

Abstract
Background
Periodontitis is an inflammatory disease caused by multiple disease‐associated bacterial species in periodontal tissues. Autophagy is known to modulate various inflammation‐driven diseases and inflammatory responses, but the role of autophagy related to the pathogenesis of periodontitis is not fully established. We investigated whether autophagic flux regulated the expression of inflammatory cytokines in the gingiva of periodontitis patients and lipopolysaccharide (LPS)‐stimulated human gingival fibroblasts (HGFs) and the underlying mechanism.
Methods
The mRNA and protein expression of proinflammatory cytokines was assessed in human gingival tissues collected from patients with periodontitis and HGFs treated with LPS. The expression of signaling molecules related to autophagy was evaluated by immunofluorescence and Western blot analyses.
Results
The expression of interleukin (IL)‐6, tumor necrosis factor‐α (TNF‐α), cyclooxygenase‐2 (COX‐2), and intercellular adhesion molecule‐1 (ICAM‐1) was increased in the gingival tissues of patients with periodontitis. LC3B‐positive cells, a typical autophagic marker, were increased in the gingival tissues of periodontitis patients and LPS‐treated HGFs. The conversion ratio of LC3‐I to LC3‐II was higher in the gingival tissues associated with periodontitis and LPS‐treated HGFs compared to the controls. The autophagy inhibitor 3‐methyladenine (3MA) significantly abrogated the LPS‐sustained inflammatory effect by reducing the expression of IL‐6, TNF‐α, COX‐2, and ICAM‐1 in HGFs. The phosphorylation of protein kinase B (AKT) and protein S6K1 (S6), signals involved in the mTOR‐dependent mechanism, was decreased in gingiva derived from periodontitis patients and LPS‐treated HGFs.
Conclusions
Autophagy augmented the production of inflammatory cytokines by mTOR inactivation via the AKT signaling pathway in the gingival tissues of patients with periodontitis and LPS‐stimulated HGFs. These findings would provide a better understanding of the mechanism by which autophagy regulates the inflammatory response associated with periodontal pathogenesis.
Keywords: AKT, autophagy, gingival fibroblast cells, inflammatory cytokines, periodontitis
1. INTRODUCTION
Periodontal disease is an inflammatory disease of the periodontium, which impacts the integrity of periodontal tissues, including gingival tissue, periodontal ligaments, and alveolar bone following the immune response of the host to bacterial pathogens. 1 The aberrant inflammation process breaks the balance between proinflammatory and anti‐inflammatory responses in the periodontium, which leads to the destruction of tooth‐supporting tissues by processes such as gingival recession and alveolar bone loss. 2 Many inflammatory cytokines give rise to periodontitis through the interaction of tissue cells and immune cells. 3 Gingival connective tissues consist of numerous gingival fibroblasts, which respond to bacterial pathogens or inflammatory cytokines, leading to periodontitis. 4 , 5 Lipopolysaccharide (LPS), which is bacterial endotoxin, induces the infiltration of polymorphonuclear leukocytes and causes edema and vasodilation in inflammatory periodontal tissues. 6 The stimulation of LPS modulates the expression levels of Toll‐like receptors (TLRs) in the host cells, which regulate the production of proinflammatory molecules could influence on the intensity of inflammation response. An excessive production of cytokines by inappropriate immune response accelerates periodontal tissue destruction. 7 Inflammatory modulators including interleukin (IL)‐6, cyclooxygenase (COX) and intercellular adhesion molecule (ICAM) are known to regulate the host response against bacterial infection. 8
Autophagy is the natural and regulated mechanism underlying the degradation of damaged organelles and the recycling of cytoplasmic materials involved in multiple pathophysiologies. 9 , 10 These damaged constituents are surrounded by a double‐membrane vesicle, also called an autophagosome, for further degradation by the hydrolytic enzymes of lysosomes, and then returned to the cytoplasm for biosynthesis and energy production. 11 , 12 Thus, autophagy plays a fundamental role in the cellular homeostasis program, which responds to various stimuli such as nutrient starvation or oxidative stress to prevent cell damage. 13 The kinase mammalian target of rapamycin (mTOR) is major modulator of autophagy and a downstream target of the phosphatidylinositol 3 kinase (PI3K) and kinase AKT (AKT) pathway. The activation of the PI3K/AKT/mTOR pathway have been found to involve in many biological processes regulated by autophagy, such as cell growth, development, and defense against pathogens. 14 , 15 Accumulating evidences suggest that autophagy plays a crucial role in innate immunity and is associated with many inflammatory diseases. 16 , 17 , 18 However, there are few studies on the significance of autophagy in the pathogenesis of periodontitis.
Recent studies reported a crosslink between autophagy and the production of proinflammatory cytokines. Autophagy increases the expression of TNF‐α and IL‐8 but suppresses the synthesis of IL‐1α, IL‐1β, and IL‐18 in macrophages and dendritic cells. 19 , 20 However, the influence of autophagy in the production of proinflammatory cytokines remains unestablished in periodontal inflammation. Finding out the mechanisms through which autophagy regulates, and is regulated by, cytokine production may provide new insight into future therapeutics for periodontitis.
Therefore, this study was aimed to clarify whether autophagy is altered in the gingival tissues of patients with periodontitis and to elucidate the mechanism underlying that autophagy regulates the production of inflammatory mediators under in vitro model with LPS‐stimulated human gingival fibroblasts (HGFs) to mimic an inflammatory milieu because periodontitis is bacterially induced disease. 21
2. MATERIALS AND METHODS
2.1. Collection of gingival tissue from the subjects
The analysis included 22 patients who visited the Department of Periodontology of Chonnam National University Dental Hospital (12 males and 10 females; mean age: 44 ± 17.4 years). Human gingival tissues, including the marginal gingival epithelium and connective tissues in the periodontal pocket, were collected during clinical crown lengthening, open flap curettage, and tooth extraction. The tissue was stored in an Eppendorf tube at –80°C immediately after harvesting. All patients met the following inclusion criteria: no consumption of antibiotics or anti‐inflammatory medications within the last 3 months, no previous periodontal treatment, and the maintenance of systemic health. The exclusion criteria included any systemic diseases that might affect periodontal disease, immunodeficiency, and previous smoking history or current smoking. All the patients were well informed and provided consent. This study was approved by the Institutional Review Board (IRB) of the Chonnam National University Dental Hospital (IRB No: CNUDH‐2016‐010). The study complied with the Declaration of Helsinki of 2013.
2.2. Clinical measurements
The periodontal pocket depth (PPD) was measured at six sites in the teeth collected using a Williams probe*, and the recorded values were rounded up to 1 mm. The sulcus bleeding index (SBI) was measured 30 seconds after the PPD measurement. 22 Based on periapical and panoramic radiographs, radiographic bone loss was defined as the distance from the cementoenamel junction to the crest of the alveolar bone. 23 The healthy group (controls) was defined by a PPD of less than 3 mm, zero SBI, and radiographic bone loss of less than 2 mm. The periodontitis group was defined by a PPD of 5 mm or more, an SBI of 1 or more, and radiographic bone loss of 4 mm or more. The human gingival tissues were classified into the control group (CG) and the periodontitis group (PG) according to the above criteria.
2.3. Measurement of the total amount of gingival crevicular fluid
The sampling site was isolated with cotton rolls and gauze, and the supragingival plaque was gently removed with a curette†. The gingival sulcus was lightly dried with an air syringe and a paper strip‡ was placed on it for 30 seconds. The paper strip was discarded if it was contaminated with blood or saliva. The gingival crevicular fluid (GCF) values measured using the Periotron 8000§ were converted to actual amounts (μL) using a standard curve. The paper strips were stored in Eppendorf tubes with 200 μL of 0.05% phosphate buffer saline‐Tween 20 (PBS‐T) at –80°C. The samples stored at –80°C were thawed and centrifuged at 12,000 rpm at 4°C for 5 minutes. A total of 200 μL of diluted GCF was prepared and stored at –20°C. 24
2.4. Hematoxylin and eosin and immunofluorescence staining
The healthy and inflamed gingival tissues were collected and fixed in 4% paraformaldehyde. Several paraffin sections were prepared (5 μm), one of which was stained with hematoxylin and eosin (H&E), and the others were used for immunofluorescence. To block a nonspecific immunoreaction, the sections were incubated with 1% bovine serum albumin in Tris‐buffered saline (TBS) for 1 hour at room temperature (RT) and incubated with the LC3 antibody** diluted 1:200 in a blocking buffer overnight at 4°C. After washing in TBS, the sections were incubated with goat anti‐rabbit IgG†† diluted 1:400 for 1 hour at RT. After washing in TBS, the specimens were counterstained with propidium iodide‡‡ to visualize the nuclei.
2.5. Cell culture and determination of cell viability
The pieces sliced from healthy gingival connective tissues were cultured in Dulbecco's modified Eagle medium (DMEM) §§, 10% fetal bovine serum (FBS),*** penicillin††† (100 U/mL), and streptomycin‡‡‡ (100 U/mL). The culture conditions were maintained at 37°C, 5% CO2, and 95% air. The isolated HGFs were sub‐cultured and the 4th to 7th generations were used for the experiment. Cell viability was measured using a 3‐(4,5‐dimethylthlazol‐2yl)‐2,5‐diphenyl‐tetrazolium bromide (MTT)‐based assay. The cells were plated on 24‐well plates (2 × 105 cells/well) and treated with LPS from Escherichia coli, followed by incubation with MTT (0.1 mg/mL) at 37°C for 3 hours. The formation of formazan was extracted in dimethyl sulfoxide and the absorbance was measured at 570 nm by a microplate reader§§§.
2.6. Reverse transcription‐polymerase chain reaction (RT‐PCR)
The HGFs were harvested after 24 hours of incubation. Total RNA extraction was performed using TRIzol reagent**** and the total RNA (2 μg) was subjected to complementary DNA (cDNA) synthesis using the Prime RT‐premix kit.†††† cDNA was amplified using the PCR‐premix kit‡‡‡‡ with specific primers: IL‐6 (sense 5′‐GAACAAGCCAGAGCTGTGCA‐3′ and antisense 5′‐TGAGGTGCCCATGCTACATT‐3′), TNF‐α (sense 5′‐TGAGGAGGACGAAC ATCCAA‐3′ and antisense 5′‐AGCTGTAGGCCCCAGTGAGT‐3′), COX‐2 sense (5′‐TCTGGTGCCTGGTCTGAT‐3′ and antisense 5′‐ CACCCCATTCAGGATGCT‐3′), and GAPDH (sense 5′‐AGTCACGGATTTGGTCGT‐3′ and antisense 5′‐ACAAGCTTCCCGTT CTCAG‐3′). The PCR products were analyzed on 1.5% agarose gels containing SYBR™§§§§ and visualized with a Fusion FX*****.
2.7. Immunofluorescence staining
The HGFs (1 × 104 cells) were cultured on slides in a glass chamber in DMEM††††† supplemented with 10% FBS‡‡‡‡‡ in the presence of LPS for 24 hours. Immunofluorescence staining was performed to evaluate the protein levels of LC3 in LPS‐treated cells according to a previously described protocol. 25
2.8. Western blot analysis
The HGFs were lysed in a radioimmunoprecipitation assay (RIPA) buffer§§§§§ containing protease inhibitor****** and phosphatase inhibitor cocktail solution††††††. The protein concentration of the cell lysates was determined by the BSA protein assay‡‡‡‡‡‡. Western blot analysis was performed as described in a previous study. 26
2.9. Statistical analysis
All data are presented as the mean ± standard deviation (SD) of three experiments. An independent t‐test was conducted to evaluate the depth of the pocket and radiographic bone loss in the study group, which satisfied normality in both groups. The statistical significance was determined using the Student's t‐test or one‐way analysis of variance (ANOVA), followed by the Student‐Newman‐Keuls test. P < 0.05 was considered statistically significant using Prism Software (GraphPad Software).
3. RESULTS
3.1. Demographic and clinical characteristics of the population
A total of 22 subjects participated in the study, including 10 in the CG group and 12 in the PG group. The mean age was 35.6 ± 17.8 years in the controls and 50.9 ± 14.3 years in the PG. The SBI, PPD, and radiographic bone loss values were higher in the PG compared to the CG (Table 1).
TABLE 1.
Demographic and clinical characteristics of the subjects
| Variable | Control | Periodontitis |
|---|---|---|
| Number of participants | 10 | 12 |
| Males/females | 4 / 6 | 8 / 4 |
| Mean age (years) | 35.6 ± 17.8 | 50.9 ± 14.3 |
| Age range (years) | 13 to 57 | 19 to 78 |
| Sulcus bleeding index | 0 | 2.59 ± 0.47 *** |
| Pocket depth (mm) | 2.3 ± 0.6 | 5.72 ± 1.61 *** |
| Radiographic bone loss (mm) | 1.35 ± 0.61 | 4.35 ± 1.56 *** |
The data are presented as the mean ± standard deviation (SD).
p < 0.001 vs. control.
3.2. GCF volume and expression of proinflammatory mediators in the gingival tissues of periodontitis patients
To identify the release of inflammatory substances from the gingival tissues of patients with periodontitis, GCF was collected from eight CG subjects and nine PG patients, because some of the paper strips among the GCFs collected from all 22 patients were contaminated with saliva or blood. The PG had a high volume of GCFs compared to the CG (Figure 1A). In addition, the expression of IL‐6, TNF‐α, COX‐2, and ICAM‐1, key inflammatory indicators, was significantly enhanced in the GCFs of the PG compared to the CG (Figure 1B).
FIGURE 1.
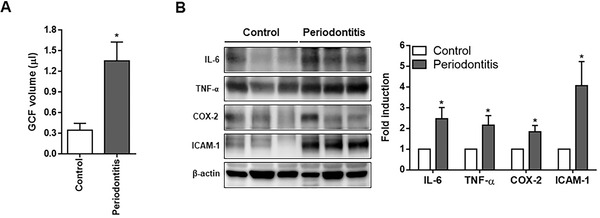
Identification of inflammation in human gingival tissues. (A) GCF was collected from eight subjects in the control group and nine in the periodontitis group. The volume of GCF was measured using a Periotron 8000 and the measured values were converted to actual amounts (μL) using a standard curve. (B) The protein levels of IL‐6, TNF‐α, COX‐2, and ICAM‐1 were determined in the gingival tissues of patients with periodontitis by Western blot analysis, using β‐actin as the internal control (control = 6, periodontitis = 6). Each protein was quantitated by densitometry and normalized to β‐actin. COX‐2, cyclooxygenase‐2; GCF, gingival crevicular fluid; ICAM‐1, intercellular adhesion molecule‐1; IL‐6, interleukin‐6; TNF‐α, tumor necrosis factor‐α. *p < 0.05 vs. control
3.3. Autophagy is increased in the gingival tissues of patients with periodontitis
The gingival tissues of the controls were collected from the clinically healthy areas of patients undergoing crown lengthening and the gingival tissues of the PG were collected from areas with a PPD of greater than 5 mm. To explore the autophagy activation in the gingival tissues of patients with periodontitis, the expression level of LC3, a protein associated with the autophagy process was assessed. The inflamed gingival tissues of the periodontitis patients displayed many inflammatory cells as well as LC3B‐positive cells compared to the healthy gingival tissues (Figure 2A). Western blot analysis was performed to evaluate the conversion of LC3‐I to LC3‐II, the hallmark of the degree of autophagy activation. LC3‐II conversion was significantly higher in the gingival tissues of the PG than in the CG (Figure 2B). These results suggest that autophagy was activated in the gingival tissues of the periodontitis patients.
FIGURE 2.
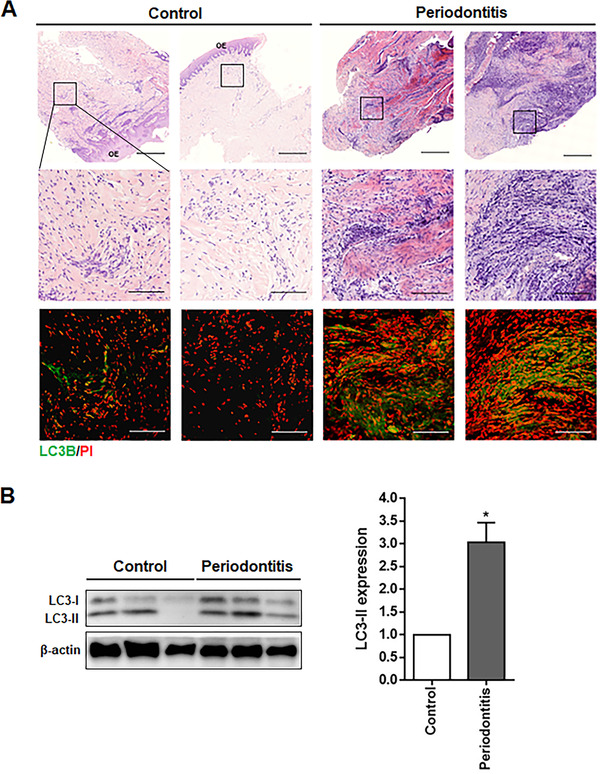
Expression of autophagy markers in the human gingival tissues of patients with periodontitis. (A) Sections of the healthy (control) or periodontitis gingival tissues were stained with H&E and subjected to immunofluorescence analysis for LC3B (green). PI (red) was used for counterstaining. (B) The protein levels of LC3‐I and LC3‐II in the gingival tissues of periodontitis patients were determined by Western blot analysis (control = 6, periodontitis = 6). LC3‐II protein levels were quantitated using densitometry and normalized to β‐actin, the loading control. Scale bars in A: 500 μm; boxed areas: 100 μm. H&E, hematoxylin and eosin; OE, oral epithelium; PI, propidium iodide. *p < 0.05 vs. control
3.4. Autophagy is enhanced in LPS‐stimulated HGFs
A cell viability assay was performed in HGFs treated with different concentrations of LPS (0.1, 1, and 10 μg/mL) for 24 hours. The cell viability was not significantly different between the LPS‐treated cells and the LPS‐untreated cells (Figure 3A).
FIGURE 3.
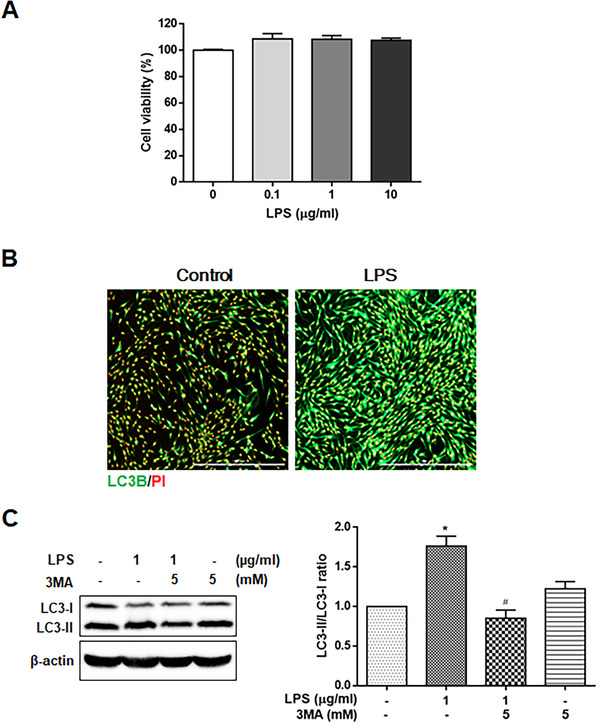
Expression of autophagy marker in LPS‐stimulated HGFs. (A) Cell viability was determined by the MTT assay in HGFs treated with 0.1, 1, and 10 μg/mL LPS. (B) Immunocytochemical analysis was performed to detect LC3B‐positive cells (green) in HGFs treated with 1 μg/mL of LPS for 24 hours. (C) The expression of LC3‐II was detected by Western blot analysis in HGFs treated with 1 μg/ml LPS for 24 hours with or without 5 mM 3MA pretreatment for 1 hour. Each protein was quantitated using densitometry and normalized to β‐actin. The data are expressed as the mean ± SD of triplicate independent experiments. Scale bars, 1000 μm. HGFs, human gingival fibroblasts; LPS, lipopolysaccharide; MTT, 3‐(4,5‐dimethylthlazol‐2yl)‐2,5‐diphenyl‐tetrazolium bromide. *p < 0.05 vs. untreated cells. # p < 0.05 vs. LPS‐treated cells
The presence of autophagy was detected in LPS‐treated HGFs by immunofluorescence staining with an LC3B antibody. The results showed an increase in LC3‐positive cells in the cytoplasm of HGFs treated with LPS (1 μg/mL) for 24 hours (Figure 3B). The autophagy inhibitor 3‐methyladenine (3MA) was used to suppress class III PI3K activity, which plays an important role as an early signal in autophagy induction. 27 The ratio of LC3‐I conversion to LC3‐II was analyzed by the relative protein abundance using Western blot analysis of LPS‐treated cells incubated for 24 hours with or without 5 mM 3MA. LPS treatment increased the ratio of LC3‐I conversion to LC3‐II, which was reduced by 3MA in the HGFs (Figure 3C). These results demonstrated that autophagy was enhanced in the LPS‐treated HGFs.
3.5. Autophagy upregulates proinflammatory mediators in LPS‐treated HGFs
We evaluated the role of autophagy in regulating the expression of proinflammatory modulators such as IL‐6, TNF‐α, COX‐2, and ICAM‐1 in the LPS‐treated HGFs using the autophagy inhibitor 3MA. As shown in Figure 4A, the mRNA expression of IL‐6, TNF‐α, and COX‐2 was significantly upregulated by LPS treatment in HGFs. However, the LPS‐elevated IL‐6, TNF‐α, COX‐2, and ICAM‐1 mRNA and protein levels were significantly decreased by the treatment of HGFs with 5 mM 3MA (Figures 4A and B). These results verified that autophagy enhanced the expression of proinflammatory mediators in the LPS‐stimulated HGFs.
FIGURE 4.
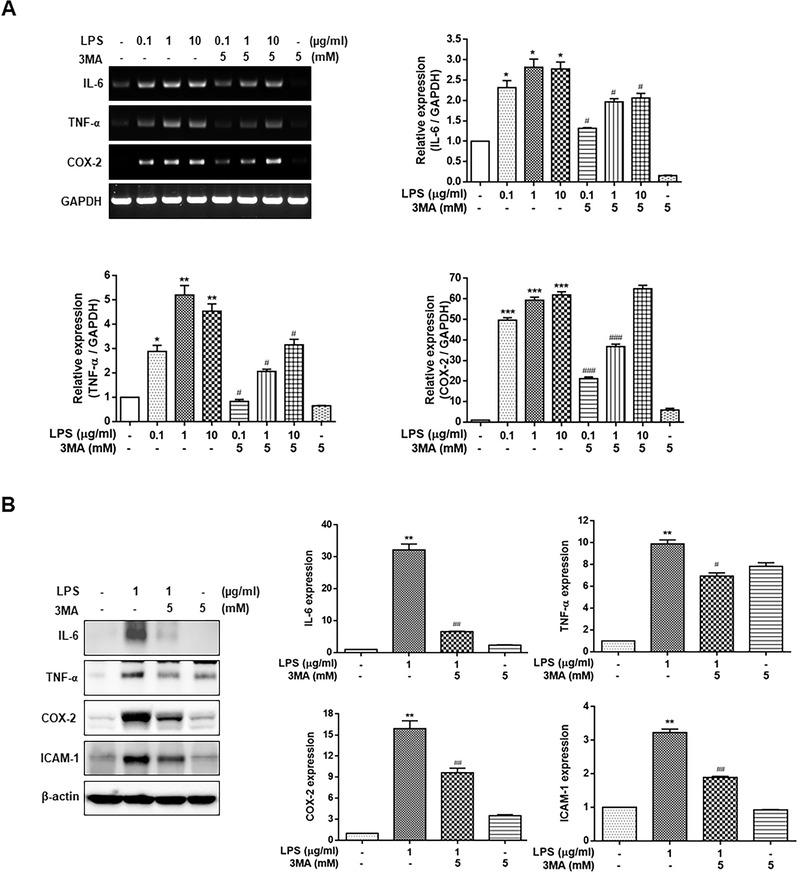
Effect of autophagy inhibition on the expression of inflammatory cytokines in LPS‐stimulated HGFs. The cells were treated with 0.1, 1, and 10 μg/mL LPS (A) or 1 μg/mL LPS (B) for 24 hours, with or without 5 mM 3MA pretreatment for 1 hour. The expression of IL‐6, TNF‐α, and COX‐2 was measured using RT‐PCR or Western blot analysis. Each protein was quantitated using densitometry and normalized to GAPDH or β‐actin. The data are expressed as the mean ± SD of triplicate independent experiments. 3MA, 3‐methyladenine; COX‐2, cyclooxygenase‐2; HGFs, human gingival fibroblasts; IL‐6, interleukin‐6; LPS, lipopolysaccharide; RT‐PCR, real‐time polymerase chain reaction; TNF‐α, tumor necrosis factor‐α. *p < 0.05, **p < 0.01, ***p < 0.001 vs. untreated cells, # p < 0.05, ## p < 0.01, ### p < 0.001 vs. LPS‐treated cells
3.6. Autophagy is augmented via the AKT/mTOR pathway in the gingival tissues of periodontitis patients and LPS‐stimulated HGFs
To identify the signaling molecules activating autophagy in the gingival tissues of patients with periodontitis and LPS‐stimulated HGFs, the expression of AKT, mTOR, and S6 ribosomal protein, important regulators related to the autophagy process, was detected by Western blot analysis. As shown in Figure 5A, the phosphorylation of AKT and S6 ribosomal protein was significantly decreased in the gingival tissues of the PG compared with the CG. The LPS‐treated HGFs also showed a significant decrease in the phosphorylation of AKT, mTOR, and S6 ribosomal protein (Figure 5B). These findings indicate that autophagy induction was associated with the AKT/mTOR pathway in the gingival tissues of periodontitis patients and LPS‐treated HGFs.
FIGURE 5.
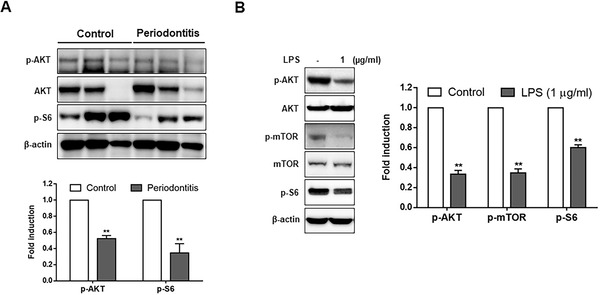
Expression of AKT, mTOR, and S6 ribosomal protein in the human gingival tissues of patients with periodontitis and LPS‐stimulated HGFs. (A) Protein levels of AKT and S6 ribosomal protein were analyzed in the healthy gingival tissues (control) or the gingival tissues of patients with periodontitis via Western blot analysis. (B) The phosphorylation of AKT, mTOR, and S6 ribosomal protein in the LPS‐treated HGFs was detected by Western blot analysis. Each protein was quantitated by densitometry and normalized to β‐actin. AKT, protein kinase B; HGFs, human gingival fibroblasts; LPS, lipopolysaccharide; S6, protein S6K1. **p < 0.01 vs. control
4. DISCUSSION
Periodontal disease is the consequence of the failed resolution of inflammation in the periodontium. Specifically, periodontitis is the result of excessive inflammation that is accompanied by alveolar bone loss, gingival recession, and bleeding. 28 In the present study, patients with periodontitis showed a severe alveolar bone loss, deep gingival pockets, and sulcus bleeding. In general, periodontitis is caused by bacteria‐derived factors and antigens that stimulate the synthesis and release of inflammatory cytokines including IL‐1β, IL‐6, IL‐12, IL‐4, and TNF‐α in gingival tissue. 29 , 30 IL‐6, IL‐4, and factor XIII‐A have been used as indirect markers of macrophage activation in chronic periodontitis. 31 Our results also showed high levels of IL‐6, TNF‐α, COX‐2, and ICAM‐1 in the gingival tissues of patients with periodontitis compared to healthy tissues.
Autophagy is the main catabolic process contributing to cell survival under stress conditions and other intrinsic and extrinsic insults. 32 However, the pathophysiology of periodontitis associated with autophagy has not been extensively investigated, although autophagy is known to be involved in various types of inflammation. 17 To investigate the relationship between autophagy and periodontitis, we examined the expression of LC3B and LC3‐II as an indicator of autophagy activation because soluble LC3‐I is converted to lipophilic LC3‐II, a specific form of autophagosomes, during autophagy activation. 33 In the present study, LC3B‐positive cells and LC3‐II protein levels were increased in the gingival tissues of patients with periodontitis compared to healthy gingival tissues, suggesting that autophagy was increased in the gingival tissues of patients with periodontitis. Our results are consistent with previous reports in which the expression of autophagy‐related genes was higher in periodontitis patients than in healthy tissue. 34 , 35 Several studies on autophagy and its role in response to periodontal pathogen have been reported. The induced autophagy after invading of periodontal pathogen enhances the survival of pathogenic bacteria by evasion of immune defense and also participates in intracellular bacteria killing by the antibacterial process. 36 , 37 Taken together, the current results strongly suggest that the autophagy induction is companied by periodontitis although the interplay between autophagy and periodontal pathogens is complex.
Recent studies have emphasized the function of autophagy associated with alterations in inflammatory responses, particularly in regulating the generation of cytokines. 38 A variety of proinflammatory cytokines has been reported to regulate the induction of autophagy. For example, IL‐1, TNF‐α, and IL‐17 induce autophagy, but IL‐13, IL‐33, IL‐10, and IL‐14 block this process. Conversely, autophagy activation can promote or inhibit the production and secretion of cytokines to modulate inflammation responses. 39 , 40 Because levels of cytokine regulated by autophagy closely influences the pathophysiology of inflammation‐related diseases, exploring the relationship between autophagic flux and proinflammatory mediators is essential.
Periodontitis is a bacterially induced inflammatory disease and gingival fibroblast responds to bacterial LPS by pathogen‐associated molecular pattern (PAMP) such as Toll‐like receptors (TLRs). 41 Induction of autophagy in LPS‐treated HGFs was confirmed by detecting LC3B protein expression and LC3‐I and LC3‐II protein levels using Western blot analysis. LPS increased the number of LC3B‐positive cells and the ratio of LC3‐II/LC3‐I. However, the autophagy inhibitor 3MA decreased the conversion ratio of LC3‐I to LC3‐II enhanced by LPS, demonstrating that LPS stimulation induced promoted autophagy in the HGFs. The periodontal bacteria‐induced enhanced autophagic flux is found in periodontal tissues and immune cells. Recently, the number of studies have reported that Porphyromonas gingivlais, a major pathogen associated with periodontal disease, and its LPS promote autophagic activity, supporting the importance of autophagy in regulating inflammatory process in periodontal disease. 34 , 42 , 43
Various bacteria and their pathogen‐associated molecules such as LPS can trigger the secretion of inflammatory cytokines IL‐6, TNF‐α, COX‐2, and the cell adhesion molecule ICAM‐1. 44 In this study, Escherichia coli LPS was chosen to simulate the effect of Gram‐negative microbiota. LPS from Escherichia coli is known to more powerful stimulator for cytokine production in gingival fibroblast and periodontal ligament cell than LPS from Porphyromonas gingivalis. 21 To elucidate the role of autophagy in the expression of inflammatory cytokines in LPS‐stimulated HGFs, we observed the expression of IL‐6, TNF‐α, COX‐2, and ICAM‐1 in LPS‐treated HGFs following treatment with 3MA. Autophagy inhibition by 3MA significantly attenuated the LPS‐upregulated IL‐6, TNF‐α, COX‐2, and ICAM‐1 mRNA and protein expression in the HGFs. These results suggest that LPS‐induced autophagy upregulates the expression of proinflammatory cytokines such as IL‐6 and TNF‐α. It is consistent with the finding that autophagy promoted the expression of TNF‐α in macrophages, demonstrating that autophagy positively regulates the production of inflammatory molecules. 45 Also, autophagy inhibitor chloroquine and 3MA attenuate periodontal inflammation in experimental. 46 Taken together, these data with our results suggest that autophagy may contribute to secretion of immune effectors during periodontal inflammation. In contrast to the current results, periodontopathogen Aggregatibater actinomycetemcomitans‐induced autophagy promotes bacterial internalization and inhibits the excessive inflammatory response by downregulating IL‐1β production in macrophages. 47 Also, rapamycin‐enhanced autophagy leads to the loss of pro‐IL‐1β expression and IL‐1β secretion in LPS‐treated dendritic cells. 48 It provides that elevated autophagy during periodontal inflammation may prevent the excessive periodontal inflammation, suppressing the secretion of proinflammatory factors and inflammation response.
Akt/mTOR pathway is a critical mediator in signal transduction pathways potentially related to autophagic process. 14 , 43 We investigated the involvement of the AKT/mTOR pathway in the mechanism of autophagy‐upregulated inflammatory cytokine production in the gingiva of an inflammatory environment. In the present study, the phosphorylation of AKT and ribosomal protein S6 was reduced in the gingival tissues of patients with periodontitis. Furthermore, the phosphorylation of AKT, mTOR, and S6 was decreased in LPS‐treated HGFs compared with the controls. These results revealed that autophagy was induced via the AKT/mTOR pathway in the gingival tissues of patients with periodontitis and LPS‐treated HGFs, consistent with a previous study demonstrating Porphyromonas gingivalis LPS‐induced autophagy via the PI3K/AKT/mTOR pathway in HGFs and cementoblast. 43 , 49 Furthermore, Porphyromonas gingivalis‐activated AKT/mTOR signaling inhibits antimicrobial autophagic process in human dendritic cells, resulting in survival of intracellular Porphyromonas gingivalis. 50 Therefore, AKT/mTOR signal axis acts as an important regulator of autophagy in periodontal inflammation process and its targeting may be an approach to prevent untoward inflammatory responses in bacterial clearance.
Periodontitis is a multifactorial disease and the complexity of the microbiota and the inflammatory response influenced on gingival fibroblast, any one cytokine or bacterial product to mimic a periodontal inflammatory milieu has limitation explaining their exact role on pathogenesis of periodontal disease. However, autophagy induced by stimulation including oral biofilms, periodontal pathogen and immune response in periodontium may play a crucial role in regulating the periodontal inflammatory response and subsequently have a protective or pathological effect in periodontitis depending on physiological or pathological responsive factors. The signal transduction mediators underlying its role may be a target for the development of new periodontal therapies.
5. CONCLUSIONS
These findings revealed that the production of inflammatory cytokines was closely associated with autophagy induction via inactivation of AKT/mTOR in the gingival tissues of patients with periodontitis and LPS‐treated HGFs. Thus, the AKT/mTOR signaling in the autophagy process may represent a therapeutic target pathway for inflammation‐driven periodontal diseases.
CONFLICT OF INTEREST
The authors report no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
All authors have made substantial contributions to conception and design for the study. Won Jae Kim, Sam Young Park and Ok Su Kim have been involved in data collection and analysis. Won Jae Kim, Sam Young Park, Ok Su Kim, Hoo Sang Park and Ji Yeon Jung have been involved in data interpretation and drafting the manuscript. Ji Yeon Jung and Won Jae Kim have critically revised the manuscript. All the authors have given final approval of the version to be published.
ACKNOWLEDGMENTS
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (Daejeon, Korea) (No. 2019R1A5A2027521 and No. 2019R1A2C1087744).
Kim WJ, Park SY, Kim OS, Park HS, Jung JY. Autophagy upregulates inflammatory cytokines in gingival tissue of patients with periodontitis and lipopolysaccharide‐stimulated human gingival fibroblasts. J Periodontol. 2022;93:380–391. 10.1002/JPER.21-0178
Won Jae Kim, Sam Young Park, and Ok Su Kim contributed equally to this study.
Footnotes
Hu‐Friedy, Chicago, IL.
Hu‐Friedy, Chicago, IL.
Pro‐flow, Bronx, NY.
Pro‐flow, Bronx, NY.
Novus Biologicals, Centennial, CO.
Life Technology, Carlsbad, CA.
Life Technology, Carlsbad, CA.
Life Technology, Carlsbad, CA.
Life Technology, Carlsbad, CA.
Life Technology, Carlsbad, CA.
Life Technology, Carlsbad, CA.
BioTek, Winooski, VT.
TaKaRa, Shiga, Japan.
Genet Bio, Cheonan, South Korea.
Genet Bio, Cheonan, South Korea.
Thermo Fisher Scientific, Waltham, MA.
Vilber Lourmat, Collégien, France.
Life Technology, Carlsbad, CA.
Life Technology, Carlsbad, CA.
Biosesang, Seongnam, South Korea.
GenDEPOT, Katy, TX.
GenDEPOT, Katy, TX.
Thermo Fisher Scientific, Waltham, MA.
REFERENCES
- 1. Könönen E, Gursoy M, Gursoy UK. Periodontitis: a multifaceted disease of tooth‐supporting tissues. J Clin Med. 2019;8:1135‐1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cekici A, Kantarci A, Hasturk H, Van Dyke TE. Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontology 2000. 2014;64:57‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Okada H, Murakami S. Cytokine expression in periodontal health and disease. Crit Rev Oral Biol Med. 1998;9:248‐266. [DOI] [PubMed] [Google Scholar]
- 4. Naruishi K, Takashiba S, Chou HH, Arai H, Nishimura F, Murayama Y. Role of soluble interleukin‐6 receptor in inflamed gingiva for binding of interleukin‐6 to gingival fibroblasts. J Periodontal Res. 1999;34:296‐300. [DOI] [PubMed] [Google Scholar]
- 5. Scheres N, Laine ML, Sipos PM, et al. Periodontal ligament and gingival fibroblasts from periodontitis patients are more active in interaction with Porphyromonas gingivalis . J Periodontal Res. 2011;46:407‐416. [DOI] [PubMed] [Google Scholar]
- 6. Page RC. The role of inflammatory mediators in the pathogenesis of periodontal disease. J Periodontal Res. 1991;26:230‐242. [DOI] [PubMed] [Google Scholar]
- 7. Van Dyke TE, Serhan CN. Resolution of inflammation: a new paradigm for the pathogenesis of periodontal diseases. J Dent Res. 2003;82:82‐90. [DOI] [PubMed] [Google Scholar]
- 8. Tsai MH, Wu CH, Lin WN, et al. Infection with Staphylococcus aureus elicits COX‐2/PGE(2)/IL‐6/MMP‐9‐dependent aorta inflammation via the inhibition of intracellular ROS production. Biomed Pharmacother. 2018;107:889‐900. [DOI] [PubMed] [Google Scholar]
- 9. Shintani T, Klionsky DJ. Autophagy in health and disease: a double‐edged sword. Science. 2004;306:990‐995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12:823‐830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li L, Michel R, Cohen J, DeCarlo A, Kozarov E. Intracellular survival and vascular cell‐to‐cell transmission of Porphyromonas gingivalis . BMC Microbiol. 2008;8:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eskelinen EL, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta. 2009;1793:664‐673. [DOI] [PubMed] [Google Scholar]
- 13. Awan MU, Deng Y. Role of autophagy and its significance in cellular homeostasis. Appl Microbiol Biotechnol. 2014;98:5319‐5328. [DOI] [PubMed] [Google Scholar]
- 14. Hou X, Hu Z, Xu H, et al. Advanced glycation endproducts trigger autophagy in cadiomyocyte via RAGE/PI3K/AKT/mTOR pathway. Cardiovasc Diabetol. 2014;13:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ravanan P, Srikumar IF, Talwar P. Autophagy: the spotlight for cellular stress responses. Life Sci. 2017;188:53‐67. [DOI] [PubMed] [Google Scholar]
- 16. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Netea‐Maier RT, Plantinga TS, van de Veerdonk FL, Smit JW, Netea MG. Modulation of inflammation by autophagy: consequences for human disease. Autophagy. 2016;12:245‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tan YQ, Zhang J, Zhou G. Autophagy and its implication in human oral diseases. Autophagy. 2017;13:225‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Crişan TO, Plantinga TS, van de Veerdonk FL, et al. Inflammasome‐independent modulation of cytokine response by autophagy in human cells. Plos One. 2011;6:e18666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harris J. Autophagy and cytokines. Cytokine. 2011;56:140‐144. [DOI] [PubMed] [Google Scholar]
- 21. Gürkan A, Emingil G, Nizam N, et al. Therapeutic efficacy of vasoactive intestinal peptide in Escherichia coli lipopolysaccharide‐induced experimental periodontitis in rats. J Periodontol. 2009;80:1655‐1664. [DOI] [PubMed] [Google Scholar]
- 22. Benamghar L, Penaud J, Kaminsky P, Abt F, Martin J. Comparison of gingival index and sulcus bleeding index as indicators of periodontal status. Bull World Health Organ. 1982;60:147‐151. [PMC free article] [PubMed] [Google Scholar]
- 23. Grossi SG, Genco RJ, Machtet EE, et al. Assessment of risk for periodontal disease. II. Risk indicators for alveolar bone loss. J Periodontol. 1995;66:23‐29. [DOI] [PubMed] [Google Scholar]
- 24. Vernal R, Dutzan N, Chaparro A, Puente J, Antonieta Valenzuela M, Gamonal J. Levels of interleukin‐17 in gingival crevicular fluid and in supernatants of cellular cultures of gingival tissue from patients with chronic periodontitis. J Clin Periodontol. 2005;32:383‐389. [DOI] [PubMed] [Google Scholar]
- 25. Park SY, Sun EG, Lee Y, et al. Autophagy induction plays a protective role against hypoxic stress in human dental pulp cells. J Cell Biochem. 2018;119:1992‐2002. [DOI] [PubMed] [Google Scholar]
- 26. Park SY, Park MY, Park HG, et al. Nitric oxide‐induced autophagy and the activation of activated protein kinase pathway protect against apoptosis in human dental pulp cells. Int Endod J. 2017;50:260‐270. [DOI] [PubMed] [Google Scholar]
- 27. Wu YT, Tan HL, Shui G, et al. Dual role of 3‐methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3‐kinase. J Biol Chem. 2010;285:10850‐10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Choi JI, Seymour GJ. Vaccines against periodontitis: a forward‐looking review. J Periodontal Implant Sci. 2010;40:153‐163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Graves DT, Cochran D. The contribution of interleukin‐1 and tumor necrosis factor to periodontal tissue destruction. J Periodontal Implant Sci. 2003;74:391‐401. [DOI] [PubMed] [Google Scholar]
- 30. Ramadan DE, Hariyani N, Indrawati R, Ridwan RD, Diyatri I. Cytokines and chemokines in periodontitis. Eur J Dent. 2020;14:483‐495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Navarrete M, García J, Dutzan N, et al. Interferon‐gamma, interleukins‐6 and ‐4, and factor XIII‐A as indirect markers of the classical and alternative macrophage activation pathways in chronic periodontitis. J Periodontol. 2014;85:751‐760. [DOI] [PubMed] [Google Scholar]
- 32. Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19:5720‐5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bullon P, Cordero MD, Quiles JL, et al. Autophagy in periodontitis patients and gingival fibroblasts: unraveling the link between chronic diseases and inflammation. Bmc Med. 2012;10:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. An Y, Liu W, Xue P, Zhang Y, Wang Q, Jin Y. Increased autophagy is required to protect periodontal ligament stem cells from apoptosis in inflammatory microenvironment. J Clin Periodontol. 2016;43:618‐625. [DOI] [PubMed] [Google Scholar]
- 36. Rodrigues PH, Belanger M, Dunn W Jr, Progulske‐Fox A. Porphyromonas gingivalis and the autophagic pathway: an innate immune interaction?. Front Biosci. 2008:178‐187. [DOI] [PubMed] [Google Scholar]
- 37. Chung J, Kim S, Lee HA, et al. Trans‐cinnamic aldehyde inhibits Aggregatibacter actinomycetemcomitans‐induced inflammation in THP‐1‐derived macrophages via autophagy activation. J Periodontol. 2018;89:1262‐1271. [DOI] [PubMed] [Google Scholar]
- 38. Singh SB, Ornatowski W, Vergne I, et al. Human IRGM regulates autophagy and cell‐autonomous immunity functions through mitochondria. Nat Cell Biol. 2010;12:1154‐1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ge Y, Huang M, Yao YM. Autophagy and proinflammatory cytokines: interactions and clinical implications. Cytokine & Cytokine Growth Factor Rev. 2018;43:38‐46. [DOI] [PubMed] [Google Scholar]
- 40. Jiang GM, Tan Y, Wang H, et al. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Cytokine Growth Factor Rev 2019;18:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Andrukhov O, Ertlschweiger S, Moritz A, Bantleon HP, Rausch‐Fan X. Different effects of P. gingivalis LPS and E. coli LPS on the expression of interleukin‐6 in human gingival fibroblasts. Acta Odontol Scand. 2014;72:337‐345. [DOI] [PubMed] [Google Scholar]
- 42. Lee K, Roberts JS, Choi CH, Atanasova KR, Yilmaz Ö. Porphyromonas gingivalis traffics into endoplasmic reticulum‐rich‐autophagosomes for successful survival in human gingival epithelial cells. Virulence. 2018;9:845‐859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu J, Wang X, Zheng M, Luan Q. Lipopolysaccharide from Porphyromonas gingivalis promotes autophagy of human gingival fibroblasts through the PI3K/Akt/mTOR signaling pathway. Life Sci. 2018;211:133‐139. [DOI] [PubMed] [Google Scholar]
- 44. Blake GJ, Ridker PM. Novel clinical markers of vascular wall inflammation. Circn Res. 2001;89:763‐771. [DOI] [PubMed] [Google Scholar]
- 45. Liu N, Meng J, Wang Z, Zhou G, Shi T, Zhao J. Autophagy mediated TiAl6V4 particle‐induced peri‐implant osteolysis by promoting expression of TNF‐alpha. Biochem Biophys Res Commun. 2016;473:133‐139. [DOI] [PubMed] [Google Scholar]
- 46. He S, Zhou Q, Luo B, Chen B, Li L, Yan F. Chloroquine and 3‐methyladenine attenuates periodontal inflammation and bone loss in experimental periodontitis. Inflammation. 2020;43:220‐230. [DOI] [PubMed] [Google Scholar]
- 47. Lee HA, Park MH, Song Y, Na HS, Chung J. Role of Aggregatibacter actinomycetemcomitans‐induced autophagy in inflammatory response. J Periodontol. 2020;91:1682‐1693. [DOI] [PubMed] [Google Scholar]
- 48. Harris J, Hartman M, Roche C, et al. Autophagy controls IL‐1beta secretion by targeting pro‐IL‐1beta for degradation. J Biol Chem. 2011;286:9587‐9597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ma L, Liu H, Wang X, et al. CXXC5 orchestrates Stat3/Erk/Akt signaling networks to modulate P. gingivalis‐elicited autophagy in cementoblasts. Biochim Biophys Acta Mol Cell Res. 2021;1868:118923. [DOI] [PubMed] [Google Scholar]
- 50. Meghil MM, Tawfik OK, Elashiry M, et al. Disruption of immune homeostasis in human dendritic cells via regulation of autophagy and apoptosis by Porphyromonas gingivalis . Front Immunol. 2019;10:2286. [DOI] [PMC free article] [PubMed] [Google Scholar]