXR; NR1I2) and constitutive androstane receptor (CAR; NR1I3). 42 While these receptors have been ascribed some modulatory effects on BA homeostasis and metabolism, experimental and clinical data on drug development or therapeutic effects directly mediated by these receptors is scarce. 43 , 44 Therefore, this review rather concentrates on BA signalling effects mediated by FXR and TGR5, since these receptors have been studied extensively in experimental and clinical settings.
4. FEEDBACK REGULATION OF BILE ACID SYNTHESIS AND ENTEROHEPATIC CIRCULATION
4.1. Gut‐liver signalling modulates bile acid synthesis
Regulation of BA synthesis marks an important inhibitory feedback mechanism that is tightly controlled by intestinal and hepatic BA signalling pathways. These regulatory cascades ensure BA homeostasis and protection from cytotoxicity occurring in supraphysiological concentrations. 45 , 46
First, intrahepatic FXR activation by BAs induces the expression of a small heterodimer partner (SHP; NR0B2) (Figures 1, 3). 47 SHP forms heterodimers with nuclear receptors, while not being able to bind DNA, resulting in transrepression of transcription factors. 48 Concordantly, SHP upregulation results in the downregulation of CYP7A1 and CYP8B1 gene expression. 47 , 49 , 50 Nevertheless, studies in mice suggest that the “intrahepatic” FXR‐SHP feedback pathway mostly regulates CYP8B1 (ie, BA pool composition) and has relatively small impact on the expression of CYP7A1 (ie, BA pool size). 51 , 52
Second, hepatic BA synthesis is regulated by an “enterohepatic” signalling pathway via fibroblast growth factor‐19 (FGF19). The term “FGF19” will be used consistently in this review to denote both human FGF19 and its murine equivalent FGF15 (when referring to animal studies). Activation of FXR in the intestines induces the expression of FGF19 (primarily in the ileum), which is subsequently released into the portal venous system. 53 FGF19 strongly suppresses CYP7A1 expression in hepatocytes by interaction with the FGF receptor 4 (FGFR4; FGFR4) 53 , 54 and its co‐receptor beta‐Klotho (KLB; KLB) 55 , 56 , 57 (Figures 1, 3). Of note, FGF19‐mediated downregulation of BA synthesis may act through a signalling cascade that is independent from SHP, 58 as Src‐mediated signalling cascades activate both SHP‐dependent and ‐independent mechanisms to suppress BA synthesis. 59 , 60
Although these two feedback mechanisms are widely accepted, some important aspects should be considered: For example, while Inagaki et al reported no detectable induction of FGF19 in mouse liver tissue upon FXR activation, 53 other studies demonstrated FXR‐dependent upregulation of FGF19 in primary human hepatocytes 58 and expression of FGF19 in liver tissue of patients with biliary atresia 61 and primary biliary cholangitis (PBC), 62 suggesting that humans also display intrahepatic auto‐/paracrine FGF19 feedback signalling. Furthermore, intestinal FGF19 release in mice is also stimulated by serum BAs (basolateral side), indicating that enterohepatic FXR‐FGF19 feedback may not exclusively rely on BAs absorbed from the intestinal lumen (apical side). 63
Interestingly, human liver cell lines displayed a continuous time‐dependent downregulation of KLB upon persistent exposure to FGF19, 64 while activation of FXR led to upregulation of KLB in mice, 65 suggesting that the FGF19‐FGFR4/KLB signalling cascade is subject to autoregulation by FXR and circulating FGF19. Studies in humans with liver disease put these experimental findings into clinical context: Byun et al reported downregulation of KLB, Src and FXR, as well as upregulation of FGFR4 in liver tissue of patients with PBC as compared to healthy liver tissue, 59 while Wunsch et al demonstrated that serum FGF19 levels and hepatic FGF19 expression positively correlated with disease severity in patients with PBC. 62 Moreover, patients with primary sclerosing cholangitis (PSC) exhibited increased hepatic FGF19 and FGFR4 gene and/or protein expression as compared to healthy liver tissue, while CYP7A1 (paradoxically) was not repressed. 66 These observations indicate that the enterohepatic BA feedback mechanism may be impaired in certain pathological conditions, particularly in advanced disease stages.
Conclusively, modulation of the FXR‐FGF19 axis emerged as therapeutic target, backed by experimental studies reporting that intestinal activation of FXR in mice with cholestasis ameliorates liver damage associated with BA toxicity, 67 and that overexpression of FGF19 mitigates alcohol‐induced liver injury in mice, 68 and has already led to application in humans with different aetiologies of chronic liver disease. Clinical trials investigating the use of FXR agonists and FGF19 analogues are discussed below.
4.2. FXR signalling determines the enterohepatic circulation of bile acids
The enterohepatic circulation of BAs and, more specifically, BA transporter expression is subject to feedback regulation. These transporters enable the circulation of BAs and determine the BA pool size by regulation of secretion and reuptake and are recognised as important cornerstones of BA homeostasis but also as potential therapeutic targets. 15
First, experimental studies in cell culture demonstrated that ASBT is downregulated by BA‐induced activation of the FXR‐SHP pathway (Figure 3). 69 , 70 Interestingly, FGF19 also downregulated expression of ASBT in a colon cancer cell line and in the intestines of mice via FGFR4‐mediated signalling. 71 Inhibition of ASBT ameliorated liver injury and downregulated the expression of proinflammatory and profibrogenic genes related to BA toxicity in a mouse model of chronic cholestasis (ie, Mdr2‐knockout). 72 , 73
Second, OST‐α and OST‐β expression is upregulated upon FXR activation, as demonstrated in human liver cell lines in vitro, 74 , 75 human ileum ex vivo, 74 as well as in the small intestine and kidney of mice in vivo. 76 OST‐α/OST‐β are present on the basolateral membrane of enterocytes and protect enterocytes from BA toxicity (Figure 3). 77 Induction of these transporters at the basolateral membrane of hepatocytes may provide an alternative efflux route under cholestatic conditions. 78 Both OST‐α/OST‐β exhibited low expression in healthy liver tissue of mice and humans, and were significantly upregulated (OST‐β»OST‐α) in bile‐duct ligated (BDL; ie, animal model for cholestasis) rats and mice as well as patients with PBC and non‐alcoholic steatohepatitis (NASH). 79 , 80 Hence, the upregulation of OST‐α/OST‐β in the liver might serve as a basolateral “pressure relieve valve” for supraphysiological intracellular BA concentrations. However, the induction of OST‐α/OST‐β alone will not eliminate BAs definitively, unless this adaptive response is accompanied by induction of metabolizing enzymes and transporters that will facilitate final elimination of BAs from the hepatocytes and the systemic circulation. The effects of PPAR‐α signalling and fibrates on the synthesis, metabolisation and excretion of BAs (in addition to FXR agonists) may therefore hold interesting perspectives for the future, as discussed recently in the context of PBC. 81
Third, FXR‐induced upregulation of SHP represses the expression of NTCP in hepatocytes in vitro, 82 which was confirmed in vivo using BDL rats and mice, indicating that this regulatory mechanism protects hepatocytes from BA toxicity (Figure 3). 83 , 84 , 85 , 86 Interestingly, a recent in vitro study suggested that endoplasmic reticulum stress may also downregulate NTCP independently from FXR, 87 which may be relevant apart from “primary” cholestatic liver diseases such as PBC, 88 since NTCP was also downregulated in mice fed with ethanol, 89 a rat model of non‐alcoholic fatty liver disease (NAFLD), 90 and patients with alcohol(ALD)‐ and hepatitis C virus‐associated cirrhosis, as compared to healthy liver tissue. 91 , 92 NTCP has emerged as an important therapeutic target since it was identified as an entry point for the hepatitis B (HBV) and D (HDV) virus. 93 Myrcludex B (bulevirtide), a selective inhibitor of NTCP, has emerged as a novel therapy for HBV/HDV and is already in clinical use, 94 but also displayed hepatoprotective effects in cholestatic mouse models. 95
Finally, studies in cell culture and FXR knockout mice showed that FXR activation induces BSEP expression (Figure 3). 96 , 97 , 98 BSEP is the rate‐limiting transporter for BA secretion into bile canaliculi and holds an important role in the enterohepatic cycle, as evident from genetic BSEP deficiencies causing cholestasis in humans. 15 , 99 However, it seems less clear whether BSEP expression is different in liver diseases: Expression and function of BSEP were preserved in rats after BDL, 100 and no significant differences of BSEP gene expression were found in (non‐cirrhotic) patients with PBC or hepatitis C, 101 as well as BSEP protein expression in advanced PBC, 88 as compared to healthy liver tissue. Conversely, patients with NAFLD exhibited downregulation of BSEP on liver histology paralleled by increasing disease activity. 102 Of note, BSEP knockout mice exhibited a milder phenotype compared to humans with BSEP deficiency, which is probably related to species‐dependent compensatory mechanisms, 103 , 104 indicating the need for further studies in humans.
4.3. Metabolic gut‐liver axis signalling by glucagon‐like peptide 1
BA signalling regulates multitudinous pathways involved in the homeostasis of cholesterol, lipid and glucose metabolism. 105 To this end, the discovery of TGR5‐mediated glucagon‐like peptide‐1 (GLP‐1) signalling marks an important example of BA signalling in the gut‐liver axis. Katsuma et al were among the first researchers to describe that the activation of TGR5 in intestinal enteroendocrine cells (L‐cells) induces the release of GLP‐1 (Figure 3). 106 The delivery of GLP‐1 to the liver and pancreas induced insulin secretion and ameliorated glucose tolerance in obese mice, which was also achieved by administration of the synthetic TGR5 agonist INT‐777. 107 Interestingly, a similar effect was achieved by the ASBT inhibitor 264W94 in rats and the BA sequestrant colesevelam in mice, presumably related to the increased luminal availability of BAs and high expression of TGR5 in the colon. 108 , 109 Naturally, these findings were considered as therapeutic options in diabetes and NAFLD (as discussed below).
5. INTESTINAL BARRIER AND BACTERIAL TRANSLOCATION
Next to the transport of nutrients, maintenance of BA homeostasis and metabolism, recent research efforts have shaped the understanding that the intestines and the liver strongly interact on an immunological level, which represents an important dimension of the gut‐liver axis. 110 This crosstalk is determined by intestinal barrier integrity and the translocation of gut bacteria and pathogen‐associated molecular patterns (PAMPs)—subsumed by the term “bacterial translocation”—that circulate to the liver via the portal venous system and trigger proinflammatory and profibrotic responses (Figure 4). 111
FIGURE 4.
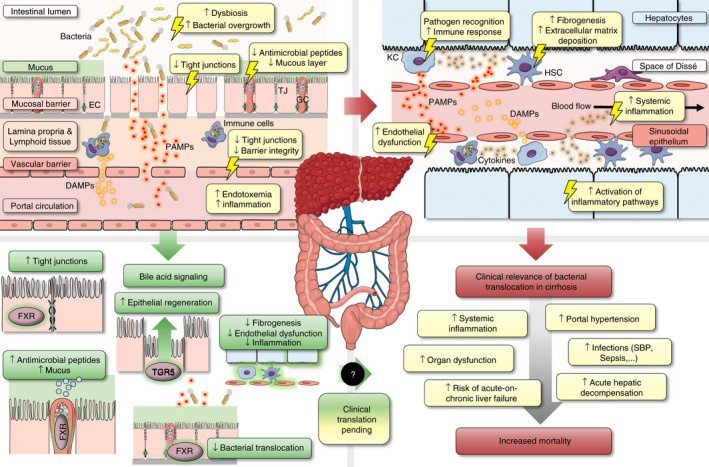
Bacterial translocation in cirrhosis: Pathophysiological aspects and therapeutic implications of bile acid signalling in the gut‐liver axis. Left upper panel: Advanced chronic liver disease (or cirrhosis) is associated with increased bacterial translocation, which is characterised by (i) dysbiosis and small intestinal bacterial overgrowth, (ii) reduced excretion of antibacterial peptides and mucus thickness, (iii) disruption of epithelial integrity associated with reduced expression of tight junction (TJ) proteins, and (iv) an impaired gut‐vascular barrier, which enables the translocation of gut bacteria and danger/pathogen associated molecular patterns (PAMPs/DAMPs) into the portal circulation. Right upper panel: Bacterial translocation induces—directly or indirectly—an intrahepatic inflammatory response, which comprises (i) activation of Kupffer cells (KC) and other immune cells associated with increased shedding of cytokines and systemic inflammation, (ii) activation of hepatic stellate cells (HSC) and induction of fibrogenesis and (iii) endothelial dysfunction of liver sinusoidal epithelial cells (LSEC) which aggravates portal hypertension. Left lower panel: Bile acid signaling may ameliorate intestinal barrier integrity and bacterial translocation via FXR‐ and TGR5‐mediated upregulation of antibacterial defense, TJ proteins, epithelial regeneration, which ultimately leads to amelioration of fibrogenesis, endothelial dysfunction and systemic inflammation in cirrhosis (data based on experimental studies in animals). Right lower panel: The clinical relevance of bacterial translocation in cirrhosis is underlined by the association of portal hypertension and systemic inflammation with the incidence of clinical complications that are associated with increased mortality. Abbreviations: DAMPs/PAMPs, danger/pathogen associated molecular patterns; EC, enterocyte; FXR, farnesoid X receptor; GC, goblet cell; HSC, hepatic stellate cell; KC, Kupffer cell; LSEC, liver sinusoidal epithelial cell; TGR5, transmembrane G protein‐coupled receptor‐5; TJ, tight junction
5.1. Pathophysiological impact and clinical relevance of bacterial translocation
Advanced chronic liver disease (ACLD), a disease spectrum characterised by advanced liver fibrosis/cirrhosis and portal hypertension, marks one of the most important clinical examples where the concept of the bacterial translocation‐induced hepatic and systemic inflammatory response has been attributed a disease‐modifying role (Figure 4). 112 In‐depth reviews on the microbiome and gut‐liver immunology have been published recently 113 , 114 , 115 and are not the focus of this review.
The paradigm of increased bacterial translocation in ACLD is supported by the observation that patients with ACLD are more prone to develop infections as compared to the general population, 116 and the associated increased mortality risk. 117 Importantly, while systemic inflammation is a common feature of distinct syndromes such as acute hepatic decompensation and acute‐on‐chronic liver failure, 118 systemic inflammation also progresses in clinically stable patients across higher ACLD disease stages without evidence of infection and predicts liver‐related complications and/or mortality. 119 The concept of bacterial translocation has led to the rationale that antibiotic prophylaxis may reduce intestinal bacterial overgrowth and, thus, diminish the hepatic and systemic exposure to bacterial translocation and protect against its detrimental impact on ACLD. 112 RCTs reported that antibiotic prophylaxis may reduce mortality in patients with ACLD, however, indicated that only certain patient subgroups benefit from this treatment. 120 , 121 Nevertheless, the long‐term prophylactic use of antibiotics is highly controversial (eg, antibiotic resistance), suggesting that other therapeutic strategies targeting the intestinal barrier and bacterial translocation are warranted.
From a pathophysiological point of view, the recognition of bacteria or PAMPs from the portal circulation by toll‐like receptors (TLR; ie, receptors of the innate immune system that recognise bacterial antigens) in the liver induces an immune response. This process is accompanied by activation of KCs (and other immune cells) and HSCs, leading to an increase of inflammation and extracellular matrix deposition (“fibrogenesis”) (Figure 4). 122 Concordantly, administration of lipopolysaccharide (LPS) activates TLR‐4 and induces hepatic fibrogenesis via transforming growth factor‐beta signalling in mice. 123 Furthermore, LPS dysregulates endothelial and inducible NO synthase (eNOS/iNOS) activity in LSECs in cell culture and rats, 124 , 125 , 126 implicating that bacterial translocation impairs the regulation of the intrahepatic vascular tone by LSECs. These effects likely aggravate portal hypertension, as supported by a study demonstrating that beta glucan‐induced activation of KCs resulted in vasoconstrictor release and increased portal pressure in rats. 127
From a clinical point of view, these mechanisms are highly relevant targets since fibrosis and portal hypertension are central drivers of disease progression in ACLD. 128 FXR agonists displayed promising effects to ameliorate fibrosis and portal hypertension in experimental studies: The FXR agonist obeticholic acid (OCA; formerly INT‐747) reduced HSC activation and liver fibrosis in rats with thioacetamide(TAA)‐induced cirrhosis. 129 This effect was linked to the inhibition of LSEC and Kupffer cell activation by LPS and tumor necrosis factor‐alpha in cell culture. 129 Moreover, OCA improved portal hypertension in rats with BDL‐ and TAA‐induced cirrhosis, which was related to elevated intrahepatic eNOS activity. 130 Concordantly, the non‐steroidal FXR agonist PX20606 (a precursor substance to cilofexor) reduced portal hypertension in rats with CCl4‐induced cirrhosis by amelioration of liver fibrosis and sinusoidal dysfunction, 131 and similar beneficial effects on portal hypertension and fibrosis were demonstrated with the non‐steroidal FXR agonist cilofexor in a rat model of NASH. 132 Of note, experimental studies suggested that PPAR‐α and PPAR‐γ, which are closely linked to FXR signalling, are essential intersections for the induction of hepatic and systemic inflammation, endothelial dysfunction, or HSC activation. 133 , 134 , 135 , 136 Intriguingly, the therapeutic effects of FXR agonists—particularly in cirrhosis and portal hypertension—may be connected to the amelioration of bacterial translocation, which underlines the significance of BA signalling in the gut‐liver axis.
5.2. Bile acid signalling modulates antibacterial defense and intestinal barrier integrity
Regarding the development of bacterial translocation, it is vital to consider that the permeability of the intestinal barrier is tightly regulated in physiological conditions. Therefore, the presence of bacterial translocation indicates the disruption of multiple mechanisms protecting against the migration of bacteria and PAMPs across the intestinal epithelium and the gut‐vascular barrier into the bloodstream, such as antimicrobial peptides, mucous layer and tight junctions (TJ) (Figure 4). 112 Concordantly, bacterial translocation was associated with the downregulation of (a) antimicrobial peptides, (b) reduced mucus thickness, (c) reduced expression of mucosal TJ proteins (primarily in ileum and colon), and (d) an impaired gut‐vascular barrier in rats and mice with CCl4‐/BDL‐induced cirrhosis. 137 , 138 , 139
Early experimental data indicated that oral BA administration ameliorated intestinal bacterial overgrowth and bacterial translocation in rats with CCl4‐induced cirrhosis that had already developed ascites. 140 Verbeke et al linked these observations to FXR signalling by demonstrating that reduced expression of TJ proteins in BDL rats with cirrhosis was associated with reduced FXR activation as evident from downregulation of SHP in the ileum. 138 Importantly, since reduced FXR activation in the ileum would be expected in the cholestatic BDL model (ie, depriving the gut from BA), treatment with OCA strongly upregulated both SHP and TJ expression in the ileum, which was paralleled by decreased bacterial translocation, immune cell recruitment and interferon‐gamma expression in the ileum. 138 Interestingly, SHP was also downregulated in the ileum of rats with CCl4‐induced cirrhosis. Treatment with OCA upregulated the expression of TJ proteins and antimicrobial peptides, leading to reduced bacterial dysbiosis and mucosal inflammation in the ileum, as well as amelioration of liver fibrogenesis. 139 Finally, Sorribas et al found that the (basolateral) gut‐vascular barrier is also impaired in mice with BDL‐/CCl4‐associated cirrhosis and confirmed that OCA and the non‐absorbable FXR agonist fexaramine reduce bacterial translocation by strengthening of the intestinal barrier. 141 A recent study in piglets subjected to BDL confirmed the beneficial effects of the FXR agonist tropifexor on intestinal inflammation and the gut barrier and indicated significant changes of the microbiome. 142
Importantly, the significance of BA signalling for intestinal mucosal integrity is not only restricted to ACLD: Sorrentino et al recently demonstrated that TGR5 activation mediates enterocyte regeneration in intestinal organoids and ameliorates epithelial injury in a mouse model of colitis (Figure 4). 143 Treatment with OCA also improved intestinal barrier integrity in mice with experimentally induced colitis. 144 Taken together, BA signalling is increasingly recognised as a therapeutic target for diseases with a compromised intestinal barrier; however, it must be recognised that confirming human data is, to the best of our knowledge, not available to date.
6. CLINICAL TRANSLATION OF BILE ACID SIGNALLING
6.1. Bile acid receptor ligands
Despite the accumulation of (mostly experimental) data on the central role of BA signalling in the gut‐liver axis, and successful therapeutic interventions with FXR and TGR5 agonists in various in vivo experiments, clinical translation towards their use in humans is still on the “test stand.” While FXR agonists are already investigated in clinical trials, the transfer of TGR5 agonists into clinical application was not successful, due to concerns regarding side effects (further discussed below). Recent and ongoing clinical trials with FXR agonists are summarised in Table 1.
TABLE 1.
Recent and ongoing clinical trials (phase 2 or higher) with steroidal and non‐steroidal FXR agonists
| Substance | Trial phase | Study population | Endpoints | Results | Status* | Reference |
|---|---|---|---|---|---|---|
| Cilofexor (GS9674) | II | PSC (< F4) | Primary: safety at W12 (randomised; vs placebo) |
|
Completed | NCT02943460 153 |
| Cilofexor (GS9674) | III | PSC (< F4) |
Primary: fibrosis progression at W96 (randomised; vs placebo) Secondary: ALP, GGT, ALT, AST, BA, hepatic collagen, histological changes, fibrosis biomarkers |
|
Recruiting | PRIMIS NCT03890120 |
| Cilofexor (GS9674) | II | NASH (< F4) | Primary: safety at W24 (randomised; vs placebo) |
|
Completed | NCT02854605 155 |
| Cilofexor (GS9674) | II | NASH (< F4) | Primary: safety at W24 (randomised; Semaglutide, Firsocostat, Cilofexor in different combinations; no placebo) |
|
Completed | NCT03987074 |
| Cilofexor (GS9674) | II | NASH (No dACLD) | Primary: safety at W12 (different combinations of selonsertib, firsocostat, cilofexor, fenofibrate, icosapent ethyl; no placebo) |
|
Completed | NCT02781584 |
| Cilofexor (GS9674) | II | NASH (F3/F4) (No dACLD) | Primary: safety, ≥1 stage fibrosis improvement without NASH worsening at W48 (randomised; Selonsertib, Firsocostat, Cilofexor vs placebo) |
|
Completed | ATLAS NCT03449446 156 |
| OCA | II | PBC (No dACLD) |
Primary: ALP change at W12 (randomised; combination with UDCA; vs placebo) Secondary: GGT, ALT, PK |
|
Completed | NCT00550862 184 |
| OCA | II | PBC (No dACLD) |
Primary: ALP change at W12 (randomised; vs placebo) Secondary: GGT, AST, ALT, bilirubin |
|
Completed | NCT00570765 185 |
| OCA | II | PBC (No dACLD) |
Primary: ALP change at W12 (randomised; in different combinations with bezafibrate) Secondary: AST, ALT, GGT, bilirubin, life quality, BA, C4 |
|
Recruiting | NCT04594694 |
| OCA | III | PBC (No dACLD) |
Primary: ALP <1.67 ULN and −15%, normal bilirubin at 12 months (UDCA non‐responders; randomised; vs placebo) Secondary: ALP, bilirubin, AST, ALT, GGT |
|
Completed | POISE NCT01473524 147 , 148 |
| OCA | IV | PBC (No dACLD) |
Primary: composite endpoint of hepatic decompensation, MELD ≥15, LT, death (randomised; vs placebo) Secondary: decompensation, death, HCC, fibrosis markers, FGF19, liver dysfunction |
|
Recruiting | COBALT NCT02308111 |
| OCA | II | PSC (No dACLD) |
Primary: ALP change at W24 (randomised; vs placebo) Secondary: AST, ALT, bilirubin, GGT, FGF19, C4 |
|
Completed | AESOP NCT02177136 152 |
| OCA | II | NASH (No dACLD) | Primary: LDL concentration and particle size at W16 (randomised; combination with statins; vs placebo) |
|
Completed | CONTROL NCT02633956 186 |
| OCA | II | NASH (< F4) |
Primary: histological NAS score at W72 (randomised; vs placebo) Secondary: NASH resolution, fibrosis improvement, histological NAS components, serum markers of glucose tolerance, systemic haemodynamics, BMI, life quality |
|
Completed | FLINT NCT01265498 187 , 188 |
| OCA | III | NASH (< F4) |
Primary: fibrosis improvement without NASH worsening, or NASH resolution without fibrosis worsening; decompensation and mortality at 18 months (randomised, vs placebo) Secondary: histological dynamics, liver biochemistry |
|
Active, not recruiting | REGENERATE NCT02548351 154 |
| OCA | II | DM type 2 |
Primary: insulin resistance at W6 (randomised; vs placebo) Secondary: liver dysfunction markers |
|
Completed | NCT00501592 189 |
| OCA | II | Obese and gallstone patients |
Primary: insulin resistance, triglycerides, expression of transport proteins and ER stress markers (randomised; vs placebo) Secondary: serum lipid levels |
|
Completed | OCABSGS NCT01625026 190 |
| OCA | II | BA diarrhea | Primary: FGF19 changes (non‐randomised, open label) |
|
Completed | OBADIAH1 NCT01585025 145 |
| OCA | II | Familial Partial Lipodystrophy | Primary: change liver triglycerides at W8 (randomised; vs placebo) |
|
Recruiting | NCT02430077 |
| OCA | II | ALD (Acute AH) |
Primary: MELD score, safety (randomised; vs placebo) Secondary: bacterial translocation, intestinal inflammation, cytokines |
|
Terminated | TREAT NCT02039219 |
| PX104 | II | NAFLD (No dACLD) |
Primary: Safety at W4; (non‐randomised) Secondary: hepatic fat (%), oGTT, FGF19, plasma BA, PK |
|
Terminated | NCT01999101 191 |
| Tropifexor (LJN452) | II | PBC (No dACLD) |
Primary: GGT, RR, HR, Temperature, ECG, Hb at W4/W12 (randomised; vs placebo) Secondary: PK, PBC‐40 score, pruritus |
|
Completed | NCT02516605 192 |
| Tropifexor (LJN452) | II | NASH (< F4) |
Primary: Safety, TA levels, hepatic fat (%) at W12 (randomised; vs placebo) Secondary: GGT, FGF19, fibrosis biomarkers, lipid profile, pruritus, BMI, serum C4 |
|
Completed | FLIGHT‐FXR NCT02855164 157 |
| Tropifexor (LJN452) | II | NASH (F2/F3) |
Primary: Safety at W48 (randomised; monotherapy vs combination with cenicriviroc) Secondary: fibrosis improvement (>1 point), NASH resolution |
|
Completed | TANDEM NCT03517540 |
| Tropifexor (LJN452) | II | NASH (F2/F3) |
Primary: fibrosis improvement without NASH worsening, NASH resolution without fibrosis worsening at W48 (randomised; monotherapy vs placebo vs combination with licogliflozin) Secondary: NASH resolution, fibrosis improvement, body weight, hepatic fat (%), AST, ALT, GGT, safety |
|
Recruiting | ELIVATE NCT04065841 |
Abbreviations: AH, alcoholic hepatitis; ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BA, bile acids; BMI, body‐mass index; c/dACLD, compensated/decompensated advanced chronic liver disease; C4, serum 7‐alpha‐hydroxy‐4‐cholesten‐3‐one; DM, diabetes mellitus; ECG, electrocardiography; FGF19, fibroblast growth factor 19; GGT, gamma‐glutamyl transferase; Hb, haemoglobin; HCC, hepatocellular carcinoma; HDL, high density lipoprotein; HR, heart rate; LT, liver transplantation; MELD, Model of End Stage Liver Disease score; NAS, NAFLD activity score; NASH, non‐alcoholic steatohepatitis; OCA, obeticholic acid; oGTT, oral glucose tolerance test; PBC, primary biliary cholangitis; PK, pharmacokinetics; PSC, primary sclerosing cholangitis; RR, blood pressure; SAE, serious adverse event; UDCA, ursodeoxycholic acid; ULN, upper limit of normal; V/LDL, (very) low density lipoprotein.
Trial status was obtained from “ClinicalTrials.gov”.
First of all, studies in humans have demonstrated that FXR agonists induce FGF19 serum levels and downregulate BA serum levels, which supports the concept of FXR‐FGF19 signalling. 145 , 146 , 147 Al‐Khaifi et al indicated that the FXR agonist PX‐102 suppresses BA synthesis at a very low dosage even in the absence of FGF19 serum level upregulation in healthy individuals, hypothesising that this effect was explained by hepatic FXR activation. 146
In patients with PBC, the central role of FXR in BA homeostasis served as a rationale for treatment with FXR agonists. In the phase 3 RCT “POISE,” OCA exhibited efficacy in PBC patients unresponsive to UDCA towards the primary endpoint defined as the reduction of alkaline phosphatase (ALP) levels (below 1.67x ULN, and −15% from baseline) and normal bilirubin levels. 147 The open‐label extension of the “POISE” study demonstrated a persistent decrease in ALP and long‐term drug safety upon OCA treatment; however, it reported no significant effect on the composite endpoint death or liver transplantation. 148 A recent study reporting histological long‐term outcomes in 17 patients from the “POISE” trial indicated that OCA improves or stabilises fibrosis and liver injury in PBC. 149 Results of the ongoing phase 4 RCT “COBALT” (NCT02308111) investigating the efficacy of OCA in PBC towards clinical endpoints are eagerly awaited. Regarding non‐steroidal FXR agonists, the phase 2 RCT assessing the efficacy of tropifexor (LJN452; NCT02516605) in PBC patients has been completed, while full results have not yet been published. Preliminary results of the phase II RCT with the non‐steroidal FXR agonist cilofexor (formerly GS‐9674; NCT02943447) showing reduced levels of ALP, gamma‐glutamyl transferase and transaminases in PBC patients after 12 weeks of treatment were presented at an international conference in 2019. 150 Interestingly, a recent observational study indicated that the combined use of UDCA, OCA and fibrates (“triple therapy”) in patients with PBC and ineffectiveness of second‐line treatment led to an improvement of liver injury markers, 151 potentially related to complementary effects of FXR and PPAR signalling. 81 Nevertheless, further studies are needed to confirm these results.
In PSC, the phase 2 RCT “AESOP” investigating the use of OCA showed a significant dose‐dependent decline of ALP levels. 152 Importantly, another phase 2 RCT found that the non‐steroidal FXR agonist cilofexor also exhibited dose‐dependent reduction of serum markers associated with cholestasis and liver injury in PSC patients without cirrhosis. Of note, pruritus (one of the main side effects of OCA) was not associated with the use of cilofexor in this study. 153 The ongoing phase 3 RCT “PRIMIS” (NCT03890120) investigates the efficacy of cilofexor in patients with PSC, with fibrosis progression after 96 weeks as the primary endpoint.
In patients with ALD, the phase 2 RCT “TREAT” (NCT02039219) designed to investigate the safety and efficacy of OCA in patients with acute alcoholic hepatitis was terminated due to hepatoxicity concerns.
In patients with NASH, the interim analysis of the phase 3 RCT “REGENERATE” displayed improved histological fibrosis (and no aggravation of NASH) upon treatment with 25 mg OCA and will continue to assess clinical endpoints. 154 Cilofexor monotherapy significantly improved hepatic fat fraction in (non‐cirrhotic) patients after 24 weeks in a phase 2 RCT. 155 The phase 2b RCT ‘ATLAS’ investigated the use of different combination regimens including cilofexor for 48 weeks in patients with compensated ACLD, however, did not report an effect on the primary endpoint (≥1‐stage improvement in fibrosis without worsening of NASH). 156 The combination of cilofexor and firsocostat led to an improvement of histological NASH activity, improved serum biochemistry, and reduction of non‐invasive fibrosis markers. 156 Furthermore, the phase 2 RCT ‘FLIGHT‐FXR’ investigates the use of tropifexor in patients with fibrosis level F2/F3 on liver histology, and reported dose‐dependent decline of hepatic fat fraction ≥30% (20% vs 64% in the placebo and tropifexor 200 µg groups respectively) and alanine aminotransferase after 12 weeks in an interim analysis, 157 however, final results are pending.
6.2. Enterokines: FGF19 and GLP‐1
Considering that FGF19 greatly determines the suppression of BA synthesis, it was hypothesised that FGF19 analogues may mitigate BA toxicity. However, concerns regarding a potential promotion of hepatocarcinogenesis with endogenous FGF19 have been raised. 158 The development of FGF19 analogues modified to lack certain hepatocarcinogenic structures, while suppressing CYP7A1, has enabled clinical trials. Recent and ongoing clinical trials with FGF19 analogues are summarised in Table 2.
TABLE 2.
Recent and ongoing clinical trials (phase 2 or higher) with FGF19 analogues.
| Substance | Trial phase | Study population | Endpoints | Results | Status* | Reference |
|---|---|---|---|---|---|---|
| NGM282 | IIb | PBC (No dACLD) |
Primary: change plasma ALP at W12 (randomised; three doses, no placebo) Secondary: change bilirubin, AST, ALT, GGT |
|
Completed | NCT02135536 |
| NGM282 | II | PBC (No dACLD) |
Primary: change plasma ALP at W4 (randomised; vs placebo) Secondary: change plasma bilirubin |
|
Completed | NCT02026401 |
| NGM282 | II | PSC (No dACLD) |
Primary: change plasma ALP at W12 (randomised; vs placebo) Secondary: AST, ALT |
|
Completed | NCT02704364 160 |
| NGM282 | II | NASH (No ACLD) |
Primary: absolute hepatic fat content at W12 (randomised; vs placebo) Secondary: relative hepatic fat content |
|
Completed | NCT02443116 161 , 162 |
| NGM282 | II | NASH (cACLD, F4) | Primary: fibrosis improvement ≥1 stage without NASH worsening, safety at W48 (randomised; vs placebo) |
|
Recruiting | ALPINE 4 NCT04210245 |
| NGM282 | II | DM type 2 |
Primary: fasting plasma glucose at W4 (randomised; vs placebo) Secondary: HbA1c, lipids |
|
Completed | NCT01943045 193 |
Abbreviations: AE, adverse event; ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; c/dACLD, compensated/decompensated advanced chronic liver disease; DM, diabetes mellitus; FGF19, fibroblast growth factor 19; GGT, gamma‐glutamyl transferase; NASH, non‐alcoholic steatohepatitis; PBC, primary biliary cholangitis; PSC, primary sclerosing cholangitis; RYGB, Roux‐en‐Y gastric bypass; W, week.
*Trial status was obtained from ‘https://ClinicalTrials.gov’.
In patients with PBC, a phase 2 RCT reported that NGM282 (a FGF19 analogue) reduced serum ALP and serum transaminases in patients unresponsive to UDCA after 12 weeks. 159 The phase 2b RCT has completed recruiting, however, final results are pending. In patients with PSC, NGM282 failed to ameliorate serum ALP levels after 12 weeks in a phase 2 RCT, however, it indicated that serum C4 levels (reflecting CYP7A1 expression) were significantly reduced. 160
In patients with NASH, NGM282 significantly reduced hepatic fat content (defined by a decline of ≥5%) in 74%/79% in the 3 mg/6 mg groups, as compared to 7% in the placebo group. 161 A recent follow‐up analysis in 43 patients from the same study indicated that NGM282 ameliorated histological disease activity and reduced fibrosis. 162
Finally, the therapeutic efficacy of GLP‐1 agonists is well‐established in patients with diabetes, and multiple phase 3 and 4 trials in patients with diabetes and/or metabolic syndrome are ongoing. A recent meta‐analysis of RCTs considering patients with NAFLD and NASH indicated that the use of GLP‐1 analogues is associated with significant reduction of hepatic fat content (mean reduction of 3.92%) and improved chances of NASH resolution without the aggravation of liver fibrosis (odds ratio 4.06, 95%CI 2.52–6.55). 163 Results of the phase 2 RCT on the use of the GLP‐1 agonist semaglutide were published recently and reported an increased rate of NASH resolution (without worsening of fibrosis) as compared to the placebo group (36%–59% in treatment groups vs 17% in the placebo group). 164 However, it remains to be investigated if the efficacy of GLP‐1 in NASH is mainly (indirectly) caused by improved diabetes control, weight loss or direct downstream signalling effects.
6.3. Enterohepatic drugs: from UDCA to inhibition of bile acid transporters
UDCA is one of the first BA‐derived drugs and has been used in patients with cholestatic liver disease for more than two decades, following the consideration that UDCA is more hydrophilic and less toxic than other BA and may exert hepatoprotective effects by the transformation of the BA pool and facilitating bile excretion. The pathophysiological mechanisms and detailed overview of studies with UDCA have been reviewed elsewhere. 165 UDCA particularly displayed efficacy in PBC, based on early reports that only 16% of patients developed oesophageal varices within 4 years of treatment as compared to 58% receiving placebo. 166 An international study comprising approximately 4000 patients with PBC showed that patients receiving UDCA exhibited decreased risk of death or liver transplantation (HR 0.46), even when considered biochemical “non‐responders” to UDCA (HR 0.56). 167
Furthermore, ASBT inhibitors have been investigated in clinical trials, considering that the increased clearance of BA via the faeces may ameliorate cholestasis. In patients with PBC and pruritus enrolled in a phase 2a RCT, the ASBT inhibitor GSK2330672 reduced pruritus severity; however, diarrhoea occurred in about one‐third of patients in the treatment group as drug‐associated side effect. 168 Conversely, also in patients with PBC, the phase 2 RCT “CLARITY” reported that the ASBT inhibitor maralixibat (LUM001), in combination with UDCA vs UDCA alone, had no significant effect on pruritus. 169 Other studies with ASBT inhibitors are ongoing, however, no results are available yet.
Finally, NTCP blockers have only been tested in the context of HBV/HDV treatment. Therefore, while Loglio et al. reported the increase of serum BA in patients with HBV/HDV treated with myrcludex B (which was well‐tolerated), 170 clinical data indicating whether targeting this step in the gut‐liver axis will have a therapeutic role besides antiviral efficacy, will hopefully become available in the future.
6.4. Side effects and safety concerns
As outlined in Table 1 and recently demonstrated in a meta‐analysis and an observational cohort study, pruritus is associated with the intake of OCA and a major cause of drug discontinuation. 171 , 172 Non‐steroidal FXR agonists seem to exhibit a better side effect profile regarding pruritus. 153 However, both steroidal and non‐steroidal FXR agonists caused a deterioration of the lipid profile in previous studies, which is particularly relevant in metabolic liver disease as patients often exhibit an increased cardiovascular risk. 173 Furthermore, reports on cases of hepatotoxicity and liver failure related to the use of OCA remain a significant safety concern, particularly in patients with ACLD or acute liver dysfunction. 174 Patients with compensated ACLD due to PBC treated with OCA showed a higher risk of hepatic decompensation in a recent retrospective study, which underlines the concerns related to the use of OCA in patients with ACLD. 175 It remains to be investigated whether non‐steroidal FXR agonists will exhibit a better safety profile and efficacy in ACLD. Furthermore, the FGF19 analogue NGM282 was associated with injection site reactions, diarrhoea, abdominal pain and nausea (Table 2). 161 , 162 The transfer of TGR5 agonists into clinical trials was not successful due to concerns towards aggravation of pruritus, blockade of gallbladder motility and increased gallstone formation, 176 as well as inhibition of apoptosis in cholangiocarcinoma cell lines. 177 , 178 Careful consideration of these aspects is important to optimise drug development, patient management and weighting the benefit‐risk‐ratio for emerging therapeutic strategies.
6.5. Perspectives and future research agenda
Although previous experimental and clinical studies indicated that modulation of BA signalling in the gut‐liver axis exerts promising therapeutic effects, future studies need to consider important issues related to methodological limitations and study designs to enable the clinical translation of novel therapies. First, experimental studies in cells and animals often display imperfect models of the human organism. For example, a landmark study on the FGF19 feedback mechanism in mice suggested that FGF19 is not expressed in the liver, 53 in contrast to studies using human hepatocytes and human liver tissue, 58 , 61 , 62 potentially explained by species‐related differences. Similarly, currently established animal models of liver disease are only mimicking (instead of imitating) human liver disease. 179 For example, BDL in rodents completely and irreversibly abrogates the excretion of BAs by the liver, which marks a relevant difference to the natural course of cholestatic liver diseases in humans. Second, mechanistic evidence on the concept of bacterial translocation and an impaired intestinal barrier in cirrhosis (or the significance of BA signalling) has not been fully addressed in humans with ACLD. Previous studies have found changes of the stool microbiome in ACLD (indicating dysbiosis), 114 or reported increased epithelial permeability by the use of duodenum biopsies; 180 however, these findings may not account for relevant changes of the microbiome along the gastrointestinal tract and the actual significance of bacterial translocation in humans, as experimental studies in animals usually indicated that the ileum is the most relevant intestinal segment for bacterial translocation. 181 , 182 , 183 Third, previous clinical trials did not assess whether experimental evidence that FXR agonists improve the intestinal epithelial barrier and bacterial translocation in cirrhosis translates into improved clinical outcomes, as patients with (particularly decompensated) ACLD were usually excluded (Table 1). Non‐steroidal or non‐absorbable FXR agonists will hopefully become effective and safe therapeutic options to address this question in the future. Finally, as indicated by the efficacy of cilofexor in combination with firsocostat (an acetyl‐CoA carboxylase inhibitor) in NASH, 156 and recent data on the combination of OCA and fibrates in PBC, 151 the use of combination treatment regimens may represent a promising approach to treat liver diseases. The use of human tissue or human organoids will be vital to close the gap between experimental data and human disease in future studies on BA signalling in the gut‐liver axis.
7. CONCLUSIONS
Modulation of BA gut‐liver signalling may exert beneficial effects on BA homeostasis and intestinal barrier integrity. Pharmacological interventions targeting the gut‐liver axis are currently focused on BA receptor ligands, FGF19, and enterohepatic drugs. Notably, the clinical use of FXR agonists in RCTs were mainly conducted in patients with non‐cirrhotic liver disease, while patients with (decompensated) ACLD were usually excluded. However, it seems justified to further explore the therapeutic effects of FXR agonism on intestinal barrier integrity to ACLD patients, since bacterial translocation in the gut‐liver axis is reportedly detrimental for the course of ACLD. Ultimately, several molecular targets of the presented BA signalling pathways remain to be explored for potential therapeutic modulations in patients with chronic liver disease.
AUTHORSHIP
Guarantor of the article: Thomas Reiberger.
Author contributions: BS and TR drafted the manuscript, which was critically revised by all authors. All authors approved the final version of the manuscript.
ACKNOWLEDGEMENT
Declaration of personal interest: BS received travel support from AbbVie and Gilead, and was supported by an international research scholar by Gilead Sciences awarded to TR. MT received speaker fees from BMS, Falk Foundation, Gilead, Intercept and MSD; advisory board fees from Albireo, BiomX, Boehringer Ingelheim, Falk Pharma GmbH, Genfit, Gilead, Intercept, MSD, Novartis, Phenex, Regulus and Shire. He further received travel grants from Abbvie, Falk, Gilead and Intercept and unrestricted research grants from Albireo, Cymabay, Falk, Gilead, Intercept, MSD and Takeda. He is also coinventor of patents on the medical use of nor‐UDCA filed by the Medical Universities of Graz and Vienna. TR received grant support from Abbvie, Boehringer‐Ingelheim, Gilead, MSD, Philips Healthcare, Gore; speaking honoraria from Abbvie, Gilead, Gore, Intercept, Roche, MSD; consulting/advisory board fee from Abbvie, Bayer, Boehringer‐Ingelheim, Gilead, MSD, Siemens; and travel support from Boehringer‐Ingelheim, Gilead and Roche.
Simbrunner B, Trauner M, Reiberger T. Review article: therapeutic aspects of bile acid signalling in the gut‐liver axis. Aliment Pharmacol Ther. 2021;54:1243–1262. 10.1111/apt.16602
The Handling Editor for this article was Professor Gideon Hirschfield, and this uncommissioned review was accepted for publication after full peer‐review.
Funding information
None.
References
- 1. de Buy M, Wenniger L, Beuers U. Bile salts and cholestasis. Dig Liver Dis. 2010;42:409–418. [DOI] [PubMed] [Google Scholar]
- 2. Hofmann AF. Chemistry and enterohepatic circulation of bile acids. Hepatology. 1984;4:4s–14s. [DOI] [PubMed] [Google Scholar]
- 3. Hofmann AF, Mysels KJ. Bile acid solubility and precipitation in vitro and in vivo: the role of conjugation, pH, and Ca2+ ions. J Lipid Res. 1992;33:617–626. [PubMed] [Google Scholar]
- 4. Thomas C, Pellicciari R, Pruzanski M, et al. Targeting bile‐acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008;7: 678–693. [DOI] [PubMed] [Google Scholar]
- 5. Matern S, Gerok W. Pathophysiology of the enterohepatic circulation of bile acids. Rev Physiol Biochem Pharmacol. 1979;85:125–204. [DOI] [PubMed] [Google Scholar]
- 6. Nimer N, Choucair I, Wang Z, et al. Bile acids profile, histopathological indices and genetic variants for non‐alcoholic fatty liver disease progression. Metabolism. 2020;116:154457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chiang JY. Bile acids: regulation of synthesis. J Lipid Res. 2009;50:1955–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou H, Hylemon PB. Bile acids are nutrient signaling hormones. Steroids. 2014;86:62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jelinek DF, Andersson S, Slaughter CA, et al. Cloning and regulation of cholesterol 7 alpha‐hydroxylase, the rate‐limiting enzyme in bile acid biosynthesis. J Biol Chem. 1990;265:8190–8197. [PMC free article] [PubMed] [Google Scholar]
- 10. Pandak WM, et al. Expression of sterol 12alpha‐hydroxylase alters bile acid pool composition in primary rat hepatocytes and in vivo. Gastroenterology. 2001;120:1801–1809. [DOI] [PubMed] [Google Scholar]
- 11. Ferrell JM, Boehme S, Li F, et al. Cholesterol 7α‐hydroxylase‐deficient mice are protected from high‐fat/high‐cholesterol diet‐induced metabolic disorders. J Lipid Res. 2016;57:1144–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fan L, Joseph JF, Durairaj P, et al. Conversion of chenodeoxycholic acid to cholic acid by human CYP8B1. Biol Chem. 2019;400:625–628. [DOI] [PubMed] [Google Scholar]
- 13. Di Ciaula A, Garruti G, Lunardi Baccetto R, et al. Bile Acid Physiology. Ann Hepatol. 2017;16:s4–s14. [DOI] [PubMed] [Google Scholar]
- 14. Hofmann AF, Hagey LR. Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol Life Sci. 2008;65(16):2461–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kunst RF, Verkade HJ, Oude Elferink RPJ, et al. Targeting the four pillars of enterohepatic bile salt cycling; lessons from genetics and pharmacology. Hepatology. 2020;73:2577–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Seol W, Choi HS, Moore DD. Isolation of proteins that interact specifically with the retinoid X receptor: two novel orphan receptors. Mol Endocrinol. 1995;9(1):72–85. [DOI] [PubMed] [Google Scholar]
- 17. Forman BM, Goode E, Chen J, et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell. 1995;81:687–693. [DOI] [PubMed] [Google Scholar]
- 18. Laffitte BA, Kast HR, Nguyen CM, et al. Identification of the DNA binding specificity and potential target genes for the farnesoid X‐activated receptor. J Biol Chem. 2000;275:10638–10647. [DOI] [PubMed] [Google Scholar]
- 19. Wang NA, Zou Q, Xu J, et al. Ligand binding and heterodimerization with retinoid X receptor α (RXRα) induce farnesoid X receptor (FXR) conformational changes affecting coactivator binding. J Biol Chem. 2018;293:18180–18191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shen H, Zhang YU, Ding H, et al. Farnesoid X receptor induces GLUT4 expression through FXR response element in the GLUT4 promoter. Cell Physiol Biochem. 2008;22:1–14. [DOI] [PubMed] [Google Scholar]
- 21. Pineda Torra I, Claudel T, Duval C, Kosykh V, Fruchart JC, Staels B. Bile acids induce the expression of the human peroxisome proliferator‐activated receptor alpha gene via activation of the farnesoid X receptor. Mol Endocrinol. 2003;17:259–272. [DOI] [PubMed] [Google Scholar]
- 22. Bookout AL, Jeong Y, Downes M, et al. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126:789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Inagaki T, Moschetta A, Lee Y‐K, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A. 2006;103:3920–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li Z, Kruijt JK, der Sluis RJV, et al. Nuclear receptor atlas of female mouse liver parenchymal, endothelial, and Kupffer cells. Physiol Genomics. 2013;45:268–275. [DOI] [PubMed] [Google Scholar]
- 25. Fickert P, Fuchsbichler A, Moustafa T, et al. Farnesoid X receptor critically determines the fibrotic response in mice but is expressed to a low extent in human hepatic stellate cells and periductal myofibroblasts. Am J Pathol. 2009;175:2392–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. De Marino S , Festa C, Sepe V, Zampella A, et al. Chemistry and pharmacology of GPBAR1 and FXR selective agonists, dual agonists, and antagonists. Handb Exp Pharmacol. 2019;256:137–165. [DOI] [PubMed] [Google Scholar]
- 27. Gege C, Hambruch E, Hambruch N, et al. Nonsteroidal FXR ligands: current status and clinical applications. Handb Exp Pharmacol. 2019;256:167–205. [DOI] [PubMed] [Google Scholar]
- 28. Maruyama T, Miyamoto Y, Nakamura T, et al. Identification of membrane‐type receptor for bile acids (M‐BAR). Biochem Biophys Res Commun. 2002;298:714–719. [DOI] [PubMed] [Google Scholar]
- 29. Kawamata Y, Fujii R, Hosoya M, et al. A G protein‐coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–9440. [DOI] [PubMed] [Google Scholar]
- 30. Watanabe M, Houten SM, Mataki C, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. [DOI] [PubMed] [Google Scholar]
- 31. Perino A, Pols TWH, Nomura M, et al. TGR5 reduces macrophage migration through mTOR‐induced C/EBPβ differential translation. J Clin Invest. 2014;124:5424–5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reich M, Deutschmann K, Sommerfeld A, et al. TGR5 is essential for bile acid‐dependent cholangiocyte proliferation in vivo and in vitro. Gut. 2016;65:487–501. [DOI] [PubMed] [Google Scholar]
- 33. Rajagopal S, Kumar DP, Mahavadi S, et al. Activation of G protein‐coupled bile acid receptor, TGR5, induces smooth muscle relaxation via both Epac‐ and PKA‐mediated inhibition of RhoA/Rho kinase pathway. Am J Physiol Gastrointest Liver Physiol. 2013;304:G527–G535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vassileva G, Golovko A, Markowitz L, et al. Targeted deletion of Gpbar1 protects mice from cholesterol gallstone formation. Biochem J. 2006;398:423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Keitel V, Cupisti K, Ullmer C, et al. The membrane‐bound bile acid receptor TGR5 is localized in the epithelium of human gallbladders. Hepatology. 2009;50:861–870. [DOI] [PubMed] [Google Scholar]
- 36. Maruyama T, Tanaka K, Suzuki J, et al. Targeted disruption of G protein‐coupled bile acid receptor 1 (Gpbar1/M‐Bar) in mice. J Endocrinol. 2006;191:197–205. [DOI] [PubMed] [Google Scholar]
- 37. Poole DP, Godfrey C, Cattaruzza F, et al. Expression and function of the bile acid receptor GpBAR1 (TGR5) in the murine enteric nervous system. Neurogastroenterol Motil. 2010;22:814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Keitel V, Donner M, Winandy S, et al. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem Biophys Res Commun. 2008;372:78–84. [DOI] [PubMed] [Google Scholar]
- 39. Keitel V, Reinehr R, Gatsios P, et al. The G‐protein coupled bile salt receptor TGR5 is expressed in liver sinusoidal endothelial cells. Hepatology. 2007;45:695–704. [DOI] [PubMed] [Google Scholar]
- 40. Holter MM, Chirikjian MK, Briere DA, et al. Compound 18 Improves Glucose Tolerance in a Hepatocyte TGR5‐dependent Manner in Mice. Nutrients. 2020;12:2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang JI, Yoon J‐H, Myung SJ, et al. Bile acid‐induced TGR5‐dependent c‐Jun‐N terminal kinase activation leads to enhanced caspase 8 activation in hepatocytes. Biochem Biophys Res Commun. 2007;361:156–161. [DOI] [PubMed] [Google Scholar]
- 42. Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol. 2014;11:55–67. [DOI] [PubMed] [Google Scholar]
- 43. Fiorucci S, Biagioli M, Zampella A, Distrutti E. Bile acids activated receptors regulate innate immunity. Front Immunol. 2018;9:1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shin DJ, Wang L. Bile acid‐activated receptors: a review on FXR and Other nuclear receptors. Handb Exp Pharmacol. 2019;256:51–72. [DOI] [PubMed] [Google Scholar]
- 45. Ðanić M, Stanimirov B, Pavlović N, et al. Pharmacological applications of bile acids and their derivatives in the treatment of metabolic syndrome. Front Pharmacol. 2018;9:1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Perez M‐J, Briz O. Bile‐acid‐induced cell injury and protection. World J Gastroenterol. 2009;15:1677–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lu TT, Makishima M, Repa JJ, et al. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell. 2000;6:507–515. [DOI] [PubMed] [Google Scholar]
- 48. Seol W, Choi HS, Moore DD. An orphan nuclear hormone receptor that lacks a DNA binding domain and heterodimerizes with other receptors. Science. 1996;272:1336–1339. [DOI] [PubMed] [Google Scholar]
- 49. Goodwin B, Jones SA, Price RR, et al. A regulatory cascade of the nuclear receptors FXR, SHP‐1, and LRH‐1 represses bile acid biosynthesis. Mol Cell. 2000;6:517–526. [DOI] [PubMed] [Google Scholar]
- 50. Brendel C, Schoonjans K, Botrugno OA, Treuter E, Auwerx J. The small heterodimer partner interacts with the liver X receptor alpha and represses its transcriptional activity. Mol Endocrinol. 2002;16:2065–2076. [DOI] [PubMed] [Google Scholar]
- 51. Kim I, Ahn S‐H, Inagaki T, et al. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res. 2007;48:2664–2672. [DOI] [PubMed] [Google Scholar]
- 52. Kong BO, Wang LI, Chiang JYL, et al. Mechanism of tissue‐specific farnesoid X receptor in suppressing the expression of genes in bile‐acid synthesis in mice. Hepatology. 2012;56:1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. [DOI] [PubMed] [Google Scholar]
- 54. Holt JA, Luo G, Billin AN, et al. Definition of a novel growth factor‐dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003;17:1581–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Goetz R, Beenken A, Ibrahimi OA, et al. Molecular insights into the klotho‐dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol Cell Biol. 2007;27:3417–3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wu X, Ge H, Gupte J, et al. Co‐receptor requirements for fibroblast growth factor‐19 signaling. J Biol Chem. 2007;282:29069–29072. [DOI] [PubMed] [Google Scholar]
- 57. Ito S, Fujimori T, Furuya A, Satoh J, Nabeshima Y, Nabeshima Y. Impaired negative feedback suppression of bile acid synthesis in mice lacking betaKlotho. J Clin Invest. 2005;115:2202–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Song KH, Li T, Owsley E, Strom S, Chiang JY. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha‐hydroxylase gene expression. Hepatology. 2009;49:297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Byun S, Kim D‐H, Ryerson D, et al. Postprandial FGF19‐induced phosphorylation by Src is critical for FXR function in bile acid homeostasis. Nat Commun. 2018;9:2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li S, Hsu D, Li B, et al. Cytoplasmic tyrosine phosphatase Shp2 coordinates hepatic regulation of bile acid and FGF15/19 signaling to repress bile acid synthesis. Cell Metab. 2014;20:320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hasegawa Y, Kawai M, Bessho K, et al. CYP7A1 expression in hepatocytes is retained with upregulated fibroblast growth factor 19 in pediatric biliary atresia. Hepatol Res. 2019;49:314–323. [DOI] [PubMed] [Google Scholar]
- 62. Wunsch E, Milkiewicz M, Wasik U, et al. Expression of hepatic fibroblast growth factor 19 is enhanced in primary biliary cirrhosis and correlates with severity of the disease. Sci Rep. 2015;5:13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Slijepcevic D, Roscam Abbing RLP, Katafuchi T, et al. Hepatic uptake of conjugated bile acids is mediated by both sodium taurocholate cotransporting polypeptide and organic anion transporting polypeptides and modulated by intestinal sensing of plasma bile acid levels in mice. Hepatology. 2017;66:1631–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lin BC, Wang M, Blackmore C, et al. Liver‐specific activities of FGF19 require Klotho beta. J Biol Chem. 2007;282:27277–27284. [DOI] [PubMed] [Google Scholar]
- 65. Fu T, Kim Y‐C, Byun S, et al. FXR primes the liver for intestinal FGF15 signaling by transient induction of β‐Klotho. Mol Endocrinol. 2016;30:92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Milkiewicz M, Klak M, Kempinska‐Podhorodecka A, et al. Impaired hepatic adaptation to chronic cholestasis induced by primary sclerosing cholangitis. Sci Rep. 2016;6:39573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Modica S, Petruzzelli M, Bellafante E, et al. Selective activation of nuclear bile acid receptor FXR in the intestine protects mice against cholestasis. Gastroenterology. 2012;142:355–365.e4. [DOI] [PubMed] [Google Scholar]
- 68. Hartmann P, Hochrath K, Horvath A, et al. Modulation of the intestinal bile acid/farnesoid X receptor/fibroblast growth factor 15 axis improves alcoholic liver disease in mice. Hepatology. 2018;67:2150–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Neimark E, Chen F, Li X, et al. Bile acid‐induced negative feedback regulation of the human ileal bile acid transporter. Hepatology. 2004;40:149–156. [DOI] [PubMed] [Google Scholar]
- 70. Chen F, Ma L, Dawson PA, et al. Liver receptor homologue‐1 mediates species‐ and cell line‐specific bile acid‐dependent negative feedback regulation of the apical sodium‐dependent bile acid transporter. J Biol Chem. 2003;278:19909–19916. [DOI] [PubMed] [Google Scholar]
- 71. Sinha J, Chen F, Miloh T, Burns RC, Yu Z, Shneider BL. beta‐Klotho and FGF‐15/19 inhibit the apical sodium‐dependent bile acid transporter in enterocytes and cholangiocytes. Am J Physiol Gastrointest Liver Physiol. 2008;295:G996–g1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Baghdasaryan A, Fuchs CD, Österreicher CH, et al. Inhibition of intestinal bile acid absorption improves cholestatic liver and bile duct injury in a mouse model of sclerosing cholangitis. J Hepatol. 2016;64:674–681. [DOI] [PubMed] [Google Scholar]
- 73. Miethke AG, Zhang W, Simmons J, et al. Pharmacological inhibition of apical sodium‐dependent bile acid transporter changes bile composition and blocks progression of sclerosing cholangitis in multidrug resistance 2 knockout mice. Hepatology. 2016;63:512–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Landrier JF, Eloranta JJ, Vavricka SR, Kullak‐Ublick GA. The nuclear receptor for bile acids, FXR, transactivates human organic solute transporter‐alpha and ‐beta genes. Am J Physiol Gastrointest Liver Physiol. 2006;290:G476–G485. [DOI] [PubMed] [Google Scholar]
- 75. Guo C, LaCerte C, Edwards JE, et al. Farnesoid X receptor agonists obeticholic acid and chenodeoxycholic acid increase bile acid efflux in sandwich‐cultured human hepatocytes: functional evidence and mechanisms. J Pharmacol Exp Ther. 2018;365:413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lee H, Zhang Y, Lee FY, Nelson SF, Gonzalez FJ, Edwards PA. FXR regulates organic solute transporters alpha and beta in the adrenal gland, kidney, and intestine. J Lipid Res. 2006;47:201–214. [DOI] [PubMed] [Google Scholar]
- 77. Ferrebee CB, Li J, Haywood J, et al. Organic solute transporter α‐β Protects ileal enterocytes from bile acid‐induced injury. Cell Mol Gastroenterol Hepatol. 2018;5:499–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Beaudoin JJ, Brouwer KLR, Malinen MM. Novel insights into the organic solute transporter alpha/beta, OSTα/β: from the bench to the bedside. Pharmacol Ther. 2020;211:107542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Boyer JL, Trauner M, Mennone A, et al. Upregulation of a basolateral FXR‐dependent bile acid efflux transporter OSTalpha‐OSTbeta in cholestasis in humans and rodents. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1124–G1130. [DOI] [PubMed] [Google Scholar]
- 80. Malinen MM, Ali I, Bezençon J, et al. Organic solute transporter OSTα/β is overexpressed in nonalcoholic steatohepatitis and modulated by drugs associated with liver injury. Am J Physiol Gastrointest Liver Physiol. 2018;314:G597–G609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gulamhusein AF, Hirschfield GM. Primary biliary cholangitis: pathogenesis and therapeutic opportunities. Nat Rev Gastroenterol Hepatol. 2020;17:93–110. [DOI] [PubMed] [Google Scholar]
- 82. Denson LA, Sturm E, Echevarria W, et al. The orphan nuclear receptor, shp, mediates bile acid‐induced inhibition of the rat bile acid transporter, ntcp. Gastroenterology. 2001;121:140–147. [DOI] [PubMed] [Google Scholar]
- 83. Gartung C, Ananthanarayanan M, Rahman MA, et al. Down‐regulation of expression and function of the rat liver Na+/bile acid cotransporter in extrahepatic cholestasis. Gastroenterology. 1996;110:199–209. [DOI] [PubMed] [Google Scholar]
- 84. Geier A, Zollner G, Dietrich CG, et al. Cytokine‐independent repression of rodent Ntcp in obstructive cholestasis. Hepatology. 2005;41:470–477. [DOI] [PubMed] [Google Scholar]
- 85. Zollner G, Wagner M, Fickert P, et al. Role of nuclear receptors and hepatocyte‐enriched transcription factors for Ntcp repression in biliary obstruction in mouse liver. Am J Physiol Gastrointest Liver Physiol. 2005;289:G798–805. [DOI] [PubMed] [Google Scholar]
- 86. Zollner G, Fickert P, Silbert D, et al. Induction of short heterodimer partner 1 precedes downregulation of Ntcp in bile duct‐ligated mice. Am J Physiol Gastrointest Liver Physiol. 2002;282:G184–G191. [DOI] [PubMed] [Google Scholar]
- 87. Robin MJD, Appelman MD, Vos HR, et al. Calnexin depletion by endoplasmic reticulum stress during cholestasis inhibits the Na(+)‐taurocholate cotransporting polypeptide. Hepatol Commun. 2018;2:1550–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zollner G, Fickert P, Silbert D, et al. Adaptive changes in hepatobiliary transporter expression in primary biliary cirrhosis. J Hepatol. 2003;38:717–727. [DOI] [PubMed] [Google Scholar]
- 89. Gong X, Zhang Q, Ruan Y, et al. Chronic alcohol consumption increased bile acid levels in enterohepatic circulation and reduced efficacy of irinotecan. Alcohol Alcohol. 2020;55:264–277. [DOI] [PubMed] [Google Scholar]
- 90. Fisher CD, Lickteig AJ, Augustine LM, et al. Experimental non‐alcoholic fatty liver disease results in decreased hepatic uptake transporter expression and function in rats. Eur J Pharmacol. 2009;613:119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Taniguchi T, Zanetti‐Yabur A, Wang P, et al. Interindividual diversity in expression of organic anion uptake transporters in normal and cirrhotic human liver. Hepatol Commun. 2020;4:739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wang L, Collins C, Kelly EJ, et al. Transporter expression in liver tissue from subjects with alcoholic or hepatitis C cirrhosis quantified by targeted quantitative proteomics. Drug Metab Dispos. 2016;44:1752–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ni YI, Lempp FA, Mehrle S, et al. Hepatitis B and D viruses exploit sodium taurocholate co‐transporting polypeptide for species‐specific entry into hepatocytes. Gastroenterology. 2014;146:1070–1083. [DOI] [PubMed] [Google Scholar]
- 94. Bogomolov P, Alexandrov A, Voronkova N, et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: First results of a phase Ib/IIa study. J Hepatol. 2016;65:490–498. [DOI] [PubMed] [Google Scholar]
- 95. Slijepcevic Davor, Roscam Abbing Reinout L P, Fuchs Claudia D, Haazen Lizette C M, Beuers Ulrich, Trauner Michael, Oude Elferink Ronald P J, van de Graaf Stan F J, Slijepcevic D, Roscam Abbing RLP, Fuchs CD, et al. Na(+) ‐taurocholate cotransporting polypeptide inhibition has hepatoprotective effects in cholestasis in mice. Hepatology. 2018;68:1057–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ananthanarayanan M, Balasubramanian N, Makishima M, et al. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J Biol Chem. 2001;276:28857–28865. [DOI] [PubMed] [Google Scholar]
- 97. Plass JRM, Mol O, Heegsma J, et al. Farnesoid X receptor and bile salts are involved in transcriptional regulation of the gene encoding the human bile salt export pump. Hepatology. 2002;35:589–596. [DOI] [PubMed] [Google Scholar]
- 98. Schuetz EG, Strom S, Yasuda K, et al. Disrupted bile acid homeostasis reveals an unexpected interaction among nuclear hormone receptors, transporters, and cytochrome P450. J Biol Chem. 2001;276:39411–39418. [DOI] [PubMed] [Google Scholar]
- 99. Stieger B. The role of the sodium‐taurocholate cotransporting polypeptide (NTCP) and of the bile salt export pump (BSEP) in physiology and pathophysiology of bile formation. Handb Exp Pharmacol. 2011;201:205–259. [DOI] [PubMed] [Google Scholar]
- 100. Lee JM, Trauner M, Soroka CJ, et al. Expression of the bile salt export pump is maintained after chronic cholestasis in the rat. Gastroenterology. 2000;118:163–172. [DOI] [PubMed] [Google Scholar]
- 101. Zollner G, Fickert P, Zenz R, et al. Hepatobiliary transporter expression in percutaneous liver biopsies of patients with cholestatic liver diseases. Hepatology. 2001;33:633–646. [DOI] [PubMed] [Google Scholar]
- 102. Okushin K, Tsutsumi T, Enooku K, et al. The intrahepatic expression levels of bile acid transporters are inversely correlated with the histological progression of nonalcoholic fatty liver disease. J Gastroenterol. 2016;51:808–818. [DOI] [PubMed] [Google Scholar]
- 103. Wang R, Sheps JA, Liu L, et al. Hydrophilic bile acids prevent liver damage caused by lack of biliary phospholipid in Mdr2(‐/‐) mice. J Lipid Res. 2019;60:85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Fuchs CD, Paumgartner G, Wahlström A, et al. Metabolic preconditioning protects BSEP/ABCB11(‐/‐) mice against cholestatic liver injury. J Hepatol. 2017;66:95–101. [DOI] [PubMed] [Google Scholar]
- 105. Chiang JYL, Ferrell JM. Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy. Am J Physiol Gastrointest Liver Physiol. 2020;318:G554–G573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Katsuma S, Hirasawa A, Tsujimoto G. Bile acids promote glucagon‐like peptide‐1 secretion through TGR5 in a murine enteroendocrine cell line STC‐1. Biochem Biophys Res Commun. 2005;329:386–390. [DOI] [PubMed] [Google Scholar]
- 107. Thomas C, Gioiello A, Noriega L, et al. TGR5‐mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Potthoff MJ, Potts A, He T, et al. Colesevelam suppresses hepatic glycogenolysis by TGR5‐mediated induction of GLP‐1 action in DIO mice. Am J Physiol Gastrointest Liver Physiol. 2013;304:G371–G380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Chen L, Yao X, Young A, et al. Inhibition of apical sodium‐dependent bile acid transporter as a novel treatment for diabetes. Am J Physiol Endocrinol Metab. 2012;302:E68–76. [DOI] [PubMed] [Google Scholar]
- 110. Wiest R, Albillos A, Trauner M, et al. Targeting the gut‐liver axis in liver disease. J Hepatol. 2017;67:1084–1103. [DOI] [PubMed] [Google Scholar]
- 111. Simbrunner B, Mandorfer M, Trauner M, et al. Gut‐liver axis signaling in portal hypertension. World J Gastroenterol. 2019;25:5897–5917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Tranah TH, Edwards LA, Schnabl B, et al. Targeting the gut‐liver‐immune axis to treat cirrhosis. Gut. 2020;70:982–994. [DOI] [PubMed] [Google Scholar]
- 113. Hartmann P, Chu H, Duan YI, et al. Gut microbiota in liver disease: too much is harmful, nothing at all is not helpful either. Am J Physiol Gastrointest Liver Physiol. 2019;316:G563–g573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wang R, Tang R, Li BO, et al. Gut microbiome, liver immunology, and liver diseases. Cell Mol Immunol. 2021;18:4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Heymann F, Tacke F. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol. 2016;13:88–110. [DOI] [PubMed] [Google Scholar]
- 116. Jalan R, Fernandez J, Wiest R, et al. Bacterial infections in cirrhosis: a position statement based on the EASL Special Conference 2013. J Hepatol. 2014;60:1310–1324. [DOI] [PubMed] [Google Scholar]
- 117. Arvaniti V, D'Amico G, Fede G, et al. Infections in patients with cirrhosis increase mortality four‐fold and should be used in determining prognosis. Gastroenterology. 2010;139:1246– 1256. [DOI] [PubMed] [Google Scholar]
- 118. Clària J, Stauber RE, Coenraad MJ, et al. Systemic inflammation in decompensated cirrhosis: characterization and role in acute‐on‐chronic liver failure. Hepatology. 2016;64:1249–1264. [DOI] [PubMed] [Google Scholar]
- 119. Costa D, Simbrunner B, Jachs M, et al. Systemic inflammation increases across distinct stages of advanced chronic liver disease and correlates with decompensation and mortality. J Hepatol. 2020;74:819–828. [DOI] [PubMed] [Google Scholar]
- 120. Fernández J, Navasa M, Planas R, et al. Primary prophylaxis of spontaneous bacterial peritonitis delays hepatorenal syndrome and improves survival in cirrhosis. Gastroenterology. 2007;133:818–824. [DOI] [PubMed] [Google Scholar]
- 121. Moreau R, Elkrief L, Bureau C, et al. Effects of long‐term norfloxacin therapy in patients with advanced cirrhosis. Gastroenterology. 2018;155:1816–1827.e9. [DOI] [PubMed] [Google Scholar]
- 122. Seki E, Schnabl B. Role of innate immunity and the microbiota in liver fibrosis: crosstalk between the liver and gut. J Physiol. 2012;590:447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Seki E, De Minicis S , Osterreicher CH, et al. TLR4 enhances TGF‐beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–1332. [DOI] [PubMed] [Google Scholar]
- 124. Kamoun WS, Karaa A, Kresge N, et al. LPS inhibits endothelin‐1‐induced endothelial NOS activation in hepatic sinusoidal cells through a negative feedback involving caveolin‐1. Hepatology. 2006;43:182–190. [DOI] [PubMed] [Google Scholar]
- 125. Merkel SM, Kamoun W, Karaa A, et al. LPS inhibits endothelin‐1‐mediated eNOS translocation to the cell membrane in sinusoidal endothelial cells. Microcirculation. 2005;12:433–442. [DOI] [PubMed] [Google Scholar]
- 126. La Mura V, Pasarín M, Rodriguez‐Vilarrupla A, et al. Liver sinusoidal endothelial dysfunction after LPS administration: a role for inducible‐nitric oxide synthase. J Hepatol. 2014;61:1321–1327. [DOI] [PubMed] [Google Scholar]
- 127. Steib CJ, Gerbes AL, Bystron M, et al. Kupffer cell activation in normal and fibrotic livers increases portal pressure via thromboxane A(2). J Hepatol. 2007;47:228–238. [DOI] [PubMed] [Google Scholar]
- 128. Bosch J, Groszmann RJ, Shah VH. Evolution in the understanding of the pathophysiological basis of portal hypertension: How changes in paradigm are leading to successful new treatments. J Hepatol. 2015;62:S121–S130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Verbeke L, Mannaerts I, Schierwagen R, et al. FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci Rep. 2016;6:33453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Verbeke L, Farre R, Trebicka J, et al. Obeticholic acid, a farnesoid X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats. Hepatology. 2014;59:2286–2298. [DOI] [PubMed] [Google Scholar]
- 131. Schwabl P, Hambruch E, Seeland BA, et al. The FXR agonist PX20606 ameliorates portal hypertension by targeting vascular remodelling and sinusoidal dysfunction. J Hepatol. 2017;66:724–733. [DOI] [PubMed] [Google Scholar]
- 132. Schwabl P, Hambruch E, Budas GR, et al. The Non‐Steroidal FXR Agonist Cilofexor Improves Portal Hypertension and Reduces Hepatic Fibrosis in a Rat NASH Model. Biomedicines. 2021;9:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Pawlak M, Baugé E, Bourguet W, et al. The transrepressive activity of peroxisome proliferator‐activated receptor alpha is necessary and sufficient to prevent liver fibrosis in mice. Hepatology. 2014;60:1593–1606. [DOI] [PubMed] [Google Scholar]
- 134. Marx N, Benéteau N, Taillandier T, et al. PPARalpha activators inhibit cytokine‐induced vascular cell adhesion molecule‐1 expression in human endothelial cells. Circulation. 1999;99:3125–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Goya K, Sumitani S, Xu X, et al. Peroxisome proliferator‐activated receptor alpha agonists increase nitric oxide synthase expression in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2004;24:658–663. [DOI] [PubMed] [Google Scholar]
- 136. Fiorucci S, Rizzo G, Antonelli E, et al. Cross‐talk between farnesoid‐X‐receptor (FXR) and peroxisome proliferator‐activated receptor gamma contributes to the antifibrotic activity of FXR ligands in rodent models of liver cirrhosis. J Pharmacol Exp Ther. 2005;315:58–68. [DOI] [PubMed] [Google Scholar]
- 137. Teltschik Z, Wiest R, Beisner J, et al. Intestinal bacterial translocation in rats with cirrhosis is related to compromised Paneth cell antimicrobial host defense. Hepatology. 2012;55:1154–1163. [DOI] [PubMed] [Google Scholar]
- 138. Verbeke L, Farre R, Verbinnen B, et al. The FXR agonist obeticholic acid prevents gut barrier dysfunction and bacterial translocation in cholestatic rats. Am J Pathol. 2015;185:409–419. [DOI] [PubMed] [Google Scholar]
- 139. Úbeda M, Lario M, Muñoz L, et al. Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J Hepatol. 2016;64:1049–1057. [DOI] [PubMed] [Google Scholar]
- 140. Lorenzo‐Zuniga V, Bartolí R, Planas R, et al. Oral bile acids reduce bacterial overgrowth, bacterial translocation, and endotoxemia in cirrhotic rats. Hepatology. 2003;37:551–557. [DOI] [PubMed] [Google Scholar]
- 141. Sorribas M, Jakob MO, Yilmaz B, et al. FxR‐modulates the gut‐vascular barrier by regulating the entry sites for bacterial translocation in experimental cirrhosis. J Hepatol. 2019;71:1126–1140. [DOI] [PubMed] [Google Scholar]
- 142. Xiao Y, Wang Y, Liu Y, et al. A nonbile acid farnesoid X receptor agonist tropifexor potently inhibits cholestatic liver injury and fibrosis by modulating the gut‐liver axis. Liver Int. 2021;41:2117–2131. [DOI] [PubMed] [Google Scholar]
- 143. Sorrentino G, Perino A, Yildiz E, et al. Bile acids signal via TGR5 to activate intestinal stem cells and epithelial regeneration. Gastroenterology. 2020;159:956–968.e8. [DOI] [PubMed] [Google Scholar]
- 144. Gadaleta RM, van Erpecum KJ, Oldenburg B, et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut. 2011;60: 463–472. [DOI] [PubMed] [Google Scholar]
- 145. Walters JRF, Johnston IM, Nolan JD, et al. The response of patients with bile acid diarrhoea to the farnesoid X receptor agonist obeticholic acid. Aliment Pharmacol Ther. 2015;41:54–64. [DOI] [PubMed] [Google Scholar]
- 146. Al‐Khaifi A, Rudling M, Angelin B. An FXR agonist reduces bile acid synthesis independently of increases in FGF19 in healthy volunteers. Gastroenterology. 2018;155:1012–1016. [DOI] [PubMed] [Google Scholar]
- 147. Nevens F, Andreone P, Mazzella G, et al. A placebo‐controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med. 2016;375:631–643. [DOI] [PubMed] [Google Scholar]
- 148. Trauner M, Nevens F, Shiffman ML, et al. Long‐term efficacy and safety of obeticholic acid for patients with primary biliary cholangitis: 3‐year results of an international open‐label extension study. Lancet Gastroenterol Hepatol. 2019;4:445–453. [DOI] [PubMed] [Google Scholar]
- 149. Bowlus CL, Pockros PJ, Kremer AE, et al. Long‐term obeticholic acid therapy improves histological endpoints in patients with primary biliary cholangitis. Clin Gastroenterol Hepatol. 2020;18:1170–1178.e6. [DOI] [PubMed] [Google Scholar]
- 150. Kowdley KV, Vuppalanchi R, Levy C, et al. The nonsteroidal farnesoid X receptor (FXR) agonist cilofexor improves liver biochemistry in patients with primary biliary cholangitis (PBC): a phase 2, randomized, placebo‐controlled trial. Hepatology. 2019;70:31–32. [Google Scholar]
- 151. Soret PA, Lam L, Carrat F et al. Combination of fibrates with obeticholic acid is able to normalise biochemical liver tests in patients with difficult‐to‐treat primary biliary cholangitis. Aliment Pharmacol Ther. 2021;53:1138–1146. [DOI] [PubMed] [Google Scholar]
- 152. Kowdley KV, Vuppalanchi R, Levy C, et al. A randomized, placebo‐controlled, phase II study of obeticholic acid for primary sclerosing cholangitis. J Hepatol. 2020;73:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Trauner M, Gulamhusein A, Hameed B, et al. The nonsteroidal Farnesoid X receptor agonist cilofexor (GS‐9674) improves markers of cholestasis and liver injury in patients with primary sclerosing cholangitis. Hepatology. 2019;70:788–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Younossi ZM, Ratziu V, Loomba R, et al. Obeticholic acid for the treatment of non‐alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo‐controlled phase 3 trial. Lancet. 2019;394:2184–2196. [DOI] [PubMed] [Google Scholar]
- 155. Patel K, Harrison SA, Elkhashab M, et al. Cilofexor, a nonsteroidal FXR agonist, in patients with noncirrhotic NASH: a phase 2 randomized controlled trial. Hepatology. 2020;72:58–71. [DOI] [PubMed] [Google Scholar]
- 156. Loomba R, Noureddin M, Kowdley KV, et al. Combination therapies including cilofexor and firsocostat for bridging fibrosis and cirrhosis attributable to NASH. Hepatology. 2021;73:625–643. [DOI] [PubMed] [Google Scholar]
- 157. Lucas KJ, Lopez P, Lawitz E, et al. Tropifexor, a highly potent FXR agonist, produces robust and dose‐dependent reductions in hepatic fat and serum alanine aminotransferase in patients with fibrotic NASH after 12 weeks of therapy: FLIGHT‐FXR Part C interim results. Dig Liver Dis. 2020;52:e38. [Google Scholar]
- 158. Nicholes K, Guillet S, Tomlinson E, et al. A mouse model of hepatocellular carcinoma: ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. Am J Pathol. 2002;160:2295–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Mayo MJ, Wigg AJ, Leggett BA, et al. NGM282 for treatment of patients with primary biliary cholangitis: a multicenter, randomized, double‐blind, placebo‐controlled trial. Hepatol Commun. 2018;2:1037–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Hirschfield GM, Chazouillères O, Drenth JP, et al. Effect of NGM282, an FGF19 analogue, in primary sclerosing cholangitis: A multicenter, randomized, double‐blind, placebo‐controlled phase II trial. J Hepatol. 2019;70:483–493. [DOI] [PubMed] [Google Scholar]
- 161. Harrison SA, Rinella ME, Abdelmalek MF, et al. NGM282 for treatment of non‐alcoholic steatohepatitis: a multicentre, randomised, double‐blind, placebo‐controlled, phase 2 trial. Lancet. 2018;391:1174–1185. [DOI] [PubMed] [Google Scholar]
- 162. Harrison SA, Rossi SJ, Paredes AH, et al. NGM282 improves liver fibrosis and histology in 12 weeks in patients with nonalcoholic steatohepatitis. Hepatology. 2020;71:1198–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Mantovani A, Petracca G, Beatrice G, et al. Glucagon‐like peptide‐1 receptor agonists for treatment of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: an updated meta‐analysis of randomized controlled trials. Metabolites. 2021;11:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Newsome PN, Buchholtz K, Cusi K, et al. A placebo‐controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med. 2021;384:1113–1124. [DOI] [PubMed] [Google Scholar]
- 165. Dyson JK, Hirschfield GM, Adams DH, et al. Novel therapeutic targets in primary biliary cirrhosis. Nat Rev Gastroenterol Hepatol. 2015;12:147–158. [DOI] [PubMed] [Google Scholar]
- 166. Lindor KD, Jorgensen RA, Therneau TM, et al. Ursodeoxycholic acid delays the onset of esophageal varices in primary biliary cirrhosis. Mayo Clin Proc. 1997;72:1137–1140. [DOI] [PubMed] [Google Scholar]
- 167. Harms MH, van Buuren HR, Corpechot C, et al. Ursodeoxycholic acid therapy and liver transplant‐free survival in patients with primary biliary cholangitis. J Hepatol. 2019;71:357–365. [DOI] [PubMed] [Google Scholar]
- 168. Hegade VS, Kendrick SFW, Dobbins RL, et al. Effect of ileal bile acid transporter inhibitor GSK2330672 on pruritus in primary biliary cholangitis: a double‐blind, randomised, placebo‐controlled, crossover, phase 2a study. Lancet. 2017;389:1114–1123. [DOI] [PubMed] [Google Scholar]
- 169. Mayo MJ, Pockros PJ, Jones D, et al. A randomized, controlled, phase 2 study of maralixibat in the treatment of itching associated with primary biliary cholangitis. Hepatol Commun. 2019;3: 365–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Loglio A, Ferenci P, Uceda Renteria SC, et al. Excellent safety and effectiveness of high‐dose myrcludex‐B monotherapy administered for 48 weeks in HDV‐related compensated cirrhosis: a case report of 3 patients. J Hepatol. 2019;71:834–839. [DOI] [PubMed] [Google Scholar]
- 171. Kulkarni AV, Tevethia HV, Arab JP, et al. Efficacy and safety of obeticholic acid in liver disease‐A systematic review and meta‐analysis. Clin Res Hepatol Gastroenterol. 2021;45:101675. [DOI] [PubMed] [Google Scholar]
- 172. D'Amato D, De Vincentis A , Malinverno F, et al. Real‐world experience with obeticholic acid in patients with primary biliary cholangitis. JHEP Rep. 2021;3:100248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Kremoser C. FXR agonists for NASH: how are they different and what difference do they make? J Hepatol. 2021;75:12–15. [DOI] [PubMed] [Google Scholar]
- 174.Obeticholic Acid, in LiverTox: Clinical and Research Information on Drug‐Induced Liver Injury. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases; 2012. [PubMed]
- 175. John BV, Schwartz K, Levy C, et al. Impact of obeticholic acid exposure on decompensation and mortality in primary biliary cholangitis and cirrhosis. Hepatol Commun. 2021;5:1426–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Holter MM, Chirikjian MK, Govani VN, et al. TGR5 signaling in hepatic metabolic health. Nutrients. 2020;12:2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Li A‐D, Xie X‐L, Qi W, et al. TGR5 promotes cholangiocarcinoma by interacting with mortalin. Exp Cell Res. 2020;389:111855. [DOI] [PubMed] [Google Scholar]
- 178. Erice O, Labiano I, Arbelaiz A, et al. Differential effects of FXR or TGR5 activation in cholangiocarcinoma progression. Biochim Biophys Acta Mol Basis Dis. 2018;1864:1335–1344. [DOI] [PubMed] [Google Scholar]
- 179. Königshofer P, Brusilovskaya K, Schwabl P, et al. Animal models of portal hypertension. Biochim Biophys Acta Mol Basis Dis. 2019;1865:1019–1030. [DOI] [PubMed] [Google Scholar]
- 180. Maccioni L, Gao B, Leclercq S, et al. Intestinal permeability, microbial translocation, changes in duodenal and fecal microbiota, and their associations with alcoholic liver disease progression in humans. Gut Microbes. 2020;12:1782157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Llovet JM, Bartolí R, March F, et al. Translocated intestinal bacteria cause spontaneous bacterial peritonitis in cirrhotic rats: molecular epidemiologic evidence. J Hepatol. 1998;28:307–313. [DOI] [PubMed] [Google Scholar]
- 182. Llovet JM, Bartoli R, Planas R, et al. Bacterial translocation in cirrhotic rats. Its role in the development of spontaneous bacterial peritonitis. Gut. 1994;35:1648–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Muñoz L, Borrero M‐J, Úbeda M, et al. Intestinal immune dysregulation driven by dysbiosis promotes barrier disruption and bacterial translocation in rats with cirrhosis. Hepatology. 2019;70:925–938. [DOI] [PubMed] [Google Scholar]
- 184. Hirschfield GM, Mason A, Luketic V, et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology. 2015;148:751–61.e8. [DOI] [PubMed] [Google Scholar]
- 185. Kowdley KV, Luketic V, Chapman R, et al. A randomized trial of obeticholic acid monotherapy in patients with primary biliary cholangitis. Hepatology. 2018;67:1890–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Pockros PJ, Fuchs M, Freilich B, et al. CONTROL: A randomized phase 2 study of obeticholic acid and atorvastatin on lipoproteins in nonalcoholic steatohepatitis patients. Liver Int. 2019;39:2082–2093. [DOI] [PubMed] [Google Scholar]
- 187. Neuschwander‐Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet. 2015;385:956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Siddiqui MS, Van Natta ML, Connelly MA, et al. Impact of obeticholic acid on the lipoprotein profile in patients with non‐alcoholic steatohepatitis. J Hepatol. 2020;72:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Mudaliar S, Henry RR, Sanyal AJ, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. 2013;145:574–82.e1. [DOI] [PubMed] [Google Scholar]
- 190. Al‐Dury S, Wahlström A, Panzitt K, et al. Obeticholic acid may increase the risk of gallstone formation in susceptible patients. J Hepatol. 2019;71:986–991. [DOI] [PubMed] [Google Scholar]
- 191. Traussnigg S, Halilbasic E, Hofer H, et al. Open‐label phase II study evaluating safety and efficacy of the non‐steroidal farnesoid X receptor agonist PX‐104 in non‐alcoholic fatty liver disease. Wien Klin Wochenschr. 2020;133:441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Schramm C, Hirschfield G, Mason AL, et al. Early assessment of safety and efficacy of tropifexor, a potent non bile‐acid FXR agonist, in patients with primary biliary cholangitis: An interim analysis of an ongoing phase 2 study. J Hepatol. 2018;68:S103. [Google Scholar]
- 193. DePaoli AM, Zhou M, Kaplan DD, et al. FGF19 analog as a surgical factor mimetic that contributes to metabolic effects beyond glucose homeostasis. Diabetes. 2019;68:1315–1328. [DOI] [PubMed] [Google Scholar]