

Abstract
Mutations in PRPH2, encoding peripherin‐2, are associated with the development of a wide variety of inherited retinal diseases (IRDs). To determine the causality of the many PRPH2 variants that have been discovered over the last decades, we surveyed all published PRPH2 variants up to July 2020, describing 720 index patients that in total carried 245 unique variants. In addition, we identified seven novel PRPH2 variants in eight additional index patients. The pathogenicity of all variants was determined using the ACMG guidelines. With this, 107 variants were classified as pathogenic, 92 as likely pathogenic, one as benign, and two as likely benign. The remaining 50 variants were classified as variants of uncertain significance. Interestingly, of the total 252 PRPH2 variants, more than half (n = 137) were missense variants. All variants were uploaded into the Leiden Open source Variation and ClinVar databases. Our study underscores the need for experimental assays for variants of unknown significance to improve pathogenicity classification, which would allow us to better understand genotype‐phenotype correlations, and in the long‐term, hopefully also support the development of therapeutic strategies for patients with PRPH2‐associated IRD.
Keywords: in silico assessment, inherited retinal disease, LOVD, molecular genetics, PRPH2
1. INTRODUCTION
PRPH2, also known as retinal degeneration slow (RDS), was first described in 1991 (Kajiwara et al., 1991). The gene encodes peripherin‐2, a 346 amino‐acid long glycoprotein that spans the membrane four times, and is located primarily in the rim regions of rod and cone outer segment (OS) discs and lamellae. Besides the four transmembrane domains, the protein contains a cytoplasmic (C) loop and two intra‐discal (D1 and D2) loops (Travis et al., 1989, 1991, 1992). Its exact molecular function inside photoreceptor cells is not yet fully understood, but it is hypothesized that the protein plays an essential role in the initiation of OS disc formation, as well as in disc stabilization, maintenance, and disc size alignment, mainly by forming oligomers with other PRPH2 molecules and/or Retinal Outer Segment Membrane Protein 1 (ROM1) (Chakraborty et al., 2020; Zulliger et al., 2018). For instance, Prph2 −/− mice failed to initiate OS disc formation, whereas Prph2 p.Cys150Ser+/− mice did not support proper OS formation, interacted abnormally with Rom1, and showed reduced Prph2 protein levels (Zulliger et al., 2018). In addition, it was shown that in Prph2 p.Cys213Tyr−/− mice, mutant Prph2 could not oligomerize with ROM1 and was mislocalized, being retained in the inner segments (Chakraborty et al., 2020). Based on these studies, PRPH2 indeed seems to be critical for proper OS formation as well as for its function.
To date, over 200 different PRPH2 variants have been described to be associated with the development of a wide variety of inherited retinal diseases (IRDs), such as retinitis pigmentosa (RP), cone‐rod dystrophy (CRD), and macular dystrophies. The group of PRPH2‐associated macular dystrophies encompasses a wide variety of phenotypes, including pseudo‐Stargardt pattern dystrophy, butterfly‐shaped pigment dystrophy (BPD), adult‐onset foveomacular vitelliform dystrophy (AOFVD), and central areolar choroidal dystrophy (CACD) (Boon et al., 2008; 2009; Boon, van Schooneveld, et al., 2007; Kersten et al., 2018). These macular dystrophy phenotypes, especially CACD, may be confused with geographic atrophy in age‐related macular degeneration (AMD), and PRPH2 mutations have been described in patients initially diagnosed with AMD (Boon et al., 2009; Kersten et al., 2018; Smailhodzic et al., 2011). PRPH2 mutations are most frequently inherited in an autosomal dominant fashion, although autosomal recessive and sporadic cases have also been reported, as well as autosomal dominant cases with reduced penetrance (Alapati et al., 2014; Birtel et al., 2018; Boon et al., 2008, 2009; Coco et al., 2010; Dryja et al., 1997; Khan et al., 2016; Manes et al., 2015). Interestingly, heterozygous mutations in both PRPH2 and ROM1 can cause digenic RP (Kajiwara et al., 1994).
In this study, we performed a systematic analysis of all 245 PRPH2 variants in IRD patients that were published to date. For this purpose, we collected all PRPH2 variants published up to July 2020 that were associated with the development of IRD. In addition, we added PRPH2 variants that were either identified via our routine diagnostics pipeline, or via a novel sequencing approach using molecular inversion probes (MIPs) (Hiatt et al., 2013; Neveling et al., 2017; Weisschuh et al., 2018), seven of which have not been described before. All variants were classified for their pathogenicity using the American College of Medical Genetics and Genomics (ACMG) guidelines, after which they were uploaded into the Leiden Open source Variation Database (LOVD) and ClinVar database for PRPH2. In addition, we attempted to establish genotype‐phenotype correlations. Several variants are reported to be associated with only one specific IRD phenotype, and these diseases can best be described as allelic disorders. However, for most variants in this study, there appears to be a high phenotypic variability between different families, as well as between members of the same family.
By performing this study, we aim to shed light on how to experimentally assess the true causality of PRPH2 variants in the future, as well as to explain the observed phenotypic variability within IRD patients. This will facilitate a better interpretation of the pathogenicity of variants that are identified in subjects with IRD, and in the long term, hopefully also support the development of therapeutic strategies for patients with PRPH2‐associated IRD.
2. MATERIAL AND METHODS
2.1. Literature search
We collected all publications up to July 2020 that report PRPH2 variants in patients with inherited retinal disease. The following PubMed search terms were used: “(retinal+degeneration+slow+OR+PRPH2+OR+peripherin)+AND+(central+areolar+choroidal+dystrophy+OR+cacd+OR+vision+disorders+OR+retinal+dystrophy)+AND+(mutation+OR+variant+OR+mutations+OR+variants)”. In addition, the HGMD professional database was used to search for variants or articles that were possibly missed with our PubMed queries. Variant detection, variant combinations, patient age, patient gender and age at onset, disease phenotype, segregation analysis, and allele frequencies (gnomAD) were collected. Obvious duplicates, in some cases following contact with the corresponding authors of the respective papers, were removed from the data set. An important point to take into consideration when retracting genetic variants from literature search tools is that it is always possible to miss certain variants. For instance, a PRPH2 variant might be present in Supporting Information data of papers that could be potentially missed by both PubMed and HGMD searches.
2.2. Subjects
This study was approved by the institutional review boards of the Radboud University Medical Center (Radboudumc) and was conducted in adherence to the tenets of the Declaration of Helsinki. All Sanger sequencing and/or whole (exome) sequencing data from the Radboudumc genome diagnostic laboratory were analyzed to determine the causative genetic defects in patients with visual impairment. PRPH2 variants were also identified by using a targeted sequencing approach based on molecular inversion probes (MIPs) (Hiatt et al., 2013; Neveling et al., 2017; Weisschuh et al., 2018). Single‐molecule MIPs were synthesized to capture and sequence overlapping 110‐nt segments of the three PRPH2 protein‐coding exons and flanking splice sites, similarly as described previously for the ABCA4 gene (Khan et al., 2020). PRPH2 variants identified with this MIPs approach were validated with Sanger sequencing.
2.3. Variant analysis
The complementary DNA (cDNA) was numbered as follows: the A of the ATG translation initiation codon of the PRPH2 reference sequence (NM_000322.5) was reported as +1 while the initiation codon was reported as codon 1. Allele frequencies of PRPH2 variants in control populations were extracted from the genome aggregation database (gnomAD: v2.1.1 and v3). This database contains both whole exome and whole genome sequencing data obtained from 213,158 healthy individuals from all over the world (https://gnomad.broadinstitute.org/).
Next, we performed statistical analysis to compare allele frequencies in the index patient group to the normal population (gnomAD). This enabled us to assess whether a specific variant is enriched in patient versus control groups. For this purpose, the Fisher's Exact test was used. To only select truly statistically significant findings, a correction by the false discovery rate (FDR) of Benjamini‐Hochberg, classical one stage method with an error margin of 5%, was carried out (Benjamini et al., 2001).
2.4. In silico predictions
For PRPH2 missense variants, Polyphen‐2 (benign = 0.000−0.150; possibly damaging = 0.150−0.850; probably damaging = 0.850−1.000), Combined Annotation Dependent Depletion (CADD, tolerated = <15; damaging = >15), and SIFT scores (tolerated = 0.050 − 1.000; damaging = 0.000–0.050) were obtained from http://genetics.bwh.harvard.edu/pph2/, http://cadd.gs.washington.edu/home (Kircher et al., 2014) and https://sift.bii.a-star.edu.sg/, respectively. For splice site variants, software to gather splicing scores available via Alamut Visual version 2.13 (Interactive Biosoftware) was used.
2.5. Variant pathogenicity classification
Pathogenicity of all reported and newly identified PRPH2 variants was predicted using the ACMG guidelines (Richards et al., 2015). These guidelines enabled us to classify each variant into one of the following categories: pathogenic, likely pathogenic, benign, likely benign, or of uncertain significance. To do so, we scored the variants based on the evidence of pathogenicity in different ACMG categories:
-
1.
Pathogenic, very strong (PVS), for example, this variant is protein‐truncating.
-
2.
Pathogenic, strong (PS), for example, this variant leads to the same amino acid being substituted compared to a previously described pathogenic variant.
-
3.
Pathogenic, moderate (PM), for example, this variant is located in a mutational hot spot and/or well‐established functional domain.
-
4.
Pathogenic, supporting (PP), for example, multiple lines of computational evidence support the variant to be pathogenic.
The scores of each category are combined to come to the final classification in one of the five pathogenicity categories that are explained in more detail in Table S1.
2.6. LOVD and ClinVar submission
The 245 reported, as well as the seven novel PRPH2 variants were uploaded to the LOVD and ClinVar database, when available, together with patient data including the description of the phenotype, patient age, patient age of onset, and segregation information. Pathogenicity scores of all variants were based on the pathogenicity assessment as described in the “In silico predictions” section.
3. PRPH2 VARIANTS
3.1. PRPH2 mutation spectrum
We collected 245 PRPH2 variants identified in 720 index patients that were described in 165 articles up to July 2020, as well as their phenotypic information (Abouelhoda et al., 2016; Abu‐Safieh et al., 2013; Ahmad et al., 2010; Alapati et al., 2014; Anand et al., 2009; Anasagasti et al., 2013; Apfelstedt‐Sylla et al., 1995; Arai et al., 2015; Avela et al., 2019, 2018; Ba‐Abbad et al., 2019, 2014; Barbazetto et al., 2007; Bareil et al., 2000, 1997; Bayés et al., 1996; Birtel et al., 2018; Boon et al., 2009; Boon, Klevering, et al., 2007; Boon, van Schooneveld, et al., 2007; Boulanger‐Scemama et al., 2015; Budu et al., 2001; Carss et al., 2017; Cheng et al., 2019; Coco‐Martin et al., 2020; Coco et al., 2010; Consugar et al., 2015; Coussa et al., 2015; Daftarian et al., 2019; de Breuk et al., 2020; Deciphering Developmental Disorders Study, 2017; de Sousa Dias et al., 2015; Donoso et al., 2003; Downes et al., 1999; Downs et al., 2007; Dryja et al., 1997; Duncan et al., 2011; Duncker et al., 2015; Ekström, Andréasson, et al., 1998; Ekström, Ponjavic, et al., 1998; Ekström, Ponjavic, Andréasson, et al., 1998; Essilfie et al., 2018; Fakin et al., 2012; Farrar et al., 1992, 1991; Feist et al., 1994; Felbor et al., 1997; Fernandez‐San Jose et al., 2015; Fishman et al., 1994, 1997; Foote et al., 2019; Francis et al., 2005; Gamundi et al., 2007; Gao et al., 2019; Glöckle et al., 2014; Gocho et al., 2016; Gorin et al., 1995; Grover et al., 2002; Grüning et al., 1994; Hanany & Sharon, 2019; Hosono et al., 2018; Hoyng et al., 1996; Huang et al., 2013; Jacobson et al., 1996, 2016; Jespersgaard et al., 2019; Jin et al., 2008; Jones et al., 2017; Kajiwara et al., 1994, 1991, 1993; Kalyanasundaram et al., 2009; Katagiri et al., 2018; Keen et al., 1994; Keilhauer, Meigen, Stöhr, et al., 2006; Keilhauer, Meigen, & Weber, 2006; Kemp et al., 1994; Kersten et al., 2018; Khan, 2019; Khan et al., 2016; Khoubian et al., 2005; Kikawa et al., 1994; C. Kim et al., 2012; R. Y. Kim et al., 1995; Kitiratschky et al., 2011; Klevering et al., 2002; Kohl et al., 1997, 2012; Lam et al., 1995; C. S. Lee & Leys, 2020; K. Lee et al., 2015; Leroy et al., 2007; Lim et al., 2009; Maertz et al., 2015; Manes et al., 2015; Martin‐Merida et al., 2018; Meins et al., 1993; Meunier et al., 2011; Michaelides et al., 2005; Miyata et al., 2018; Moshfeghi et al., 2006; Nakazawa et al., 1994, 1995, 1996; Nanda et al., 2019; Neveling et al., 2012; Nichols, Drack, et al., 1993; Nichols, Sheffield, et al., 1993; Oishi et al., 2014; Pajic et al., 2006; Palma et al., 2019; Passerini et al., 2007; Patel et al., 2016; Payne et al., 1998; Poloschek et al., 2010; Ramkumar et al., 2017; Reeves et al., 2020; Reig et al., 1995; Renner et al., 2004, 2009; Richards & Creel, 1995; Saga et al., 1993; Sallevelt et al., 2017; Schatz et al., 2003; Schorderet et al., 2014; Sears et al., 2001; Shankar et al., 2015, 2016; Simonelli et al., 2007; Smailhodzic et al., 2011; Sohocki et al., 2001; Souied et al., 1998; Stone et al., 2017; Strafella et al., 2019; Strom et al., 2012; Sullivan et al., 2006, 2013; Sun et al., 2015; Testa et al., 2005; Trujillo et al., 1998, 2001; Trujillo Tiebas et al., 2002; Vaclavik et al., 2012; Van Cauwenbergh et al., 2017; van Lith‐Verhoeven et al., 2003; X. Wang et al., 2013; J. Wang et al., 2014; H. Wang et al., 2015; Wawrocka et al., 2018; Weleber et al., 1993; Wells et al., 1993; Wolock et al., 2019; Wroblewski et al., 1994a; 1994b; Xiang et al., 2012; Xu et al., 2014; Xue et al., 2014; Yanagihashi et al., 2003; H. Yang et al., 2000; Z. Yang et al., 2003; Z. Yang et al., 2004; Yeoh et al., 2010; Yoon et al., 2015; Zaneveld et al., 2015; Zhang et al., 2002; Zhao et al., 2015; Zhou et al., 2018; Zhuk & Edwards, 2006). Additionally, 139 index cases from either the Radboudumc genome diagnostic laboratory or that were studied via MIPs analysis, carried PRPH2 variants that were published before (Table 1). Finally, we also identified seven novel variants in eight additional index patients that, to our knowledge, were never identified (Table 1; variants depicted in bold lettering). Of the collective 252 PRPH2 variants, 137 were missense, 85 were protein‐truncating, 10 were splice sites, 15 were in‐frame amino acid changes, three were synonymous, and two were located in the 5′‐ or 3′‐untranslated regions (UTRs) (Figure S1A; Table 2). Of the total 720 previously reported index patients, 686 patients carried heterozygous variants, 8 patients carried compound heterozygous variants, 14 patients carried homozygous variants, and 11 patients had variants in two different genes. There was one patient that carried variants in three different genes (PRPH2, ROM1, ABCA4). The authors speculated trigenic inheritance; however, it still needs to be elucidated to determine whether this case is truly trigenic. The first homozygous PRPH2 variants were identified by Wang and colleagues. A cohort of 179 index cases was genetically screened leading to the identification of two unrelated patients carrying the homozygous p.Leu185Pro and p.Cys213Trp PRPH2 variants, respectively (X. Wang et al., 2013). These patients were both suffering from Leber congenital amaurosis (LCA), suggesting that homozygous PRPH2 variants are associated with more severe phenotypes compared to heterozygous PRPH2 mutation carriers. Of the 139 index cases identified by the Radboudumc genome diagnostic laboratory or our MIPs analysis, two patients also carried homozygous variants, while the remaining 137 index cases were all heterozygous carriers. The two index cases that were found to be homozygous both carried the PRPH2‐p.Arg142Trp variant, and were diagnosed with CACD. To our knowledge, this is the first time this particular variant was present in a homozygous state. For one of the patients, we were able to genetically screen both parents. The father was also affected by CACD. Interestingly, the mother did not show apparent signs of retinal disease upon ophthalmological examination. Both parents were proven to carry the variant in a heterozygous state. When studying the phenotype of both patients in more detail, we could observe that it resembles the phenotype we observe in heterozygous p.Arg142Trp carriers, pointing toward a semi‐dominant inheritance with a dominant‐negative effect on the wildtype allele. Finally, the eight index patients carrying the seven novel PRPH2 variants, were all heterozygous for the respective variants. All variants, together with a description of the phenotype and, when available, segregation analysis, were uploaded to the LOVD and ClinVar database (https://databases.lovd.nl/shared/genes/PRPH2).
Table 1.
Previously unreported PRPH2 variants identified in index cases by MIPs or the Radboudumc genome diagnostic laboratory
| PID | cDNA change | Protein change | Type of mutation | Identified by | Phenotype |
|---|---|---|---|---|---|
| 1 | c.2T>C | p.? | Fail‐to‐start | MIPs | Pseudo‐STGD |
| 2 | c.63G>A | p.Trp21* | Nonsense | Exome seq. | MD/PD |
| 3 | c.63G>A | p.Trp21* | Nonsense | Sanger seq. | MD/PD |
| 4 | c.63G>A | p.Trp21* | Nonsense | Sanger seq. | MD/PD |
| 5 | c.94A>G | p.Ile32Val | Missense | MIPs | AMD |
| 6 | c.112G>T | p.Gly38* | Nonsense | MIPs | Pseudo‐STGD |
| 7 | c.122T>C | p.Leu41Pro | Missense | Sanger seq. | MD/PD |
| 8 | c.133C>T | p.Leu45Phe | Missense | MIPs | AMD |
| 9 | c.136C>T | p.Arg46* | Nonsense | MIPs | Pseudo‐STGD |
| 10 | c.209dup | p.Ser71Ilefs*106 | Frameshift | MIPs | Pseudo‐STGD |
| 11 | c.253G>A | p.Ala85Thr | Missense | Sanger seq. | MD/PD |
| 12 | c.271T>A | p.Tyr91Asn | Missense | MIPs | Pseudo‐STGD |
| 13 | c.281G>A | p.Trp94* | Nonsense | MIPs | Pseudo‐STGD |
| 14 | c.303C>G | p.Tyr101* | Nonsense | Exome seq. | MD/PD |
| 15 | c.367C>T | p.Arg123Trp | Missense | Exome seq. | RP |
| 16 | c.377T>C | p.Leu126Pro | Missense | Exome seq. | RP |
| 17 | c.415_417del | p.Lys139del | Deletion | MIPs | Pseudo‐STGD |
| 18 | c.415_417del | p.Lys139del | Deletion | MIPs | Pseudo‐STGD |
| 19 | c.423C>A | p.Tyr141* | Nonsense | MIPs | Pseudo‐STGD |
| 20 | c.424C>T | p.Arg142Trp | Missense | MIPs | Pseudo‐STGD |
| 21 | c.424C>T | p.Arg142Trp | Missense | Exome seq. | MD |
| 22 | c.424C>T | p.Arg142Trp | Missense | Exome seq. | RD |
| 23 | c.424C>T | p.Arg142Trp | Missense | Exome seq. | RCD |
| 24 | c.424C>T | p.Arg142Trp | Missense | Exome seq. | MD |
| 25 | c.424C>T | p.Arg142Trp | Missense | Exome seq. | MD |
| 26 | c.424C>T | p.Arg142Trp | Missense | Exome seq. | MD |
| 27 | c.424C>T | p.Arg142Trp | Missense | Exome seq. | MD |
| 28 | c.424C>T | p.Arg142Trp | Missense | Exome seq. | MD |
| 29 | c.424C>T | p.Arg142Trp | Missense | Exome seq. | MD |
| 30 | c.424C>T | p.Arg142Trp | Missense | Exome seq. | CACD |
| 31 | c.424C>T | p.Arg142Trp | Missense | Exome seq. | MD/CD |
| 32 | c.424C>T | p.Arg142Trp | Missense | Exome seq. | MD |
| 33 | c.424C>T | p.Arg142Trp | Missense | Exome seq. | MD/PD |
| 34 | c.424C>T | p.Arg142Trp | Missense | Exome seq. | MD |
| 35 | c.424C>T | p.Arg142Trp | Missense | Exome seq. | MD |
| 36 | c.424C>T | p.Arg142Trp | Missense | Exome seq. | CACD |
| 37 | c.424C>T | p.Arg142Trp | Missense | Exome seq. | MD |
| 38 | c.424C>T | p.Arg142Trp | Missense | Exome seq. | RD |
| 39 | c.424C>T | p.Arg142Trp | Missense | Sanger seq. | CACD |
| 40 | c.424C>T | p.Arg142Trp | Missense | Sanger seq. | CACD |
| 41 | c.424C>T | p.Arg142Trp | Missense | Sanger seq. | MD/PD |
| 42 | c.424C>T | p.Arg142Trp | Missense | Sanger seq. | CACD |
| 43 | c.424C>T | p.Arg142Trp | Missense | Sanger seq. | CACD |
| 44 | c.424C>T | p.Arg142Trp | Missense | Sanger seq. | CACD |
| 45 | c.424C>T | p.Arg142Trp | Missense | Sanger seq. | CACD |
| 46 | c.424C>T | p.Arg142Trp | Missense | Sanger seq. | CACD |
| 47 | c.424C>T | p.Arg142Trp | Missense | Sanger seq. | MD/PD |
| 48 | c.424C>T | p.Arg142Trp | Missense | Sanger seq. | MD/PD |
| 49 | c.424C>T | p.Arg142Trp | Missense | Sanger seq. | CACD |
| 50 | c.424C>T | p.Arg142Trp | Missense | Sanger seq. | CACD |
| 51 | c.424C>T | p.Arg142Trp | Missense | Sanger seq. | CACD |
| 52 | c.424C>T | p.Arg142Trp | Missense | Sanger seq. | MD/PD |
| 53 | c.424C>T | p.Arg142Trp | Missense | Sanger seq. | CACD |
| 54 | c.424C>T | p.Arg142Trp | Missense | Sanger seq. | CACD |
| 55 | c.433_434del | p.Asp145Hisfs*31 | Frameshift | Sanger seq. | CACD |
| 56 | c.441del | p.Gly148Alafs*5 | Frameshift | MIPs | Pseudo‐STGD |
| 57 | c.441del | p.Gly148Alafs*5 | Frameshift | MIPs | Pseudo‐STGD |
| 58 | c.441del | p.Gly148Alafs*5 | Frameshift | Sanger seq. | MD/PD |
| 59 | c.441del | p.Gly148Alafs*5 | Frameshift | Sanger seq. | MD/PD |
| 60 | c.441del | p.Gly148Alafs*5 | Frameshift | Exome seq. | MD |
| 61 | c.458A>G | p.Lys153Arg | Missense | Asper | RP |
| 62 | c.469G>A | p.Asp157Asn | Missense | MIPs | Pseudo‐STGD |
| 63 | c.499G>A | p.Gly167Ser | Missense | Sanger seq. | MD/PD |
| 64 | c.499G>A | p.Gly167Ser | Missense | MIPs | Pseudo‐STGD |
| 65 | c.505_507del | p.Asn169del | Deletion | Sanger seq. | MD/PD |
| 66 | c.505_507del | p.Asn169del | Deletion | Exome seq. | RP |
| 67 | c.514C>T | p.Arg172Trp | Missense | MIPs | Pseudo‐STGD |
| 68 | c.514C>T | p.Arg172Trp | Missense | MIPs | Pseudo‐STGD |
| 69 | c.514C>T | p.Arg172Trp | Missense | MIPs | Pseudo‐STGD |
| 70 | c.514C>T | p.Arg172Trp | Missense | Asper | RD |
| 71 | c.514C>T | p.Arg172Trp | Missense | Sanger seq. | MD/PD |
| 72 | c.514C>T | p.Arg172Trp | Missense | Sanger seq. | MD/PD |
| 73 | c.514C>T | p.Arg172Trp | Missense | Exome seq. | CD |
| 74 | c.514C>T | p.Arg172Trp | Missense | Exome seq. | MD |
| 75 | c.514C>T | p.Arg172Trp | Missense | Exome seq. | MD |
| 76 | c.515G>A | p.Arg172Gln | Missense | MIPs | Pseudo‐STGD |
| 77 | c.515G>A | p.Arg172Gln | Missense | Exome seq. | MD |
| 78 | c.515G>A | p.Arg172Gln | Missense | MIPs | Pseudo‐STGD |
| 79 | c.520T>A | p.Trp174Arg | Missense | Sanger seq. | MD/PD |
| 80 | c.520T>A | p.Trp174Arg | Missense | Exome seq. | MD |
| 81 | c.522G>C | p.Trp174Cys | Missense | MIPs | Pseudo‐STGD |
| 82 | c.581+1G>A | p.? | Splice site | MIPs | Pseudo‐STGD |
| 83 | c.581+4dup | p.? | Splice site | Sanger seq. | ? |
| 84 | c.582‐1G>A | p.? | Splice site | MIPs | Pseudo‐STGD |
| 85 | c.582‐1G>A | p.? | Splice site | Sanger seq. | CACD |
| 86 | c.582‐2A>T | p.? | Splice site | Sanger seq. | CACD |
| 87 | c.582_828del | p.Asp194Glufs*2 | Frameshift | MIPs | Pseudo‐STGD |
| 88 | c.583C>T | p.Arg195* | Nonsense | MIPs | Pseudo‐STGD |
| 89 | c.583C>T | p.Arg195* | Nonsense | MIPs | Pseudo‐STGD |
| 90 | c.584G>T | p.Arg195Leu | Missense | MIPs | Pseudo‐STGD |
| 91 | c.584G>A | p.Arg195Gln | Missense | Sanger seq. | CACD |
| 92 | c.584G>A | p.Arg195Gln | Missense | MIPs | Pseudo‐STGD |
| 93 | c.614T>C | p.Leu205Pro | Missense | Sanger seq. | CACD |
| 94 | c.623G>A | p.Gly208Asp | Missense | MIPs | Pseudo‐STGD |
| 95 | c.623G>A | p.Gly208Asp | Missense | Sanger seq. | MD/PD |
| 96 | c.623G>A | p.Gly208Asp | Missense | Sanger seq. | MD/PD |
| 97 | c.623G>A | p.Gly208Asp | Missense | MIPs | AMD |
| 98 | c.628C>T | p.Pro210Ser | Missense | MIPs | Pseudo‐STGD |
| 99 | c.633C>A | p.Phe211Leu | Missense | Asper | RP |
| 100 | c.646C>T | p.Pro216Ser | Missense | Asper | RP |
| 101 | c.646C>T | p.Pro216Ser | Missense | Exome seq. | RCD |
| 102 | c.646C>T | p.Pro216Ser | Missense | Exome seq. | RP |
| 103 | c.646C>T | p.Pro216Ser | Missense | Exome seq. | RP |
| 104 | c.646C>T | p.Pro216Ser | Missense | Sanger seq. | RP |
| 105 | c.646C>T | p.Pro216Ser | Missense | Exome seq. | RP |
| 106 | c.656C>G | p.Pro219Arg | Missense | Sanger seq. | CACD |
| 107 | c.657_662del | p.Arg220_Pro221del | Deletion | Sanger seq. | MD/PD |
| 108 | c.658C>T | p.Arg220Trp | Missense | Sanger seq. | MD/PD |
| 109 | c.658del | p.Arg220fs*34 | Frameshift | Exome seq. | RCD/CRD |
| 110 | c.658del | p.Arg220fs*34 | Frameshift | Sanger seq. | MD/PD |
| 111 | c.659G>A | p.Arg220Gln | Missense | Sanger seq. | MD/PD |
| 112 | c.665G>A | p.Cys222Try | Missense | MIPs | CRD |
| 113 | c.715C>T | p.Gln239* | Missense | MIPs | AMD |
| 114 | c.736T>C | p.Trp246Arg | Missense | MIPs | AMD |
| 115 | c.746del | p.Gly249Alafs*7 | Frameshift | Exome seq. | MD/PD |
| 116 | c.749G>A | p.Cys250Tyr | Missense | MIPs | Pseudo‐STGD |
| 117 | c.754G>C | p.Ala252Pro | Missense | MIPs | AMD |
| 118 | c.781C>T | p.Leu261Phe | Missense | MIPs | AMD |
| 119 | c.809_810del | p.Leu270Profs*30 | Frameshift | MIPs | Pseudo‐STGD |
| 120 | c.850C>T | p.Arg284Cys | Missense | MIPs | AMD |
| 121 | c.866C>T | p.Ser289Leu | Missense | MIPs | AMD |
| 122 | c.866C>T | p.Ser289Leu | Missense | MIPs | AMD |
| 123 | c.866C>T | p.Ser289Leu | Missense | MIPs | AMD |
| 124 | c.866C>T | p.Ser289Leu | Missense | MIPs | AMD |
| 125 | c.866C>T | p.Ser289Leu | Missense | MIPs | AMD |
| 126 | c.866C>T | p.Ser289Leu | Missense | MIPs | AMD |
| 127 | c.866C>T | p.Ser289Leu | Missense | MIPs | AMD |
| 128 | c.866C>T | p.Ser289Leu | Missense | MIPs | AMD |
| 129 | c.866C>T | p.Ser289Leu | Missense | MIPs | AMD |
| 130 | c.866C>T | p.Ser289Leu | Missense | MIPs | AMD |
| 131 | c.866C>T | p.Ser289Leu | Missense | MIPs | AMD |
| 132 | c.866C>T | p.Ser289Leu | Missense | MIPs | AMD |
| 133 | c.866C>T | p.Ser289Leu | Missense | MIPs | AMD |
| 134 | c.866C>T | p.Ser289Leu | Missense | MIPs | AMD |
| 135 | c.866C>T | p.Ser289Leu | Missense | MIPs | AMD |
| 136 | c.866C>T | p.Ser289Leu | Missense | MIPs | AMD |
| 137 | c.866C>T | p.Ser289Leu | Missense | MIPs | AMD |
| 138 | c.866C>T | p.Ser289Leu | Missense | Sanger seq. | MD/PD |
| 139 | c.923T>A | p.Leu308Gln | Missense | MIPs | AMD |
| 140 | c.923T>A | p.Leu308Gln | Missense | MIPs | AMD |
| 141 | c.938C>T | p.Pro313Leu | Missense | MIPs | AMD |
| 142 | c.938C>T | p.Pro313Leu | Missense | MIPs | AMD |
| 143 | c.938C>T | p.Pro313Leu | Missense | MIPs | AMD |
| 144 | c.828+1G>A | p.? | Splice site | Sanger seq. | MD/PD |
| 145 | c.829‐3_829‐1del | p.? | Splice site | MIPs | Pseudo‐STGD |
| 146 | c.897_898del | p.Ser301Argfs*90 | Frameshift | Exome seq. | MD |
| 147 | c.946T>G | p.Trp316Gly | Missense | Exome seq. | MD/PD |
Note: Novel variants identified via MIPs are depicted in bold.
Abbreviations phenotypes: AMD, age‐related macular degeneration; CACD, central areolar choroidal dystrophy; CD, cone dystrophy; CRD, cone‐rod dystrophy; MD, macular dystrophy; PD, pattern dystrophy; pseudo‐STGD, pseudo‐Stargardt disease; RD, retinal dystrophy; RP, retinitis pigmentosa; RCD, rod‐cone dystrophy.
Table 2.
Distribution of PRPH2 variants found in IRD patients
| Variant type | Number of unique variants | Number of alleles |
|---|---|---|
| Missense | 137 | 605 |
| Protein‐truncating | 85 | 180 |
| Splice site | 10 | 77 |
| In‐frame amino acid insertions/deletions | 15 | 24 |
| Synonymous | 3 | 3 |
| 5′‐ or 3′‐UTR | 2 | 2 |
| Total | 252 | 891 |
Abbreviation: UTR, untranslated region.
3.2. Recurrent PRPH2 variants
The most recurrent PRPH2 variant among IRD patients is p.Arg142Trp. This variant was reported in 95 out of 867 index patients, of which 93 were heterozygous carriers and two were homozygous (Table S2). The variant was exclusively reported in individuals with Caucasian ancestry, and in the Netherlands, this variant is mainly associated with the development of CACD. Analysis of single nucleotide polymorphisms (SNPs) in close proximity of the p.Arg142Trp variant in three Dutch families revealed the presence of a shared chromosomal segment of approximately 519 kb, strongly suggesting that this particular variant represents a founder mutation in the Netherlands (Boon et al., 2009). As mentioned in the introduction section, CACD may be confused with AMD, and besides retinal imaging modalities (optical coherence tomography and fundus autofluorescence), screening for the p.Arg142Trp may help to better discriminate between these two phenotypes (Kersten et al., 2018; Smailhodzic et al., 2011). One interesting observation in large Dutch CACD families harboring this variant is that there seems to be reduced penetrance (Boon et al., 2009). Within these families, some individuals were significantly less severely affected compared to age‐matched family members also carrying the variant. The molecular mechanisms behind this phenomenon still need to be elucidated. Possible explanations could be: (1) reduced expression of the mutant allele; (2) increased expression of the wild‐type allele; (3) the influence of modifier alleles located in other genes; or (4) influence of environmental factors (e.g., smoking, nutrition).
Other recurrently reported variants (reported in >10 index patients) are p.Arg46*, p.Tyr141Cys, p.Gly148Alafs*5, p.Arg172Trp, p.Arg172Gln, p.Leu185Pro, p.Arg195Leu, p.Gly208Asp, p.Pro210Arg, p.Pro216Ser, p.Pro216Leu, p.Gln239*, p.Ser289Leu, and c.828+3A>T. The p.Arg172Trp variant was reported in 60 index patients and was mainly found in Caucasians with British ancestry. There was only one Japanese index patient carrying this variant. Of the total 60 cases, 58 were heterozygous for this variant (Table S2). Payne and colleagues performed haplotype analysis in multiple British families carrying the stand‐alone p.Arg172Trp variant and revealed that in Britain, this is a founder mutation (Payne et al., 1998). A German index patient carried an additional pathogenic heterozygous ROM1 missense variant (p.Arg229His), and another patient carried two additional heterozygous missense variants, one in ROM1 (p.Arg229His) and one in ABCA4 (p.Val2050Leu (Table S2) (Poloschek et al., 2010). The authors argued that the ROM1 and ABCA4 variants act as a moderator, worsening the pattern dystrophy phenotype compared to individuals that only carry the p.Arg172Trp variant in PRPH2. However, when applying the ACMG criteria to both the ROM1 and ABCA4 variants, the p.Val2050Leu variant in ABCA4 was classified as likely benign, which makes it unlikely to be a modifier that worsens the phenotype.
The splice site variant c.828+3A>T was recurrently found in 57 index patients. This variant is predicted to result in aberrant splicing of the PRPH2 messenger RNA, and consequently in the formation of a nonfunctional truncated protein (Shankar et al., 2015, 2016). The remaining 237 variants were reported in only a single or a few index patients, which clearly demonstrates the enormous allelic heterogeneity among patients with PRPH2‐associated IRD.
4. PATHOGENICITY ASSESSMENT OF ALL PRPH2 VARIANTS
The pathogenicity of all PRPH2 variants was assessed using the ACMG classification system, as described in the Materials and Methods section. According to our analysis, 107 variants were classified as pathogenic, 92 as likely pathogenic, one as benign, and two as likely benign (Figure S1B and Table S2). The remaining 50 variants were classified as variants of uncertain significance (Tables S2 and S3). Of the collective 199 (likely) pathogenic variants, 93 were missense, 85 were protein‐truncating, 8 were splice‐site, and 13 were in‐frame amino acid insertions/deletions (Figure 1A and Table S2).
Figure 1.
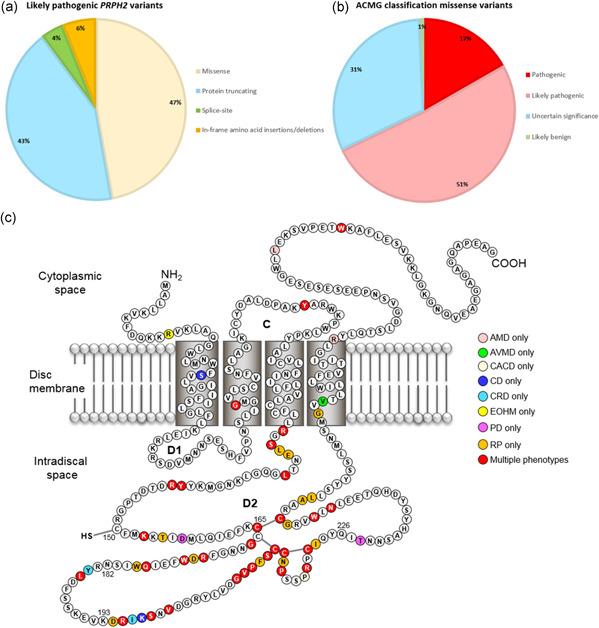
(a) Pie‐chart showing the distribution of (likely) pathogenic PRPH2 variants in IRD patients. (b) Pie‐chart showing the ACMG pathogenicity assessment of missense variants. About two‐thirds of the missense variants were classified as pathogenic or likely pathogenic. (c) Location likely pathogenic missense variants relative to the protein structure. The vast majority of the likely pathogenic missense variants are located in the D2 loop. AMD, age‐related macular degeneration; AVMD, adult vitelliform macular dystrophy; CD, cone dystrophy; CRD, cone‐rod dystrophy; EOHM, early‐onset high myopia; IRD, inherited retinal disease; PD, pattern dystrophy; RP, retinitis pigmentosa
4.1. Missense variants
The vast majority of PRPH2 variants reported in IRD patients represent missense variants. In total, 137 missense variants have been reported, corresponding to 605 alleles (Figure S1B and Table 2). Before assessing the pathogenicity of these variants, in silico predictions were performed using PolyPhen2 (benign = 0.000−0.150; possibly damaging = 0.150–0.850; probably damaging = 0.850–1.000), SIFT (tolerated = 0.050–1.000; damaging = 0.000–0.050), and CADD (tolerated = <15; damaging = >15). To consider a variant as pathogenic (supporting evidence; PP3), two out of three in silico prediction programs should predict a damaging effect. When combining all information for the final ACMG pathogenicity assessment, of the 137 missense variants, 93 were classified as pathogenic/likely pathogenic (Figure 1B; Tables 3 and S2). One variant was classified as likely benign. The remaining 43 were considered as variants of uncertain significance (Tables S2 and S3), mainly due to lack of family history and segregation analysis, or because the variant did not co‐segregate with the disease.
Table 3.
Likely pathogenic and pathogenic missense variants
| cDNA change | Protein change | Heterozygous | Compound heterozygous | Homozygous | Digenic | Trigenic | Protein domain |
|---|---|---|---|---|---|---|---|
| c.38G>A | p.Arg13Gln | 2 | 0 | 0 | 0 | 0 | N‐terminus |
| c.80C>T | p.Ser27Phe | 1 | 0 | 0 | 0 | 0 | 1st TMD |
| c.202G>C | p.Gly68Arg | 2 | 0 | 0 | 0 | 0 | 2nd TMD |
| c.271T>A | p.Tyr91Asn | 1 | 2 | 0 | 0 | 0 | C‐loop |
| c.271T>C | p.Tyr91His | 1 | 0 | 0 | 0 | 0 | C‐loop |
| c.367C>T | p.Arg123Trp | 2 | 0 | 0 | 0 | 0 | D2‐loop |
| c.374C>T | p.Ser125Leu | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.377T>C | p.Leu126Pro | 3 | 0 | 0 | 0 | 0 | D2‐loop |
| c.377T>G | p.Leu126Arg | 2 | 0 | 0 | 0 | 0 | D2‐loop |
| c.380A>G | p.Glu127Gly | 2 | 0 | 0 | 0 | 0 | D2‐loop |
| c.389T>C | p.Leu130Pro | 6 | 0 | 0 | 0 | 0 | D2‐loop |
| c.421T>C | p.Tyr141His | 2 | 0 | 0 | 0 | 0 | D2‐loop |
| c.422A>G | p.Tyr141Cys | 24 | 0 | 0 | 0 | 0 | D2‐loop |
| c.424C>T | p.Arg142Trp | 93 | 0 | 2 | 0 | 0 | D2‐loop |
| c.425G>A | p.Arg142Gln | 2 | 0 | 0 | 0 | 0 | D2‐loop |
| c.457A>G | p.Lys153Glu | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.458A>G | p.Lys153Arg | 3 | 0 | 0 | 0 | 0 | D2‐loop |
| c.464C>T | p.Thr155Ile | 0 | 0 | 0 | 1 | 0 | D2‐loop |
| c.469G>A | p.Asp157An | 5 | 0 | 0 | 0 | 0 | D2‐loop |
| c.494G>T | p.Cys165Pe | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.494G>A | p.Cys165Tyr | 4 | 0 | 0 | 0 | 0 | D2‐loop |
| c.493T>C | p.Cys165Ag | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.499G>A | p.Gly167Ser | 10 | 0 | 0 | 0 | 0 | D2‐loop |
| c.500G>A | p.Gly167Asp | 3 | 0 | 0 | 0 | 0 | D2‐loop |
| c.515G>A | p.Arg172Gln | 13 | 0 | 0 | 0 | 0 | D2‐loop |
| c.514C>G | p.Arg172Gly | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.514C>T | p.Arg172Trp | 58 | 0 | 0 | 1 | 1 | D2‐loop |
| c.518A>T | p.Asp173Val | 4 | 0 | 0 | 0 | 0 | D2‐loop |
| c.520T>A | p.Trp174Arg | 2 | 0 | 0 | 0 | 0 | D2‐loop |
| c.533A>G | p.Gln178Arg | 2 | 0 | 0 | 0 | 0 | D2‐loop |
| c.536G>T | p.Trp179Leu | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.535T>C | p.Trp179Arg | 4 | 0 | 0 | 0 | 0 | D2‐loop |
| c.537G>T | p.Trp179Cys | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.551A>C | p.Tyr184Ser | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.554T>C | p.Leu185Pro | 6 | 0 | 2 | 4 | 0 | D2‐loop |
| c.582T>A | p.Asp194Gu | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.583C>G | p.Arg195Gly | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.584G>A | p.Arg195Gln | 6 | 0 | 0 | 0 | 0 | D2‐loop |
| c.584G>T | p.Arg195Leu | 12 | 0 | 0 | 0 | 0 | D2‐loop |
| c.587T>A | p.Ile196Asn | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.589A>G | p.Lys197Glu | 2 | 0 | 0 | 0 | 0 | D2‐loop |
| c.592A>C | p.Ser198Arg | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.594C>G | p.Ser198Arg | 3 | 0 | 0 | 0 | 0 | D2‐loop |
| c.599T>A | p.Val200Glu | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.599T>G | p.Val200Gly | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.623G>A | p.Gly208Asp | 12 | 0 | 1 | 0 | 0 | D2‐loop |
| c.625G>A | p.Val209Ile | 2 | 0 | 0 | 0 | 0 | D2‐loop |
| c.625G>T | p.Val209Phe | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.626T>A | p.Val209Asp | 2 | 0 | 0 | 0 | 0 | D2‐loop |
| c.628C>T | p.Pro210Ser | 2 | 0 | 0 | 0 | 0 | D2‐loop |
| c.629C>G | p.Pro210Arg | 20 | 0 | 1 | 0 | 0 | D2‐loop |
| c.629C>T | p.Pro210Leu | 2 | 0 | 0 | 0 | 0 | D2‐loop |
| c.631T>C | p.Phe211Leu | 6 | 0 | 0 | 0 | 0 | D2‐loop |
| c.634A>G | p.Ser212Gly | 7 | 0 | 0 | 0 | 0 | D2‐loop |
| c.635G>C | p.Ser212Thr | 4 | 0 | 0 | 0 | 0 | D2‐loop |
| c.636 T>A | p.Cys213Ser | 2 | 0 | 0 | 0 | 0 | D2‐loop |
| c.637T>C | p.Cys213Arg | 1 | 0 | 1 | 0 | 0 | D2‐loop |
| c.638G>T | p.Cys213Phe | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.638G>A | p.Cys213Tyr | 6 | 0 | 0 | 0 | 0 | D2‐loop |
| c.639C>G | p.Cys213Trp | 3 | 0 | 0 | 0 | 0 | D2‐loop |
| c.641G>A | p.Cys214Tyr | 2 | 0 | 0 | 0 | 0 | D2‐loop |
| c.641G>C | p.Cys214Ser | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.643A>T | p.Asn215Tyr | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.646C>T | p.Pro216Ser | 21 | 0 | 0 | 0 | 0 | D2‐loop |
| c.646C>G | p.Pro216Ala | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.647C>G | p.Pro216Arg | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.656C>G | p.Pro219Arg | 2 | 0 | 0 | 0 | 0 | D2‐loop |
| c.658C>T | p.Arg220Trp | 8 | 0 | 0 | 0 | 0 | D2‐loop |
| c.659G>A | p.Arg220Gln | 3 | 0 | 1 | 0 | 0 | D2‐loop |
| c.659G>C | p.Arg220Pro | 2 | 0 | 0 | 0 | 0 | D2‐loop |
| c.664T>C | p.Cys222Arg | 2 | 0 | 0 | 0 | 0 | D2‐loop |
| c.665G>C | p.Cys222Ser | 2 | 0 | 0 | 0 | 0 | D2‐loop |
| c.665G>A | p.Cys222Tyr | 2 | 0 | 0 | 0 | 0 | D2‐loop |
| c.668T>A | p.Ile223Asn | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.683C>T | p.Thr228Ile | 5 | 0 | 0 | 0 | 0 | D2‐loop |
| c.730A>C | p.Asn244His | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.732C>A | p.Asn244Lys | 2 | 0 | 0 | 0 | 0 | D2‐loop |
| c.732C>G | p.Asn244Lys | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.736T>C | p.Trp246Arg | 2 | 0 | 0 | 0 | 0 | D2‐loop |
| c.738G>C | p.Trp246Cys | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.745G>A | p.Gly249Ser | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.748T>C | p.Cys250Arg | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.748T>G | p.Cys250Gly | 3 | 1 | 0 | 1 | 0 | D2‐loop |
| c.748T>A | p.Cys250Ser | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.749G>A | p.Cys250Tyr | 2 | 0 | 0 | 0 | 0 | D2‐loop |
| c.749G>T | p.Cys250Phe | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.758C>A | p.Ala253Glu | 1 | 0 | 0 | 0 | 0 | D2‐loop |
| c.761T>A | p.Leu254Gln | 5 | 0 | 1 | 0 | 0 | D2‐loop |
| c.797G>A | p.Gly266Asp | 4 | 0 | 0 | 0 | 0 | 4th TMD |
| c.802G>A | p.Val268Ile | 1 | 0 | 0 | 0 | 0 | 4th TMD |
| c.850C>T | p.Arg284Cys | 1 | 0 | 0 | 0 | 0 | C‐terminus |
| c.923T>A | p.Leu308Gln | 2 | 0 | 0 | 0 | 0 | C‐terminus |
| c.946T>G | p.Trp316Gly | 5 | 0 | 0 | 0 | 0 | C‐terminus |
Note: Novel missense variants are depicted in bold.
Abbreviations: 1st TMD, first transmembrane domain; 2nd TMD, second transmembrane domain; 4th TMD, fourth transmembrane domain; C‐loop, cytoplasmic loop; D2‐loop, second intradiscal loop.
Next, we compared the position of (likely) pathogenic PRPH2 missense variants relative to the protein structure of PRPH2, to identify regions that may be more prone to harbor disease‐causing variants (Figure 1c). This analysis demonstrated that most missense variants (83 out of 93; Table 3; Figure 1c) are located within the D2 loop of the protein. It is hypothesized that the D2 loop is important for complex assembly with ROM1 and/or dimerization with other PRPH2 molecules, to both initiate and stabilize OS disc formation (Chakraborty et al., 2009, 2016; Goldberg et al., 1998). Furthermore, the D2 loop contains numerous highly conserved cysteine residues (Cys165, Cys166, Cys213, Cys214, Cys222, Cys250) that form disulfide bonds to maintain the structure of the loop and to regulate photoreceptor folding (Goldberg et al., 1998). For this reason, variants affecting amino acid residues within this loop will likely disrupt the structure or function of this loop, in turn interfering with PRPH2‐PRPH2 and/or PRPH2‐ROM1 interactions. Therefore, moderate evidence (PM1) was assigned if a variant was predicted to change the amino acid within this loop. Moderate evidence (PM1) was also assigned when missense variants were located in one of the transmembrane domains or C‐terminus (Boon et al., 2008; Salinas et al., 2013).
For most of the missense variants, patient ethnicity was not always mentioned in the studied papers, which made it difficult to determine whether some variants might be specific for patients of a particular ancestry. Papers that did mention the ethnicity showed that most variants were reported in patients of Caucasian or Asian ancestry. Some variants seem to be specific for a certain ethnicity; for instance, the p.Cys250Gly variant was exclusively reported in patients with Asian ancestry, whereas p.Arg142Trp and p.Arg172Gln were only identified in patients with Caucasian ancestry. The p.Arg172Trp variant was almost exclusively reported in Caucasians but also reported once in a Japanese family (Nakazawa et al., 1995).
4.2. Protein‐truncating variants
In total, 180 out of 891 PRPH2 alleles (20.2%) can be considered as protein‐truncating, represented by 85 unique variants (Table 2). Of these, 55 were frameshift, 27 were nonsense, two were fail‐to‐start, and one was a deletion of exon 1. Most of these variants are considered rare, and only four of them were present in the gnomAD database. We classified a protein‐truncating variant as damaging if the variant induces a premature stop codon before amino acid 331 since the p.Gln331* variant was reported to be pathogenic (Grover et al., 2002). Moreover, it was determined that the valine residue at position 332 is critical for targeting PRPH2 toward the OS of photoreceptor cells (Salinas et al., 2013). Based on this knowledge, we classified all protein‐truncating variants as pathogenic or likely pathogenic (Table S2). One aspect to take into account is that the annotations of p.Tyr140ins 1bp, p.Ser217_dup16bp, and p.224ins‐37bp, as described in the original articles, are not correct. We used Alamut to check whether we were able to identify the correct cDNA annotation, but this could not be deduced. For this reason, we kept the protein annotation used in the corresponding articles and put a question mark for the cDNA change annotation, although each is categorized as a protein‐truncating variant. These variants are indicated in red in Table S2.
4.3. Splice variants
Variants that were known to affect the canonical di‐nucleotides of the splice acceptor (AG) or splice donor (GT) site were assigned to be pathogenic (PVS1). For all noncanonical splice site variants, four different splice prediction tools in Alamut were used to predict pathogenicity. A variant was classified as pathogenic when an increase or decrease of >10% in splice scores was observed for all four programs, as described previously (Messchaert et al., 2018). Based on this, together with the ACMG classification, almost all splice variants were classified as (likely) pathogenic (Table S2). There were two exceptions, namely c.581+4dupA and c.829‐4C>G. For these variants, there was not enough robust evidence for their pathogenicity, and these were thus classified as variants of uncertain significance (Tables S2 and S3).
4.4. In‐frame amino acid insertions/deletions
Of the 15 in‐frame insertions/deletions, 13 were classified as pathogenic or likely pathogenic. All in‐frame amino acid insertions/deletions were assigned with PM4, indicating they might affect normal protein length and function. In addition, none of these variants were present in gnomAD, indicating that these variants in fact might be disease‐causing (Table S2). Two variants, namely p.Met67del and p.Met67_Gly68delinsArgHisArg, were classified as variants of uncertain significance (Table S3).
4.5. Synonymous variants
Three synonymous variants were reported, namely p.Tyr101Tyr (c.303T>C), p.Tyr236Tyr (c.708T>C), and p.Glu335Glu (c.1005G>A). These variants were exclusively found in patients with macular dystrophy or pattern dystrophy. All variants were first classified as likely benign due to the fact that in silico prediction programs defined them as benign/tolerated. Moreover, no evidence for putative splice defects was predicted. However, the p.Tyr101Tyr (c.303T>C) and p.Glu335Glu (c.1005G>A) variants were not reported in the gnomAD database, indicating that they are not commonly found in control individuals (Table S2). For this reason, we classified these variants to be of uncertain significance (Tables S2 and S3). The p.Tyr236Tyr (c.708T>C) variant was reported in gnomAD, and statistical analysis showed that the variant was significantly enriched in the healthy control group when compared to index patients. Therefore, this variant was classified as likely benign (Table S2).
4.6. Digenic and trigenic variants
In Section 3.2, it was shown that in a large German family segregating pattern dystrophy, p.Arg172Trp was present in patients that also carried a ROM1 (p.Arg229His) and/or an ABCA4 (p.Val2050Leu) variant (Poloschek et al., 2010). When looking at the severity of each individual's phenotype, it appeared that patients carrying an additional ROM1 (digenic carriers) variant were more severely affected compared to patients that only carried the PRPH2 p.Arg172Trp variant. The phenotype was even more severe in patients carrying PRPH2, ROM1, and ABCA4 variants (trigenic carriers). The authors hypothesized that the additional ROM1 and ABCA4 variants function as genetic modifiers that worsen the pattern dystrophy phenotype. However, the p.Val2050Leu variant in ABCA4 on itself was classified as likely benign, which makes it unlikely to be a modifier worsening the disease phenotype.
The p.Leu185Pro variant in PRPH2 was also reported together with ROM1 variants (p.Gly80Gly, p.Gly113Glu, or p.Leu114Leu in four large digenic RP families (Dryja et al., 1997; Kajiwara et al., 1994). In these families, individuals only carrying the p.Leu185Pro variant seemed to remain unaffected by disease, while individuals that also carried one of the ROM1 variants showed clear phenotypic characteristics of RP. The p.Leu270del variant in PRPH2 was found in combination with the p.Met318Alafs*17 variant in ROM1 in another digenic RP patient (Dryja et al., 1997). Recently, PRPH2 p.Arg46* in combination with the complex p.[(Leu2027Phe);(Gly1977Ser)] ABCA4 variant, was identified in a patient with cone‐rod dystrophy (Coco‐Martin et al., 2020). Finally, the same group also reported a patient with pattern dystrophy carrying both p.Lys154del in PRPH2 and p.Arg2030Gln in ABCA4 (Coco‐Martin et al., 2020). These findings indicate that besides RP, also cone‐rod and pattern dystrophy might be caused by digenic variants (i.e. one in PRH2 and one in another gene). However, mutations in IRD associated genes are relatively frequent. Nishiguchi and Rivolta screened high‐quality genome sequences of control individuals from various ancestries and estimated that ~1 in 4–5 individuals from the general population carry variants that are associated with IRD (Nishiguchi & Rivolta, 2012). Furthermore, Hanany and colleagues created a database containing 276,921 sequence variants that were identified in 187 autosomal recessive IRD genes and found that 2.7 billion people worldwide (36% of the population) are being healthy carriers of at least one IRD variant (Hanany et al., 2020). Thus, patients may carry variants in multiple IRD genes without any clinical consequences. For this reason, caution is warranted, and studies of large cohorts are required to determine if the disease in a patient is truly inherited in a di‐or trigenic fashion.
4.7. Reclassified variants
The evaluation of the pathogenicity for some variants resulted in a different outcome when compared to the original publication. This is partly due to the fact that more specific variant classification tools have become available. For example, the p.Tyr101Tyr (c.303T>C) and p.Glu335Glu (c.1005G>A) variants were previously classified to be benign by Dryja et al. (1997), mainly since they do not change the amino acid sequence, and thus are predicted not to have a deleterious effect on the final protein. However, we have classified these variants as being of uncertain significance since they were not present in normal controls reported in gnomAD, and thus in fact may be disease‐causing. Experimental assays are needed to truly determine the potential causality of these variants. Another example is the c.−11A>C variant, which is located in the 5′‐UTR region. This variant was classified as likely pathogenic because it was not present in single‐nucleotide polymorphism databases, and was not found in 92 controls (Boon, Klevering, et al., 2007). However, we have classified the variant as benign since when comparing the allele frequency in gnomAD to the allele frequency in the reported index patients, the variant was found to be enriched in the control population (gnomAD).
There were also some variants that were considered to be of uncertain significance in initial publications but were classified as (likely) pathogenic following our classification. For instance, the p.Gly167Ser variant (Meunier et al., 2011; Strom et al., 2012) is located in the D2 loop (mutational hotspot; PM1), was absent in controls (gnomAD; PM2), was predicted to be damaging by all three in silico prediction programs (PP3; Table S2), and co‐segregated with disease (PP1). Another mis‐classified variant was p.Gly68Arg, originally considered to be of uncertain significance (Dryja et al., 1997), and found in patients that were suspected to have digenic RP. However, analysis of the segregation of ROM1 alleles was uninformative. For this reason, the authors could not determine whether the p.Gly68Arg defect is pathogenic or that it represents a rare benign variant. Upon our pathogenicity classification, the variant was classified as likely pathogenic since it was significantly enriched in the patient population compared to controls (gnomAD; PS4), was located in the first TMD (PM1), and was predicted to be damaging by all three in silico prediction programs (PP3; Table S2). These findings demonstrate that pathogenicity classification tools have improved, enabling reclassification of certain variants that were reported many years ago.
5. GENOTYPE‐PHENOTYPE CORRELATIONS
To define genotype‐phenotype correlations, we collected information about disease phenotype, age at onset, and, when available, segregation data. However, most of the PRPH2 variants show high phenotypic variability, both between different families and within the same family, which made it difficult to draw proper and well‐defined conclusions regarding the relationship between PRPH2 variants and disease phenotype. For instance, in an Italian family, the p.Cys165Arg variant resulted in clinically different phenotypes (fundus flavimaculatus or butterfly‐shaped pattern dystrophy) within the same family (Simonelli et al., 2007). The mechanism behind this intrafamilial phenotypic variability remains to be elucidated but the authors suggested the following explanation; since PRPH2 interacts with other adhesion proteins, the inheritance of genes encoding such proteins may—to some extent—explain the enormous phenotypic variability observed within families. A family described by Daftarian et al. carries both the p.Gln239* and the p.Ile32Val variant (Daftarian et al., 2019). The proband carried both variants in a homozygous state, indicating that these two variants are on the same allele, which resulted in the more severe phenotype LCA. Family members carrying the variants in a heterozygous state developed much milder phenotypes with a later age of onset. The authors concluded that homozygous variants result in more severe phenotypes compared to heterozygous PRPH2 variants. A family with even higher phenotypic variability is a large Dutch family (family E) described by Boon et al. Here, similarly‐aged family members showed phenotypes ranging from retinitis pigmentosa, pseudo‐Stargardt pattern dystrophy, to only mild foveal pigmentary changes, despite carrying the same frameshift mutation (p.Gly148Alafs*5) (Boon, van Schooneveld, et al., 2007). The broad spectrum of phenotypical variability associated with PRPH2 variants is further highlighted by a reduction in penetrance described in several papers. For example, Michaelides et al. described a five‐generation family in which two individuals carrying the p.Arg172Trp mutation, a mother (49 years old) and her daughter (24 years old), had an entirely normal phenotype upon detailed testing (Michaelides et al., 2005). As previously mentioned, Boon et al. also described a normal phenotype in patients carrying the p.Arg142Trp mutation, even up until the age of 64 years (Boon et al., 2009), illustrating the reduced penetrance for (at least some) PRPH2 mutations. Another point to address is that we did not observe obvious indications that individuals with protein‐truncating variants had a more severe phenotype than, for instance, individuals carrying missense variants or in‐frame amino acid insertions/deletions. Finally, when looking at the location of likely pathogenic PRPH2 variants, it was striking that these variants seem to cluster in the D2 loop (Figure 1c), suggesting that this loop is a mutational hot spot for amino acid changes associated with PRPH2‐associated IRD. Taken together, additional genetic data from IRD patients would be of great help to determine genotype‐phenotype correlations and to study the often observed intra‐ and inter‐familiar phenotypic variability.
6. DISCUSSION
6.1. Improvement of in silico analysis
Before classifying the pathogenicity of PRPH2 missense variants according to ACMG, in silico predictions were performed using Polyphen‐2, SIFT, and CADD. Polyphen2 is a software program and is accessible via a Web server. The program predicts the possible effects of non‐synonymous single‐nucleotide variants (SNVs) on the stability and function of human proteins. It uses both structural and comparative evolutionary considerations. These properties are then combined to estimate the probability of a particular missense variant being damaging to the protein of interest (Adzhubei et al., 2013). SIFT uses datasets of functionally related protein sequences that are obtained via a protein database. The algorithm scans each position in the sequence of interest, after which the probabilities for all possible 20 amino acids at that position are calculated, resulting in one final SIFT score (Kumar et al., 2009). The CADD in silico prediction software is also available via a webserver. The program integrates multiple lineages of data, including genomic features within the surrounding sequence of interest, gene model annotations, evolutionary data from multiple species, epigenetic measurements, as well as functional predictions. In contrast to many other in silico prediction programs, CADD is not limited to the number of genomic variants of which either a pathogenic or benign status is already known (Rentzsch et al., 2019). Instead, CADD bases its score on less biased and much larger data sets and thus might have an advantage over Polyphen2 and SIFT.
Some missense variants, including p.Ala2Ser, p.Lys15Arg, p.Ser27Phe, p.Arg123Trp, p.Ser125Leu, p.Glu127Gly, p.Arg142Gln, p.Thr155Ile, p.Arg172Gln, p.Asp186Asn, p.Asp194Glu, p.Ile196Phe, p.Lys197Glu, p.Asn199Asp, p.Gly208Asp, p.Ser212Thr, p.Pro216Leu, p.Thr228Ile, p.Glu242Gly, p.Ala252Pro, p.Leu261Phe, p.Trp316Gly, and p.Ala337Thr, gave contradictory outcomes upon in silico pathogenicity predictions. Other variants, including p.Ile32Val, p.Ala85Thr, p.Ala116Ser, p.Met152Val, p.Lys154Gln, p.Ile161Met, p.Ser217Gly, p.Leu261Phe, p.Val268Ile, p.Pro313Ser, and p.Pro313Leu were predicted to be benign. While different algorithms may rely on the same type of data to predict pathogenicity, it is known that for some genes, similar algorithms can have a significantly different outcome (Richards et al., 2015). Only when at least two out of three of the in silico programs used give the same prediction output, the evidence can be counted as supporting (PP3) (Richards et al., 2015). Based on this, the in silico predictions for the p.Lys15Arg, p.Ile32Val, p.Ala85Thr, p.Ala116Ser, p.Glu127Gly, p.Met152Val, p.Lys154Gln, p.Ile161Met, p.Asp186Asn, p.ASp194Glu, p.Ile196Phe, p.Pro216Leu, p.Ser217Gly, p.Thr228Ile, p.Leu261Phe, p.Val268Ile, p.Pro313Ser, and p.Pro313Leu, p.Trp316Gly, and p.Ala337Thr variants were not considered to be of supporting evidence (PP3). It would therefore be very helpful if there were more experimental data available that could show whether these variants have a deleterious effect on normal protein morphology and/or function. This will be explained in more detail in the next section.
6.2. Experimental assessment of variants of unknown significance
Approximately one‐third of the missense variants were classified as variants of unknown significance (Figure 1b). This was due to the fact that these variants did not robustly meet important ACMG pathogenicity criteria. Furthermore, two splice‐site, two in‐frame amino acid insertions/deletions, two synonymous variants, and one 3′‐UTR variant also needed to be classified as variants of uncertain significance for the same reasons as for the missense variants. Experimental models would be of great help to classify such variants. However, experimental data are extremely limited. This is mainly due to the fact that PRPH2 expression is highly retina‐specific. This makes it difficult to use widely used immortalized lymphoblastoid, skin fibroblast, or blood cell lines. However, depending on the type and location of each variant, this issue may be circumvented. For instance, when studying missense variants, one could clone the coding region of PRPH2 (~1.1 kb) into specific expression vectors to create a PRPH2 wildtype vector. Next, site‐directed mutagenesis can be applied to insert the desired mutation. Wildtype and mutant vectors can be transfected into widely used cell lines to study, for example, expression patterns (western blots) or interacting proteins (yeast‐two‐hybrid, co‐immunoprecipitation), and compare mutant and wild‐type conditions. These vectors can even be administered to neonatal mice to study in vivo localization of wildtype or mutant PRPH2. Chakraborty and colleagues created vectors containing wild‐type PRPH2 or the PRPH2 p.Cys213Tyr variant and electroporated these into neonatal mice. Four weeks after injection, retinas were collected and it was seen that the p.Cys213Tyr construct mislocalized to the inner segments and perinuclear region (Chakraborty et al., 2020). Another research group showed that constructs containing the p.Pro210Leu and p.Cys214Ser missense variants mislocalized to the inner segments upon administration to wildtype mice (Becirovic et al., 2016). Interestingly, results differed between rods and cones, indicating that PRPH2 might have a different function in the two different photoreceptor cell types.
Second, when one wants to study splice site variants, minigene or midigene splice assays can be used, as described previously (Sangermano et al., 2018). Minigenes or midigenes represent plasmids in which the desired PRPH2 splice site variant can be cloned between two exons of interest. The plasmids are transfected into, for example, HEK293T cells, and splice effects can be analyzed using reverse‐transcription polymerase chain reaction (RT‐PCR) to determine whether there are differences between RNAs transcribed from wild‐type and mutant minigenes. Becirovic and colleagues constructed PRPH2 wild‐type minigenes as well as minigenes containing the IRD‐associated p.Arg195Leu, p.Ser198Arg, p.Val209Ile, p.Pro210Leu, p.Ser212Thr, p.Cys214Ser, p.Arg220Trp, p.Arg220Gln, p.Trp246Arg, and p.Gly249Ser missense variants (Becirovic et al., 2016). RT‐PCR analysis of PRPH2 transcripts in murine retinas transduced with the wild‐type or the mutant PRPH2 minigene identified three different PRPH2 splice isoforms in rods and cones. These splice isoforms consisted of the unspliced transcript, a transcript in which intron 1 was retained, and the correctly spliced PRPH2. The p.Gly249Ser variant created a new splice donor site, resulting in aberrant splicing of the protein. This suggests that also point mutations in coding regions might affect splicing (Becirovic et al., 2016).
Finally, for studying all types of variants, one could ideally make use of induced pluripotent stem cell (iPSC) technology. In short, somatic cells are extracted from PRPH2‐associated IRD patients or from healthy controls, which can then be reprogrammed toward iPSCs. Next, these iPSCs can be differentiated toward retina‐like cells thus carrying the variant of interest (Giacalone et al., 2016; Öner, 2018), after which a variety of functional studies can be performed, including expression (western blot), localization (immunohistochemistry), and morphological (microscopy) studies. A drawback of this approach is that it is very labor‐ and time‐consuming, as well as expensive.
Taken together, these experimental studies are crucial to improve the classification of PRPH2 variants and thereby aid in the molecular diagnostics of IRDs.
6.3. Phenotypic variability and genotype‐phenotype correlations
As detailed in the introduction and genotype‐phenotype correlation section, there is significant variability in clinical phenotypes, with regard to age at onset, severity, and incomplete penetrance. Some variants show a clear genotype‐phenotype correlation like p.Leu126Pro, p.Leu126Arg, p.Glu127Gly, p.Gly137Asp, p.Arg142QGln, p.Met152Val, p.Cys165Tyr, p.Asp173Val, p.Trp179Arg, p.Ser198Arg, p.Pro210Ser, p.Phe211Leu, and p.Gly266Asp, and that all seem to be associated with the development of RP only (Anasagasti et al., 2013; Bareil et al., 2000; de Sousa Dias et al., 2015; Dryja et al., 1997; Gao et al., 2019; Grüning et al., 1994; Jin et al., 2008; Kemp et al., 1994; Manes et al., 2015; Reeves et al., 2020; Renner et al., 2009; Sohocki et al., 2001; Souied et al., 1998; Stone et al., 2017; Sullivan et al., 2013; Van Cauwenbergh et al., 2017; Yoon et al., 2015). Other examples are p.Asp157Asn and p.Cys213Trp, which are exclusively associated with PD (Boon, van Schooneveld, et al., 2007; Jacobson et al., 1996; Reeves et al., 2020). Thus, these disorders can best be described as allelic disorders due to the fact that there is a clear variant‐phenotype relationship. However, for most of the variants detailed in this study, there appears to be a high phenotypic variability between different families, as well as between members within the same family. A clear example of this is explained by the c.828+3 A>T variant described by Shankar and colleagues. This particular variant results in the development of multiple distinct IRD phenotypes, including RP, CRD, PD, and macular dystrophy (Shankar et al., 2015, 2016). The multiple phenotypes that are seen in these families can best be referred to as variable expressivity as was also concluded by the authors of the respective paper. Another example also showing variable expressivity is p.Arg142Trp. This variant is associated with multiple IRD phenotypes that, in some cases, also show incomplete penetrance.
6.3.1. Insights from the molecular pathophysiology of the PRPH2 p.Arg172Trp variant
When looking at the subtype of IRD in patients carrying the p.Arg172Trp variant, it becomes apparent that these subjects have a relatively young mean age of onset, and an overall cone‐dominant phenotype (Reeves et al., 2020; Wells et al., 1993; Wroblewski et al., 1994a). Most of the patients display a significant decrease in cone function, while rod function seems to remain unaffected (Conley et al., 2007). In a rare number of cases, however, usually observed in later stages of the disease, rods become affected as well. Expression of the human‐specific p.Arg172Trp variant in mice resulted in the rescue of the rod but not cone phenotypes. This finding strongly indicates that multiple variant‐specific mechanisms exist (Ding et al., 2004).
Co‐expression of the p.Arg172Trp mutation and a full complement of wildtype Prph2 initially resulted in normal rod function and structure (Conley et al., 2014). For cones, however, something different was observed. The retina of mice carrying the Arg172Trp variant exhibited a significant loss in the total number of blue and green cones, as well as a decrease in photopic ERG responses. Furthermore, defects in Prph2‐Rom1 complex formation were observed (Conley et al., 2014). In another study, the p.Arg172Trp variant was expressed on a rod dominant (Prph2+/−) background and led to a late‐onset reduction in the number of rod cells as well as scotopic responses. The authors speculated that this was highly likely due to the haploinsufficiency of Prph2 (Conley et al., 2014). Expression of the Arg172Trp mutation in the cone‐dominant (Nrl−/−) retina resulted in a functional decline of the photopic responses, as well as disrupted cone OS structures, due to the formation of abnormal high molecular weight Prph2‐Rom1 aggregates (Conley et al., 2014).
Based on the aforementioned, we can conclude that the p.Arg172Trp variant is most frequently associated with cone‐dominant phenotypes and that multiple variant‐specific mechanisms underlying the development of IRD exist. Together, the exact mechanisms by which PRHP2 mutations can lead to one or more distinct clinical subtypes of IRD for a large part remain elusive.
6.4. Development of therapeutic strategies for PRPH2‐associated IRD
Proper classification of possible disease‐causing variants is of great importance, not only to determine the true causal genetic defect in IRD patients but also for developing therapeutic strategies. PRPH2 has been a target for gene therapy for over two decades now, mainly due to the disease burden associated with variants in this gene, as well as the availability of several extremely well‐characterized animal models mimicking important phenotypic characteristics of patients with PRPH2‐associated IRD (Conley & Naash, 2014). Currently, at least three therapeutic strategies can be distinguished: (1) gene replacement therapy; (2) gene knockdown therapy; and (3) delivery of neurotrophic factors. To develop such therapeutic strategies, it is important to not only know the genetic defect but also the underlying pathophysiological mechanism (e.g., dominant‐negative vs. haploinsufficiency).
6.4.1. Gene replacement therapy
The first proof‐of‐principle studies regarding gene replacement therapy attempted to correct IRD phenotypes in the Prph2 −/− and Prph2 +/− mice. For this purpose, a wild‐type Prph2 transgene was delivered, and the results were highly promising since in Prph2 −/− mice, the structure of the OS of rod photoreceptor cells was preserved (Travis et al., 1992). Furthermore, in Prph2 +/− mice, the expression of a wildtype Prph2 transgene rescued rod and cone OS structure and function (Nour et al., 2004). In mice harboring the recurrent p.Arg172Trp variant, which is considered a dominant‐negative variant, expression of wildtype PRPH2 also caused structural and functional improvements (Conley et al., 2007; Nour et al., 2008).
6.4.2. Gene knockdown therapy
As some mutations in PRPH2‐associated IRD are believed to act in a dominant‐negative manner, such as p.Arg172Trp (Conley et al., 2007), and the fact that gene replacement did not completely correct the dominant phenotype in mice carrying this specific variant, alternative approaches are needed to eliminate the mutant allele. One such approach is the gene knockdown approach. For PRPH2, the usage of short hairpin RNA (shRNA) to knock down the mutant allele seems to be the most promising. For example, an shRNA that was shown to knock down Prph2 in vitro was packaged into an rAAV vector. Upon injection into the subretinal space of wild‐type mice, wild‐type Prph2 levels were reduced by 75% (Petrs‐Silva et al., 2012). Next, they created a vector containing wildtype Prph2 that was proven to be resistant to the aforementioned shRNA. Co‐delivery of these vectors resulted in the rescue of functional defects caused by the shRNA knockdown and partial recovery of total Prph2 levels (Petrs‐Silva et al., 2012). Similarly, using electroporation instead of a virus, coinjection of both an shRNA vector that is able to knock down both wildtype and mutant Prph2 and an shRNA‐resistant copy of wild‐type Prph2 resulted in degradation of endogenous Prph2 and stabilized expression of the exogenously delivered Prph2 in mouse retinal explants (Palfi et al., 2006). Although the efficacy of this kind of therapy has not yet been evaluated in IRD disease models, these studies show that allele‐independent knockdown in combination with gene supplementation represents a potential therapy to counteract genetic defects in PRPH2‐associated diseases.
6.4.3. Delivery of neurotrophic factors
Unlike gene replacement and gene knockdown therapies, also more general therapeutic strategies are considered, for example, the delivery of neurotrophic factors. An advantage of such a strategy is that it can be applied to multiple genetic subtypes of IRD. The first proof‐of‐principle came from a mouse study in which ciliary neurotrophic factor (CNTF) was injected into the intravitreal space of Prph2 −/− mice (Cayouette et al., 1998), and lead to an improvement in OS structure. However, at the functional level, only a small improvement in rods and no improvement in cones was observed. In a similar study performed by another group, CTNF was delivered to p.Pro216Leu Prph2 +/− mice. These mice showed improved OS structure, but the authors also observed dose‐dependent abnormalities in photoreceptor nuclei as well as a decrease in both rod and cone function (Bok et al., 2002). Follow‐up studies revealed that CNTF alters retinal signaling pathways. Furthermore, they observed a downregulation of critical phototransduction genes, such as cone opsins (Rhee et al., 2007). Although these adverse findings have led to the preclusion to use CNTF to treat PRPH2‐associated IRD, other neurotrophic factors were investigated. For example, lentiviruses carrying either fibroblast growth factor‐2 (FGF‐2) or human pigment epithelial‐derived factor (PEDF) were reported to significantly improve both scotopic a‐ and b‐waves in Prph2 −/− mice. However, these agents did not restore photoreceptor survival (Miyazaki et al., 2008). Other nontraditional neurotrophic factors, such as hormones, also have been shown to result in neuroprotection. Administration of some of these agents, including erythropoietin (hormone) (Rex et al., 2004, 2009), and nilvadipine (calcium channel blocker) (Takeuchi et al., 2008), significantly improved photoreceptor function in Prph2 −/− and Prph2 +/− mouse models. However, more studies are required to determine both the safety and efficacy of such particular therapeutic approaches.
7. CONCLUDING REMARKS
Taken together, in this study, we describe 245 reported and seven novel PRPH2 variants identified in 891 alleles in 867 index cases and uploaded these to the LOVD and ClinVar database for PRPH2. This study thereby provides an important step toward a complete overview of all PRPH2 variants in a single database. Continuous addition of genetic data from newly identified patients with PRPH2 variants is of great importance for more robust classification of pathogenic variants. Furthermore, additional data regarding phenotypes might aid the identification of genotype‐phenotype correlations. Our analysis resulted in the in silico classification of all the 245 reported, as well as the seven novel identified PRPH2 variants. More importantly, our study illustrates the need for experimental assays to identify the true causality of the many PRPH2 variants that are now still assigned to be variants of uncertain significance. This will help to improve molecular diagnostics and, in the long‐term, hopefully also support the development of therapeutic strategies for patients with PRPH2‐associated IRD.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
WEB RESOURCES
https://gnomad.broadinstitute.org/ http://genetics.bwh.harvard.edu/pph2/ http://cadd.gs.washington.edu/home https://sift.bii.a-star.edu.sg/ https://databases.lovd.nl/shared/genes/PRPH2 https://www.ncbi.nlm.nih.gov/clinvar/?term=PRPH2[all]
Supporting information
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
ACKNOWLEDGMENTS
We gratefully acknowledge Stéphanie S. Cornelis for the assistance with the statistical analysis and LOVD submission, as well as Bjorn Bakker for technical assistance. Members of the PRPH2 study group (Drs. C. Ayuso; S. Banfi; T. Barakat; N. Bax; T. Ben‐Yosef, M. Breukink; J. de Hoog; J. de Roach; A. Delbecq; J. Ferraz Sallum; K. Fujinami; M. Gorin; W. Hartstra; Y. Hettinga; S. Ijzer; J. Keunen, A. Kievit; T. Kleefstra; T. Lamey; P. Liskova; M. Oldak; J. Ossewaarde ‐ van Norel; M. Phan; O. Podhajcer; P. Rump; M. Sinnema; D. Smailhodzic; C. Stumpel; A. Thiadens; J. van de Ven; R. van Leeuwen; S. Vermeer; J. Verheij and B. Weber) who submitted DNA samples in which PRHP2 variants were identified. This study was supported by an internal RadboudUMC PhD grant provided by the Donders Institute for Brain, Cognition and Behavior, as well as by the Foundation Fighting Blindness USA, grant BR‐GE‐0120‐0775‐LUMC.
Peeters, M. H. , Khan, M. , Rooijakkers, A. A. , Mulders, T. , Haer‐Wigman, L. , Boon, C. J. , Klaver, C. C. , van den Born, L. I. , Hoyng, C. B. , Cremers, F. P. , den Hollander, A. I. , Dhaenens, C.‐M. , & Collin, R. W. (2021). PRPH2 mutation update: In silico assessment of 245 reported and 7 novel variants in patients with retinal disease. Human Mutation, 42, 1521–1547. 10.1002/humu.24275
DATA AVAILABILITY STATEMENT
All variant data described in this study are freely available via the LOVD and ClinVar database for PRPH2 (https://databases.lovd.nl/shared/variants/PRPH2; PRPH2[all] ‐ ClinVar ‐ NCBI (nih.gov)). Other data that support the findings detailed in this study are available from the corresponding author, upon reasonable request.
REFERENCES
- Abouelhoda, M. , Faquih, T. , El‐Kalioby, M. , & Alkuraya, F. S. (2016). Revisiting the morbid genome of Mendelian disorders. Genome Biology, 17(1), 235. 10.1186/s13059-016-1102-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu‐Safieh, L. , Alrashed, M. , Anazi, S. , Alkuraya, H. , Khan, A. O. , Al‐Owain, M. , Al‐Zahrani, J. , Al‐Abdi, L. , Hashem, M. , Al‐Tarimi, S. , Sebai, M. A. , Shamia, A. , Ray‐Zack, M. D. , Nassan, M. , Al‐Hassnan, Z. N. , Rahbeeni, Z. , Waheeb, S. , Alkharashi, A. , Abboud, E. , … Alkuraya, F. S. (2013). Autozygome‐guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Research, 23(2), 236–247. 10.1101/gr.144105.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei, I. , Jordan, D. M. , & Sunyaev, S. R. (2013). Predicting functional effect of human missense mutations using PolyPhen‐2. Current Protocols in Human Genetics, 20. 10.1002/0471142905.hg0720s76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad, O. R. , Ayyagari, R. , & Zacks, D. N. (2010). A novel missense mutation in the rds/peripherin gene associated with retinal pattern dystrophy. Retinal Cases & Brief Reports, 4(1), 84–85. 10.1097/ICB.0b013e318198d8f7 [DOI] [PubMed] [Google Scholar]
- Alapati, A. , Goetz, K. , Suk, J. , Navani, M. , Al‐Tarouti, A. , Jayasundera, T. , Tumminia, S. J. , Lee, P. , & Ayyagari, R. (2014). Molecular diagnostic testing by eyeGENE: Analysis of patients with hereditary retinal dystrophy phenotypes involving central vision loss. Investigative Ophthalmology and Visual Science, 55(9), 5510–5521. 10.1167/iovs.14-14359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand, S. , Sheridan, E. , Cassidy, F. , Inglehearn, C. , Williams, G. , Springell, K. , Allgar, V. , Kelly, T. L. , & McKibbin, M. (2009). Macular dystrophy associated with the Arg172Trp substitution in peripherin/RDS: Genotype‐phenotype correlation. Retina, 29(5), 682–688. 10.1097/IAE.0b013e318198dbed [DOI] [PubMed] [Google Scholar]
- Anasagasti, A. , Barandika, O. , Irigoyen, C. , Benitez, B. A. , Cooper, B. , Cruchaga, C. , López de Munain, A. , & Ruiz‐Ederra, J. (2013). Genetic high throughput screening in Retinitis Pigmentosa based on high resolution melting (HRM) analysis. Experimental Eye Research, 116, 386–394. 10.1016/j.exer.2013.10.011 [DOI] [PubMed] [Google Scholar]
- Apfelstedt‐Sylla, E. , Theischen, M. , Rüther, K. , Wedemann, H. , Gal, A. , & Zrenner, E. (1995). Extensive intrafamilial and interfamilial phenotypic variation among patients with autosomal dominant retinal dystrophy and mutations in the human RDS/peripherin gene. The British Journal of Ophthalmology, 79, 28–34. 10.1136/bjo.79.1.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai, Y., Maeda, A. , Hirami, Y. , Ishigami, C. , Kosugi, S. , Mandai, M. , Kurimoto, Y. , & Takahashi, M. (2015). Retinitis Pigmentosa with EYS mutations is the most prevalent inherited retinal dystrophy in Japanese populations. Journal of Ophthalmology, 2015, 819760. 10.1155/2015/819760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avela, K. , Salonen‐Kajander, R. , Laitinen, A. , Ramsden, S. , Barton, S. , & Rudanko, S. L. (2019). The genetic aetiology of retinal degeneration in children in Finland—New founder mutations identified. Acta Ophthalmologica, 97(8), 805–814. 10.1111/aos.14128 [DOI] [PubMed] [Google Scholar]
- Avela, K. , Sankila, E. M. , Seitsonen, S. , Kuuluvainen, L. , Barton, S. , Gillies, S. , & Aittomäki, K. (2018). A founder mutation in CERKL is a major cause of retinal dystrophy in Finland. Acta Ophthalmologica, 96(2), 183–191. 10.1111/aos.13551 [DOI] [PubMed] [Google Scholar]
- Ba‐Abbad, R. , Robson, A. G. , MacPhee, B. , Webster, A. R. , & Michaelides, M. (2019). Rod‐cone dystrophy associated with the Gly167Asp variant in PRPH2. Ophthalmic Genetics, 40(2), 188–189. 10.1080/13816810.2019.1605393 [DOI] [PubMed] [Google Scholar]
- Ba‐Abbad, R. , Robson, A. G. , Yap, Y. C. , Moore, A. T. , Webster, A. R. , & Holder, G. E. (2014). Prph2 mutations as a cause of electronegative ERG. Retina, 34(6), 1235–1243. 10.1097/iae.0000000000000052 [DOI] [PubMed] [Google Scholar]
- Barbazetto, I. A. , Yannuzzi, N. A. , Klais, C. M. , Merriam, J. E. , Zernant, J. , Peiretti, E. , Yannuzzi, L. A. , & Allikmets, R. (2007). Pseudo‐vitelliform macular detachment and cuticular drusen: Exclusion of 6 candidate genes. Ophthalmic Genetics, 28(4), 192–197. 10.1080/13816810701538596 [DOI] [PubMed] [Google Scholar]
- Bareil, C. , Delague, V. , Arnaud, B. , Demaille, J. , Hamel, C. , & Claustres, M. (2000). W179R: A novel missense mutation in the peripherin/RDS gene in a family with autosomal dominant retinitis pigmentosa. Human Mutation, 15(6), 583–584. [DOI] [PubMed] [Google Scholar]
- Bareil, C. , Hamel, C. , Arnaud, B. , Demaille, J. , & Claustres, M. (1997). A complex allele (1064delTC and IVS2 + 22ins7) in the peripherin/RDS gene in retinitis pigmentosa with macular dystrophy. Ophthalmic Genetics, 18(3), 129–138. 10.3109/13816819709057126 [DOI] [PubMed] [Google Scholar]
- Bayés, M. , Martínez‐Mir, A. , Valverde, D. , del Río, E. , Vilageliu, L. , Grinberg, D. , Balcells, S. , Ayuso, C. , Baiget, M. , & Gonzàlez‐Duarte, R. (1996). Autosomal recessive retinitis pigmentosa in Spain: Evaluation of four genes and two loci involved in the disease. Clinical Genetics, 50(5), 380–387. 10.1111/j.1399-0004.1996.tb02392.x [DOI] [PubMed] [Google Scholar]
- Becirovic, E. , Böhm, S. , Nguyen, O. N. , Riedmayr, L. M. , Koch, M. A. , Schulze, E. , Kohl, S. , Borsch, O. , Santos‐Ferreira, T. , Ader, M. , Michalakis, S. , & Biel, M. (2016). In vivo analysis of disease‐associated point mutations unveils profound differences in mRNA splicing of Peripherin‐2 in rod and cone photoreceptors. PLOS Genetics, 12(1), e1005811. 10.1371/journal.pgen.1005811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini, Y. , Drai, D. , Elmer, G. , Kafkafi, N. , & Golani, I. (2001). Controlling the false discovery rate in behavior genetics research. Behavioural Brain Research, 125(1–2), 279–284. 10.1016/s0166-4328(01)00297-2 [DOI] [PubMed] [Google Scholar]
- Birtel, J. , Eisenberger, T. , Gliem, M. , Müller, P. L. , Herrmann, P. , Betz, C. , Zahnleiter, D. , Neuhaus, C. , Lenzner, S. , Holz, F. G. , Mangold, E. , Bolz, H. J. , & Charbel Issa, P. (2018). Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone‐rod dystrophy. Scientific Reports, 8(1), 4824. 10.1038/s41598-018-22096-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bok, D. , Yasumura, D. , Matthes, M. T. , Ruiz, A. , Duncan, J. L. , Chappelow, A. V. , Zolutukhin, S. , Hauswirth, W. , & LaVail, M. M. (2002). Effects of adeno‐associated virus‐vectored ciliary neurotrophic factor on retinal structure and function in mice with a P216L rds/peripherin mutation. Experimental Eye Research, 74(6), 719–735. 10.1006/exer.2002.1176 [DOI] [PubMed] [Google Scholar]
- Boon, C. J. , den Hollander, A. I. , Hoyng, C. B. , Cremers, F. P. , Klevering, B. J. , & Keunen, J. E. (2008). The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Progress in Retina and Eye Research, 27(2), 213–235. 10.1016/j.preteyeres.2008.01.002 [DOI] [PubMed] [Google Scholar]
- Boon, C. J. , Klevering, B. J. , Cremers, F. P. , Zonneveld‐Vrieling, M. N. , Theelen, T. , Den Hollander, A. I. , & Hoyng, C. B. (2009). Central areolar choroidal dystrophy. Ophthalmology, 116(4), 771–782. 10.1016/j.ophtha.2008.12.019 [DOI] [PubMed] [Google Scholar]
- Boon, C. J. , Klevering, B. J. , den Hollander, A. I. , Zonneveld, M. N. , Theelen, T. , Cremers, F. P. , & Hoyng, C. B. (2007). Clinical and genetic heterogeneity in multifocal vitelliform dystrophy. Archives of Ophthalmology, 125(8), 1100–1106. 10.1001/archopht.125.8.1100 [DOI] [PubMed] [Google Scholar]
- Boon, C. J., van Schooneveld, M. J. , den Hollander, A. I. , van Lith‐Verhoeven, J. J. , Zonneveld‐Vrieling, M. N. , Theelen, T. , Cremers, F. P. , Hoyng, C. B. , & Klevering, B. J. (2007). Mutations in the peripherin/RDS gene are an important cause of multifocal pattern dystrophy simulating STGD1/fundus flavimaculatus. British Journal of Ophthalmology, 91(11), 1504–1511. 10.1136/bjo.2007.115659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulanger‐Scemama, E. , El Shamieh, S. , Démontant, V. , Condroyer, C. , Antonio, A. , Michiels, C. , Boyard, F. , Saraiva, J. P. , Letexier, M. , Souied, E. , Mohand‐Saïd, S. , Sahel, J. A. , Zeitz, C. , & Audo, I. (2015). Next‐generation sequencing applied to a large French cone and cone‐rod dystrophy cohort: Mutation spectrum and new genotype‐phenotype correlation. Orphanet Journal of Rare Diseases, 10, 85. 10.1186/s13023-015-0300-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budu, H. S. , Matsumoto, M. , Yamada, T. , Zhang, X. Y. , & Hayasaka, Y. (2001). Peripherin/RDS gene mutation (Pro210Leu) and polymorphisms in Japanese patients with retinal dystrophies. Japanese Journal of Ophthalmology, 45(4), 355–358. 10.1016/s0021-5155(01)00334-3 [DOI] [PubMed] [Google Scholar]
- Carss, K. J. , Arno, G. , Erwood, M. , Stephens, J. , Sanchis‐Juan, A. , Hull, S. , Megy, K. , Grozeva, D. , Dewhurst, E. , Malka, S. , Plagnol, V. , Penkett, C. , Stirrups, K. , Rizzo, R. , Wright, G. , Josifova, D. , Bitner‐Glindzicz, M. , Scott, R. H. , Clement, E. , … Raymond, F. L. (2017). Comprehensive rare variant analysis via whole‐genome sequencing to determine the molecular pathology of inherited retinal disease. American Journal of Human Genetics, 100(1), 75–90. 10.1016/j.ajhg.2016.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayouette, M. , Behn, D. , Sendtner, M. , Lachapelle, P. , & Gravel, C. (1998). Intraocular gene transfer of ciliary neurotrophic factor prevents death and increases responsiveness of rod photoreceptors in the retinal degeneration slow mouse. Journal of Neuroscience, 18(22), 9282–9293. 10.1523/jneurosci.18-22-09282.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty, D. , Conley, S. M. , Zulliger, R. , & Naash, M. I. (2016). The K153Del PRPH2 mutation differentially impacts photoreceptor structure and function. Human Molecular Genetics, 25(16), 3500–3514. 10.1093/hmg/ddw193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty, D. , Ding, X. Q. , Conley, S. M. , Fliesler, S. J. , & Naash, M. I. (2009). Differential requirements for retinal degeneration slow intermolecular disulfide‐linked oligomerization in rods versus cones. Human Molecular Genetics, 18(5), 797–808. 10.1093/hmg/ddn406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty, D. , Strayve, D. G. , Makia, M. S. , Conley, S. M. , Kakahel, M. , Al‐Ubaidi, M. R. , & Naash, M. I. (2020). Novel molecular mechanisms for Prph2‐associated pattern dystrophy. FASEB Journal, 34(1), 1211–1230. 10.1096/fj.201901888R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, J. , Fu, J. , Zhou, Q. , Xiang, X. , Wei, C. , Yang, L. , Fu, S. , Khan, M. A. , Lv, H. , & Fu, J. (2019). A novel splicing mutation in the PRPH2 gene causes autosomal dominant retinitis pigmentosa in a Chinese pedigree. Journal of Cellular and Molecular Medicine, 23(5), 3776–3780. 10.1111/jcmm.14278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coco, R. M. , Tellería, J. J. , Sanabria, M. R. , Rodríguez‐Rúa, E. , & García, M. T. (2010). PRPH2 (Peripherin/RDS) mutations associated with different macular dystrophies in a Spanish population: A new mutation. European Journal of Ophthalmology, 20(4), 724–732. 10.1177/112067211002000413 [DOI] [PubMed] [Google Scholar]
- Coco‐Martin, R. M. , Sanchez‐Tocino, H. T. , Desco, C. , Usategui‐Martín, R. , & Tellería, J. J. (2020). PRPH2‐related retinal diseases: Broadening the clinical spectrum and describing a new mutation. Genes (Basel), 11(7), 773. 10.3390/genes11070773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley, S. , Nour, M. , Fliesler, S. J. , & Naash, M. I. (2007). Late‐onset cone photoreceptor degeneration induced by R172W mutation in Rds and partial rescue by gene supplementation. Investigative Ophthalmology and Visual Science, 48(12), 5397–5407. 10.1167/iovs.07-0663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley, S. M. , & Naash, M. I. (2014). Gene therapy for PRPH2‐associated ocular disease: Challenges and prospects. Cold Spring Harbor Perspectives in Medicine, 4(11), a017376. 10.1101/cshperspect.a017376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley, S. M. , Stuck, M. W. , Burnett, J. L. , Chakraborty, D. , Azadi, S. , Fliesler, S. J. , & Naash, M. I. (2014). Insights into the mechanisms of macular degeneration associated with the R172W mutation in RDS. Human Molecular Genetics, 23(12), 3102–3114. 10.1093/hmg/ddu014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consugar, M. B. , Navarro‐Gomez, D. , Place, E. M. , Bujakowska, K. M. , Sousa, M. E. , Fonseca‐Kelly, Z. D. , Taub, D. G. , Janessian, M. , Wang, D. Y. , Au, E. D. , Sims, K. B. , Sweetser, D. A. , Fulton, A. B. , Liu, Q. , Wiggs, J. L. , Gai, X. , & Pierce, E. A. (2015). Panel‐based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genetics in Medicine, 17(4), 253–261. 10.1038/gim.2014.172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussa, R. G. , Chakarova, C. , Ajlan, R. , Taha, M. , Kavalec, C. , Gomolin, J. , Khan, A. , Lopez, I. , Ren, H. , Waseem, N. , Kamenarova, K. , Bhattacharya, S. S. , & Koenekoop, R. K. (2015). Genotype and phenotype studies in autosomal dominant Retinitis Pigmentosa (adRP) of the French Canadian Founder Population. Investigative Ophthalmology and Visual Science, 56(13), 8297–8305. 10.1167/iovs.15-17104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daftarian, N. , Mirrahimi, M. , Sabbaghi, H. , Moghadasi, A. , Zal, N. , Dehghan Banadaki, H. , Ahmadieh, H. , & Suri, F. (2019). PRPH2 mutation as the cause of various clinical manifestations in a family affected with inherited retinal dystrophy. Ophthalmic Genetics, 40(5), 436–442. 10.1080/13816810.2019.1678178 [DOI] [PubMed] [Google Scholar]
- de Breuk, A. , Acar, I. E. , Kersten, E. , Schijvenaars, M. , Colijn, J. M. , Haer‐Wigman, L. , Bakker, B. , de Jong, S. , Meester‐Smoor, M. A. , Verzijden, T. , Missotten, T. , Monés, J. , Biarnés, M. , Pauleikhoff, D. , Hense, H. W. , Silva, R. , Nunes, S. , Melo, J. B. , Fauser, S. , … The EYE‐RISK Consortium (2020). Development of a genotype assay for age‐related macular degeneration: The EYE‐RISK Consortium. Ophthalmology. 10.1016/j.ophtha.2020.07.037 [DOI] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders Study . (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature, 542(7642), 433–438. 10.1038/nature21062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sousa Dias, M. , Hernan, I. , Delás, B. , Pascual, B. , Borràs, E. , Gamundi, M. J. , Mañé, B. , Fernández‐San José, P. , Ayuso, C. , & Carballo, M. (2015). New COL6A6 variant detected by whole‐exome sequencing is linked to break points in intron 4 and 3'‐UTR, deleting exon 5 of RHO, and causing adRP. Molecular Vision, 21, 857–870. [PMC free article] [PubMed] [Google Scholar]
- Ding, X. Q. , Nour, M. , Ritter, L. M. , Goldberg, A. F. , Fliesler, S. J. , & Naash, M. I. (2004). The R172W mutation in peripherin/rds causes a cone‐rod dystrophy in transgenic mice. Human Molecular Genetics, 13(18), 2075–2087. 10.1093/hmg/ddh211 [DOI] [PubMed] [Google Scholar]
- Donoso, L. A. , Hageman, G. , Frost, A. , Sheffield, V. , Beck, J. , Hébert, M. , & MacDonald, I. M. (2003). Autosomal dominant macular dystrophy in a large Canadian family. Canadian Journal of Ophthalmology, 38(1), 33–40. 10.1016/s0008-4182(03)80006-6 [DOI] [PubMed] [Google Scholar]
- Downes, S. M. , Fitzke, F. W. , Holder, G. E. , Payne, A. M. , Bessant, D. A. , Bhattacharya, S. S. , & Bird, A. C. (1999). Clinical features of codon 172 RDS macular dystrophy: Similar phenotype in 12 families. Archives of Ophthalmology, 117(10), 1373–1383. 10.1001/archopht.117.10.1373 [DOI] [PubMed] [Google Scholar]
- Downs, K. , Zacks, D. N. , Caruso, R. , Karoukis, A. J. , Branham, K. , Yashar, B. M. , Haimann, M. H. , Trzupek, K. , Meltzer, M. , Blain, D. , Richards, J. E. , Weleber, R. G. , Heckenlively, J. R. , Sieving, P. A. , & Ayyagari, R. (2007). Molecular testing for hereditary retinal disease as part of clinical care. Archives of Ophthalmology (Chicago, Ill.: 1960), 125(2), 252–258. 10.1001/archopht.125.2.252 [DOI] [PubMed] [Google Scholar]
- Dryja, T. P. , Hahn, L. B. , Kajiwara, K. , & Berson, E. L. (1997). Dominant and digenic mutations in the peripherin/RDS and ROM1 genes in retinitis pigmentosa. Investigative Ophthalmology and Visual Science, 38(10), 1972–1982. [PubMed] [Google Scholar]
- Duncan, J. L. , Talcott, K. E. , Ratnam, K. , Sundquist, S. M. , Lucero, A. S. , Day, S. , Zhang, Y. , & Roorda, A. (2011). Cone structure in retinal degeneration associated with mutations in the peripherin/RDS gene. Investigative Ophthalmology and Visual Science, 52(3), 1557–1566. 10.1167/iovs.10-6549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncker, T. , Tsang, S. H. , Woods, R. L. , Lee, W. , Zernant, J. , Allikmets, R. , Delori, F. C. , & Sparrow, J. R. (2015). Quantitative fundus autofluorescence and optical coherence tomography in PRPH2/RDS‐ and ABCA4‐associated disease exhibiting phenotypic overlap. Investigative Ophthalmology and Visual Science, 56(5), 3159–3170. 10.1167/iovs.14-16343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekström, U. , Andréasson, S. , Ponjavic, V. , Abrahamson, M. , Sandgren, O. , Nilsson‐Ehle, P. , & Ehinger, B. (1998). A Swedish family with a mutation in the peripherin/RDS gene (Arg‐172‐Trp) associated with a progressive retinal degeneration. Ophthalmic Genetics, 19(3), 149–156. 10.1076/opge.19.3.149.2186 [DOI] [PubMed] [Google Scholar]
- Ekström, U. , Ponjavic, V. , Abrahamson, M. , Nilsson‐Ehle, P. , Andrëasson, S. , Stenström, I. , & Ehinger, B. (1998). Phenotypic expression of autosomal dominant retinitis pigmentosa in a Swedish family expressing a Phe‐211‐Leu variant of peripherin/RDS. Ophthalmic Genetics, 19(1), 27–37. 10.1076/opge.19.1.27.2179 [DOI] [PubMed] [Google Scholar]
- Ekström, U. , Ponjavic, V. , Andréasson, S. , Ehinger, B. , Nilsson‐Ehle, P. , & Abrahamson, M. (1998). Detection of alterations in all three exons of the peripherin/RDS gene in Swedish patients with retinitis pigmentosa using an efficient DGGE system. Molecular Pathology, 51(5), 287–291. 10.1136/mp.51.5.287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essilfie, J. O. , Sanfilippo, C. J. , & Sarraf, D. (2018). BULL'S eye maculopathy with mutations in RDS/PRPH2 and ROM‐1. Retinal Cases & Brief Reports, 12(Suppl 1), S55–S58. 10.1097/icb.0000000000000669 [DOI] [PubMed] [Google Scholar]
- Fakin, A. , Zupan, A. , Glavač, D. , & Hawlina, M. (2012). Combination of retinitis pigmentosa and hearing loss caused by a novel mutation in PRPH2 and a known mutation in GJB2: Importance for differential diagnosis of Usher syndrome. Vision Research, 75, 71–76. 10.1016/j.visres.2012.07.011 [DOI] [PubMed] [Google Scholar]
- Farrar, G. J. , Kenna, P. , Jordan, S. A. , Kumar‐Singh, R. , Humphries, M. M. , Sharp, E. M. , Sheils, D. , & Humphries, P. (1992). Autosomal dominant retinitis pigmentosa: A novel mutation at the peripherin/RDS locus in the original 6p‐linked pedigree. Genomics, 14(3), 805–807. 10.1016/s0888-7543(05)80193-4 [DOI] [PubMed] [Google Scholar]
- Farrar, G. J. , Kenna, P. , Jordan, S. A. , Kumar‐Singh, R. , Humphries, M. M. , Sharp, E. M. , Sheils, D. M. , & Humphries, P. (1991). A three‐base‐pair deletion in the peripherin‐RDS gene in one form of retinitis pigmentosa. Nature, 354(6353), 478–480. 10.1038/354478a0 [DOI] [PubMed] [Google Scholar]
- Feist, R. M. , White, M. F, Jr. , Skalka, H. , Stone, & E. M. (1994). Choroidal neovascularization in a patient with adult foveomacular dystrophy and a mutation in the retinal degeneration slow gene (Pro 210 Arg). American Journal of Ophthalmology, 118(2), 259–260. 10.1016/s0002-9394(14)72913-7 [DOI] [PubMed] [Google Scholar]
- Felbor, U. , Schilling, H. , & Weber, B. H. (1997). Adult vitelliform macular dystrophy is frequently associated with mutations in the peripherin/RDS gene. Human Mutation, 10(4), 301–309. [DOI] [PubMed] [Google Scholar]
- Fernandez‐San Jose, P. , Corton, M. , Blanco‐Kelly, F. , Avila‐Fernandez, A. , Lopez‐Martinez, M. A. , Sanchez‐Navarro, I. , Sanchez‐Alcudia, R. , Perez‐Carro, R. , Zurita, O. , Sanchez‐Bolivar, N. , Lopez‐Molina, M. I. , Garcia‐Sandoval, B. , Riveiro‐Alvarez, R. , & Ayuso, C. (2015). Targeted next‐generation sequencing improves the diagnosis of autosomal dominant Retinitis Pigmentosa in Spanish patients. Investigative Ophthalmology and Visual Science, 56(4), 2173–2182. 10.1167/iovs.14-16178 [DOI] [PubMed] [Google Scholar]
- Fishman, G. A. , Stone, E. , Gilbert, L. D. , Vandenburgh, K. , Sheffield, V. C. , & Heckenlively, J. R. (1994). Clinical features of a previously undescribed codon 216 (proline to serine) mutation in the peripherin/retinal degeneration slow gene in autosomal dominant retinitis pigmentosa. Ophthalmology, 101(8), 1409–1421. 10.1016/s0161-6420(94)31156-0 [DOI] [PubMed] [Google Scholar]
- Fishman, G. A. , Stone, E. M. , Alexander, K. R. , Gilbert, L. D. , Derlacki, D. J. , & Butler, N. S. (1997). Serine‐27‐phenylalanine mutation within the peripherin/RDS gene in a family with cone dystrophy. Ophthalmology, 104(2), 299–306. 10.1016/s0161-6420(97)30320-0 [DOI] [PubMed] [Google Scholar]
- Foote, K. G. , De la Huerta, I. , Gustafson, K. , Baldwin, A. , Zayit‐Soudry, S. , Rinella, N. , Porco, T. C. , Roorda, A. , & Duncan, J. L. (2019). Cone spacing correlates with retinal thickness and microperimetry in patients with inherited retinal degenerations. Investigative Ophthalmology and Visual Science, 60(4), 1234–1243. 10.1167/iovs.18-25688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis, P. J. , Schultz, D. W. , Gregory, A. M. , Schain, M. B. , Barra, R. , Majewski, J. , Ott, J. , Acott, T. , Weleber, R. G. , & Klein, M. L. (2005). Genetic and phenotypic heterogeneity in pattern dystrophy. British Journal of Ophthalmology, 89(9), 1115–1119. 10.1136/bjo.2004.062695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamundi, M. J. , Hernan, I. , Muntanyola, M. , Trujillo, M. J. , García‐Sandoval, B. , Ayuso, C. , Baiget, M. , & Carballo, M. (2007). High prevalence of mutations in peripherin/RDS in autosomal dominant macular dystrophies in a Spanish population. Molecular Vision, 13, 1031–1037. [PMC free article] [PubMed] [Google Scholar]
- Gao, F. J. , Li, J. K. , Chen, H. , Hu, F. Y. , Zhang, S. H. , Qi, Y. H. , Xu, P. , Wang, D. D. , Wang, L. S. , Chang, Q. , Zhang, Y. J. , Liu, W. , Li, W. , Wang, M. , Chen, F. , Xu, G. Z. , & Wu, J. H. (2019). Genetic and clinical findings in a large cohort of Chinese patients with suspected Retinitis Pigmentosa. Ophthalmology, 126(11), 1549–1556. 10.1016/j.ophtha.2019.04.038 [DOI] [PubMed] [Google Scholar]
- Giacalone, J. C. , Wiley, L. A. , Burnight, E. R. , Songstad, A. E. , Mullins, R. F. , Stone, E. M. , & Tucker, B. A. (2016). Concise review: Patient‐specific stem cells to interrogate inherited eye disease. Stem Cells Translational Medicine, 5(2), 132–140. 10.5966/sctm.2015-0206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glöckle, N. , Kohl, S. , Mohr, J. , Scheurenbrand, T. , Sprecher, A. , Weisschuh, N. , Bernd, A. , Rudolph, G. , Schubach, M. , Poloschek, C. , Zrenner, E. , Biskup, S. , Berger, W. , Wissinger, B. , & Neidhardt, J. (2014). Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. European Journal of Human Genetics, 22(1), 99–104. 10.1038/ejhg.2013.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gocho, K. , Akeo, K. , Itoh, N. , Kameya, S. , Hayashi, T. , Katagiri, S. , Gekka, T. , Ohkuma, Y. , Tsuneoka, H. , & Takahashi, H. (2016). High‐resolution adaptive optics retinal image analysis at early stage central areolar choroidal dystrophy with PRPH2 mutation. Ophthalmic Surgery, Lasers and Imaging Retina, 47(12), 1115–1126. 10.3928/23258160-20161130-05 [DOI] [PubMed] [Google Scholar]
- Goldberg, A. F. , Loewen, C. J. , & Molday, R. S. (1998). Cysteine residues of photoreceptor peripherin/rds: Role in subunit assembly and autosomal dominant retinitis pigmentosa. Biochemistry, 37(2), 680–685. 10.1021/bi972036i [DOI] [PubMed] [Google Scholar]
- Gorin, M. B. , Jackson, K. E. , Ferrell, R. E. , Sheffield, V. C. , Jacobson, S. G. , Gass, J. D. , Mitchell, E. , & Stone, E. M. (1995). A peripherin/retinal degeneration slow mutation (Pro‐210‐Arg) associated with macular and peripheral retinal degeneration. Ophthalmology, 102(2), 246–255. 10.1016/s0161-6420(95)31029-9 [DOI] [PubMed] [Google Scholar]
- Grover, S. , Fishman, G. A. , & Stone, E. M. (2002). Atypical presentation of pattern dystrophy in two families with peripherin/RDS mutations. Ophthalmology, 109(6), 1110–1117. 10.1016/s0161-6420(02)01029-1 [DOI] [PubMed] [Google Scholar]
- Grüning, G. , Millan, J. M. , Meins, M. , Beneyto, M. , Caballero, M. , Apfelstedt‐Sylla, E. , Bosch, R. , Zrenner, E. , Prieto, F. , & Gal, A. (1994). Mutations in the human peripherin/RDS gene associated with autosomal dominant retinitis pigmentosa. Human Mutation, 3(3), 321–323. 10.1002/humu.1380030326 [DOI] [PubMed] [Google Scholar]
- Hanany, M. , Rivolta, C. , & Sharon, D. (2020). Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proceedings of the National Academy of Sciences of the United States of America, 117(5), 2710–2716. 10.1073/pnas.1913179117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanany, M. , & Sharon, D. (2019). Allele frequency analysis of variants reported to cause autosomal dominant inherited retinal diseases question the involvement of 19% of genes and 10% of reported pathogenic variants. Journal of Medical Genetics, 56(8), 536–542. 10.1136/jmedgenet-2018-105971 [DOI] [PubMed] [Google Scholar]
- Hiatt, J. B. , Pritchard, C. C. , Salipante, S. J. , O'Roak, B. J. , & Shendure, J. (2013). Single molecule molecular inversion probes for targeted, high‐accuracy detection of low‐frequency variation. Genome Research, 23(5), 843–854. 10.1101/gr.147686.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosono, K. , Nishina, S. , Yokoi, T. , Katagiri, S. , Saitsu, H. , Kurata, K. , Miyamichi, D. , Hikoya, A. , Mizobuchi, K. , Nakano, T. , Minoshima, S. , Fukami, M. , Kondo, H. , Sato, M. , Hayashi, T. , Azuma, N. , & Hotta, Y. (2018). Molecular diagnosis of 34 Japanese families with Leber congenital amaurosis using targeted next generation sequencing. Scientific Reports, 8(1), 8279. 10.1038/s41598-018-26524-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyng, C. B. , Heutink, P. , Testers, L. , Pinckers, A. , Deutman, A. F. , & Oostra, B. A. (1996). Autosomal dominant central areolar choroidal dystrophy caused by a mutation in codon 142 in the peripherin/RDS gene. American Journal of Ophthalmology, 121(6), 623–629. 10.1016/s0002-9394(14)70627-0 [DOI] [PubMed] [Google Scholar]
- Huang, L. , Li, S. , Xiao, X. , Jia, X. , Wang, P. , Guo, X. , & Zhang, Q. (2013). Screening for variants in 20 genes in 130 unrelated patients with cone‐rod dystrophy. Molecular Medicine Reports, 7(6), 1779–1785. 10.3892/mmr.2013.1415 [DOI] [PubMed] [Google Scholar]
- Jacobson, S. G. , Cideciyan, A. V. , Maguire, A. M. , Bennett, J. , Sheffield, V. C. , & Stone, E. M. (1996). Preferential rod and cone photoreceptor abnormalities in heterozygotes with point mutations in the RDS gene. Experimental Eye Research, 63(5), 603–608. 10.1006/exer.1996.0152 [DOI] [PubMed] [Google Scholar]
- Jacobson, S. G. , McGuigan, D. B., 3rd , Sumaroka, A. , Roman, A. J. , Gruzensky, M. L. , Sheplock, R. , Palma, J. , Schwartz, S. B. , Aleman, T. S. , & Cideciyan, A. V. (2016). Complexity of the Class B phenotype in autosomal dominant Retinitis Pigmentosa due to rhodopsin mutations. Investigative Ophthalmology and Visual Science, 57(11), 4847–4858. 10.1167/iovs.16-19890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jespersgaard, C. , Fang, M. , Bertelsen, M. , Dang, X. , Jensen, H. , Chen, Y. , Bech, N. , Dai, L. , Rosenberg, T. , Zhang, J. , Møller, L. B. , Tümer, Z. , Brøndum‐Nielsen, K. , & Grønskov, K. (2019). Molecular genetic analysis using targeted NGS analysis of 677 individuals with retinal dystrophy. Scientific Reports, 9(1), 1219. 10.1038/s41598-018-38007-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, Z. B. , Mandai, M. , Yokota, T. , Higuchi, K. , Ohmori, K. , Ohtsuki, F. , Takakura, S. , Itabashi, T. , Wada, Y. , Akimoto, M. , Ooto, S. , Suzuki, T. , Hirami, Y. , Ikeda, H. , Kawagoe, N. , Oishi, A. , Ichiyama, S. , Takahashi, M. , Yoshimura, N. , & Kosugi, S. (2008). Identifying pathogenic genetic background of simplex or multiplex retinitis pigmentosa patients: A large scale mutation screening study. Journal of Medical Genetics, 45(7), 465–472. 10.1136/jmg.2007.056416 [DOI] [PubMed] [Google Scholar]
- Jones, K. D. , Wheaton, D. K. , Bowne, S. J. , Sullivan, L. S. , Birch, D. G. , Chen, R. , & Daiger, S. P. (2017). Next‐generation sequencing to solve complex inherited retinal dystrophy: A case series of multiple genes contributing to disease in extended families. Molecular Vision, 23, 470–481. [PMC free article] [PubMed] [Google Scholar]
- Kajiwara, K. , Berson, E. L. , & Dryja, T. P. (1994). Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science, 264(5165), 1604–1608. 10.1126/science.8202715 [DOI] [PubMed] [Google Scholar]
- Kajiwara, K. , Hahn, L. B. , Mukai, S. , Travis, G. H. , Berson, E. L. , & Dryja, T. P. (1991). Mutations in the human retinal degeneration slow gene in autosomal dominant retinitis pigmentosa. Nature, 354(6353), 480–483. 10.1038/354480a0 [DOI] [PubMed] [Google Scholar]
- Kajiwara, K. , Sandberg, M. A. , Berson, E. L. , & Dryja, T. P. (1993). A null mutation in the human peripherin/RDS gene in a family with autosomal dominant retinitis punctata albescens. Nature Genetics, 3(3), 208–212. 10.1038/ng0393-208 [DOI] [PubMed] [Google Scholar]
- Kalyanasundaram, T. S. , Black, G. C. , O'Sullivan, J. , & Bishop, P. N. (2009). A novel peripherin/RDS mutation resulting in a retinal dystrophy with phenotypic variation. Eye (Lond), 23(1), 237–239. 10.1038/eye.2008.33 [DOI] [PubMed] [Google Scholar]
- Katagiri, S. , Hayashi, T. , Mizobuchi, K. , Yoshitake, K. , Iwata, T. , & Nakano, T. (2018). Autosomal dominant retinitis pigmentosa with macular involvement associated with a disease haplotype that included a novel PRPH2 variant (p.Cys250Gly). Ophthalmic Genetics, 39(3), 357–365. 10.1080/13816810.2018.1459737 [DOI] [PubMed] [Google Scholar]
- Keen, T. J. , Inglehearn, C. F. , Kim, R. , Bird, A. C. , & Bhattacharya, S. (1994). Retinal pattern dystrophy associated with a 4 bp insertion at codon 140 in the RDS‐peripherin gene. Human Molecular Genetics, 3(2), 367–368. 10.1093/hmg/3.2.367 [DOI] [PubMed] [Google Scholar]
- Keilhauer, C. N. , Meigen, T. , Stöhr, H. , & Weber, B. H. (2006). Late‐onset central areolar choroidal dystrophy caused by a heterozygous frame‐shift mutation affecting codon 307 of the peripherin/RDS gene. Ophthalmic Genetics, 27(4), 139–144. 10.1080/13816810600976822 [DOI] [PubMed] [Google Scholar]
- Keilhauer, C. N. , Meigen, T. , & Weber, B. H. (2006). Clinical findings in a multigeneration family with autosomal dominant central areolar choroidal dystrophy associated with an Arg195Leu mutation in the peripherin/RDS gene. Archives of Ophthalmology, 124(7), 1020–1027. 10.1001/archopht.124.7.1020 [DOI] [PubMed] [Google Scholar]
- Kemp, C. M. , Jacobson, S. G. , Cideciyan, A. V. , Kimura, A. E. , Sheffield, V. C. , & Stone, E. M. (1994). RDS gene mutations causing retinitis pigmentosa or macular degeneration lead to the same abnormality in photoreceptor function. Investigative Ophthalmology and Visual Science, 35(8), 3154–3162. [PubMed] [Google Scholar]
- Kersten, E. , Geerlings, M. J. , Pauper, M. , Corominas, J. , Bakker, B. , Altay, L. , Fauser, S. , de Jong, E. K. , Hoyng, C. B. , & den Hollander, A. I. (2018). Genetic screening for macular dystrophies in patients clinically diagnosed with dry age‐related macular degeneration. Clinical Genetics, 94(6), 569–574. 10.1111/cge.13447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, A. O. (2019). Recessive pediatric‐onset cone‐rod dysfunction or dominant maculopathy in a consanguineous family harboring the peripherin mutation p.Arg220Gln. Ophthalmic Genetics, 40(1), 60–63. 10.1080/13816810.2019.1579346 [DOI] [PubMed] [Google Scholar]
- Khan, A. O. , Al Rashaed, S. , Neuhaus, C. , Bergmann, C. , & Bolz, H. J. (2016). Peripherin mutations cause a distinct form of recessive Leber congenital amaurosis and dominant phenotypes in asymptomatic parents heterozygous for the mutation. British Journal of Ophthalmology, 100(2), 209–215. 10.1136/bjophthalmol-2015-306844 [DOI] [PubMed] [Google Scholar]
- Khan, M., Cornelis, S. S. , Pozo‐Valero, M. D. , Whelan, L. , Runhart, E. H. , Mishra, K. , Bults, F. , AlSwaiti, Y. , AlTalbishi, A. , De Baere, E. , Banfi, S. , Banin, E. , Bauwens, M. , Ben‐Yosef, T. , Boon, C. , van den Born, L. I. , Defoort, S. , Devos, A. , Dockery, A. , … Cremers, F. (2020). Resolving the dark matter of ABCA4 for 1054 Stargardt disease probands through integrated genomics and transcriptomics. Genetics in Medicine, 22(7), 1235–1246. 10.1038/s41436-020-0787-4 [DOI] [PubMed] [Google Scholar]
- Khoubian, F. J. , Shakin, E. P. , Tantri, A. , Kim, D. Y. , Edwards, A. O. , & Donoso, L. A. (2005). Autosomal dominant pattern dystrophy: Identification of a novel splice site mutation in the peripherin/RDS gene. Retina, 25(8), 999–1004. 10.1097/00006982-200512000-00008 [DOI] [PubMed] [Google Scholar]
- Kikawa, E. , Nakazawa, M. , Chida, Y. , Shiono, T. , & Tamai, M. (1994). A novel mutation (Asn244Lys) in the peripherin/RDS gene causing autosomal dominant retinitis pigmentosa associated with bull's‐eye maculopathy detected by nonradioisotopic SSCP. Genomics, 20(1), 137–139. 10.1006/geno.1994.1142 [DOI] [PubMed] [Google Scholar]
- Kim, C. , Kim, K. J. , Bok, J. , Lee, E. J. , Kim, D. J. , Oh, J. H. , Park, S. P. , Shin, J. Y. , Lee, J. Y. , & Yu, H. G. (2012). Microarray‐based mutation detection and phenotypic characterization in Korean patients with retinitis pigmentosa. Molecular Vision, 18, 2398–2410. [PMC free article] [PubMed] [Google Scholar]
- Kim, R. Y. , Dollfus, H. , Keen, T. J. , Fitzke, F. W. , Arden, G. B. , Bhattacharya, S. S. , & Bird, A. C. (1995). Autosomal dominant pattern dystrophy of the retina associated with a 4‐base pair insertion at codon 140 in the peripherin/RDS gene. Archives of Ophthalmology, 113(4), 451–455. 10.1001/archopht.1995.01100040067029 [DOI] [PubMed] [Google Scholar]
- Kircher, M. , Witten, D. M. , Jain, P. , O'Roak, B. J. , Cooper, G. M. , & Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics, 46(3), 310–315. 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitiratschky, V. B. , Glöckner, C. J. , & Kohl, S. (2011). Mutation screening of the GUCA1B gene in patients with autosomal dominant cone and cone rod dystrophy. Ophthalmic Genetics, 32(3), 151–155. 10.3109/13816810.2011.559650 [DOI] [PubMed] [Google Scholar]
- Klevering, B. J. , van Driel, M. , van Hogerwou, A. J. , van De Pol, D. J. , Deutman, A. F. , Pinckers, A. J. , Cremers, F. P. , & Hoyng, C. B. (2002). Central areolar choroidal dystrophy associated with dominantly inherited drusen. The British Journal of Ophthalmology, 86(1), 91–96. 10.1136/bjo.86.1.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl, S. , Christ‐Adler, M. , Apfelstedt‐Sylla, E. , Kellner, U. , Eckstein, A. , Zrenner, E. , & Wissinger, B. (1997). RDS/peripherin gene mutations are frequent causes of central retinal dystrophies. Journal of Medical Genetics, 34(8), 620–626. 10.1136/jmg.34.8.620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl, S. , Kitiratschky, V. , Papke, M. , Schaich, S. , Sauer, A. , & Wissinger, B. (2012). Genes and mutations in autosomal dominant cone and cone‐rod dystrophy. Advances in Experimental Medicine and Biology, 723, 337–343. 10.1007/978-1-4614-0631-0_44 [DOI] [PubMed] [Google Scholar]
- Kumar, P. , Henikoff, S. , & Ng, P. C. (2009). Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nature Protocols, 4(7), 1073–1081. 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- Lam, B. L. , Vandenburgh, K. , Sheffield, V. C. , & Stone, E. M. (1995). Retinitis pigmentosa associated with a dominant mutation in codon 46 of the peripherin/RDS gene (arginine‐46‐stop). American Journal of Ophthalmology, 119(1), 65–71. 10.1016/s0002-9394(14)73815-2 [DOI] [PubMed] [Google Scholar]
- Lee, C. S. , & Leys, M. (2020). A family affected by novel C213W mutation in PRPH2: Long‐term follow‐up of CNV secondary to pattern dystrophy. Ophthalmic Surg Lasers Imaging Retina, 51(6), 354–362. 10.3928/23258160-20200603-06 [DOI] [PubMed] [Google Scholar]
- Lee, K. , Berg, J. S. , Milko, L. , Crooks, K. , Lu, M. , Bizon, C. , Owen, P. , Wilhelmsen, K. C. , Weck, K. E. , Evans, J. P. , & Garg, S. (2015). High diagnostic yield of whole exome sequencing in participants with retinal dystrophies in a clinical ophthalmology setting. American Journal of Ophthalmology, 160(2), 354–363. 10.1016/j.ajo.2015.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy, B. P. , Kailasanathan, A. , De Laey, J. J. , Black, G. C. , & Manson, F. D. (2007). Intrafamilial phenotypic variability in families with RDS mutations: Exclusion of ROM1 as a genetic modifier for those with retinitis pigmentosa. British Journal of Ophthalmology, 91(1), 89–93. 10.1136/bjo.2006.101915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, K. P. , Yip, S. P. , Cheung, S. C. , Leung, K. W. , Lam, S. T. , & To, C. H. (2009). Novel PRPF31 and PRPH2 mutations and co‐occurrence of PRPF31 and RHO mutations in Chinese patients with retinitis pigmentosa. Archives of Ophthalmology, 127(6), 784–790. 10.1001/archophthalmol.2009.112 [DOI] [PubMed] [Google Scholar]
- Maertz, J. , Gloeckle, N. , Nentwich, M. , & Rudolph, G. (2015). [Genotype‐phenotype correlation in patients with PRPH2‐mutations]. Klinische Monatsblätter für Augenheilkunde, 232, 266–274. 10.1055/s-0035-1545702 [DOI] [PubMed] [Google Scholar]
- Manes, G. , Guillaumie, T. , Vos, W. L. , Devos, A. , Audo, I. , Zeitz, C. , Marquette, V. , Zanlonghi, X. , Defoort‐Dhellemmes, S. , Puech, B. , Said, S. M. , Sahel, J. A. , Odent, S. , Dollfus, H. , Kaplan, J. , Dufier, J. L. , Le Meur, G. , Weber, M. , Faivre, L. , … Hamel, C. P. (2015). High prevalence of PRPH2 in autosomal dominant retinitis pigmentosa in france and characterization of biochemical and clinical features. American Journal of Ophthalmology, 159(2), 302–314. 10.1016/j.ajo.2014.10.033 [DOI] [PubMed] [Google Scholar]
- Martin‐Merida, I. , Aguilera‐Garcia, D. , Fernandez‐San Jose, P. , Blanco‐Kelly, F. , Zurita, O. , Almoguera, B. , Garcia‐Sandoval, B. , Avila‐Fernandez, A. , Arteche, A. , Minguez, P. , Carballo, M. , Corton, M. , & Ayuso, C. (2018). Toward the mutational landscape of autosomal dominant Retinitis Pigmentosa: A comprehensive analysis of 258 Spanish families. Investigative Ophthalmology and Visual Science, 59(6), 2345–2354. 10.1167/iovs.18-23854 [DOI] [PubMed] [Google Scholar]
- Meins, M. , Grüning, G. , Blankenagel, A. , Krastel, H. , Reck, B. , Fuchs, S. , Schwinger, E. , & Gal, A. (1993). Heterozygous 'null allele' mutation in the human peripherin/RDS gene. Human Molecular Genetics, 2(12), 2181–2182. 10.1093/hmg/2.12.2181 [DOI] [PubMed] [Google Scholar]
- Messchaert, M. , Haer‐Wigman, L. , Khan, M. I. , Cremers, F. P. M. , & Collin, R. W. J. (2018). EYS mutation update: In silico assessment of 271 reported and 26 novel variants in patients with retinitis pigmentosa. Human Mutation, 39(2), 177–186. 10.1002/humu.23371 [DOI] [PubMed] [Google Scholar]
- Meunier, I. , Sénéchal, A. , Dhaenens, C. M. , Arndt, C. , Puech, B. , Defoort‐Dhellemmes, S. , Manes, G. , Chazalette, D. , Mazoir, E. , Bocquet, B. , & Hamel, C. P. (2011). Systematic screening of BEST1 and PRPH2 in juvenile and adult vitelliform macular dystrophies: A rationale for molecular analysis. Ophthalmology, 118(6), 1130–1136. 10.1016/j.ophtha.2010.10.010 [DOI] [PubMed] [Google Scholar]
- Michaelides, M. , Holder, G. E. , Bradshaw, K. , Hunt, D. M. , & Moore, A. T. (2005). Cone‐rod dystrophy, intrafamilial variability, and incomplete penetrance associated with the R172W mutation in the peripherin/RDS gene. Ophthalmology, 112(9), 1592–1598. 10.1016/j.ophtha.2005.04.004 [DOI] [PubMed] [Google Scholar]
- Miyata, M. , Oishi, A. , Oishi, M. , Hasegawa, T. , Ikeda, H. O. , & Tsujikawa, A. (2018). Long‐term efficacy and safety of anti‐VEGF therapy in retinitis pigmentosa: A case report. BMC Ophthalmology, 18(1), 248. 10.1186/s12886-018-0914-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki, M. , Ikeda, Y. , Yonemitsu, Y. , Goto, Y. , Kohno, R. , Murakami, Y. , Inoue, M. , Ueda, Y. , Hasegawa, M. , Tobimatsu, S. , Sueishi, K. , & Ishibashi, T. (2008). Synergistic neuroprotective effect via simian lentiviral vector‐mediated simultaneous gene transfer of human pigment epithelium‐derived factor and human fibroblast growth factor‐2 in rodent models of retinitis pigmentosa. Journal of Gene Medicine, 10(12), 1273–1281. 10.1002/jgm.1257 [DOI] [PubMed] [Google Scholar]
- Moshfeghi, D. M. , Yang, Z. , Faulkner, N. D. , Karan, G. , Thirumalaichary, S. , Pearson, E. , Zhao, Y. , Tsai, T. , & Zhang, K. (2006). Choroidal neovascularization in patients with adult‐onset foveomacular dystrophy caused by mutations in the RDS/peripherin gene. Advances in Experimental Medicine and Biology, 572, 35–40. 10.1007/0-387-32442-9_6 [DOI] [PubMed] [Google Scholar]
- Nakazawa, M. , Kikawa, E. , Kamio, K. , Chida, Y. , Shiono, T. , & Tamai, M. (1994). Ocular findings in patients with autosomal dominant Retinitis Pigmentosa and transversion mutation in codon 244 (Asn244Lys) of the Peripherin/RDS gene. Archives of Ophthalmology, 112(12), 1567–1573. 10.1001/archopht.1994.01090240073028 [DOI] [PubMed] [Google Scholar]
- Nakazawa, M. , Naoi, N. , Wada, Y. , Nakazaki, S. , Maruiwa, F. , Sawada, A. , & Tamai, M. (1996). Autosomal dominant cone‐rod dystrophy associated with a Val200Glu mutation of the peripherin/RDS gene. Retina, 16(5), 405–410. 10.1097/00006982-199616050-00007 [DOI] [PubMed] [Google Scholar]
- Nakazawa, M. , Wada, Y. , & Tamai, M. (1995). Macular dystrophy associated with monogenic Arg172Trp mutation of the peripherin/RDS gene in a Japanese family. Retina, 15(6), 518–523. 10.1097/00006982-199515060-00011 [DOI] [PubMed] [Google Scholar]
- Nanda, A. , McClements, M. E. , Clouston, P. , Shanks, M. E. , & MacLaren, R. E. (2019). The location of exon 4 mutations in RP1 raises challenges for genetic counseling and gene therapy. American Journal of Ophthalmology, 202, 23–29. 10.1016/j.ajo.2019.01.027 [DOI] [PubMed] [Google Scholar]
- Neveling, K. , Collin, R. W. , Gilissen, C. , van Huet, R. A. , Visser, L. , Kwint, M. P. , Gijsen, S. J. , Zonneveld, M. N. , Wieskamp, N. , de Ligt, J. , Siemiatkowska, A. M. , Hoefsloot, L. H. , Buckley, M. F. , Kellner, U. , Branham, K. E. , den Hollander, A. I. , Hoischen, A. , Hoyng, C. , Klevering, B. J. , … Scheffer, H. (2012). Next‐generation genetic testing for retinitis pigmentosa. Human Mutation, 33(6), 963–972. 10.1002/humu.22045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neveling, K. , Mensenkamp, A. R. , Derks, R. , Kwint, M. , Ouchene, H. , Steehouwer, M. , van Lier, B. , Bosgoed, E. , Rikken, A. , Tychon, M. , Zafeiropoulou, D. , Castelein, S. , Hehir‐Kwa, J. , Tjwan Thung, D. , Hofste, T. , Lelieveld, S. H. , Bertens, S. M. , Adan, I. B. , Eijkelenboom, A. , … Hoischen, A. (2017). BRCA testing by single‐molecule molecular inversion probes. Clinical Chemistry, 63(2), 503–512. 10.1373/clinchem.2016.263897 [DOI] [PubMed] [Google Scholar]
- Nichols, B. E. , Drack, A. V. , Vandenburgh, K. , Kimura, A. E. , Sheffield, V. C. , & Stone, E. M. (1993). A 2 base pair deletion in the RDS gene associated with butterfly‐shaped pigment dystrophy of the fovea. Human Molecular Genetics, 2(8), 1347. 10.1093/hmg/2.8.1347-a [DOI] [PubMed] [Google Scholar]
- Nichols, B. E. , Sheffield, V. C. , Vandenburgh, K. , Drack, A. V. , Kimura, A. E. , & Stone, E. M. (1993). Butterfly‐shaped pigment dystrophy of the fovea caused by a point mutation in codon 167 of the RDS gene. Nature Genetics, 3(3), 202–207. 10.1038/ng0393-202 [DOI] [PubMed] [Google Scholar]
- Nishiguchi, K. M. , & Rivolta, C. (2012). Genes associated with retinitis pigmentosa and allied diseases are frequently mutated in the general population. PLOS One, 7(7), e41902. 10.1371/journal.pone.0041902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nour, M. , Ding, X. Q. , Stricker, H. , Fliesler, S. J. , & Naash, M. I. (2004). Modulating expression of peripherin/rds in transgenic mice: Critical levels and the effect of overexpression. Investigative Ophthalmology and Visual Science, 45(8), 2514–2521. 10.1167/iovs.04-0065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nour, M. , Fliesler, S. J. , & Naash, M. I. (2008). Genetic supplementation of RDS alleviates a loss‐of‐function phenotype in C214S model of retinitis pigmentosa. Advances in Experimental Medicine and Biology, 613, 129–138. 10.1007/978-0-387-74904-4_14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi, M. , Oishi, A. , Gotoh, N. , Ogino, K. , Higasa, K. , Iida, K. , Makiyama, Y. , Morooka, S. , Matsuda, F. , & Yoshimura, N. (2014). Comprehensive molecular diagnosis of a large cohort of Japanese retinitis pigmentosa and Usher syndrome patients by next‐generation sequencing. Investigative Ophthalmology and Visual Science, 55(11), 7369–7375. 10.1167/iovs.14-15458 [DOI] [PubMed] [Google Scholar]
- Öner, A. (2018). Stem cell treatment in retinal diseases: Recent developments. Turkish Journal of Ophthalmology, 48(1), 33–38. 10.4274/tjo.89972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajic, B. , Weigell‐Weber, M. , Schipper, I. , Kryenbühl, C. , Büchi, E. R. , Spiegel, R. , & Hergersberg, M. (2006). A novel complex mutation event in the peripherin/RDS gene in a family with retinal pattern dystrophy. Retina, 26(8), 947–953. 10.1097/01.iae.0000250010.60908.e3 [DOI] [PubMed] [Google Scholar]
- Palfi, A. , Ader, M. , Kiang, A. S. , Millington‐Ward, S. , Clark, G. , O'Reilly, M. , Mcmahon, H. P. , Kenna, P. F. , Humphries, P. , & Farrar, G. J. (2006). RNAi‐based suppression and replacement of rds‐peripherin in retinal organotypic culture. Human Mutation, 27(3), 260–268. 10.1002/humu.20287 [DOI] [PubMed] [Google Scholar]
- Palma, M. M. D. , Martin, D. , Salles, M. V. , Motta, F. L. T. , Abujamra, S. , & Sallum, J. M. F. (2019). Retinal dystrophies and variants in PRPH2. Arquivos Brasileiros de Oftalmologia, 82(2), 158–160. 10.5935/0004-2749.20190033 [DOI] [PubMed] [Google Scholar]
- Passerini, I. , Sodi, A. , Giambene, B. , Menchini, U. , & Torricelli, F. (2007). Phenotypic intrafamilial variability associated with S212G mutation in the RDS/peripherin gene. European Journal of Ophthalmology, 17(6), 1000–1003. 10.1177/112067210701700624 [DOI] [PubMed] [Google Scholar]
- Patel, N. , Aldahmesh, M. A. , Alkuraya, H. , Anazi, S. , Alsharif, H. , Khan, A. O. , Sunker, A. , Al‐Mohsen, S. , Abboud, E. B. , Nowilaty, S. R. , Alowain, M. , Al‐Zaidan, H. , Al‐Saud, B. , Alasmari, A. , Abdel‐Salam, G. M. , Abouelhoda, M. , Abdulwahab, F. M. , Ibrahim, N. , Naim, E. , … Alkuraya, F. S. (2016). Expanding the clinical, allelic, and locus heterogeneity of retinal dystrophies. Genetics in Medicine, 18(6), 554–562. 10.1038/gim.2015.127 [DOI] [PubMed] [Google Scholar]
- Payne, A. M. , Downes, S. M. , Bessant, D. A. , Bird, A. C. , & Bhattacharya, S. S. (1998). Founder effect, seen in the British population, of the 172 peripherin/RDS mutation‐and further refinement of genetic positioning of the peripherin/RDS gene. American Journal of Human Genetics, 62(1), 192–195. 10.1086/301679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrs‐Silva, H. , Yasumura, D. , Matthes, M. T. , LaVail, M. M. , Lewin, A. S. , & Hauswirth, W. W. (2012). Suppression of rds expression by siRNA and gene replacement strategies for gene therapy using rAAV vector. Advances in Experimental Medicine and Biology, 723, 215–223. 10.1007/978-1-4614-0631-0_29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poloschek, C. M. , Bach, M. , Lagrèze, W. A. , Glaus, E. , Lemke, J. R. , Berger, W. , & Neidhardt, J. (2010). ABCA4 and ROM1: Implications for modification of the PRPH2‐associated macular dystrophy phenotype. Investigative Ophthalmology and Visual Science, 51(8), 4253–4265. 10.1167/iovs.09-4655 [DOI] [PubMed] [Google Scholar]
- Ramkumar, H. L. , Gudiseva, H. V. , Kishaba, K. T. , Suk, J. J. , Verma, R. , Tadimeti, K. , Thorson, J. A. , & Ayyagari, R. (2017). A report on molecular diagnostic testing for inherited retinal dystrophies by targeted genetic analyses. Genetic Testing and Molecular Biomarkers, 21(2), 66–73. 10.1089/gtmb.2016.0251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves, M. J. , Goetz, K. E. , Guan, B. , Ullah, E. , Blain, D. , Zein, W. M. , Tumminia, S. J. , & Hufnagel, R. B. (2020). Genotype‐phenotype associations in a large PRPH2‐related retinopathy cohort. Human Mutation, 41, 1528–1539. 10.1002/humu.24065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reig, C., Serra, A. , Gean, E. , Vidal, M. , Arumí, J. , De la Calzada, M. D. , Antich, J. , & Carballo, M. (1995). A point mutation in the RDS‐peripherin gene in a Spanish family with central areolar choroidal dystrophy. Ophthalmic Genetics, 16(2), 39–44. 10.3109/13816819509056911 [DOI] [PubMed] [Google Scholar]
- Renner, A. B. , Fiebig, B. S. , Weber, B. H. , Wissinger, B. , Andreasson, S. , Gal, A. , Cropp, E. , Kohl, S. , & Kellner, U. (2009). Phenotypic variability and long‐term follow‐up of patients with known and novel PRPH2/RDS gene mutations. American Journal of Ophthalmology, 147(3), 518–530. e511. 10.1016/j.ajo.2008.09.007 [DOI] [PubMed] [Google Scholar]
- Renner, A. B. , Tillack, H. , Kraus, H. , Kohl, S. , Wissinger, B. , Mohr, N. , Weber, B. H. , Kellner, U. , & Foerster, M. H. (2004). Morphology and functional characteristics in adult vitelliform macular dystrophy. Retina, 24(6), 929–939. 10.1097/00006982-200412000-00014 [DOI] [PubMed] [Google Scholar]
- Rentzsch, P. , Witten, D. , Cooper, G. M. , Shendure, J. , & Kircher, M. (2019). CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Research, 47(D1), D886–d894. 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex, T. S. , Allocca, M. , Domenici, L. , Surace, E. M. , Maguire, A. M. , Lyubarsky, A. , Cellerino, A. , Bennett, J. , & Auricchio, A. (2004). Systemic but not intraocular Epo gene transfer protects the retina from light‐and genetic‐induced degeneration. Molecular Therapy, 10(5), 855–861. 10.1016/j.ymthe.2004.07.027 [DOI] [PubMed] [Google Scholar]
- Rex, T. S. , Wong, Y. , Kodali, K. , & Merry, S. (2009). Neuroprotection of photoreceptors by direct delivery of erythropoietin to the retina of the retinal degeneration slow mouse. Experimental Eye Research, 89(5), 735–740. 10.1016/j.exer.2009.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee, K. D. , Ruiz, A. , Duncan, J. L. , Hauswirth, W. W. , Lavail, M. M. , Bok, D. , & Yang, X. J. (2007). Molecular and cellular alterations induced by sustained expression of ciliary neurotrophic factor in a mouse model of retinitis pigmentosa. Investigative Ophthalmology and Visual Science, 48(3), 1389–1400. 10.1167/iovs.06-0677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & ACMG Laboratory Quality Assurance, C. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. C. , & Creel, D. J. (1995). Pattern dystrophy and retinitis pigmentosa caused by a peripherin/RDS mutation. Retina, 15(1), 68–72. 10.1097/00006982-199515010-00013 [DOI] [PubMed] [Google Scholar]
- Saga, M. , Mashima, Y. , Akeo, K. , Oguchi, Y. , Kudoh, J. , & Shimizu, N. (1993). A novel Cys‐214‐Ser mutation in the peripherin/RDS gene in a Japanese family with autosomal dominant retinitis pigmentosa. Human Genetics, 92(5), 519–521. 10.1007/bf00216463 [DOI] [PubMed] [Google Scholar]
- Salinas, R. Y. , Baker, S. A. , Gospe, S. M., 3rd , & Arshavsky, V. Y. (2013). A single valine residue plays an essential role in peripherin/rds targeting to photoreceptor outer segments. PLOS One, 8(1), e54292. 10.1371/journal.pone.0054292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallevelt, S. , de Koning, B. , Szklarczyk, R. , Paulussen, A. D. C. , de Die‐Smulders, C. E. M. , & Smeets, H. J. M. (2017). A comprehensive strategy for exome‐based preconception carrier screening. Genetics in Medicine, 19(5), 583–592. 10.1038/gim.2016.153 [DOI] [PubMed] [Google Scholar]
- Sangermano, R. , Khan, M. , Cornelis, S. S. , Richelle, V. , Albert, S. , Garanto, A. , Elmelik, D. , Qamar, R. , Lugtenberg, D. , van den Born, L. I. , Collin, R. , & Cremers, F. (2018). ABCA4 midigenes reveal the full splice spectrum of all reported noncanonical splice site variants in Stargardt disease. Genome Research, 28(1), 100–110. 10.1101/gr.226621.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz, P. , Abrahamson, M. , Eksandh, L. , Ponjavic, V. , & Andréasson, S. (2003). Macular appearance by means of OCT and electrophysiology in members of two families with different mutations in RDS (the peripherin/RDS gene). Acta Ophthalmologica Scandinavica, 81(5), 500–507. 10.1034/j.1600-0420.2003.00134.x [DOI] [PubMed] [Google Scholar]
- Schorderet, D. F. , Bernasconi, M. , Tiab, L. , Favez, T. , & Escher, P. (2014). IROme, a new high‐throughput molecular tool for the diagnosis of inherited retinal dystrophies—A price comparison with Sanger sequencing. Advances in Experimental Medicine and Biology, 801, 171–176. 10.1007/978-1-4614-3209-8_22 [DOI] [PubMed] [Google Scholar]
- Sears, J. E. , Aaberg, T. A., Sr. , Daiger, S. P. , & Moshfeghi, D. M. (2001). Splice site mutation in the peripherin/RDS gene associated with pattern dystrophy of the retina. American Journal of Ophthalmology, 132(5), 693–699. 10.1016/s0002-9394(01)01179-5 [DOI] [PubMed] [Google Scholar]
- Shankar, S. P. , Birch, D. G. , Ruiz, R. S. , Hughbanks‐Wheaton, D. K. , Sullivan, L. S. , Bowne, S. J. , Stone, E. M. , & Daiger, S. P. (2015). Founder effect of a c.828+3A>T splice site mutation in Peripherin 2 (PRPH2) causing autosomal dominant retinal dystrophies. JAMA Ophthalmol, 133(5), 511–517. 10.1001/jamaophthalmol.2014.6115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar, S. P. , Hughbanks‐Wheaton, D. K. , Birch, D. G. , Sullivan, L. S. , Conneely, K. N. , Bowne, S. J. , Stone, E. M. , & Daiger, S. P. (2016). Autosomal dominant retinal dystrophies caused by a founder splice site mutation, c.828+3A>T, in PRPH2 and protein Haplotypes in trans as modifiers. Investigative Ophthalmology and Visual Science, 57(2), 349–359. 10.1167/iovs.15-16965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonelli, F. , Testa, F. , Marini, V. , Interlandi, E. , Rossi, S. , Pognuz, D. R. , Virgili, G. , Garrè, C. , & Bandello, F. (2007). Intrafamilial clinical heterogeneity associated with a novel mutation of the retinal degeneration slow/peripherin gene. Ophthalmic Research, 39(5), 255–259. 10.1159/000108118 [DOI] [PubMed] [Google Scholar]
- Smailhodzic, D. , Fleckenstein, M. , Theelen, T. , Boon, C. J. , van Huet, R. A. , van de Ven, J. P. , Den Hollander, A. I. , Schmitz‐Valckenberg, S. , Hoyng, C. B. , Weber, B. H. , Holz, F. G. , & Klevering, B. J. (2011). Central areolar choroidal dystrophy (CACD) and age‐related macular degeneration (AMD): Differentiating characteristics in multimodal imaging. Investigative Ophthalmology and Visual Science, 52(12), 8908–8918. 10.1167/iovs.11-7926 [DOI] [PubMed] [Google Scholar]
- Sohocki, M. M. , Daiger, S. P. , Bowne, S. J. , Rodriquez, J. A. , Northrup, H. , Heckenlively, J. R. , Birch, D. G. , Mintz‐Hittner, H. , Ruiz, R. S. , Lewis, R. A. , Saperstein, D. A. , & Sullivan, L. S. (2001). Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Human Mutation, 17(1), 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souied, E. H. , Rozet, J. M. , Gerber, S. , Dufier, J. L. , Soubrane, G. , Coscas, G. , Munnich, A. , & Kaplan, J. (1998). Two novel missense mutations in the peripherin/RDS gene in two unrelated French patients with autosomal dominant retinitis pigmentosa. European Journal of Ophthalmology, 8(2), 98–101. [DOI] [PubMed] [Google Scholar]
- Stone, E. M. , Andorf, J. L. , Whitmore, S. S. , DeLuca, A. P. , Giacalone, J. C. , Streb, L. M. , Braun, T. A. , Mullins, R. F. , Scheetz, T. E. , Sheffield, V. C. , & Tucker, B. A. (2017). Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology, 124(9), 1314–1331. 10.1016/j.ophtha.2017.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strafella, C. , Caputo, V. , Pagliaroli, G. , Iozzo, N. , Campoli, G. , Carboni, S. , Peconi, C. , Galota, R. M. , Zampatti, S. , Minozzi, G. , Novelli, G. , Giardina, E. , & Cascella, R. (2019). NGS analysis for molecular diagnosis of Retinitis Pigmentosa (RP): Detection of a novel variant in PRPH2 gene. Genes (Basel), 10(10). 10.3390/genes10100792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom, S. P. , Gao, Y. Q. , Martinez, A. , Ortube, C. , Chen, Z. , Nelson, S. F. , Nusinowitz, S. , Farber, D. B. , & Gorin, M. B. (2012). Molecular diagnosis of putative Stargardt Disease probands by exome sequencing. BMC Medical Genetics, 13, 67. 10.1186/1471-2350-13-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan, L. S. , Bowne, S. J. , Birch, D. G. , Hughbanks‐Wheaton, D. , Heckenlively, J. R. , Lewis, R. A. , Garcia, C. A. , Ruiz, R. S. , Blanton, S. H. , Northrup, H. , Gire, A. I. , Seaman, R. , Duzkale, H. , Spellicy, C. J. , Zhu, J. , Shankar, S. P. , & Daiger, S. P. (2006). Prevalence of disease‐causing mutations in families with autosomal dominant retinitis pigmentosa: A screen of known genes in 200 families. Investigative Ophthalmology and Visual Science, 47(7), 3052–3064. 10.1167/iovs.05-1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan, L. S. , Bowne, S. J. , Reeves, M. J. , Blain, D. , Goetz, K. , Ndifor, V. , Vitez, S. , Wang, X. , Tumminia, S. J. , & Daiger, S. P. (2013). Prevalence of mutations in eyeGENE probands with a diagnosis of autosomal dominant retinitis pigmentosa. Investigative Ophthalmology and Visual Science, 54(9), 6255–6261. 10.1167/iovs.13-12605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, W. , Huang, L. , Xu, Y. , Xiao, X. , Li, S. , Jia, X. , Gao, B. , Wang, P. , Guo, X. , & Zhang, Q. (2015). Exome sequencing on 298 probands with early‐onset high myopia: Approximately one‐fourth show potential pathogenic mutations in RetNet genes. Investigative Ophthalmology and Visual Science, 56(13), 8365–8372. 10.1167/iovs.15-17555 [DOI] [PubMed] [Google Scholar]
- Takeuchi, K. , Nakazawa, M. , & Mizukoshi, S. (2008). Systemic administration of nilvadipine delays photoreceptor degeneration of heterozygous retinal degeneration slow (rds) mouse. Experimental Eye Research, 86(1), 60–69. 10.1016/j.exer.2007.09.008 [DOI] [PubMed] [Google Scholar]
- Testa, F. , Marini, V. , Rossi, S. , Interlandi, E. , Nesti, A. , Rinaldi, M. , Varano, M. , Garré, C. , & Simonelli, F. (2005). A novel mutation in the RDS gene in an Italian family with pattern dystrophy. British Journal of Ophthalmology, 89(8), 1066–1068. 10.1136/bjo.2004.064188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis, G. H. , Brennan, M. B. , Danielson, P. E. , Kozak, C. A. , & Sutcliffe, J. G. (1989). Identification of a photoreceptor‐specific mRNA encoded by the gene responsible for retinal degeneration slow (rds). Nature, 338(6210), 70–73. 10.1038/338070a0 [DOI] [PubMed] [Google Scholar]
- Travis, G. H. , Christerson, L. , Danielson, P. E. , Klisak, I. , Sparkes, R. S. , Hahn, L. B. , Dryja, T. P. , & Sutcliffe, J. G. (1991). The human retinal degeneration slow (RDS) gene: Chromosome assignment and structure of the mRNA. Genomics, 10(3), 733–739. 10.1016/0888-7543(91)90457-p [DOI] [PubMed] [Google Scholar]
- Travis, G. H. , Groshan, K. R. , Lloyd, M. , & Bok, D. (1992). Complete rescue of photoreceptor dysplasia and degeneration in transgenic retinal degeneration slow (rds) mice. Neuron, 9(1), 113–119. 10.1016/0896-6273(92)90226-4 [DOI] [PubMed] [Google Scholar]
- Trujillo, M. J. , Bueno, J. , Osorio, A. , Sanz, R. , Garcia‐Sandoval, B. , Ramos, C. , & Ayuso, C. (1998). Three novel RDS‐peripherin mutations (689delT, 857del17, G208D) in Spanish families affected with autosomal dominant retinal degenerations. Mutations in brief no. 147. Online. Human Mutation, 12(1), 70. [DOI] [PubMed] [Google Scholar]
- Trujillo, M. J. , Martinez‐Gimeno, M. , Giménez, A. , Lorda, I. , Bueno, J. , García‐Sandoval, B. , Ramos, C. , Carballo, M. , & Ayuso, C. (2001). Two novel mutations (Y141H; C214Y) and previously published mutation (R142W) in the RDS‐peripherin gene in autosomal dominant macular dystrophies in Spanish families. Human Mutation, 17(1), 80. [DOI] [PubMed] [Google Scholar]
- Trujillo Tiebas, M. J. , Giménez Pardo, A. , García Sandoval, B. , & Ayuso García, C. (2002). [Phenotypic variation in a family affected by autosomal dominant retinal dystrophy caused by the Gly208Asp mutation in the RDS peripherin gene]. Medicina Clínica (Barc), 118(18), 716. 10.1016/s0025-7753(02)72505-0 [DOI] [PubMed] [Google Scholar]
- Vaclavik, V. , Tran, H. V. , Gaillard, M. C. , Schorderet, D. F. , & Munier, F. L. (2012). Pattern dystrophy with high intrafamilial variability associated with Y141C mutation in the peripherin/RDS gene and successful treatment of subfoveal CNV related to multifocal pattern type with anti‐VEGF (ranibizumab) intravitreal injections. Retina, 32(9), 1942–1949. 10.1097/IAE.0b013e31824b32e4 [DOI] [PubMed] [Google Scholar]
- Van Cauwenbergh, C. , Coppieters, F. , Roels, D. , De Jaegere, S. , Flipts, H. , De Zaeytijd, J. , Walraedt, S. , Claes, C. , Fransen, E. , Van Camp, G. , Depasse, F. , Casteels, I. , de Ravel, T. , Leroy, B. P. , & De Baere, E. (2017). Mutations in splicing factor genes are a major cause of autosomal dominant Retinitis Pigmentosa in Belgian families. PLOS One, 12(1), e0170038. 10.1371/journal.pone.0170038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Lith‐Verhoeven, J. J. , van den Helm, B. , Deutman, A. F. , Bergen, A. A. , Cremers, F. P. , Hoyng, C. B. , & de Jong, P. T. (2003). A peculiar autosomal dominant macular dystrophy caused by an asparagine deletion at codon 169 in the peripherin/RDS gene. Archives of Ophthalmology, 121(10), 1452–1457. 10.1001/archopht.121.10.1452 [DOI] [PubMed] [Google Scholar]
- Wang, H., Wang, X. , Zou, X. , Xu, S. , Li, H. , Soens, Z. T. , Wang, K. , Li, Y. , Dong, F. , Chen, R. , & Sui, R. (2015). Comprehensive molecular diagnosis of a large Chinese Leber congenital Amaurosis cohort. Investigative Ophthalmology and Visual Science, 56(6), 3642–3655. 10.1167/iovs.14-15972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., Zhang, V. W. , Feng, Y. , Tian, X. , Li, F. Y. , Truong, C. , Wang, G. , Chiang, P. W. , Lewis, R. A. , & Wong, L. J. (2014). Dependable and efficient clinical utility of target capture‐based deep sequencing in molecular diagnosis of retinitis pigmentosa. Investigative Ophthalmology and Visual Science, 55(10), 6213–6223. 10.1167/iovs.14-14936 [DOI] [PubMed] [Google Scholar]
- Wang, X., Wang, H. , Sun, V. , Tuan, H. F. , Keser, V. , Wang, K. , Ren, H. , Lopez, I. , Zaneveld, J. E. , Siddiqui, S. , Bowles, S. , Khan, A. , Salvo, J. , Jacobson, S. G. , Iannaccone, A. , Wang, F. , Birch, D. , Heckenlively, J. R. , Fishman, G. A. , … Chen, R. (2013). Comprehensive molecular diagnosis of 179 Leber congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next generation sequencing. Journal of Medical Genetics, 50(10), 674–688. 10.1136/jmedgenet-2013-101558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wawrocka, A. , Skorczyk‐Werner, A. , Wicher, K. , Niedziela, Z. , Ploski, R. , Rydzanicz, M. , Sykulski, M. , Kociecki, J. , Weisschuh, N. , Kohl, S. , Biskup, S. , Wissinger, B. , & Krawczynski, M. R. (2018). Novel variants identified with next‐generation sequencing in Polish patients with cone‐rod dystrophy. Molecular Vision, 24, 326–339. [PMC free article] [PubMed] [Google Scholar]
- Weisschuh, N. , Feldhaus, B. , Khan, M. I. , Cremers, F. P. M. , Kohl, S. , Wissinger, B. , & Zobor, D. (2018). Molecular and clinical analysis of 27 German patients with Leber congenital amaurosis. PLOS One, 13(12), e0205380. 10.1371/journal.pone.0205380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weleber, R. G. , Carr, R. E. , Murphey, W. H. , Sheffield, V. C. , & Stone, E. M. (1993). Phenotypic variation including retinitis pigmentosa, pattern dystrophy, and fundus flavimaculatus in a single family with a deletion of codon 153 or 154 of the peripherin/RDS gene. Archives of Ophthalmology, 111(11), 1531–1542. 10.1001/archopht.1993.01090110097033 [DOI] [PubMed] [Google Scholar]
- Wells, J. , Wroblewski, J. , Keen, J. , Inglehearn, C. , Jubb, C. , Eckstein, A. , Jay, M. , Arden, G. , Bhattacharya, S. , & Fitzke, F. (1993). Mutations in the human retinal degeneration slow (RDS) gene can cause either retinitis pigmentosa or macular dystrophy. Nature Genetics, 3(3), 213–218. 10.1038/ng0393-213 [DOI] [PubMed] [Google Scholar]
- Wolock, C. J. , Stong, N. , Ma, C. J. , Nagasaki, T. , Lee, W. , Tsang, S. H. , Kamalakaran, S. , Goldstein, D. B. , & Allikmets, R. (2019). A case‐control collapsing analysis identifies retinal dystrophy genes associated with ophthalmic disease in patients with no pathogenic ABCA4 variants. Genetics in Medicine, 21(10), 2336–2344. 10.1038/s41436-019-0495-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wroblewski, J. J. , Wells, J. A., 3rd , Eckstein, A. , Fitzke, F. , Jubb, C. , Keen, T. J. , Inglehearn, C. , Bhattacharya, S. , Arden, G. B. , Jay, M. , & Bird, A. C. (1994a). Macular dystrophy associated with mutations at codon 172 in the human retinal degeneration slow gene. Ophthalmology, 101(1), 12–22. 10.1016/s0161-6420(94)31377-7 [DOI] [PubMed] [Google Scholar]
- Wroblewski, J. J. , Wells, J. A., 3rd , Eckstein, A. , Fitzke, F. W. , Jubb, C. , Keen, T. J. , Inglehearn, C. F. , Bhattacharya, S. S. , Arden, G. B. , & Jay, M. R. (1994b). Ocular findings associated with a 3 base pair deletion in the peripherin‐RDS gene in autosomal dominant retinitis pigmentosa. British Journal of Ophthalmology, 78(11), 831–836. 10.1136/bjo.78.11.831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang, F. , Yan, M. , Song, G. , & Zheng, F. (2012). Gene mapping and mutation screening in candidate genes in a Chinese family of autosomal dominant retinitis pigmentosa. Genetika, 48(1), 125–129. [PubMed] [Google Scholar]
- Xu, Y. , Guan, L. , Shen, T. , Zhang, J. , Xiao, X. , Jiang, H. , Li, S. , Yang, J. , Jia, X. , Yin, Y. , Guo, X. , Wang, J. , & Zhang, Q. (2014). Mutations of 60 known causative genes in 157 families with retinitis pigmentosa based on exome sequencing. Human Genetics, 133(10), 1255–1271. 10.1007/s00439-014-1460-2 [DOI] [PubMed] [Google Scholar]
- Xue, Y. , Zhang, Y. , Wang, M. , Liu, W. , & Xu, G. (2014). [Gene mutations and clinical features of adult vitelliform macular dystrophy in 5 patients]. Zhonghua yan ke za zhi, 50(7), 523–528. [PubMed] [Google Scholar]
- Yanagihashi, S. , Nakazawa, M. , Kurotaki, J. , Sato, M. , Miyagawa, Y. , & Ohguro, H. (2003). Autosomal dominant central areolar choroidal dystrophy and a novel Arg195Leu mutation in the peripherin/RDS gene. Archives of Ophthalmology, 121(10), 1458–1461. 10.1001/archopht.121.10.1458 [DOI] [PubMed] [Google Scholar]
- Yang, H. , Luo, C. , Zhou, J. , Yan, M. , Chen, D. , & Huang, Q. (2000). [The study of RDS gene mutation and clinical phenotype in a family with primary retinitis pigmentosa]. Zhonghua Yan Ke Za Zhi, 36(1), 52–57. [PubMed] [Google Scholar]
- Yang, Z. , Li, Y. , Jiang, L. , Karan, G. , Moshfeghi, D. , O'Connor, S. , Li, X. , Yu, Z. , Lewis, H. , Zack, D. , Jacobson, S. , & Zhang, K. (2004). A novel RDS/peripherin gene mutation associated with diverse macular phenotypes. Ophthalmic Genetics, 25(2), 133–145. 10.1080/13816810490514388 [DOI] [PubMed] [Google Scholar]
- Yang, Z. , Lin, W. , Moshfeghi, D. M. , Thirumalaichary, S. , Li, X. , Jiang, L. , Zhang, H. , Zhang, S. , Kaiser, P. K. , Traboulsi, E. I. , & Zhang, K. (2003). A novel mutation in the RDS/Peripherin gene causes adult‐onset foveomacular dystrophy. American Journal of Ophthalmology, 135(2), 213–218. 10.1016/s0002-9394(02)01815-9 [DOI] [PubMed] [Google Scholar]
- Yeoh, J. , Rahman, W. , Chen, F. , Hooper, C. , Patel, P. , Tufail, A. , Webster, A. R. , Moore, A. T. , & Dacruz, L. (2010). Choroidal imaging in inherited retinal disease using the technique of enhanced depth imaging optical coherence tomography. Graefes Archive for Clinical and Experimental Ophthalmology, 248(12), 1719–1728. 10.1007/s00417-010-1437-3 [DOI] [PubMed] [Google Scholar]
- Yoon, C. K. , Kim, N. K. , Joung, J. G. , Shin, J. Y. , Park, J. H. , Eum, H. H. , Lee, H. O. , Park, W. Y. , & Yu, H. G. (2015). The diagnostic application of targeted re‐sequencing in Korean patients with retinitis pigmentosa. BMC Genomics, 16(1), 515. 10.1186/s12864-015-1723-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaneveld, J. , Siddiqui, S. , Li, H. , Wang, X. , Wang, H. , Wang, K. , Li, H. , Ren, H. , Lopez, I. , Dorfman, A. , Khan, A. , Wang, F. , Salvo, J. , Gelowani, V. , Li, Y. , Sui, R. , Koenekoop, R. , & Chen, R. (2015). Comprehensive analysis of patients with Stargardt macular dystrophy reveals new genotype‐phenotype correlations and unexpected diagnostic revisions. Genetics in Medicine, 17(4), 262–270. 10.1038/gim.2014.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, K. , Garibaldi, D. C. , Li, Y. , Green, W. R. , & Zack, D. J. (2002). Butterfly‐shaped pattern dystrophy: A genetic, clinical, and histopathological report. Archives of Ophthalmology, 120(4), 485–490. 10.1001/archopht.120.4.485 [DOI] [PubMed] [Google Scholar]
- Zhao, L., Wang, F. , Wang, H. , Li, Y. , Alexander, S. , Wang, K. , Willoughby, C. E. , Zaneveld, J. E. , Jiang, L. , Soens, Z. T. , Earle, P. , Simpson, D. , Silvestri, G. , & Chen, R. (2015). Next‐generation sequencing‐based molecular diagnosis of 82 retinitis pigmentosa probands from Northern Ireland. Human Genetics, 134(2), 217–230. 10.1007/s00439-014-1512-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, L. , Xiao, X. , Li, S. , Jia, X. , & Zhang, Q. (2018). Frequent mutations of RetNet genes in eoHM: Further confirmation in 325 probands and comparison with late‐onset high myopia based on exome sequencing. Experimental Eye Research, 171, 76–91. 10.1016/j.exer.2018.02.007 [DOI] [PubMed] [Google Scholar]
- Zhuk, S. A. , & Edwards, A. O. (2006). Peripherin/RDS and VMD2 mutations in macular dystrophies with adult‐onset vitelliform lesion. Molecular Vision, 12, 811–815. [PubMed] [Google Scholar]
- Zulliger, R. , Conley, S. M. , Mwoyosvi, M. L. , Al‐Ubaidi, M. R. , & Naash, M. I. (2018). Oligomerization of Prph2 and Rom1 is essential for photoreceptor outer segment formation. Human Molecular Genetics, 27(20), 3507–3518. 10.1093/hmg/ddy240 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Data Availability Statement
All variant data described in this study are freely available via the LOVD and ClinVar database for PRPH2 (https://databases.lovd.nl/shared/variants/PRPH2; PRPH2[all] ‐ ClinVar ‐ NCBI (nih.gov)). Other data that support the findings detailed in this study are available from the corresponding author, upon reasonable request.