

Abstract
Selumetinib (ARRY‐142886), an oral, potent and highly selective allosteric mitogen‐activated protein kinase kinase 1/2 inhibitor, is approved by the US Food and Drug Administration for the treatment of pediatric patients aged ≥2 years with neurofibromatosis type 1 with symptomatic, inoperable plexiform neurofibromas. A physiologically based pharmacokinetic (PBPK) model was constructed to predict plasma concentration–time profiles of selumetinib, and to evaluate the impact of coadministering moderate cytochrome P450 (CYP) 3A4/2C19 inhibitors/inducers. The model was also used to extrapolate pharmacokinetic exposures from older children with different body surface area to guide dosing in younger children. This model was built based on physiochemical data and clinical in vivo drug‐drug interaction (DDI) studies with itraconazole and fluconazole, and verified against data from an in vivo rifampicin DDI study and an absolute bioavailability study. The pediatric model was updated by changing system‐specific input parameters using the Simcyp pediatric module. The model captured the observed selumetinib pharmacokinetic profiles and the interactions with CYP inhibitors/inducers. The predictions from the PBPK model showed a DDI effect of 30% to 40% increase or decrease in selumetinib exposure when coadministered with moderate CYP inhibitors or inducers, respectively, which was used to inform dose management and adjustments. The pediatric PBPK model was applied to simulate exposures in specific body surface area brackets that matched those achieved with a 25 mg/m2 dose in SPRINT clinical trials. The pediatric PBPK model was used to guide the dose for younger patients in a planned pediatric clinical study.
Keywords: drug‐drug interactions, MEK inhibitor, neurofibromatosis type 1, PBPK, pediatric, prediction and simulation, selumetinib
Selumetinib (ARRY‐142886) is an oral, potent, and highly selective allosteric mitogen‐activated protein kinase kinase (MEK) 1/2 inhibitor with a short half‐life. 1 Selumetinib is approved for the treatment of pediatric patients aged ≥2 years, who have neurofibromatosis type 1 with symptomatic and inoperable plexiform neurofibromas. 2
Selumetinib pharmacokinetics (PK) are comparable in healthy volunteers, adult patients with various tumors, and pediatric patients with neurofibromatosis type 1. 3 , 4 , 5 In adults, absorption is rapid with a time to reach maximum plasma concentration (Cmax) of approximately 1.0 to 1.6 hours and terminal elimination half‐life of ≈5.0 to 9.0 hours. 4 , 6 , 7 The selumetinib PK profile is approximately dose proportional from 25 mg up to 100 mg in adult patients with cancer and 25 to 75 mg in healthy volunteers. 4 , 6 , 8 A 75‐mg dose has been established as the maximum tolerated dose in adults with solid tumors. In the pediatric SPRINT phase 1 study, 20, 25, and 30 mg/m2 twice‐daily dose levels were evaluated for safety, and 25 mg/m2 was established as the maximum tolerated dose. 5 In pediatric patients, following a single oral dose, selumetinib was rapidly absorbed with a median time to reach Cmax of ≈1 hour and terminal elimination half‐life of ≈6 hours. 5
In vitro, selumetinib undergoes phase 1 metabolism including oxidation of the side chain, N‐demethylation, and loss of the side chain to form amide and acid metabolites. 2 , 9 , 10 The isoform predominantly responsible for selumetinib oxidative metabolism is cytochrome P450 (CYP) 3A4, while CYP2C19, CYP1A2, CYP2C9, CYP2E1, and CYP3A5 are involved to a lesser extent. 2 , 9 In vitro studies indicate that selumetinib also undergoes direct phase 2 metabolic reactions to form glucuronide conjugates principally involving the enzymes uridine 5′‐diphospho‐glucuronosyltransferase (UGT) family member 1 A1 and UGT1A3. 2 Glucuronidation is a significant route of elimination for selumetinib phase 1 metabolites involving several UGT isoforms. 2 , 7 Based on in vitro studies, selumetinib and N‐desmethyl selumetinib are unlikely to contribute to clinically relevant drug interactions with CYP or transporter substrates. 2
Assessment of the potential for a drug as a perpetrator or victim of drug‐drug interactions (DDIs) is a standard element of the drug development process. 11 , 12 A recent analysis showed that inhibition and induction of CYP3A explained most of the drug interactions involving recently approved drugs. 13 As selumetinib is metabolized by CYP3A, it is clinically important to evaluate the potential for PK‐based DDIs when it is administered with strong CYP3A inhibitors, such as itraconazole, and strong CYP3A inducers, such as rifampicin. 14 Data from such DDI studies provide insight into the potential for interaction with a wide range of potential inhibitors and inducers. Drugs considered in the development and verification of the model described in this article include, in addition to itraconazole and rifampicin, fluconazole, erythromycin, diltiazem, fluoxetine, and efavirenz, all of which are commonly used in real‐world clinical practice.
In this analysis, the physiologically based pharmacokinetic (PBPK) model development and verification based on results from the clinical DDI studies and in vitro data, to characterize the PK of selumetinib and its interaction with CYP inhibitors/inducers, is presented. The application of the model to predict the potential impact of selumetinib PK when coadministered with moderate CYP3A and CYP2C19 inhibitors as well as moderate and weak CYP3A inducers is also discussed. Furthermore, we discuss the use of the model to support dose recommendations for selumetinib in younger children.
Methods
All studies were conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization/Good Clinical Practice. The final protocol and informed consent form were approved by the institutional review boards at the respective study sites. Informed consent was obtained from all volunteers before any study procedures were conducted.
The quantitative mass balance diagram of selumetinib is depicted in Figure 1. The overall modeling strategy is shown in Figure 2. The PBPK model for selumetinib was developed using internally generated physiochemical data and 2 DDI studies, 9 which were used to define fraction of metabolism by CYP3A and CYP2C19, respectively. The model was then verified with another DDI study (NCT02046850) and an absolute bioavailability clinical study (NCT02238782) to confirm that the final adult PBPK model could describe the selumetinib PK profile, as well as potential drug interactions. Finally, the verified final adult PBPK model was applied to predict the potential impact of coadministering other CYP3A/CYP2C19 inhibitors/inducers on the PK of selumetinib in healthy volunteers. The model was developed in Simcyp software version 17 (Certara, Sheffield, UK).
Figure 1.
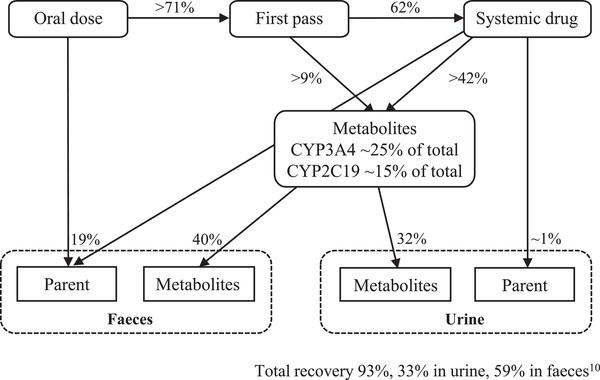
Quantitative mass balance diagram of selumetinib. CL, clearance; CYP, cytochrome P450; UGT, uridine 5′‐diphospho‐glucuronosyltransferase family member 1.
Figure 2.
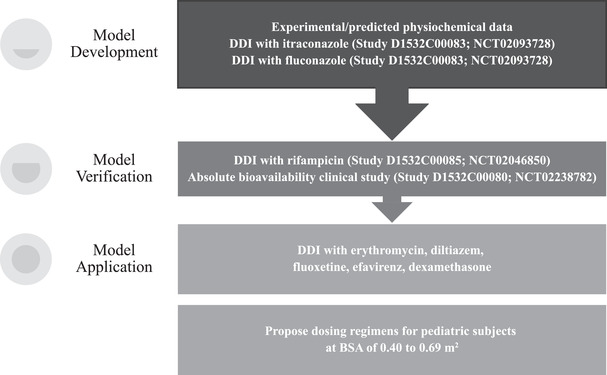
Overall modeling strategy. BSA, body surface area; DDI, drug‐drug interaction.
The first‐order absorption model, which treats the gut as a single compartment associated with a single first‐order absorption rate constant (ka) and fraction absorbed (fa), was used. The fa and ka were predicted from the effective permeability derived from the permeability measured in the Madin‐Darby Canine Kidney II cell system. A minimal PBPK model, together with a single adjusting compartment (SAC) was applied to selumetinib. The minimal PBPK model can be described as a PBPK model that, in its simplest form, has only 4 compartments, predicting the systemic, portal vein, and liver concentrations. The SAC is a nonphysiological compartment that permits adjustment to the drug concentration profile in the systemic compartment, where all tissues were collapsed into 1 compartment excluding the liver and portal vein. Clearance of selumetinib is mainly mediated by hepatic metabolism with significant contributions from CYP3A and CYP2C19, and other metabolic pathways, including UGT, which were grouped together as “the rest” in the model. The retrograde model was used to calculate the intrinsic hepatic clearance of selumetinib, based on information from adult human in vivo oral clearance values, hepatic uptake factor, and the elimination pathways involved.
Clinical Studies
Clinical studies were conducted to evaluate the potential DDI for selumetinib as a victim of CYP inhibition/induction. More specifically, the impact of itraconazole (a strong CYP3A inhibitor), fluconazole (a strong CYP2C19 and moderate CYP3A inhibitor) and rifampicin (a strong inducer of multiple enzymes including CYP3A/CYP2C19) on selumetinib exposure were evaluated (NCT02093728 and NCT02046850). 9 , 15 Itraconazole coadministration increased selumetinib exposure compared to administration of selumetinib alone with a geometric least‐squares mean ratio (90% confidence interval [CI]) for area under the plasma concentration–time curve (AUC) of 1.49 (1.40‐1.59) and Cmax of 1.19 (1.04‐1.35). Fluconazole coadministration increased selumetinib exposure compared to administration of selumetinib alone with a geometric least‐squares mean ratio (90%CI) for AUC of 1.53 (1.44‐1.63) and Cmax of 1.26 (1.10‐1.43). Coadministration with rifampicin decreased selumetinib exposure (AUC was decreased by 51% [90%CI, 47.4‐54.1], and Cmax was reduced by 26% [90%CI, 16.7‐34.1]) compared to administration of selumetinib alone. 9 In addition, an absolute bioavailability study (NCT02238782) of a single oral dose of selumetinib in healthy adult subjects showed it to be 62% (90%CI, 60.1%‐64.1%). 16
Model Development
The values and sources of all parameters for selumetinib are summarized in Table 1. The partition coefficient, acid dissociation constant, and blood‐to‐plasma ratio in the selumetinib model were measured experimentally and reported in the internal database. With respect to the extent of plasma protein binding, the fraction unbound was set at 0.016 based on experimental observations. 2 The Madin‐Darby Canine Kidney II permeability measurement in the study report KMN036 was used to predict human intestinal effective permeability, fa, ka, and nominal flow through the gut. The volume of distribution at steady state value of 1.37 L/kg was predicted by the Rodgers and Rowland Method within Simcyp. 17 A minimal PBPK model with a SAC (intercompartmental clearance = 8 L/h; volume of SAC = 1 L/kg) was applied to capture the distribution phase of selumetinib.
Table 1.
Input Parameters Used to Simulate the Kinetics of Selumetinib
| Parameter | Value | Reference |
|---|---|---|
| MW | 457.7 | Internal database |
| logP | 3.88 | Internal database |
| Compound type | Diprotic base | Internal database |
| pKa | 8.2, 2.7 | Internal database |
| B:P | 0.625 | 0.556‐0.714, Report KPJ003 |
| fu | 0.016 | Report KPJ003 |
| MDCK II, 10–6 cm/s | 37.7 | Report KMN036 |
| Peff,man , ×10–4 cm/s | 4.82 | Predicted |
| Q, L/h | 8 | Population PK model predicted |
| Vss, L/kg | 1.37 | Predicted (Method 2: after Rodgers and Rowland 17 ) |
| Vsac, L/kg | 1 | Estimated based on observed Cmax and PK profile (Study NCT02093728) |
| CYP3A CLint, μL/min/pmol | 0.568 | Retrograde: CLpo = 19.6 L/h (pooled CL/F from 9 studies), fm = 25% |
| CYP2C19 CLint, μL/min/pmol | 3.335 | Retrograde: CLpo = 19.6 L/h (pooled CL/F from 9 studies), fm = 15% |
| Additional liver HLM, μL/min/mg | 186.8 | Retrograde: CLpo = 19.6 L/h (pooled CL/F from 9 studies) |
B:P, blood‐to‐plasma ratio; CL/F, clearance/bioavailability = oral clearance; CLint, intrinsic clearance; CLpo, oral clearance; Cmax, maximum plasma concentration; fm, fraction metabolized; fu, unbound fraction in plasma; HLM, human liver microsome; logP, partition coefficient; MDCK, Madin‐Darby Canine Kidney; MW, molecular weight; Peff,man , human intestinal effective permeability; PK, pharmacokinetic; pKa, acid dissociation constant (logarithmic scale); Q, intercompartmental clearance; Vsac, volume of single adjusting compartment; Vss, volume of distribution at steady state.
The average oral clearance of 19.6 L/h pooled from 9 clinical studies (NCT01635023, NCT01974349, NCT02056392, NCT02322749, NCT02238782, NCT02063204, NCT02063230, NCT02093728, and NCT02046850) was applied to define metabolic clearance of selumetinib with the retrograde method (a built‐in feature of Simcyp). The percentages of selumetinib metabolized (fraction metabolized, fm) by CYP3A and CYP2C19 were assigned at 25% and 15%, according to a DDI study with itraconazole and fluconazole, respectively. 9 Therefore, the intrinsic clearance values of 0.568 and 3.335 μL/min/pmol were estimated for CYP3A and CYP2C19, respectively. The rest of the metabolism/elimination was estimated as additional human liver microsome clearance (186.8 μL/min/mg). Renal elimination was minor elimination pathway of selumetinib (<1% of dose eliminated as unchanged in urine) and was ignored in this analysis.
In the simulations for model development, age and sex were matched with the demographic information reported in the corresponding clinical studies. The dosing regimens were consistent with the actual clinical trial designs.
In the model development procedure, 2 sets of 10 virtual trials were simulated in which 10 healthy volunteers in each trial aged 18 to 44 years (7.7% women) received a single oral dose of selumetinib 25 mg in the presence or absence of an 11‐day oral regimen of itraconazole dosed at 200 mg twice daily, and fluconazole dosed at 400 mg on day 1 and then 200 mg once daily on day 2 to day 11. The single oral dose of selumetinib was administered on day 8 in both of these series of trials. Predicted plasma concentration–time profiles of selumetinib were compared with those observed in NCT02093728. 9 The file for the capsule formulation of itraconazole was selected from the Simcyp library for use in this analysis, with an adjusted fa of 0.45 and a ka of 0.6 hour−1 (to match the predicted minimum plasma concentration) with the observed itraconazole concentrations at day 6 to day 8 from NCT02093728. 9 In NCT02093728, fluconazole and itraconazole were administered after a light breakfast, whereas selumetinib was administered 4 hours later in a fasted state. In the PBPK model simulations, itraconazole was administered with a meal and selumetinib was administered 4 hours later in a fasted state; whereas both fluconazole and selumetinib were administered in a fasted state (fluconazole was administered 4 hours before selumetinib). 9 The US Prescribing Information for fluconazole states that exposure to fluconazole is not affected by food. 18 Therefore, it is not expected that simulations of administration of fluconazole in either a fed or fasted state will alter exposure of fluconazole.
Model Verification
Data from the study of single 75‐mg selumetinib dosing on day 9 alone or in presence of rifampicin 600 mg once daily given from day 1 through day 12 (NCT02046850) was used in the model verification procedure. 9 In the simulations for model verification, age and sex were matched with the demographic information reported in the corresponding clinical studies. The dosing regimens were consistent with the actual trial design.
Similar to the model development procedure described above, model verification involved 10 virtual trials that were simulated in which 10 healthy volunteers in each trial aged 22 to 44 years (4.5% women) received a single oral dose of 75 mg of selumetinib in the absence or presence of an 11‐day oral regimen of rifampicin 600 mg once daily. For simulations involving coadministration of selumetinib and rifampicin, the single oral dose of selumetinib was administered on day 9.
In addition, the final adult PBPK model was also applied to predict selumetinib PK after intravenous (IV) dosing by mimicking NCT02238782. A 12 × 10 trial design was applied where each trial has 12 male subjects between the ages of 23 and 59 years at a dose of 80 μg of selumetinib infused over 15 minutes; the concentration of selumetinib after IV infusion was measured with radiolabeling. Predicted plasma concentration–time profiles of selumetinib were compared with those observed in NCT02046850 and NCT02238782. 9
Sensitivity Analysis
In vitro studies indicated that selumetinib is a substrate of P‐glycoprotein (P‐gp) transporters. 2 Two additional models (models f1 and f2) were explored by incorporating P‐gp in the intestine and liver to the final adult PBPK model to evaluate the potential effect of P‐gp on selumetinib PK. By using the approach presented by Troutman and Thakker 19 and assuming one‐site Michaelis‐Menten saturable kinetics, maximal carrier‐mediated flux (Jmax) and Michaelis‐Menten constant (Km) can be estimated as 9.9 pmol/min and 15.8 μM, resulting in an intrinsic clearance of 0.62 μL/min. The estimated Jmax and Km values were incorporated as part of the following 2 models:
Model f1: The Advanced Dissolution Absorption and Metabolism (ADAM) model was applied to replace first‐order absorption, and the rest of model parameters were kept the same as current model (minimal PBPK with ADAM absorption). The estimated Jmax and Km values of P‐gp were incorporated at the intestinal apical site.
Model f2: Model f1 was further developed as a full PBPK model with ADAM absorption to evaluate the impact of P‐gp at hepatic site (full PBPK with ADAM absorption). To recover the selumetinib PK profile, a tissue:plasma partition coefficient of 0.4 was applied in this full PBPK model, and the rest of model parameters were kept the same. The estimated Jmax and Km values were incorporated, at a hepatic canalicular site.
For both models f1 and f2, a sensitivity analysis was evaluated by changing Km from 1.58 to 158 μM in 20 steps.
Additional sensitivity analyses were applied by changing fm of CYP3A or CYP2C19 of the final adult PBPK model. In the sensitivity analysis of CYP3A, fm of CYP2C19 was kept at 15%, and fm of CYP3A was changed from 5%, 15%, 25%, 35%, 45%, 55%, 65%, 75%, to 85% in both itraconazole and rifampicin interactions. In the sensitivity analysis of CYP2C19, fm of CYP3A was kept at 25%, and fm of CYP2C19 was changed from 5%, 15%, 25%, 35%, 45%, 55%, 65%, to 75% in fluconazole interaction.
Model Application
Drug‐Drug Interactions
Various clinical DDI scenarios were simulated using the final adult PBPK model. Ten virtual trials with 10 healthy volunteers in each trial aged between 20 and 50 years (50% women) receiving a single oral dose of 25 mg of selumetinib in the absence or presence of moderate CYP3A inhibitor (erythromycin or diltiazem) or CYP2C19 inhibitor (fluoxetine) dosing for 10 days. The single oral dose of selumetinib was administered on day 8.
In addition, 10 virtual trials with 10 healthy volunteers in each trial aged 20 to 50 years (50% women) receiving a single oral dose of 75 mg of selumetinib in the absence or presence of moderate or weak CYP3A inducer dosing for 10 days. The single oral dose of selumetinib was administered on day 8. Compound files were used as provided by the Simcyp software for all CYP3A inhibitors (itraconazole, erythromycin, diltiazem, fluoxetine), and inducers (rifampicin, efavirenz). Doses used in these simulations correspond to those commonly used in real‐world clinical practice.
Pediatric Predictions
The final adult PBPK model was extrapolated to children using the pediatric module in Simcyp, where the system‐specific inputs were modified for children and drug‐specific inputs remained unaltered. This pediatric PBPK model was first verified using the clinical study data from NCT01362803. The verified pediatric PBPK model was then applied to provide prospective estimates of body surface area (BSA)‐specific PK, from which proposed dosing regimens for BSA‐specific groups were determined by matching the simulated pediatric PK exposures with the reference exposures. The reference exposures included the pediatric PBPK model predicted exposure after a single oral dose of 25 mg/m2 in patients aged 3 to 17 years old and the population PK post hoc estimation of individual exposure of pediatric patients in the phase 2 study (NCT01362803).
Results
Model Development
A list of PBPK models applied for the studied simulation scenarios is provided in Table S1. Simulated plasma concentration–time profiles and observed concentrations of selumetinib in the absence and presence of itraconazole, fluconazole are displayed in Figure 3A and B (log scale) and Figure S1 (linear scale). The associated geometric mean Cmax and AUC values for selumetinib from these models are listed in Table S2. For itraconazole, the model generally recovered the PK profiles of selumetinib. The predicted selumetinib AUC values were 1185 and 1801 ng•h/mL in the absence and presence of itraconazole, respectively, which were similar to the observed values of 1180 and 1760 ng•h/mL, respectively. The predicted AUC ratio was 1.52 in the presence of itraconazole, which is similar to the observed ratio of 1.49, indicating that the fm assignment of 25% for CYP3A was reasonable.
Figure 3.
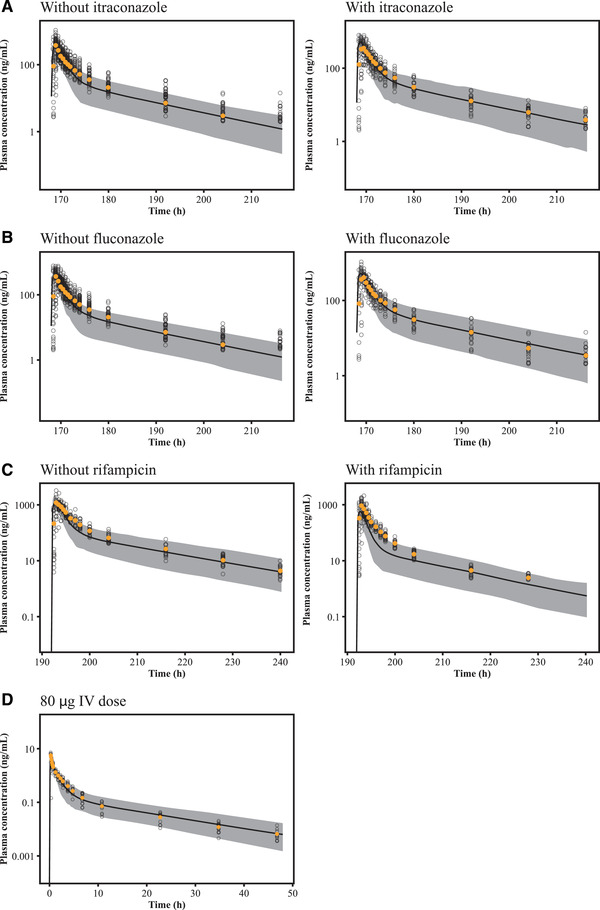
Plasma concentrations of selumetinib after a single oral dose of 25 mg of selumetinib to healthy volunteers in the absence or presence of repeated doses of itraconazole (A), fluconazole (B), rifampicin (C), and after a single IV infusion of 80 μg (D) selumetinib on log scale. The solid lines represent the predicted median plasma concentration of selumetinib and the shaded areas show the 90% prediction interval of the simulations. The yellow closed circles represent the observed mean plasma concentration of selumetinib, and the open circles show the observed individual plasma concentrations. IV, intravenous.
For fluconazole, the model generally recovered the PK profiles of selumetinib. The predicted selumetinib AUC values were 1190 and 1985 ng•h/mL in the absence and presence of fluconazole respectively, which were similar to the observed values 1180 and 1770 ng•h/mL, respectively. The predicted AUC ratio was 1.67 in the presence of fluconazole, which is also similar to the observed ratio of 1.53, indicating the fm assignment of 15% for CYP2C19 was reasonable.
Model Verification
Simulated plasma concentration–time profiles and observed concentrations of selumetinib in the absence and presence of rifampicin and after IV are displayed in Figure 3C and D (log scale) and Figure S1 (linear scale). The associated geometric mean Cmax and AUC values for selumetinib from these models are listed in Table S2. For rifampicin, the model generally recovered the range of selumetinib PK profiles, although slightly underpredicted the mean PK profile in the presence of rifampicin. The predicted selumetinib AUC values were 3616 and 1082 ng•h/mL in the absence and presence of rifampicin, respectively. The predicted AUC ratio was 0.30 in the presence of rifampicin, which is lower than the observed ratio of 0.49. But prediction is still within reasonable range of the observed value, suggesting the developed final adult PBPK model could be used to predict untested clinical situations for potential DDI assessment.
For the absolute bioavailability study, the final adult PBPK model reasonably recovered the selumetinib PK profile after IV and oral dose, the model slightly underestimated the mean Cmax value after IV dose, but the 90% prediction interval still covers the majority of the individual Cmax values. The predicted terminal phase of selumetinib PK matched well with those observed after both IV and oral doses, suggesting that selumetinib clearance was also well characterized by the current PBPK model. As selumetinib clearance and oral PK are key parameters, the model was able to predict them reasonably well to be appropriate for further simulations and predictions.
Sensitivity Analysis
The final adult PBPK model was further explored via sensitivity analysis. The minimal PBPK with ADAM absorption model (model f1) could still reasonably recover the PK of selumetinib. The model f1 predicted Cmax and AUC of selumetinib with P‐gp effect at intestine are presented in Table S3. A sensitivity analysis was evaluated by changing Km from 1.58 to 158 μM in 20 steps (Figure S2). There was an approximate 20% decrease in selumetinib Cmax at a Km of 1.58 μM as compared with a Km of 158 μM, and a negligible change in AUC. In addition, there was no change in Cmax or AUC ratio (in the presence of itraconazole) in the Km range of 1.58 to 158 μM. The sensitivity analysis results suggest that the P‐gp impact on selumetinib absorption is minimal.
Similarly, model f2‐predicted Cmax and AUC of selumetinib with inclusion of the P‐gp effect at hepatic site are presented in Table S3. A sensitivity analysis was also applied by changing the Km from 1.58 to 158 μM in 20 steps (Figure S3). There was little change in selumetinib Cmax or AUC over the Km range of 1.58 to 158 μM. In addition, there was no change in Cmax or AUC ratio (in the presence of itraconazole) in the Km range of 1.58 to 158 μM, suggesting that the P‐gp impact on the biliary excretion is minimal.
Figure S4 presents the fm sensitivity analysis results; the observed Cmax and AUC ratio are also included for comparison. As the fm of CYP3A or CYP2C19 increases, the AUC and Cmax ratios change accordingly (increase for inhibition and decrease for induction), suggesting fm is a sensitive parameter in the model. An fm of 25% for CYP3A provides a reasonable AUC ratio prediction for itraconazole interaction (1.52 predicted vs 1.49 observed), while an fm of ≤15% provides a reasonable AUC ratio prediction for rifampicin interaction (0.41 predicted vs 0.49 observed). In addition, an fm of 15% for CYP3A provides a reasonable Cmax ratio prediction for itraconazole interaction, while an fm of 5% provides a reasonable Cmax ratio prediction for rifampicin interaction. Taken together, these data indicate that an fm ranging from 15% to 25% could be a reasonable assessment for CYP3A contribution to selumetinib metabolism. An fm of 25% was applied in the current PBPK to consider the “worst”‐case scenario. For CYP2C19 contribution, an fm of 15% could provide reasonable predictions for both the AUC and Cmax ratios for fluconazole interaction, and an fm of 15% was used in the current model.
Model Application
After verification, the final adult PBPK model was applied to predict untested clinical outcomes for DDIs with other CYP3A and CYP2C19 inhibitors/inducers. The moderate CYP3A inhibitors erythromycin and diltiazem were predicted to potentially increase selumetinib AUC values by 41% and 32%, respectively. The model predicted the multiple doses of moderate CYP2C19 inhibitor, fluoxetine, could potentially increase selumetinib AUC by 33%. The model predicted the multiple doses of moderate CYP3A inducer, efavirenz, could potentially decrease selumetinib AUC by 38%, while the weak CYP3A inducer, dexamethasone, had minimal impact on selumetinib exposure. The geometric means of ratios in DDI observations and simulations are summarized in Figure 4. The associated geometric mean Cmax and AUC values for selumetinib from all these simulations are listed in Table S4.
Figure 4.
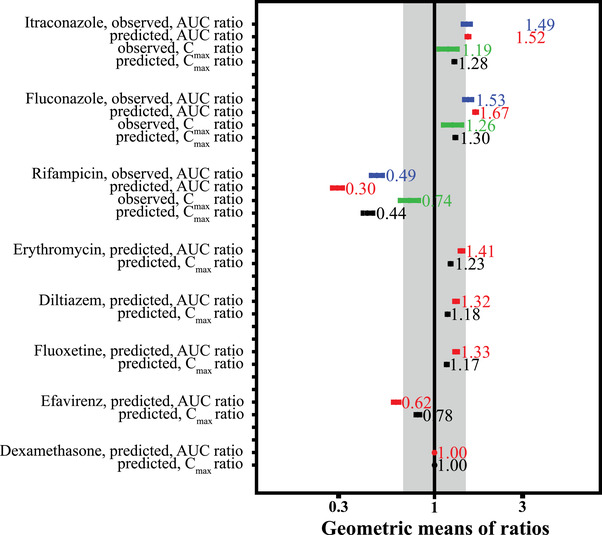
Geometric means of ratios in DDI observations and simulations. The gray area shows the 0.67‐ to 1.5‐fold range. AUC, area under the plasma concentration–time curve; CI, confidence interval; Cmax, maximum plasma concentration; DDI, drug‐drug interaction.
Pediatric Application
The pediatric PBPK model was used to predict geometric means of area under the concentration–time curve from time 0 to 12 hours and the observed exposure from noncompartmental analysis after 20, 25, or 30 mg/m2 single oral dose (Table S5). The simulated plasma concentration–time profiles and the corresponding individual PK observations were overlaid in Figure 5 and Figure S5. The pediatric PBPK model generally recovered the range of selumetinib PK profiles in pediatric subjects. The overall prediction suggested the final adult PBPK model could be used to predict pediatric clinical situations by applying the pediatric population module in Simcyp.
Figure 5.
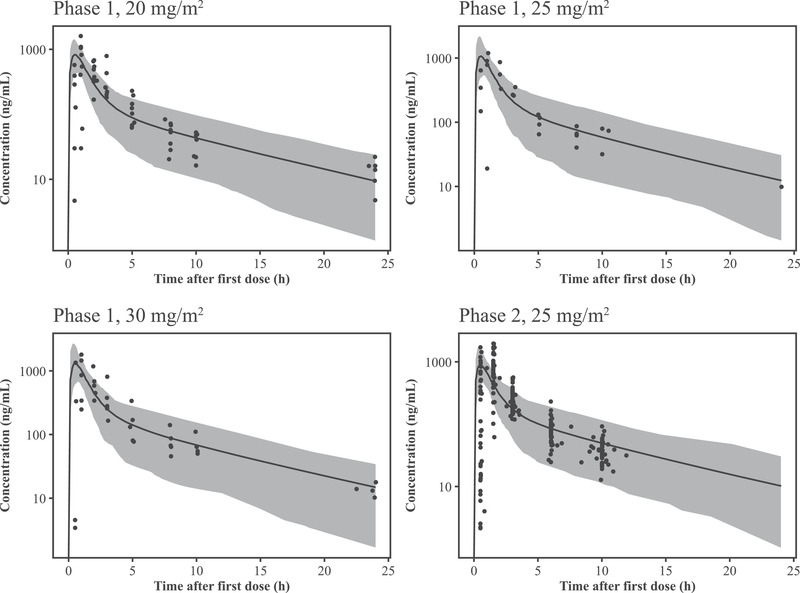
Plasma concentrations of selumetinib after a single oral dose of 20, 25, or 30 mg/m2 in pediatric patients (semilog scale). The solid lines represent the predicted median plasma concentration of selumetinib and the shaded areas show the 90% prediction interval of the simulations. The black closed circles show the observed individual plasma concentrations.
Doses were identified by matching exposures in specific pediatric BSA brackets to the reference exposure in pediatric patients. A single oral dose of 25 mg/m2 selumetinib in the pediatric PBPK model produced a simulated geometric mean (geometric coefficient of variation) AUC from time 0 to 12 hours of 2204 (38.4%) ng•h/mL (median, 2211 ng•h/mL). This prediction is in agreement with exposure data observed in cohorts treated with 25 mg/m2 selumetinib in the SPRINT phase 1/2 study (2199 and 2009 ng•h/mL, respectively). Based on the simulation results for AUC‐based exposure matching, BSA‐based dosing regimens of 7.5, 10, and 12.5 mg are proposed for pediatric subjects in the respective BSA brackets of 0.40 to 0.49, 0.50 to 0.59, 0.60, to 0.69 m2. The boxplots were applied to visualize the exposure matching, as shown in Figure 6.
Figure 6.
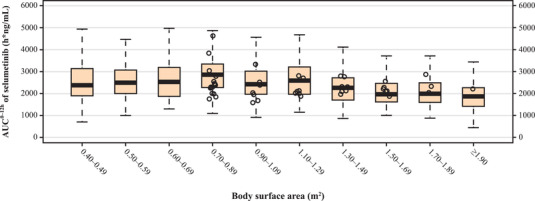
Exposure comparison between model predicted selumetinib exposure and observed values in the BSA bracket range of 0.40 to 1.9 m2. The PBPK model predicted AUC0‐12h of subjects at BSA 0.40 to 0.69 m2 and at 1 to 7 years old with PBPK recommended nomogram (7.5, 10 and 12.5 mg for BSA brackets of 0.40 to 0.49, 0.50 to 0.59, 0.60 to 0.69 m2, respectively) as well as at BSA of 0.70 to 2.20 m2 with the nomogram from the package insert. The boxplots displayed first quartile, median, third quartile, and 1.5‐time interquartile range of model predicted AUC0‐12h in each BSA bracket. The population pharmacokinetic post hoc estimation of individual AUC0‐12h of patients at 3 to 17 years old in study D1532C00057 (NCT01362803) are plotted as open circles. AUC0‐12h, area under the plasma concentration–time curve from time 0 to 12 hours; BSA, body surface area; PBPK, physiologically based pharmacokinetic.
Discussion
In this analysis, the selumetinib PBPK model in healthy volunteers was developed using physiochemical, in vitro, and PK data from a DDI clinical study (NCT02093728). 9 The model was then verified with another DDI clinical study in healthy volunteers (NCT02046850) 9 and an absolute bioavailability study (NCT02238782). 16 The final adult PBPK model was also extended to a pediatric population and verified with clinical study data (NCT01362803). 20
The final adult PBPK model was able to recover the observed plasma concentrations of selumetinib following single oral doses of 25 or 75 mg of selumetinib in healthy volunteers. The predicted selumetinib AUC ratios were 1.52 and 1.67 in the presence and absence of itraconazole and fluconazole, respectively, which were generally consistent with the observed AUC ratios (1.49 and 1.53 in the presence of itraconazole and fluconazole) and were within the 0.8 to 1.25 limits, indicating that the assigned fm values of 25% and 15% for CYP3A and CYP2C19 were reasonable. There was an overprediction for the rifampicin interaction in the final adult PBPK model (predicted ratio of 0.30 compared to the observed value of 0.49); however, it should be noted rifampicin is a pleiotropic inducer, and in general, the clinical observations vs the predictions for rifampicin can vary depending on factors other than CYP3A induction, which are difficult to model. 21 The sensitivity analysis suggested that an fm of 15% for CYP3A would provide better prediction for rifampicin interaction, while an fm of 25% is considered a worst‐case scenario in the interaction with CYP3A inhibitors. Moreover, the predicted AUC or Cmax changes in the presence of various CYP3A or CYP2C19 modulators were similar using both fm models, and only numeric differences were observed between the 2 fm models. Both models suggest selumetinib is unlikely to be a sensitive substrate of CYP3A or CYP2C19, and DDI predictions from either model would provide the same recommendation for potential dose adjustment of selumetinib when coadministrated with moderate CYP3A/CYP2C19 inhibitors or with a CYP3A inducer. Overall, these predictions are consistent with recommendations and guidance, and are considered appropriate to use for DDI predictions. 22
Both the minimal PBPK model with ADAM absorption (model f1) and full PBPK model with ADAM absorption (model f2) were used to further evaluate the impact of P‐gp on selumetinib exposure based on the in vitro results. The simulations underestimated the Cmax to a small degree, and both models were able to predict the AUC reasonably well and could be applied to estimate the impact of P‐gp on absorption and biliary excretion of selumetinib. The sensitivity analysis suggests that the impact of P‐gp is likely to be negligible on the PK of selumetinib. This is consistent with the high fraction of absorption (>71%) 10 and oral bioavailability (62%). 16 Therefore, the simplest model without P‐gp transport is considered as the final adult PBPK model.
The pediatric PBPK model reasonably recovered the selumetinib PK profiles in pediatric subjects, suggesting that the developed final adult PBPK model could be extrapolated to predict pediatric clinical situations by applying the pediatric population module. The simulations from this pediatric PBPK model were used to support and guide dose recommendation in the planned pediatric patient clinical trials.
This study has certain limitations. We assume that absorption of selumetinib is a first‐order process. CYP3A is the major CYP isoform involved in the metabolism of selumetinib; however, the magnitude of the contribution made by CYP2C19 is not well defined. Should the assumptions regarding these or other parameters not be accurate, then the model predictions would be expected to diverge from clinical trial data. Similarly, if the absorption or elimination processes in children differ markedly from those in the adults used to inform the model, then the predictions in children would be less accurate. Some data used in the model were obtained from healthy volunteers. The application of these data to patients, especially those with organ dysfunction, may introduce some degree of error. Continuous evaluation of this model as data continue to accumulate will allow us to determine the accuracy of the assumptions that support the model. Despite these limitations, we believe that the application of the final adult PBPK model is considered appropriate for addressing the DDI potential and to support dose recommendations for selumetinib in younger children.
Conclusion
In conclusion, the final adult PBPK model predicts systemic exposure to selumetinib with reasonable accuracy when coadministered with itraconazole, fluconazole, or rifampicin, and is thus considered suitable for use in the prospective prediction of changes in selumetinib exposure when coadministered with other inhibitors and inducers of CYP3A and CYP2C19. Additionally, the pediatric PBPK model was used to guide dosing regimen selection for younger pediatric patients.
Conflicts of Interest
Sarit Cohen‐Rabbie and Li Zhou report employment with AstraZeneca. Karthick Vishwanathan, Martin Wild, Stein Schalkwijk, Helen Tomkinson, and Diansong Zhou report employment with and stock ownership in AstraZeneca. Lokesh Jain, Sherrie Xu, and Tomoko Freshwater report employment with Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, New Jersey, and stock ownership in Merck & Co., Inc.
Funding
This work was funded by AstraZeneca, the manufacturer of selumetinib.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
The authors thank Preeyah Purang, BSc, of Ashfield Healthcare Communications, Macclesfield, UK, part of UDG Healthcare plc; and Robyn Fowler, PhD, of OPEN Health Communications, London, UK, for editorial assistance that was funded by AstraZeneca in accordance with Good Publications Practice guidelines (http://www.ismpp.org/gpp3).
Data Availability Statement
Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca's data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
References
- 1. Yeh TC, Marsh V, Bernat BA, et al. Biological characterization of ARRY‐142886 (AZD6244), a potent, highly selective mitogen‐activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13(5):1576‐1583. [DOI] [PubMed] [Google Scholar]
- 2. US Food and Drug Administration . Koselugo (selumetinib) prescribing information. AstraZeneca Pharmaceuticals LP. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213756s000lbl.pdf. Accessed October 5, 2020.
- 3. Tong X, Xu H, Carlile DJ, Tomkinson H, Al‐Huniti N, Zhou D. Population pharmacokinetic and exposure‐response analysis of selumetinib and its N‐desmethyl metabolite in patients with non‐small cell lung cancer. J Clin Pharmacol. 2019;59(1):112‐122. [DOI] [PubMed] [Google Scholar]
- 4. Tomkinson H, McBride E, Martin P, et al. Comparison of the pharmacokinetics of the phase II and phase III capsule formulations of selumetinib and the effects of food on exposure: results from two randomized crossover trials in healthy male subjects. Clin Ther. 2017;39(11):2260‐2275.e2261. [DOI] [PubMed] [Google Scholar]
- 5. Dombi E, Baldwin A, Marcus LJ, et al. Activity of selumetinib in neurofibromatosis type 1‐related plexiform neurofibromas. N Engl J Med. 2016;375(26):2550‐2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Banerji U, Camidge DR, Verheul HM, et al. The first‐in‐human study of the hydrogen sulfate (Hyd‐sulfate) capsule of the MEK1/2 inhibitor AZD6244 (ARRY‐142886): a phase I open‐label multicenter trial in patients with advanced cancer. Clin Cancer Res. 2010;16(5):1613‐1623. [DOI] [PubMed] [Google Scholar]
- 7. Leijen S, Soetekouw PM, Jeffry Evans TR, et al. A phase I, open‐label, randomized crossover study to assess the effect of dosing of the MEK 1/2 inhibitor selumetinib (AZD6244; ARRY‐142866) in the presence and absence of food in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2011;68(6):1619‐1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Patel P, Howgate E, Martin P, Carlile DJ, Aarons L, Zhou D. Population pharmacokinetics of the MEK inhibitor selumetinib and its active N‐desmethyl metabolite: data from 10 phase I trials. Br J Clin Pharmacol. 2018;84(1):52‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dymond AW, So K, Martin P, et al. Effects of cytochrome P450 (CYP3A4 and CYP2C19) inhibition and induction on the exposure of selumetinib, a MEK1/2 inhibitor, in healthy subjects: results from two clinical trials. Eur J Clin Pharmacol. 2017;73(2):175‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dymond AW, Howes C, Pattison C, et al. Metabolism, excretion, and pharmacokinetics of selumetinib, an MEK1/2 inhibitor, in healthy adult male subjects. Clin Ther. 2016;38(11):2447‐2458. [DOI] [PubMed] [Google Scholar]
- 11.US Food and Drug Administration. Center for Drug Evaluation and Research. Clinical drug interaction studies—cytochrome P450 enzyme‐ and transporter‐mediated drug interactions guidance for industry. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions. Published January 2020. Accessed July 13, 2021.
- 12. US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) . Clinical drug interaction studies—cytochrome P450 enzyme‐ and transporter‐mediated drug interactions. Guidance for Industry: Clinical Pharmacology; 2020.
- 13. Yu J, Petrie ID, Levy RH, Ragueneau‐Majlessi I. Mechanisms and clinical significance of pharmacokinetic‐based drug‐drug interactions with drugs approved by the U.S. Food and Drug Administration in 2017. Drug Metab Dispos. 2019;47(2):135‐144. [DOI] [PubMed] [Google Scholar]
- 14. US Food and Drug Administration . Drug development and drug interactions: Table of substrates, inhibitors and inducers. https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers#table3-1. Published October 2020. Accessed March 2021.
- 15. Bruggemann RJ, Alffenaar JW, Blijlevens NM, et al. Clinical relevance of the pharmacokinetic interactions of azole antifungal drugs with other coadministered agents. Clin Infect Dis. 2009;48(10):1441‐1458. [DOI] [PubMed] [Google Scholar]
- 16. ClinicalTrials.gov Identifier: NCT02238782. A study to assess theabsolute bioavailability of oral selumetinib in healthy male volunteers. https://clinicaltrials.gov/ct2/show/NCT02238782. Accessed July 13, 2021.
- 17. Rodgers T, Rowland M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci. 2006;95(6):1238‐1257. [DOI] [PubMed] [Google Scholar]
- 18. Zimmermann T, Yeates RA, Laufen H, Pfaff G, Wildfeuer A. Influence of concomitant food intake on the oral absorption of two triazole antifungal agents, itraconazole and fluconazole. Eur J Clin Pharmacol. 1994;46(2):147‐150. [DOI] [PubMed] [Google Scholar]
- 19. Troutman MD, Thakker DR. Efflux ratio cannot assess P‐glycoprotein–mediated attenuation of absorptive transport: asymmetric effect of P‐glycoprotein on absorptive and secretory transport across Caco‐2 cell monolayers. Pharm Res. 2003;20(8):1200‐1209. [DOI] [PubMed] [Google Scholar]
- 20. Gross AM, Wolters PL, Dombi E, et al. Selumetinib in children with inoperable plexiform neurofibromas. N Engl J Med. 2020;382(15):1430‐1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rae JM, Johnson MD, Lippman ME, Flockhart DA. Rifampin is a selective, pleiotropic inducer of drug metabolism genes in human hepatocytes: studies with cDNA and oligonucleotide expression arrays. J Pharmacol Exp Ther. 2001;299(3):849‐857. [PubMed] [Google Scholar]
- 22. Shebley M, Sandhu P, Emami Riedmaier A, et al. Physiologically based pharmacokinetic model qualification and reporting procedures for regulatory submissions: a consortium perspective. Clin Pharmacol Ther. 2018;104(1):88‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca's data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.