

Abstract
Background
Comparative‐effectiveness studies using real‐world data (RWD) can be susceptible to surveillance bias. In solid tumor oncology studies, analyses of endpoints such as progression‐free survival (PFS) are based on progression events detected by imaging assessments. This study aimed to evaluate the potential bias introduced by differential imaging assessment frequency when using electronic health record (EHR)‐derived data to investigate the comparative effectiveness of cancer therapies.
Methods
Using a nationwide de‐identified EHR‐derived database, we first analyzed imaging assessment frequency patterns in patients diagnosed with advanced non‐small cell lung cancer (aNSCLC). We used those RWD inputs to develop a discrete event simulation model of two treatments where disease progression was the outcome and PFS was the endpoint. Using this model, we induced bias with differential imaging assessment timing and quantified its effect on observed versus true treatment effectiveness. We assessed percent bias in the estimated hazard ratio (HR).
Results
The frequency of assessments differed by cancer treatment types. In simulated comparative‐effectiveness studies, PFS HRs estimated using real‐world imaging assessment frequencies differed from the true HR by less than 10% in all scenarios (range: 0.4% to −9.6%). The greatest risk of biased effect estimates was found comparing treatments with widely different imaging frequencies, most exaggerated in disease settings where time to progression is very short.
Conclusions
This study provided evidence that the frequency of imaging assessments to detect disease progression can differ by treatment type in real‐world patients with cancer and may induce some bias in comparative‐effectiveness studies in some situations.
Keywords: cancer, comparative‐effectiveness analysis, imaging assessment timing, measurement bias, progression‐free survival (PFS), real‐word data (RWD), scan timing, simulation modeling
KEY POINTS.
Frequency of outcome assessments in real‐world oncology care is lower than in clinical trial protocols, and variable according to treatment.
Based on the differences observed in a real‐world cohort of patients with aNSCLC, differences in outcome assessment timing may introduce bias in comparative‐ effectiveness studies.
The magnitude of the bias introduced by differences in assessment timing appears to be minor in generally clinically plausible scenarios, and rarely leads to false conclusions.
1. INTRODUCTION
Real‐world data (RWD) captured during the course of routine clinical care, and real‐world evidence (RWE) generated through RWD analyses, have emerged as a complement to traditional prospective trials. 1 Electronic health records (EHRs) have become a key RWD source, providing information about patient populations larger and more inclusive than those in clinical trials. Unlocking the full value of EHR‐derived data, requires determining the reliability of endpoint metrics obtained from them, especially considering how routine care differs from clinical trials.
Progression‐free survival (PFS), or the time lapsed from an index date to disease progression (or death) for a specified population, as well as response rate, or the proportion of patients with tumor‐burden reductions, are acceptable efficacy measures in support of regulatory drug approvals and health technology assessment in oncology research. 2 , 3 , 4 , 5 In clinical trials in solid tumors, progression and response are identified by applying the Response Evaluation Criteria in Solid Tumors (RECIST) 6 to imaging assessments timed according to protocol specifications. In routine clinical practice, clinical judgment is combined with imaging evaluations, and assessment timing is flexible. These are challenges to the evaluation of tumor burden endpoints from RWD sources such as EHRs. While prior work developed a reliable method to identify progression events from EHR sources 7 , 8 and generate real‐world (rw)PFS estimates consistent with clinical trial results, 9 , 10 accurate quantification of time‐to‐event endpoints also depends on the frequency of the originating imaging assessments.
Progression events in patients with solid tumors are anchored on the dates of imaging tests and subsequent clinic visits. Variability in assessment frequency causes surveillance bias that impacts comparative PFS estimates (i.e., treatment effects, Figure 1). 11 , 12 , 13 , 14 , 15 , 16 A number of studies have simulated PFS estimations under assessment schedules in clinical trials. 12 , 13 , 17 Little has been reported on the potential influence of real‐world variability in assessment timing and frequency when estimating PFS.
FIGURE 1.
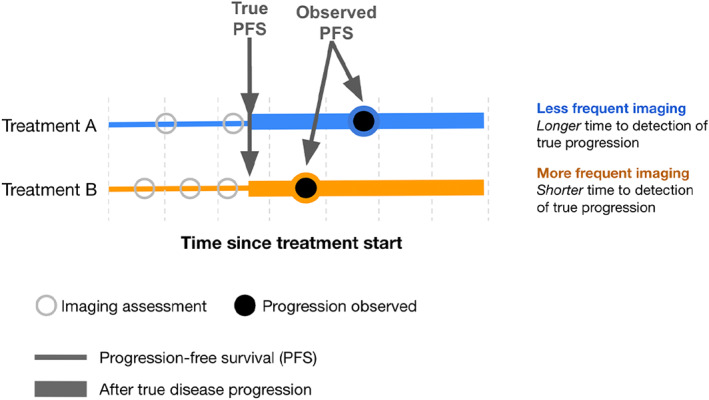
Conceptual diagram of the effect of differential timing in assessments when comparing two treatment groups. Even in theoretical cases where the progression free survival times are the same, more frequent assessments can bias the detection of progression towards shorter times
This study aims to characterize the variability in imaging assessment timing in a cohort of real‐world patients with advanced or metastatic non‐small cell lung cancer (aNSCLC) and to evaluate through simulation the degree of bias that differential assessment frequency between treatment groups may introduce in comparative‐effectiveness studies.
2. REAL‐WORLD DESCRIPTIVE ANALYSIS
2.1. Methods
We utilized Flatiron Health's nationwide longitudinal de‐identified EHR‐derived database comprised of patient‐level structured and unstructured data, curated via technology‐enabled abstraction. 18 , 19 During the study period, the data originated from approximately 280 US cancer clinics (~800 sites of care). Institutional Review Board approval of the study protocol was obtained prior to study conduct and included a waiver of informed consent. Analyses were conducted in R version 3.6.1.
A database of patients who had undergone next generation sequencing (NGS) testing of their tumor samples was used to source those with at least two clinic visits on or after January 1, 2011, a confirmed diagnosis of advanced or metastatic aNSCLC on or before December 31, 2018, and documentation of receiving at least one line of systemic therapy. In order to optimize information availability, patients had to have at least one radiographic imaging assessment during the therapy line of interest indicated in their EHR, with the corresponding documentation manually abstracted. Assessment time points indexed to a line of treatment were captured retrospectively. Starting from the first imaging test, imaging tests performed up to 14 days later were considered one single assessment time point at the date of first imaging, since unique assessments may encompass a multiple‐test succession (e.g., a chest CT followed by a brain MRI 1 week later) documented in a single synthesized entry. Any tests in the first 30 days after treatment start date were excluded, as well as those from anatomic sites without prior or new disease involvement, from baseline onward (those scans are likely not intended to assess disease burden changes following therapy). We excluded patients without structured EHR activity within 90 days after the advanced diagnosis date.
The primary outcome of interest was imaging assessment frequency during the observation period, defined as the time from start date of the line of therapy until either the first radiographic disease progression event, death within 90 days of treatment discontinuation, or censoring (patients without an event were censored at the date of the last imaging assessment prior to the treatment discontinuation/switch [as commonly specified in clinical trials]; Table S1). Imaging assessment frequency was measured using two methods: (1) we calculated the mean time (weeks) between scans during the observation period (reported as the observation period divided by the number of assessments time points) and (2) we defined observation windows, mimicking imaging assessment cadences common in clinical trials and intended to reflect clinically meaningful, unique assessment opportunities, 20 , 21 , 22 consisting of six‐week periods for the first 36 weeks and nine‐week periods thereafter, as in the POPLAR trial. 21 Based on this schema, we calculated the proportion of observation windows with at least one documented imaging assessment at the individual patient level. The last observation window for each patient is the last complete window, including the end date of the observation period. At a cohort level, we calculated the mean, median, and variance in patients' probability of receiving an imaging assessment in any observation window, stratified by treatment group (ALK inhibitors, anti‐VEGF–containing therapies, EGFR TKIs, PD‐1/PD‐L1–based therapies, platinum‐based chemotherapy combination, single agent chemotherapies, and other). A sensitivity analysis examined imaging assessment frequency in second‐line treatment compared to first‐line treatment (oncologist‐defined, rule‐based lines of therapy as defined in prior studies 10 ).
2.2. Results
The real‐world analysis included 3118 patients (Table 1; Figure S1; comparisons between study and parent cohorts, Tables S2–S4). The frequency of imaging assessments was greater for patients who had an initial diagnosis of Stage IV aNSCLC (compared to Stage III), and those diagnosed in 2018 (compared to earlier years).
TABLE 1.
Characteristics of the real‐world cohort of patients with aNSCLC included in this study, overall and by tertile of scan frequency
| Total | Frequency of imaging assessment tertile a | |||
|---|---|---|---|---|
| Low | Medium | High | ||
| N = 3118 | n = 1034 | n = 1027 | n = 1057 | |
| Age at advanced diagnosis, median [IQR] | 67.0 [60.0;74.0] | 69.0 [61.0;75.0] | 67.0 [59.0;74.0] | 66.0 [59.0;73.0] |
| Year of advanced diagnosis | ||||
| <2014 | 686 (22.0%) | 278 (26.9%) | 224 (21.8%) | 184 (17.4%) |
| 2015–2017 | 1999 (64.1%) | 663 (64.1%) | 667 (64.9%) | 669 (63.3%) |
| 2018 | 433 (13.9%) | 93 (9.0%) | 136 (13.2%) | 204 (19.3%) |
| Sex | ||||
| Female | 1607 (51.5%) | 554 (53.6%) | 513 (50.0%) | 540 (51.1%) |
| Male | 1511 (48.5%) | 480 (46.4%) | 514 (50.0%) | 517 (48.9%) |
| Race | ||||
| White | 2257 (72.4%) | 731 (70.7%) | 741 (72.2%) | 785 (74.3%) |
| Black, Afr. Am | 204 (6.5%) | 75 (7.3%) | 61 (5.9%) | 68 (6.4%) |
| Asian | 109 (3.5%) | 54 (5.2%) | 30 (2.9%) | 25 (2.4%) |
| Other | 290 (9.3%) | 96 (9.3%) | 97 (9.4%) | 97 (9.2%) |
| Not reported | 258 (8.3%) | 78 (7.5%) | 98 (9.5%) | 82 (7.8%) |
| Practice type | ||||
| Academic | 95 (3.0%) | 30 (2.9%) | 34 (3.3%) | 31 (2.9%) |
| Community | 3023 (97.0%) | 1004 (97.1%) | 993 (96.7%) | 1026 (97.1%) |
| Smoking history | ||||
| Yes | 2498 (80.1%) | 798 (77.2%) | 824 (80.2%) | 876 (82.9%) |
| No | 609 (19.5%) | 233 (22.5%) | 199 (19.4%) | 177 (16.7%) |
| Unknown/Not doc. | 11 (0.4%) | 3 (0.3%) | 4 (0.4%) | 4 (0.4%) |
| Disease stage | ||||
| Stage I | 262 (8.4%) | 112 (10.8%) | 79 (7.7%) | 71 (6.7%) |
| Stage II | 184 (5.9%) | 72 (7.0%) | 52 (5.1%) | 60 (5.7%) |
| Stage III | 578 (18.5%) | 256 (24.8%) | 183 (17.8%) | 139 (13.2%) |
| Stage IV | 2044 (65.6%) | 573 (55.4%) | 694 (67.6%) | 777 (73.5%) |
| Other | 50 (1.6%) | 21 (2.0%) | 19 (1.9%) | 10 (0.9%) |
| Histology | ||||
| Non‐squamous | 2432 (78.0%) | 811 (78.4%) | 806 (78.5%) | 815 (77.1%) |
| Squamous | 566 (18.2%) | 184 (17.8%) | 181 (17.6%) | 201 (19.0%) |
| NOS | 120 (3.8%) | 39 (3.8%) | 40 (3.9%) | 41 (3.9%) |
| Therapy class in first‐line | ||||
| Platinum‐based | 1150 (36.9%) | 320 (30.9%) | 374 (36.4%) | 456 (43.1%) |
| PD‐1/PD‐L1‐based | 761 (24.4%) | 240 (23.2%) | 239 (23.3%) | 282 (26.7%) |
| Anti‐VEGF‐containing | 602 (19.3%) | 183 (17.7%) | 212 (20.6%) | 207 (19.6%) |
| EGFR TKIs | 423 (13.6%) | 222 (21.5%) | 132 (12.9%) | 69 (6.5%) |
| ALK inhibitors | 95 (3.0%) | 38 (3.7%) | 37 (3.6%) | 20 (1.9%) |
| Single agent chemother. | 63 (2.0%) | 25 (2.4%) | 26 (2.5%) | 12 (1.1%) |
| Other | 24 (0.8%) | 6 (0.6%) | 7 (0.7%) | 11 (1.1%) |
Abbreviations: ALK, anaplastic lymphoma kinase; aNSCLC, advanced non‐small cell lung cancer; EGFR, epidermal growth factor receptor; IQR, interquartile range; NOS, not otherwise specified; PD‐(L)1, programmed death (ligand) 1; TKI, tyrosine kinase inhibitor; VEGF, vascular endothelial growth factor.
Imaging frequency tertile of low, medium, or high corresponds to mean weeks between assessment time points being >11.9, 8.6–11.9, and 3.5–8.5 respectively.
Imaging assessment frequency was highest for patients receiving platinum‐based chemotherapy (every 10.4 weeks on average; mean probability for assessment within observation windows, 0.57 [sd = 0.20]), and lowest for EGFR TKIs (every 14.9 weeks; mean probability for assessment within observation windows, 0.47 [sd = 0.17]; Figure 2). A sensitivity analysis found similar assessment frequencies and patterns in first‐ and second‐line therapy (Tables S5 and S6, Figure S2).
FIGURE 2.
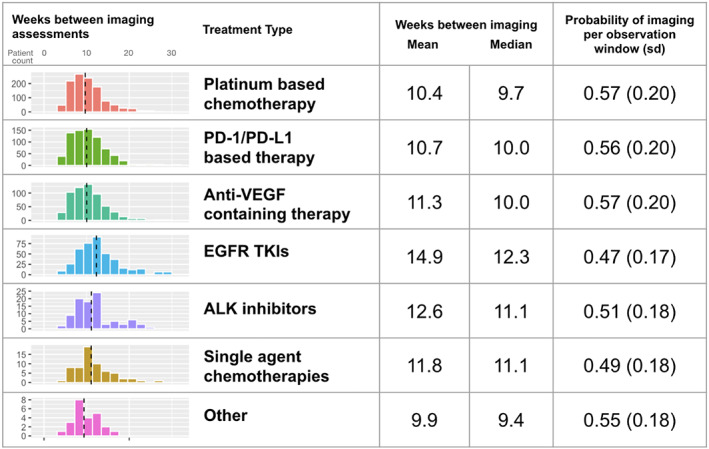
Descriptive statistics for the frequency of imaging assessments among real‐world patients with aNSCLC on first‐line therapy, both for the length of the interval between assessments and the probability of assessment in pre‐defined observation windows. Dashed vertical lines represent the median. “Other” category includes other minor therapy classes with less than 20 patients
3. SIMULATION STUDY
3.1. Methods
The assessment probabilities per observation window from the real‐world cohort analysis were used as inputs in a simulation to evaluate how differences in the frequency of imaging assessments by therapy class could bias estimates in comparative‐effectiveness studies. Simulation modeling allows for generation of the “true” disease progression event for each individual within a hypothetical study group, generation of a set of plausible imaging assessment dates dependent on the treatment arm, and then calculation of the date when disease progression would be observed; these simulated data were devoid of confounders (Figure 1).
Key model parameters are provided in Table S7. The event time of true disease progression and death for each individual was probabilistically drawn from an exponential distribution with constant hazard (i.e., Weibull with shape parameter of 1). The overall beta distributions for each cohort were aggregated based on “individual patient journey” simulations. The series of imaging dates for each individual were generated by adding random noise to the scan date schedule and then defining for each observation window whether the scan occurred, based on a probabilistic draw from the treatment type‐specific parametric distribution of the probability for a patient to receive a scan in the window (see Data S1). By assigning the true hazard ratio (HR true ) for Treatment A compared to B as an input to the model, we could explore the influence of other model parameters to understand the related effect on the observed hazard ratio (HR observed ) in comparison to HR true . The main outcome measure of interest was percent bias, defined here as (HR true ‐ HR observed ) /HR true to capture the magnitude and direction towards or away from a null hypothesis. A secondary outcome measure was the percent of simulated trials where conclusions differ between the HR true and HR observed . Conclusions differ in the simulated study if the HR observed was not statistically significant, having a 95% CI for the HR observed crossing the null hypothesis value of 1.0.
The primary analysis simulated a set of 1000 comparative‐effectiveness studies with 500 patients in each treatment group (total of 1000 patients per trial), true median PFS of 4 months with Treatment A, HR true of 0.80 of Treatment B relative to A, and the probability of an imaging assessment in each observation window was 0.50 for Treatment A and 0.65 for Treatment B. This primary simulation was conceived as an extreme scenario in that the HR true is not that far from one (small treatment effect), the PFS is short, and there is a very large difference in the frequency of imaging assessments between the treatment groups.
We conducted a one‐way sensitivity analysis to investigate key factors influencing bias (magnitude and direction), and altering treatment effectiveness conclusions: the size of the differential in imaging frequency between treatment groups (small or large), direction of the difference in treatment imaging frequency (i.e., intervention assessed more or less often than reference), patient cohort size (small or large), expected reference progression‐free time (short or long), and treatment effect size (small or large). We recalculated the model using plausible upper and lower ranges for these parameter values, one at a time with all other inputs fixed, and documenting percent bias and percent of altered conclusions for each simulation.
In addition, three clinically meaningful and practically plausible comparative‐effectiveness case studies were simulated to ascertain potential bias away from corresponding randomized controlled trial results with identical imaging assessments across arms (Table 2, Table S9). Comparisons incorporated real‐world assessment frequencies in scenarios corresponding to the Keynote‐024 20 trial (a phase III trial comparing two infusional therapies, the immunotherapeutic agent [PD‐L1 inhibitor] pembrolizumab to investigator's choice of platinum‐based chemotherapy, in previously untreated patients with advanced NSCLC; featured in the main Results) and the EURTAC 23 trial (a phase III trial comparing an oral agent erlotinib [an EGFR TKI] to infusional platinum‐based chemotherapy, in patients with NSCLC and tumors harboring EGFR mutations; featured in Data S1). We used published trial results to define treatment arms, true median PFS, and true efficacy input values for case studies. Two case studies simulated possible outcomes using RWD for each arm (main Results and Supplement Section 2.5 in Data S1), and a third (Supplement Section 2.5 in Data S1) simulated outcomes of a clinical‐trial arm compared to a real‐world cohort.
TABLE 2.
Results from simulation model primary analysis and case study a
| Primary simulation | Case study | |
|---|---|---|
| Hypothetical base case | RWD versus RWD simulation | |
| INPUT | ||
| Treatment Group A (reference) | Drug A | Chemotherapy |
| True median PFS (95% CI), months | 4.0 (3.5, 4.5) | 6.0 (5.3, 6.8) b |
| Imaging frequency, median weeks between scans | 12.0 | 10.6 |
| Treatment Group B | Drug B | PD‐L1inh. |
| True median PFS (95% CI), months | 5.0 (4.4, 5.7) | 12.0 (10.5, 13.6) b |
| Imaging frequency, median weeks between scans | 9.2 | 10.8 |
| True difference in median PFS, months | 1.0 | 6.0 |
| True HR, (95% CI) | 0.80 (0.71, 0.91) | 0.50 (0.44–0.57) b |
| RESULT | ||
| Observed difference in median PFS, months | 0.64 | 6.6 |
| Observed HR, (95% CI) | 0.86 (0.76, 0.97) | 0.51 (0.44–0.57) |
| Bias in HR, mean relative % (95% CI) | −7.0% (−20.6, 5.7) | −1.3% (−14.0, 11.7) |
| Conclusions differ, c % of 1000 simulations | 30% | 0 |
Abbreviations: CI, confidence interval; EGFR, epidermal growth factor receptor; HR, hazard ratio; PD‐(L)1, programmed death (ligand) 1; RWD, real‐world data; TKI, tyrosine kinase inhibitor.
EHR‐derived data analysis stratified by treatment class. Note: Each case study and the main analysis simulate 500 patients in each treatment group and 1000 comparative‐effectiveness trials.
Based on the Keynote‐024 study. 20
In the trials simulations where conclusions differed, the 95% CI of the observed HR crossed 1.0 and the null hypothesis could not be rejected.
The model was coded in R version 3.6.1. Uncertainty in the observed HR and percent bias is reflected by 95% confidence intervals (CI) from parametric bootstrapping with 1000 simulated datasets (see Data S1 for parametric distributions). Detailed methods and functions for replication are provided in Data S1.
3.2. Results
In the primary analysis simulation of 1000 studies with 1000 patients per study, where the probability of undergoing imaging assessment during an observation window was 0.50 for Treatment A and 0.65 for Treatment B, the HR true was fixed to 0.80 and the mean HR observed was 0.86 (95% CI: 0.76 to 0.97). These conditions resulted in a surveillance bias of −7% (95% CI: −21 to 6%). In 30% of the replicated studies, conclusions differed because of biased HR with 95% CIs crossing 1.0, therefore the null hypothesis could not be rejected, leading to false null results. The true difference in median PFS between treatments A and B was 1.0 month and the observed incremental gain from Treatment B was 0.6 months (Table 2).
In our case studies, the assessment frequency differences across treatment arms introduced a smaller amount of bias and no conclusions differed from truth (Table 2, Table S9, Figure S4), that is, HR observed was in the same direction as HR true and the null hypothesis was correctly rejected. The main case study compared pembrolizumab versus chemotherapy, with HR true of 0.50 based on clinical trial Keynote‐024. 20 Using the real world frequency of imaging from our descriptive analysis for the corresponding therapies (both simulation arms), the difference in the assessment frequency was 1.8%. This difference introduced a −1.3% bias in the HR analysis, for an HR observed of 0.51, which did not lead to false conclusions in any of the 1000 simulations performed (Table 2 and Figure 3).
FIGURE 3.
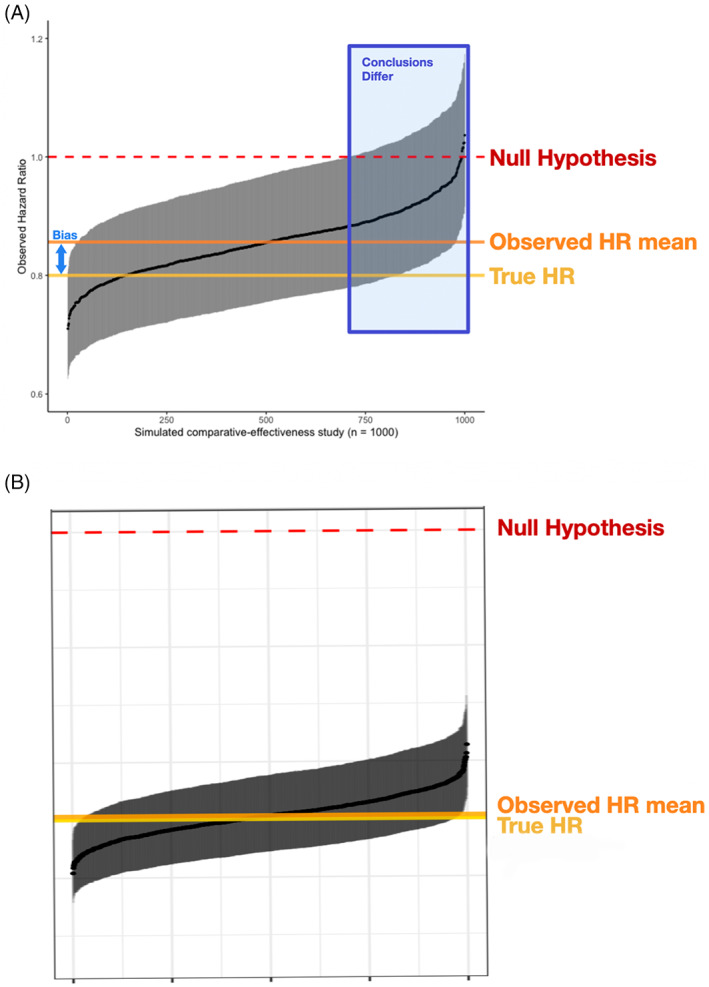
Results from simulated comparative‐effectiveness studies for A, the primary simulation and B, a case study based on real‐world assessment frequencies. Black points represent the observed HR for each study with 95% CI bars in gray, horizontal orange line compared to true HR at yellow line. The 1000 studies are ordered by observed HR along the x‐axis
In both the primary simulation and the case study, the more effective treatment arm had similar or greater imaging assessment frequency. The bias attributed to that differential shifted the effectiveness estimate towards the null hypothesis, yielding a higher HR estimate. However, the shift was larger in the pre‐specified primary theoretical simulation and smaller in the case study based on actual observations from real‐world practice (Figure 3).
Our one‐way sensitivity analysis dissected key inputs one by one, evaluating the impact that surveillance differences may have on the ultimate results according to a range of values for each input (drivers of bias, Figure 4). The key drivers of percent bias were the size of absolute and relative differences in frequency of imaging assessments between treatments, with greater differences causing greater bias. Results were also sensitive to the true PFS in the reference group, where longer PFS time in the reference treatment group reduced susceptibility to bias from imaging frequency.
FIGURE 4.
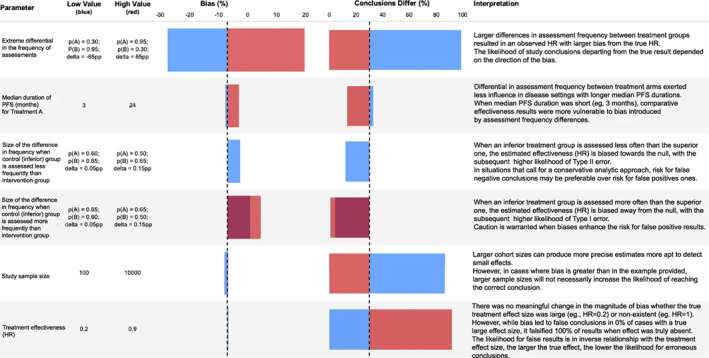
Key considerations influencing susceptibility to surveillance bias from differential imaging assessment frequency in comparative‐effectiveness studies, based on one‐way sensitivity analysis results. The one‐way sensitivity analysis calculates results for the upper and lower range of a parameter while holding all other parameters fixed. The horizontal axis dashed line represents the value estimated in the main analysis. HR, hazard ratio; PFS, progression free survival; pp, percentage points of mean relative percent bias in observed versus true hazard ratio
4. DISCUSSION
We found imaging assessments in routine practice to be usually less frequent than clinical trial schedules for patients with aNSCLC, 19 , 20 , 21 and consistent with NCCN Guidelines®. 24 The comparative‐effectiveness study simulation quantified the magnitude and direction of bias in the HR for PFS introduced by differences in assessment frequencies. Having an inferior comparator arm with fewer assessments than an intervention arm biased results towards the null. In a theoretical scenario, a comparison of a single‐arm clinical trial (novel therapy arm) with an external control RWD arm under extremely divergent assessment conditions, would yield false negative results (erroneous rejections of an effective therapy) nearly a third of the time, but would not lead to false positive results with erroneous benefit claims for the investigational therapy. However, our descriptive results indicated that real‐world variability may not necessarily reach the span we theorized for our pre‐specified simulation, which allowed us to craft clinically plausible case studies; in those cases, the bias was smaller and did not change the conclusions in any of the simulations performed.
Understanding factors driving surveillance bias and potentially false conclusions is important, considering the growing interest in using RWE to contextualize clinical trial results. 25 , 26 , 27 , 28 , 29 , 30 According to our univariate sensitivity analysis (Figure 4), the extent of relative bias remained similar for small and large effect sizes, but incorrect conclusions became more likely with smaller effect sizes. Therefore, the potential impact of differential assessment frequencies may become a relevant limitation in RWD studies with small treatment effects. The risk for false negative results also depended on the direction of the assessment frequency differential (whether the arm with more/less frequent assessments is the one with superior/inferior outcomes), becoming greater when the direction of surveillance bias opposes the true difference direction (i.e., the arm with more true events has fewer observed events), as when an inferior treatment arm has less frequent assessments. When the surveillance bias has the same direction as the true difference (i.e., the arm with more true events has more observed events), estimates tend to shift away from the null (the larger the differences in assessment frequency, the greater the shift), making this the only setting risking false positive findings and overestimated benefit. Therefore, the risk for false results rests on both the magnitude and the direction of differential scan frequency, in the context of the effectiveness differences between arms. Researchers should consider these factors, particularly when facing binary decisions (i.e., yes/no regulatory decisions or go/no‐go in clinical development).
The recent study by Kapetanakis et al 31 explored a similar question in the context of comparisons of separate single‐arm clinical trials. That setting has the advantage of processing information from two treatment arms where imaging assessment timing may be different, but modifiable in a uniform manner. Our simulation study used real‐world inputs to investigate the potential bias that could be introduced by imaging assessment frequency variations; therefore the inter‐cohort differences (between real‐world cohorts, or clinical trial cohorts and real‐world control arms) were non‐modifiable, and exhibited intrinsic variability.
This study has some limitations. Our originating data source for the descriptive analysis is EHR‐derived information for a specific patient cohort who has received NGS testing and has EHR documentation of imaging assessments. We focused on this cohort to filter potentially incomplete data in our EHR‐derived database since data missingness is a known issue in RWD sources (Ma et al 18 reported a comprehensive comparison of our master data source to other standard observational databases in oncology). Yet, the entry requirements could be exerting some cohort selection bias, which we tried to contextualize by comparing baseline characteristics of our study cohort to patients with no documented scans (see Data S1). Another limitation is the potential unmeasured bias related to insurance coverage, which may be an important determinant of assessment practices, for which, unfortunately, our databases lack comprehensive information. We made some specific research choices: focus on one single disease (aNSCLC), a simulation model structure with a unique likelihood of imaging assessment per window for each individual, maintained constant over time as a simplification, and we presented idealized comparisons devoid of other confounders. Subsequent research on actual cohorts should investigate the generalizability across tumor types and disease stages and will likely have to face more complex scenarios where surveillance bias may be compounded by real‐world confounding factors. Finally, we assigned relatively large sample sizes to our simulated studies; in cases with smaller sample sizes, true differences may become more difficult to detect.
Imaging assessment in real‐world oncology practice is likely to be less frequent and less consistent than in clinical trials and can systematically differ by treatment type. The potential influence of differences in the timing of outcome measurement should be considered during the design stage when assessing the feasibility and validity of real‐world comparisons. When differential scan timing may impact results, more guidance on best practices to correct and/or minimize bias would be beneficial. Possible approaches for debate could include statistical adjustment or interval censoring. The scientific community must define and develop these methods for surveillance bias correction in order to rigorously learn from the experiences of patients with cancer and generate valid evidence for decision‐making.
CONFLICT OF INTEREST
At the time of the study, all authors are employees of Flatiron Health, Inc., which is an independent subsidiary of the Roche Group. BSJA, XM, SDG, SS and ABB report stock ownership in Roche. SDG, SS and ABB report equity ownership in Flatiron Health.
ETHICS STATEMENT
Institutional Review Board approval of the study protocol was obtained prior to study conduct, and included a waiver of informed consent.
AUTHOR CONTRIBUTIONS
Study design/concept: Blythe J. S. Adamson, Xinran Ma, Elizabeth M. Sweeney, Somnath Sarkar, Sandra D. Griffith, Ariel B. Bourla. Data collection: Flatiron Health. Data interpretation and analysis: Blythe J. S. Adamson, Xinran Ma, Somnath Sarkar, Sandra D. Griffith, Ariel B. Bourla. Manuscript writing/review and approval: All Authors.
Supporting information
Data S1. Supporting Information.
ACKNOWLEDGMENTS
Koen Degeling, Andrew Briggs, Aracelis Torres, Michael Vasconcelles, Katherine Tan, Meghna Samant, Paul You for helpful discussions and comments during the drafting of the manuscript. Kellie Ciofalo, Alex Vance for administrative support. Julia Saiz‐Shimosato for editorial support. This study was sponsored by Flatiron Health, Inc., which is an independent subsidiary of the Roche Group.
Adamson BJS, Ma X, Griffith SD, Sweeney EM, Sarkar S, Bourla AB. Differential frequency in imaging‐based outcome measurement: Bias in real‐world oncology comparative‐effectiveness studies. Pharmacoepidemiol Drug Saf. 2022;31(1):46‐54. 10.1002/pds.5323
Funding information Flatiron Health, Inc.
REFERENCES
- 1. Schurman B. The framework for FDA's real‐world evidence program. Appl Clin Trials. 2019;28:15‐17. [Google Scholar]
- 2. Chen EY, Raghunathan V, Prasad V. An overview of cancer drugs approved by the US Food and Drug Administration based on the surrogate end point of response rate. JAMA Intern Med. 2019;179:915‐921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kemp R, Prasad V. Surrogate endpoints in oncology: when are they acceptable for regulatory and clinical decisions, and are they currently overused? BMC Med. 2017;15:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen EY, Haslam A, Prasad V. FDA acceptance of surrogate end points for cancer drug approval: 1992‐2019. JAMA Intern Med. 2020;180:912‐914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wilson MK, Karakasis K, Oza AM. Outcomes and endpoints in trials of cancer treatment: the past, present, and future. Lancet Oncol. 2015;16:e32‐e42. [DOI] [PubMed] [Google Scholar]
- 6. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228‐247. [DOI] [PubMed] [Google Scholar]
- 7. Griffith SD, Miksad RA, Calkins G, et al. Characterizing the feasibility and performance of real‐world tumor progression end points and their association with overall survival in a large advanced non‐small‐cell lung cancer data set. JCO Clin Cancer Inform. 2019;3:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Griffith SD, Tucker M, Bowser B, et al. Generating real‐world tumor burden endpoints from electronic health record data: comparison of RECIST, radiology‐anchored, and clinician‐anchored approaches for abstracting real‐world progression in non‐small cell lung cancer. Adv Ther. 2019;36:2122‐2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang Bartlett C, Mardekian J, Cotter MJ, et al. Concordance of real‐world versus conventional progression‐free survival from a phase 3 trial of endocrine therapy as first‐line treatment for metastatic breast cancer. PLoS One. 2020;15:e0227256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Khozin S, Miksad RA, Adami J, et al. Real‐world progression, treatment, and survival outcomes during rapid adoption of immunotherapy for advanced non‐small cell lung cancer. Cancer. 2019;125:4019‐4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Magder LS, Hughes JP. Logistic regression when the outcome is measured with uncertainty. Am J Epidemiol. 1997;146:195‐203. [DOI] [PubMed] [Google Scholar]
- 12. Marten K, Auer F, Schmidt S, Kohl G, Rummeny EJ, Engelke C. Inadequacy of manual measurements compared to automated CT volumetry in assessment of treatment response of pulmonary metastases using RECIST criteria. Eur Radiol. 2006;16:781‐790. [DOI] [PubMed] [Google Scholar]
- 13. Panageas KS, Ben‐Porat L, Dickler MN, Chapman PB, Schrag D. When you look matters: the effect of assessment schedule on progression‐free survival. J Natl Cancer Inst. 2007;99:428‐432. [DOI] [PubMed] [Google Scholar]
- 14. Zeng L, Cook RJ, Wen L, Boruvka A. Bias in progression‐free survival analysis due to intermittent assessment of progression. Stat Med. 2015;34:3181‐3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stone A, Wheeler C, Carroll K, Barge A. Optimizing randomized phase II trials assessing tumor progression. Contemp Clin Trials. 2007;28:146‐152. [DOI] [PubMed] [Google Scholar]
- 16. Qi Y, Allen Ziegler KL, Hillman SL, et al. Impact of disease progression date determination on progression‐free survival estimates in advanced lung cancer. Cancer. 2012;118:5358‐5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Broglio KR, Berry DA. Detecting an overall survival benefit that is derived from progression‐free survival. J Natl Cancer Inst. 2009;101:1642‐1649. 10.1093/jnci/djp369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ma X, Long L, Moon S, Adamson BJS, Baxi SS. Comparison of population characteristics in real‐world clinical oncology databases in the US: flatiron health, SEER, and NPCR. medRxiv 2020: 2020.03.16.20037143 .
- 19. Birnbaum B, Nussbaum N, Seidl‐Rathkopf K, et al. Model‐assisted cohort selection with bias analysis for generating large‐scale cohorts from the EHR for oncology research. arXiv preprint arXiv:2001.09765. January 13, 2020
- 20. Reck M, Rodriguez‐Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD‐L1‐positive non‐small‐cell lung cancer. N Engl J Med. 2016;375:1823‐1833. [DOI] [PubMed] [Google Scholar]
- 21. Fehrenbacher L, Spira A, Ballinger M, et al. Atezolizumab versus docetaxel for patients with previously treated non‐small‐cell lung cancer (POPLAR): a multicentre, open‐label, phase 2 randomised controlled trial. Lancet. 2016;387(10030):1837‐1846. [DOI] [PubMed] [Google Scholar]
- 22. Ramalingam SS, Dahlberg SE, Belani CP, et al. Pemetrexed, bevacizumab, or the combination as maintenance therapy for advanced nonsquamous non‐small‐cell lung cancer: ECOG‐ACRIN 5508. J Clin Oncol. 2019;37:2360‐2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first‐line treatment for European patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (EURTAC): a multicentre, open‐label, randomised phase 3 trial. Lancet Oncol. 2012;13:239‐246. [DOI] [PubMed] [Google Scholar]
- 24. Ettinger D. Wood DE, Aisner DL. NCCN clinical practice guidelines in oncology: non‐small cell lung cancer. National Comprehensive Cancer Network. 2020.
- 25. US Food & Drug Administration . Framework for FDA'S Real‐World Evidence Program. 2018.
- 26. Plueschke K, McGettigan P, Pacurariu A, Kurz X, Cave A. EU‐funded initiatives for real world evidence: descriptive analysis of their characteristics and relevance for regulatory decision‐making. BMJ Open. 2018;8:e021864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ventz S, Lai A, Cloughesy TF, Wen PY, Trippa L, Alexander BM. Design and evaluation of an external control arm using prior clinical trials and real‐world data. Clin Cancer Res. 2019;25:4993‐5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chau I, Le DT, Ott PA, et al. Developing real‐world comparators for clinical trials in chemotherapy‐refractory patients with gastric cancer or gastroesophageal junction cancer. Gastric Cancer. 2020;23:133‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carrigan G, Whipple S, Capra WB, et al. Using electronic health records to derive control arms for early phase single‐arm lung cancer trials: proof‐of‐concept in randomized controlled trials. Clin Pharmacol Ther. 2020;107:369‐377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schmidli H, Häring DA, Thomas M, Cassidy A, Weber S, Bretz F. Beyond randomized clinical trials: use of external controls. Clin Pharmacol Ther. 2020;107:806‐816. [DOI] [PubMed] [Google Scholar]
- 31. Kapetanakis V, Prawitz T, Schlichting M, et al. Assessment‐schedule matching in unanchored indirect treatment comparisons of progression‐free survival in cancer studies. Pharmacoeconomics. 2019;37:1537‐1551. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information.