

Abstract
Increased interest in real‐world evidence (RWE) for clinical and regulatory decision making and the need to evaluate long‐term benefits and risks of pharmaceutical products raise the importance of understanding the use of external controls (ECs) for uncontrolled extensions of randomized controlled trials (RCTs). We searched clinicaltrials.gov from 2009 to 2019 for uncontrolled extensions and assessed the use of ECs in the trial protocol registry and PubMed. We present characteristics of identified uncontrolled extensions, their adoption of ECs, and a qualitative appraisal of published uncontrolled extensions with ECs according to good pharmacoepidemiologic practice. The number of uncontrolled extensions increased slightly across the study period, resulting in a total of 1,115 studies. Most originated from phase III RCTs (62.2%) and specified safety outcomes (61.9% among those with specified outcomes). Most uncontrolled extensions incorporated no control group with only 7 out of 1,115 (0.6%) employing ECs. For those studies with ECs, all involved treatments for rare conditions and assessment of effectiveness. Attempts to balance comparison groups varied from none mentioned to propensity score matching. We noted consistent deficiencies in outcome ascertainment methods and approaches to address attrition bias. The contrast of the large and growing number of uncontrolled extensions with the small number of studies that utilized ECs showed clear opportunities for enhancement in design, measurement, and analysis of uncontrolled extensions to allow causal inferences on long‐term treatment effects. As extensions continue to expand within RWE regulatory frameworks, development of guidelines for use of EC with uncontrolled extensions is needed.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ External control cohorts have long been used with single‐arm clinical trials to support regulatory decision making, but less is known about their use in uncontrolled extensions of clinical trials.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study provided an overview of uncontrolled extensions of clinical trials over the past decade, their use of external controls, and the methodological challenges of such designs.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ This study found a sizeable and growing number of uncontrolled extensions, but little use of external controls. Several opportunities for methodological improvements in the design, measurement, and reporting of studies with external controls were uncovered.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ This study highlighted the missed opportunity for use of external controls to provide context for observed safety and effectiveness outcomes in uncontrolled extensions, and the need for adoption of best pharmacoepidemiologic practices to support causal inferences. The described opportunities for methodological improvement can inform planning and conduct of uncontrolled extensions and advance the development of real‐world evidence approaches.
Open‐label extensions following completion of phase II/III randomized controlled trials (RCTs) are often used to collect long‐term safety and effectiveness data. 1 The extension period of the trial follows well‐described populations recruited for the “parent trial” beyond the time needed to evaluate the primary efficacy outcome. These extension studies provide valuable information on long‐term use of the study product in the preauthorization period, often required by health authorities, and consistent with guidelines put forward by the International Council for Harmonization (ICH) of Technical Requirements for Pharmaceuticals for Human Use. 2 For example, the combined safety data from phase III trials and a long‐term extension for brodalumab, which targets interleukin 17 (IL‐17) receptor and is indicated for moderate–severe plaque psoriasis, showed a potential increase in suicidal ideation and behavior (SIB), despite that no imbalance of SIB was seen within the 12‐week placebo controlled and 12–52 week active‐controlled phases of the trials. As a result, a boxed warning for SIB is added into the label upon US Food and Drug Administration (FDA) approval. 3 In those situations where extensions continue beyond the data cutoff for the original registration package, they can provide additional safety and effectiveness information even in the postlaunch period.
Extension studies may contain only an active treatment arm without a randomized comparator. There are multiple pragmatic reasons that sponsors have increasingly moved to follow RCTs with long‐term, uncontrolled extensions. 4 First, it is well recognized that a placebo‐controlled (or an active controlled) trial may require a substantially shorter duration to demonstrate efficacy based on health authority recognized primary end points than is often needed to obtain adequate demonstration of safety. In some diseases, for example type 2 diabetes, it is rare to see a placebo‐controlled comparison last longer than 6 months, given the availability of many approved alternatives. Thus, continuing the placebo group, even with “escape” options, often does not meet the equipoise principle, particularly when the parent trial demonstrated benefit. Second, to adequately demonstrate both safety and durability of effect, a longer period of observation is desired or required, based on ICH guidelines, 2 as is a well‐defined number of patients with specifically defined durations of treatment. Clearly, a trial can enroll more patients meeting the required distribution of duration of treatment by moving all patients from the control group in the parent RCT to the novel agent in a long‐term extension. Such a transition of patient allocation also permits a cost and time efficient utilization of all enrolled patients vs. switching the placebo‐controlled patients to an active control (assuming such were available) to ensure equipoise, which would require enrolling many more patients to meet the ICH requirements. Worth noting is that it is not uncommon that there is no available, approved comparator agent for such a long‐term extension. In the case of a limited set of alternatives, which might include relatively newer biologics, inclusion of such a comparator can drive up the cost of drug development considerably. Moreover, the prospect for participation in uncontrolled extensions also provides incentives for patients to participate in the parent trials, especially considering the opportunity to switch from placebo to the active treatment group, and thus obtain early access to a promising new product for an extended period. Despite these benefits, uncontrolled extensions face the dilemma that without a control group it is generally not possible to draw causal inferences about effectiveness and safety. New safety signals that emerge during the extension period need to be interpreted in the context of background risk of the target population, which may not be available. For example, in the case of sirukumab, an IL‐6 inhibitor submitted for regulatory approval for treatment of moderate to severe rheumatoid arthritis, mortality during the uncontrolled extension phase was found to be higher than that during the placebo‐controlled period. Because of the missing contemporaneous control arm in the extension study, the FDA advisory committee was unable to discern whether the excess mortality risk was an artifact of selection bias related to the trial design or a true long‐term safety signal. The advisory committee voted 12 to 1 against drug approval. 5 , 6
Problems regarding appropriate data on background rates for effectiveness or safety outcomes have long been recognized for single‐arm trials, which may be conducted and considered adequate for approval if patient recruitment for randomized designs is infeasible. 7 Context for the interpretation of single‐arm trials is sometimes provided via external control groups, which can be established from placebo or active arms of previous clinical trials with similar selection criteria or other cohorts of untreated patients or patients with other active treatments and similar characteristics as the trial sample. Particular promise lies in the increasing availability of real‐world data sources to identify external controls, though comparability of patients’ baseline risk, medical practice, use of similar diagnostic criteria and follow‐up procedures need to be addressed. 8 , 9
Theoretically, as with single‐arm trials, the same approach can be used for uncontrolled extensions of RCTs, although additional challenges exist. For example, while selection criteria of external control groups for single‐arm trials may be guided by the original trial selection criteria, the same process for extension studies is complicated by attrition primarily due to lack of efficacy or adverse events over the course of the parent RCT, leaving a subgroup transiting into the extension that may not be representative of the original trial enrollees (Figure 1 ).
Figure 1.
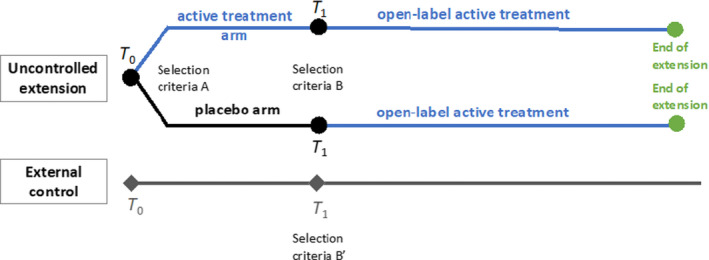
Design of uncontrolled extension with external controls. T 0 represents the start of parent trial and T 1 represents the start of uncontrolled extension. Participants who received placebo in the parent trial cross over to active treatment during the extension period, while participants who received active treatment remain on active treatment during the extension period. [Colour figure can be viewed at wileyonlinelibrary.com]
In light of rapidly increasing interest in and use of real‐world evidence for clinical and regulatory decision making, 9 , 10 it is important to understand whether and how external controls for uncontrolled extensions of RCTs are used. This project aimed to systematically identify and describe characteristics of uncontrolled extensions following randomized controlled phase II and III parent trials, including use of external controls. We also reviewed methods in the identified uncontrolled extensions that employed external controls to inform future recommendations regarding good pharmacoepidemiologic practices for the conduct of and communication about the design and results of such studies.
Methods
Systematic Search for uncontrolled extensions of RCTs
We conducted a search in the clinicaltrials.gov database to identify uncontrolled extensions using the following search strategy: keyword: extension or continuation; filter: phase II or III; and time span: May 14, 2009 to May 14, 2019. The process used by clinicaltrials.gov to register clinical studies has been described previously. 11 , 12 In brief, clinicaltrials.gov is a web‐based repository of information about clinical studies and their results. Initiated by the US Food and Drug Administration and maintained by the National Library of Medicine at the National Institutes of Health, clinicaltrials.gov is one of the largest public databases of clinical studies in the world. It includes both publicly and privately supported clinical studies in all 50 US states and 220 countries. Trial information including both mandatory and optional data elements are self‐reported by sponsors and investigators. A primary reviewer (C.‐Y.W.) screened all identified studies to determine inclusion. Studies with ambiguous information that did not allow explicit application of the inclusion criteria were forwarded to three secondary reviewers (N.S., B.G., and J.A.B.), and disagreements were resolved via discussion. Final inclusion and exclusion criteria were derived during review of a preliminary sample of studies and then applied in the subsequent selection process. Parent trials had to be phase II or III RCTs, and the extension had to lack a control group originating from the parent trial; combined extensions where participants from more than one trial entered an uncontrolled extension were included, if any of the parent trials was an RCT. Studies that were terminated but indicated initial intent for an extension were included. Extensions with additional experimental components, such as additional crossover treatment assignments, were excluded. Finally, single‐arm treatment group extensions where treatment was discontinued during the extension were included, given the potential benefits of employing external controls.
Whenever the information provided in clininicaltrials.gov was not adequate to apply these criteria, an attempt was made to contact the listed principal investigators or the sponsor. The following information was extracted for all included studies: phase of the parent RCT, sample size, main condition (grouped into nine therapeutic categories), duration of uncontrolled extension, whether there were multiple dosing arms, and primary outcomes assessed in the uncontrolled extension. Primary outcomes were grouped into four categories: safety only; effectiveness only; safety and effectiveness; or not reported. Study duration was based on the longest follow‐up time regardless of treatment duration.
Search for uncontrolled extensions with applied external controls
To assess the use of external controls, a second search was conducted in the clinicaltrials.gov database by expanding the original search strategy with the requirement for the terms external control or historical control. To identify relevant publications, we also conducted a search in PubMed using the following search strategy: (active ingredient) AND (extension* or continuation*) AND (study completion date (Date ‐ Publication): 3000 (Date ‐ Publication)) (filters: Clinical Trial; Humans). For example, for the Open Label Extension Study for the Long‐term Efficacy and Safety of FG‐4592 in Dialysis and Non‐dialysis Chronic Kidney Disease Patients (NCT01630889), which was started in May 2012 and was completed in December 2019, the following search strategy was adopted: FG‐4592 AND (extension* or continuation*) AND (2019/12/01(Date ‐ Publication) : 3000 (Date ‐ Publication)) in which 3000 (Date ‐ Publication) represents the present in PubMed. With this search strategy, all articles indexed in PubMed with the term “FG‐4592” and “extension” or “continuation” published between study completion date and the date of the search were identified. The same strategy was adopted for all other extension studies identified in the clinicaltrials.gov database. Articles were included if the data from extensions were compared with external controls. The following information was extracted: publication year, title, condition, active ingredient, duration of parent trial, treatment arms for parent trial, duration of extension study, treatment arms for extension study, outcome assessed in extension study, outcome assessed for external control comparative analysis, data source for external control, sample size for extension study group and external control group, and whether there were multiple dosing arms.
Appraisal of uncontrolled extension studies with external controls
Because the application of external controls in uncontrolled extensions to RCTs is a new field in observational study design, we conducted a qualitative appraisal of the identified published studies based on application of best pharmacoepidemiologic practices. To focus on specific methodological challenges for the design and analysis of uncontrolled extensions with external controls, we reviewed best practices from common published pharmacoepidemiologic standards and two recent publications with specific focus on this topic (Table 1 ). 4 , 13 , 14 , 15 We selected best practice criteria for studies that explicitly aimed to conduct a formal comparison between the extension and the external controls. The criteria were revised and refined by consensus among authors. Studies listed only on clinicaltrials.gov but without any full publication were excluded from appraisal due to the limited information available.
Table 1.
Condensed criteria for design and analysis of uncontrolled extensions with external controls
| Category | Criteria |
|---|---|
| Data source | Rationale for selection of data source (fitness for purpose) is provided; setting and standard of care are similar to extension |
| Sampling |
|
| Outcome |
|
| Exposure |
|
| Attrition |
|
ITT, intention to treat.
Results
Systematic search for uncontrolled extensions of RCTs
Among 216,944 clinical trials registered in clinicaltrials.gov between May 14, 2009 and May 14, 2019, 3,262 were identified by the online search and 1,115 uncontrolled extension studies met the inclusion criteria (Figure 2 ). More than half (62.2%) originated from phase III RCTs. Therapeutic categories were diverse, including 348 (31.2%) trials with treatments for central nervous system / movement disorders, 283 (25.4%) for autoimmune/inflammatory disease, 131 (11.8%) for metabolic/endocrinologic disease, and 106 (9.5%) with indications in hematology/oncology (Figure S1 ). Most uncontrolled extensions (82.2%) had only one active treatment arm without dose comparisons. Sample size was right‐skewed, with a median of 158 and a maximum of 27,395 enrollees. Among all 1,115 uncontrolled extension studies, 919 (82.4%) specified their primary outcomes, including 569 (51%) that identified a safety outcome (61.9% of those that specified an outcome), followed by 206 (18.5%) with an effectiveness outcome and 114 (12.9%) specifying both safety and effectiveness outcomes. The mean study duration was around 2 years, and 89 (8%) uncontrolled extensions did not report study durations. The number of uncontrolled extensions increased over the past 10 years, so did the proportion of uncontrolled extensions to all phase II and III clinical studies registered in clinicaltrials.gov, with about 4.5 additional studies per year for the absolute number or 0.07% increase per year for the proportion (Figure S2 ).
Figure 2.
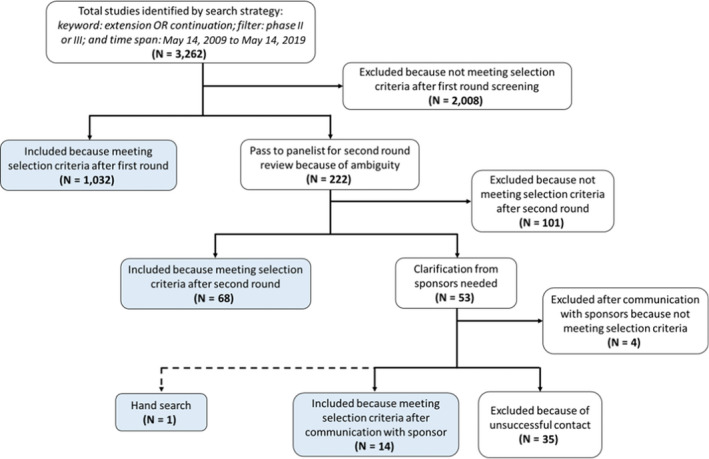
Uncontrolled extension study selection. Blue boxes represent studies that were included. [Colour figure can be viewed at wileyonlinelibrary.com]
Search for uncontrolled extensions with applied external controls
Our second search in clinicaltrials.gov added the keywords “external control or historical control,” resulting in 107 listings. Upon review, only three actually incorporated external controls. Many false positives arose due to the search terms being used in a different context, or it was an indication that planned comparators had not been used. None of the three studies that used external controls was published.
For the PubMed search, we identified 760 uncontrolled extensions that were completed by the time of the search (May 14, 2020). The search strategies identified 1,465 studies in PubMed of which 8 studies were uncontrolled extensions with external controls. These eight publications originated from four extension studies. Therefore, we identified 7 (0.6%) RCT extensions that employed external controls (4 from PubMed and 3 from clinicaltrials.gov) out of all 1,115 uncontrolled extensions (Table 2 ). All seven studies involved drugs indicated for rare conditions. The study duration ranged from 24 weeks to 168 weeks. Five out of seven extension studies had more than one dosing arm, and two used more than one external control group. Five out of a total of nine control groups were untreated patients or patients receiving placebo from other clinical trials; one study enrolled external controls who received another specified treatment; and three enrolled patients with unspecified treatments. Data sources for external controls were prior clinical trials or disease registries. The majority of studies evaluated effectiveness, with only one reporting both safety and effectiveness end points.
Table 2.
Summary of seven uncontrolled extensions with external controls
| Study # / NCT number | Title of extension trial (clinicaltrials.gov) | Condition | Active ingredient | Duration of extension trial | Treatment arms for extension | Treatment of External control | Data source for external control | Outcomes assessed in external control analysis (effectiveness/safety) |
|---|---|---|---|---|---|---|---|---|
| # 1 / 01931839 | A Phase 3 Rollover Study of Lumacaftor in Combination With Ivacaftor in Subjects 12 Years and Older With Cystic Fibrosis | Cystic fibrosis |
Lumacaftor/ ivacaftor combination therapy |
96 weeks |
Two dosing strategies: lumacaftor 400–600 mg/ivacaftor 250 mg |
Unspecified | Cystic Fibrosis Foundation Patient Registry |
Change in predicted forced expiratory volume; weight; BMI (effectiveness) |
| # 2 / 01415427 | Long‐Term Efficacy and Safety Extension Study of BMN 110 in Patients With Mucopolysaccharidosis IVA (Morquio A Syndrome) |
Morquio A (mucopolysaccharidosis IVA) |
Elosulfase alfa | 96 weeks |
Two dosing strategies: 2 mg/kg/week; 2 mg/kg/every other week with transition to 2 mg/kg/week between week 36 and week 96 |
Untreated | Morquio A natural history study; MOR‐001; NCT00787995 |
Various physical function / ADL assessments; urine keratan sulfate levels; forced vital capacity; forced expiratory volume; maximum voluntary ventilation (effectiveness) |
| # 3 / 01540409 | Efficacy, Safety, and Tolerability Rollover Study of Eteplirsen in Subjects With Duchenne Muscular Dystrophy | Duchenne muscular dystrophy | Eteplirsen (AVI‐4658) | 156 weeks |
Two dosing strategies: 30–50 mg/kg/week |
Untreated | Baseline samples from untreated control arm from separate phase III eteplirsen study (PROMOVI) | Dystrophin expression (Means of PDPF, Bioquant relative fluorescence intensity, and Western blot) (effectiveness) |
|
2 natural history cohorts from the Leuven Neuromuscular Reference Center (LNMRC) and the Italian Telethon registry |
6‐Minute Walk Test; pulmonary function (effectiveness) |
|||||||
| # 4 / 01214421 | Open‐Label Tolvaptan Study in Subjects with ADPKD (TEMPO 4/4) | autosomal dominant polycystic kidney disease | Tolvaptan | 2 year | Tolvaptan | Unspecified | CRISP cohort (NCT01039987) and the HALT PKD Study B clinical trial (NCT01885559) | eGFR (effectiveness) |
| # 5 (unpublished)/02760277 | An Extension Study to Assess Vamorolone in Boys With Duchenne Muscular Dystrophy | Duchenne muscular dystrophy | Vamorolone | 24 weeks | Four dosing strategies from 0.25 to 6.0 mg/day/day |
(1) untreated (2) prednisone |
unspecified prior studies |
|
| # 6 (unpublished)/03167255 | Extension Study of NS‐065/NCNP‐01 in Boys With Duchenne Muscular Dystrophy | Duchenne muscular dystrophy | NS‐065/NCNP‐01 | 168 weeks |
Two dosing strategies: 40 and 80 mg/kg |
N/S (matched historical controls) | N/S | Various physical function (e.g., walking) assessments; muscle strength (effectiveness) |
| # 7 (unpublished)/03759379 | HELIOS‐A: A Study of Vutrisiran (ALN‐TTRSC02) in Patients With Hereditary Transthyretin Amyloidosis | Hereditary transthyretin amyloidosis | Vutrisiran (ALN‐TTRSC02) | N/S | Vutrisiran | Placebo | prior clinical trial (APOLLO study) | N/S |
ADL, activity of daily living; BMI, body mass index; eGFR, estimated glomerular filtration rate; N/S, not specified; PDPF, percentage of dystrophin positive fibers.
Appraisal of uncontrolled extension studies with external controls
In our appraisal of study methods several themes emerged. First, because the evaluation of most disease conditions involved in the identified studies relied on functional assessments (e.g., 6‐minute walk tests) or specific assays that are not typically performed and/or systematically recorded during routine clinical care, all studies had to resort to other prospective cohorts (including prior clinical trials and disease registries) for the identification of external controls. Because such data are limited, especially in rare diseases, the authors may not have felt compelled to discuss their rationale for selection of a particular external data source or to assess fitness for purpose. Only one study, with a focus on muscular dystrophy, included a statement that the identified sources for external controls were the only natural history data sets that provided similar standardized assessments of their primary outcome, had similar inclusion criteria as the uncontrolled extension and similar patient care standards. 16 We also noted that most manuscripts had a strong focus on the uncontrolled extensions, oftentimes with more details devoted to the presentation of single‐arm assessments in the extension rather than the comparison between the uncontrolled extensions and the external controls. Accordingly, there were disparities in descriptions of the enrollment criteria, outcomes ascertainment methods, and attrition when compared with the external controls for which such detail was commonly omitted. We elaborate on examples in the following sections and Table 3 .
Table 3.
Qualitative methodological assessment of four published uncontrolled extensions with external controls
| Study # | Matching inclusion criteria | Similar outcomes ascertainment | Changes in exposure | Attrition |
|---|---|---|---|---|
| # 1 |
|
Similar standard assessments and similar minimum number of assessments for rate of change analyses | Not considered for extension; ignored external control treatments | Insufficient data to assess; differences in the number and timing of assessments is acknowledged |
| # 2 |
|
Similar standard assessments and similar criteria for measure timing, though assessments for external control occurred systematically later; inadequate detail on administration of PRO surveys to assess potential for bias | ITT and per protocol analysis for extension; external control was untreated | Limited attrition among extension subjects but significant attrition in external control; detail for attrition not provided; baseline characteristics of initial and remaining external controls are compared; ITT stops analysis at point of follow‐up |
| # 3.A |
|
|
Extension subjects received weekly infusions and no problems with treatment discontinuation are reported; external control was untreated and no further detail on relevant changes during follow‐up is reported | It is unclear how and when external controls were sampled from the control arm of another trial and thus attrition cannot be assessed; reason for loss to follow‐up among extension subjects is provided |
| # 3.B |
|
|
Detail on extension treatment was minimal but based on companion manuscript was consistent; no detail on treatment of external controls | Detailed report on attrition in both groups; sensitivity analysis included patients lost to follow‐up by carrying the last observation forward |
| # 4 |
|
Unclear when and how eGFR was measured for external controls; definition of baseline relative to start of the extension was not provided | Treatment duration for extension during follow‐up was unclear; no detail on treatments for external controls | Attrition and reasons for attrition are described for the extension but not the external controls; follow‐up ends at last outcomes measure ascertained during treatment; attrition bias cannot be assessed due to missing information |
ANCOVA, analysis of covariance; eGFR, estimated glomerular filtration rate; ITT, intention to treat; PRO, patient reported outcomes.
Sampling and selection bias: Similarity of enrollment criteria and between‐group balance of risk factors for the study outcomes
We found it difficult to evaluate the similarity of selection criteria between uncontrolled extensions and external control groups because criteria were not commonly reported side by side. Most studies reported inclusion and exclusion criteria for entry into the extension phase, but such criteria for the external control cohorts were often unspecified. Sometimes replicating inclusion criteria that were employed for the extensions was infeasible because the relevant data were not available in the external control data source, especially when implicit criteria based on investigator or clinician assessments were used. For example, in the cystic fibrosis study conducted by Konstan et al., exclusion criteria for enrollment in the extension phase included “any comorbidity … or laboratory abnormality that, in the opinion of the investigator, might confound the results of the study or pose an additional risk in administering study drug to the participant.” 17 In addition, certain exclusion criteria, such as history of drug intolerance and poor compliance with treatment during the initial trial phase, may intuitively enhance the success of the extension studies (e.g., by decreasing risk for attrition), but may lead to the risk of selection bias for the comparison to the external control if they were not highly correlated with other disease risk factors available in both data sources.
Most studies presented a table that compared baseline characteristics between enrollees in the uncontrolled extensions and external controls. However, these characteristics were not necessarily incorporated in the outcome analysis. For example, while the above‐referenced cystic fibrosis study 17 used propensity score matching based on an exhaustive list of covariates, the Morquio A syndrome studies limited adjusted covariates to age, height, and the baseline value of the respective outcome. 18 , 19 , 20 , 21 These studies used explicit inclusion and exclusion criteria for the extension that were replicated with the external control data source and showed similar baseline characteristics in regard to these criteria. It should be noted that no publication discussed the potential for residual selection bias by highlighting important risk factors for the outcome that could not be assessed for balance because they were not available in the data sources.
We also encountered problems in determining the timepoint when baseline characteristics were measured relative to follow‐up. This was complicated by varying definitions of study entry, e.g., start at the initial trial enrollment vs. start of the extension and duration of treatment in the parent trial. The definition of baseline for extension patients who had crossed over from placebo to treatment assignment was sometimes unclear, thus compromising the ability to assess presence of selection bias. This issue was further exacerbated by the retrospective nature of the external control group data: Even when these data were originally ascertained following a predefined protocol, the time frames for ascertainment may have been different, thus complicating the definition of a common baseline period among extension and external control patients.
Outcome ascertainment and measurement bias
We noted that detail about the ascertainment of clinical measures was typically more comprehensive than for patient‐reported outcomes, which in fact may have greater susceptibility for measurement bias. Detail about outcomes ascertainment, especially frequency thereof, was generally more comprehensive for extensions than for external controls. Some authors pointed to the similarity of specific standards in outcomes ascertainment across comparison groups. For example, in one muscular dystrophy study, the authors state that “the assessment of the 6‐minute walk test in the historical control was performed by specifically trained physiotherapists according to the same established standards used for the eteplirsen‐treated patients.” 16 In addition, explicit rules for the selection and temporal alignment of tests for longitudinal analysis were reported. In contrast, in the Morquio A syndrome study, while aiming to align measurement intervals, the authors arrived at an appreciably delayed assessment pattern for the external controls. For example, visits intended to be at a 12‐month follow‐up in the external controls included assessments collected between days 270 and 609 with a mean of 446 days and standard deviation of 74 days. 18
Exposure during follow‐up and related biases
Some studies reported details on the treatment modalities for the external controls or imposed explicit restrictions to ensure homogeneous (e.g., truly untreated) comparison groups, while others provided no such details. Details regarding adherence and persistence on treatment were typically more complete in uncontrolled extensions, though approaches to address treatment changes varied.
For the uncontrolled extension arms, most studies noted that patients received different treatment (active treatment or placebo/standard of care) during the parent trial period, though they adopted different approaches to address the differences in earlier treatments. For example, in the cystic fibrosis study, participants in the extension trial represented a mix of 1/3 former placebo and 2/3 former treatment patients. 17 The outcome, rate of change in percent predicted forced expiratory volume from baseline, was analyzed in two mixed‐effects models for repeated measures. Baseline was defined differently in these two models, one as the baseline in the parent study and another as the baseline before the first active treatment. In contrast, the Morquio A syndrome studies included only enrollees who had been in the active treatment group of the parent trial. Evidently, these decisions were influenced by assumptions about disease progression and the relative impact of the active treatment in the parent trial on the observed outcomes in the extension phase. Of note, some studies with different start of follow‐up for previous treatment and placebo patients failed to provide group‐specific detail on whether the reported baseline characteristics originated from the entry in the parent trial or extension period.
The challenges in appropriate lineup of comparison groups are illustrated in the following. Figure 3 illustrates a design where follow‐up time begins at start of treatment, resulting in different entry times for previous treatment and placebo group. Follow‐up begins at the start of parent trial (T 0) for the former treatment group and begins at the start of extension (T 1) for the former placebo group. Selection criteria A represent the selection criteria for the parent trial and selection criteria B represent the selection criteria that were further applied when participants entered the extension phase. This design maximizes the number of enrollees and follow‐up time available for analysis. However, the validity of the design relies on the assumption that significant disease progression is not expected during the parent trial period and that baseline characteristics of the treatment group at T 0 and the placebo group at T 1 are similar. It should also be noted that potential differences in ascertainment of risk factors at T 0 and T 1 will complicate this design and the alignment of a suitable external control group.
Figure 3.
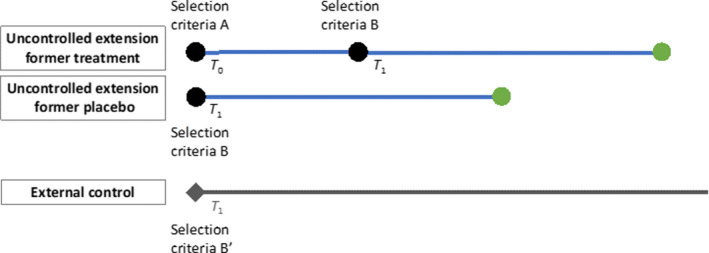
Follow‐up for former treatment group starts at parent trial enrollment, while former placebo group starts at the extension phase. T 0 represents the start of parent trial and T 1 represents the start of uncontrolled extension. [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 4 illustrates alternatives concerning how potential biases of this approach could be addressed. In scenario 1 follow‐up begins at the start of the extension, resulting in a single point for both former parent trial groups when disease risk factors are assessed and aligned with the external control. However, the observed treatment effect during the extension will not only be attributable to new drug exposure during the extension, but for the previous treatment arm patients, also to carryover effects from previous treatment during the parent trial. This is especially concerning when evaluating safety end points because patients in the former treatment group who remain for the extension are most likely those who tolerate the drug. These patients can be fundamentally different from patients in the former placebo group, especially in terms of their risk of developing adverse events. Scenario 2 recognizes potential differences in disease progression and other risk factors and constructs separate external comparisons for the former treatment and placebo groups, and thus minimizes selection bias. Scenarios 3 and 4 represent the same approach but the comparison is restricted to one of the former parent trial arms. It should be noted that while scenario 4 maximizes the available follow‐up during treatment, the additional selection criteria at the time of extension create a subgroup, which may not be representative of the parent trial treatment arm. Thus, both sets of risk factors at T 0 and T 1 may need to be considered when selecting an external control group.
Figure 4.

Alternative scenarios to address differences in study entry and follow‐up among former parent trial treatment and placebo groups. T 0 represents the start of parent trial and T 1 represents the start of uncontrolled extension. PBO, placebo; TX, treatment; UE, uncontrolled extension. [Colour figure can be viewed at wileyonlinelibrary.com]
Attrition and related biases
Differences in data sources may lead to differences in follow‐up time and informative censoring. For example, if the extension trial loses patients for reasons that are associated with the study outcome (e.g., patients susceptible to drug side effects), whereas external controls from registries or real‐world data may remain available, the differential composition of baseline risk among comparison groups, even if originally balanced upon study entry, leads to bias. If this type of bias presents, statistical methods that address informative censoring and/or data missingness are essential. Neither detail on attrition that allowed assessment of bias nor methods to address related bias were consistently reported in the published studies.
For example, in the cystic fibrosis study, while patient attrition and reasons for loss to follow‐up for extension were reported, this detail was not described for the external control group. 17 In the Morquio A syndrome studies, attrition from the uncontrolled extension was limited, but the registry used to develop the external control cohort experienced significant loss to follow‐up (11% vs. 59%). Reasons for loss to follow‐up were provided for the extension but not the external control group, limiting assessment of attrition bias, though comparisons of baseline characteristics of the initial and remaining external control cohort were provided in the supplementary material of the published article. 20 In this study, which reported results from an intention‐to‐treat and per‐protocol analysis on longitudinal changes in outcome measures, no attempt was made to address loss‐to‐follow‐up in the intention‐to‐treat analysis. Instead, patients who discontinued treatment were excluded from further analyses. Traditional methods to evaluate bias imposed by such exclusions would be imputation approaches but given the observational nature of the study design, formal epidemiologic approaches to capture attrition bias such as use of censoring weights could also be considered. 22 , 23
Discussion
In this review, we identified a sizeable and growing number of uncontrolled extensions to phase II and III clinical trials but only a small proportion (7 out of 1,115, 0.6%) that attempted to employ an external control to add context for observed outcomes and enhance the study’s ability to draw safety and/or effectiveness inferences. Though we may have missed some studies using external controls, our small overall yield emphasizes the strong discrepancy between the interest in long‐term extensions and the reported use of external controls in such settings. We commonly found that results from extension studies were reported descriptively in simple case series or with comparison to reference data with unknown applicability to the trial population. Considering the increasing adoption of pharmacoepidemiologic methods in evaluating effectiveness to support both new indications and new drug/biologic applications, 24 , 25 , 26 in addition to these methods’ long‐established use in drug safety evaluations, we noted the limited consideration that has been given to these methods in the design enhancement of uncontrolled extensions. Use of external controls that are sampled from data sources that are fit‐for‐purpose and analyzed with prespecified protocol and appropriate causal inference methods do not only provide context for emerging safety concerns but can also support inferences about long‐term effectiveness and/or safety, considering background risk and disease progression.
Review of the methods in published uncontrolled extensions with external controls solidified the impression of a focus on the uncontrolled extension on its own rather than the value added by an external control. In this paper we described examples of enrollment criteria that were not reproducible with real‐world data sets, thus limiting the ability to establish balanced comparison groups. We noted disparities in the description of sample characteristics, exposure, measurement detail, and attrition. Lacking key information about the external control cohorts did oftentimes preclude assessment for potential bias. Perhaps compounded by small sample sizes, which limited the range of statistical adjustments that would have been possible, several studies showed only a small number of baseline characteristics for comparison, which was mirrored by limited assessments for attrition bias. We noted several opportunities where the review and integration of best pharmacoepidemiologic practices in the a priori design of uncontrolled extensions with external controls at the conception of the extension would have enhanced the rigor of the reviewed studies. We do acknowledge that all uncontrolled extension studies that included an external control in our defined sample focused on rare diseases with great challenges in patient recruitment and availability of adequate data sources for external controls. We should note that we cannot comment on the growth of uncontrolled extension studies in the context of similar extensions that did use randomized controls. If feasible, retention of the original randomized exposure assignment would circumvent some of the problems in balancing baseline risk of different populations and aligning different data sources, though challenges with attrition, especially once efficacy results are available, may create similar issues.
Although extension studies are common, whether the extension was controlled or uncontrolled and whether an external control was planned were often unclear or lacking in clinicaltrials.gov. Given the growing prominence of these studies within an increased interest in real‐world evidence, we suggest amendments to the clinicaltrials.gov database that would enhance such detail, including requirements for a detailed description of the extension phase of a trial with designation of whether controlled or uncontrolled; capture of the intent to employ external controls and the data sources for such controls; and cross‐reference of uncontrolled extension protocols, if registered separately, to the parent trial.
Our study has limitations. First, the search for uncontrolled extensions was limited to the clinicaltrials.gov database. While the goal of the clinicaltrials.gov database is to provide a comprehensive listing of all clinical studies, it does not contain information about all clinical studies because not all studies are required by law to be registered. Thus, the accuracy of our search relied on capture of uncontrolled extensions in the database as well as the sensitivity of the adopted search strategy, and some studies may have been missed. Moreover, because clinicaltrials.gov allows sponsors to update the information at any time, studies identified by our search strategy can only represent planned extension studies at the time of the search. Second, despite the capability to update the trial information, many records were not updated in a timely manner by the sponsors or investigators when there is no incentive to do so. This influenced our search for uncontrolled extension employing external control in PubMed. For example, if drug names changed over time (e.g., from the company’s number designations to the compound name) and were not updated in clinicaltrials.gov, our PubMed search strategy would have failed to identify relevant publications. Additionally, even though we attempted to find all extension studies employing external controls by conducting further searches in PubMed, the comparison between extensions and external controls may be included in product filings to provide regulators with safety/effectiveness context but are not recorded in clinicaltrials.gov or available in PubMed. Our searches would fail to identify such information if this is the case. Third, although we observed an increasing trend in the number of uncontrolled extensions from 2009 to 2018, this could also represent improvement in reporting of uncontrolled extensions over the years. The passage of FDA Amendments Act (FDAAA) section 801 in 2007 requires more types of trials to be registered and additional trial registration information to be submitted. FDAAA section 801 also established penalties for failing to register or submit the results of trials. 27 This regulation which took effect in 2017 along with other statements and policies (including International Committee of Medical Journal Editors 28 and NIH policy on the dissemination of NIH‐funded clinical trial information) 29 have increased the registration of clinical studies in clinicaltrials.gov over time. 30 Lastly, the condensed criteria for design and analysis of uncontrolled extensions with external controls presented in Table 1 were derived by the authors based on their own experience and in response to the methodological issues encountered in the included studies. Future work should consider the development of consensus‐based standards utilizing a broader group of experts in this area and integrating others’ work, such as another recent compilation of potential biases in the use of external controls for long‐term trial extensions. 31 Despite these limitations, our study is the first to quantify the number of uncontrolled extensions and the application of external controls. To the best of our knowledge, it is also the first study that qualitatively evaluated extension studies applying external controls based on use of good pharmacoepidemiologic practices.
We conclude that the large and growing number of identified uncontrolled extensions was in contrast to the small number of studies that employed external controls, highlighting missed opportunities for more rigorous designs and appropriate contextualization of safety and effectiveness outcomes. The small number of published uncontrolled extensions with external controls exhibited several opportunities for enhancement in design, measurement, and analysis to allow causal inferences on long‐term effects. Actions to keep advancing the science through increased engagement of pharmacoepidemiology best practice guidance at the design level and prespecifying the methods and analysis plan to address biases are needed.
Funding
Funding to support this manuscript development was provided by the International Society for Pharmacoepidemiology (ISPE). This manuscript is endorsed by the International Society for Pharmacoepidemiology (ISPE).
Conflict of Interest
J.A.B. is a full‐time employee of Johnson & Johnson and holds stock in the company; B.G. is a full‐time employee of Blackstone Life Sciences and holds stock in Blackstone; K.D. is a full‐time employee of Janssen and holds stock in the company (J&J); N.A.D. is a full‐time employee of IQVIA and holds stock in the company; W.Z. is a full‐time employee of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, and holds stock in Merck & Co., Inc., Kenilworth, NJ; J.D.S. is a full‐time employee of UnitedHealth Group and holds stock in the company; A.G.W. has current research funding from the NIH, AHRQ, FDA, PCORI, the State of Florida, and Merck, Sharp & Dohme. All other authors declared no competing interests for this work.
Author Contributions
C.‐Y.W., and A.G.W. wrote the manuscript. All authors designed the research. C.‐Y.W., J.A.B., B.G., and N.S. performed the research. C.‐Y.W. analyzed the data.
Disclaimer
The opinions expressed in this manuscript are those of the authors and should not be interpreted as the position of the US Food and Drug Administration.
Supporting information
Figure S1
Figure S2
Acknowledgment
The authors would like to thank the contribution of Michelle Iannacone for reviewing the manuscript and providing her comments.
References
- 1. Day, R.O. & Williams, K.M. Open‐label extension studies: do they provide meaningful information on the safety of new drugs? Drug. Saf. 30, 93–105 (2007). [DOI] [PubMed] [Google Scholar]
- 2. International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH). Harmonized tripartite guideline . The extent of population exposure to assess clinical safety for drugs intended for long‐term treatment of non‐life‐threatening conditions; E1 <https://database.ich.org/sites/default/files/E1_Guideline.pdf> (1994).
- 3. Slides for the July 19, 2016 Meeting of the Dermatologic and Ophthalmic Drugs Advisory Committee (DODAC) <https://www.fda.gov/advisory‐committees/dermatologic‐and‐ophthalmic‐drugs‐advisory‐committee/slides‐july‐19‐2016‐meeting‐dermatologic‐and‐ophthalmic‐drugs‐advisory‐committee‐dodac> (2016). Accessed May 18, 2021.
- 4. Seeger, J.D. et al. Methods for external control groups for single arm trials or long‐term uncontrolled extensions to randomized clinical trials. Pharmacoepidemiol. Drug Saf. 29, 1382–1392 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sutter, S. US FDA panel weighs design of a pre‐approval safety study for Janssen's Sirukumab <https://pink.pharmaintelligence.informa.com/PS121280/US‐FDA‐Panel‐Weighs‐Design‐Of‐A‐Preapproval‐Safety‐Study‐For‐Janssens‐Sirukumab> (2017). Accessed July 22, 2020.
- 6. Janssen's Sirukumab falls on mortality concerns at US FDA panel <https://pink.pharmaintelligence.informa.com/PS121249/Janssens‐Sirukumab‐Falls‐On‐Mortality‐Concerns‐At‐US‐FDA‐Panel> (2017). Accessed July 22, 2020.
- 7. Tenhunen, O. , Lasch, F. , Schiel, A. & Turpeinen, M. Single‐arm clinical trials as pivotal evidence for cancer drug approval: a retrospective cohort study of centralized European marketing authorizations between 2010 and 2019. Clin. Pharmacol. Ther. 108, 653–660 (2020). [DOI] [PubMed] [Google Scholar]
- 8. Eckert J, Wahl K, Lederer NM, et al. Virtual Workshop Series: The science of similarity – using RWD‐generated external comparators for single‐arm trials. Meeting Summary. Duke Margolis Center for Health Policy Research, Washington, DC. September 8, 2020. [Google Scholar]
- 9. US Food and Drug Administration . Framework for FDA's real world evidence program <https://www.fda.gov/media/120060/download> (2018). Accessed July 22, 2020.
- 10. US Food and Drug Administration . Guidance for industry and Food and Drug Administration staff: Use of real‐world evidence to support regulatory decision‐making for medical devices. <https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/use‐real‐world‐evidence‐support‐regulatory‐decision‐making‐medical‐devices> (2017). Accessed July 22, 2020.
- 11. Gillen, J.E. , Tse, T. , Ide, N.C. & McCray, A.T. Design, implementation and management of a web‐based data entry system for ClinicalTrials.gov. Stud. Health Technol. Inform. 107, 1466–1470 (2004). [PubMed] [Google Scholar]
- 12. Zarin, D.A. , Ide, N.C. , Tse, T. , Harlan, W.R. , West, J.C. & Lindberg, D.A.B. Issues in the registration of clinical trials. JAMA 297, 2112–2120 (2007). [DOI] [PubMed] [Google Scholar]
- 13. Burcu, M. et al. Real‐world evidence to support regulatory decision‐making for medicines: considerations for external control arms. Pharmacoepidemiol. Drug Saf. 29, 1228–1235 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Public Policy Committee, International Society for Pharmacoepidemiology . Guidelines for good pharmacoepidemiology practice (GPP). Pharmacoepidemiol. Drug Saf. 25, 2–10 (2016). [DOI] [PubMed] [Google Scholar]
- 15. The European Network of Centres for Pharmacoepidemiology and Pharmacovigilance (ENCePP) . Guide on Methodological Standards in Pharmacoepidemiology ‐ Revision 9. EMA/95098/2010 <http://www.encepp.eu/standards_and_guidances/documents/1.ENCePPMethodsGuideRev.9.pdf> (2021). [Google Scholar]
- 16. Mendell, J.R. et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 79, 257–271 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Konstan, M.W. et al. Assessment of safety and efficacy of long‐term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del‐CFTR mutation (PROGRESS): a phase 3, extension study. Lancet. Respir. Med. 5, 107–118 (2017). [DOI] [PubMed] [Google Scholar]
- 18. Hendriksz, C.J. et al. Long‐term endurance and safety of elosulfase alfa enzyme replacement therapy in patients with Morquio A syndrome. Mol. Genet. Metab. 119, 131–143 (2016). [DOI] [PubMed] [Google Scholar]
- 19. Hendriksz, C.J. et al. Impact of long‐term elosulfase alfa treatment on respiratory function in patients with Morquio A syndrome. J. Inherit. Metab. Dis. 39, 839–847 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hendriksz, C.J. et al. Impact of long‐term elosulfase alfa on activities of daily living in patients with Morquio A syndrome in an open‐label, multi‐center, phase 3 extension study. Mol. Genet. Metab. 123, 127–134 (2018). [DOI] [PubMed] [Google Scholar]
- 21. Hughes, D. et al. Clinical outcomes in a subpopulation of adults with Morquio A syndrome: results from a long‐term extension study of elosulfase alfa. Orphanet. J. Rare Dis. 12, 98 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Robins, J.M. & Finkelstein, D.M. Correcting for noncompliance and dependent censoring in an AIDS Clinical Trial with inverse probability of censoring weighted (IPCW) log‐rank tests. Biometrics 56, 779–788 (2000). [DOI] [PubMed] [Google Scholar]
- 23. McCoy, C.E. Understanding the intention‐to‐treat principle in randomized controlled trials. West. J. Emerg. Med. 18, 1075–1078 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dreyer, N.A. Advancing a framework for regulatory use of real‐world evidence: when real is reliable. Ther. Innov. Regul. Sci. 52, 362–368 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Girman, C.J. , Ritchey, M.E. , Zhou, W. & Dreyer, N.A. Considerations in characterizing real‐world data relevance and quality for regulatory purposes: a commentary. Pharmacoepidemiol. Drug Saf. 28, 439–442 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dreyer, N.A. , Hall, M. & Christian, J.B. Modernizing regulatory evidence with trials and real‐world studies. Ther. Innov. Regul. Sci. 54, 1112–1115 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tse, T. , Fain, K.M. & Zarin, D.A. How to avoid common problems when using ClinicalTrials.gov in research: 10 issues to consider. BMJ 361, k1452 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. De Angelis, C.D. et al. Is this clinical trial fully registered? A statement from the International Committee of Medical Journal Editors. N. Engl. J. Med. 352, 2436‐2438 (2005). [DOI] [PubMed] [Google Scholar]
- 29. National Institute of Health . NIH policy on the dissemination of NIH‐Funded clinical trial information <https://grants.nih.gov/policy/clinical‐trials/reporting/understanding/nih‐policy.htm> (2016).
- 30. US National Library of Medicine . Trends, charts, and maps. Clinicaltrials.gov <https://clinicaltrials.gov/ct2/resources/trends#RegisteredStudiesOverTime> (2021). Accessed May 20, 2021.
- 31. Burger, H.U. et al. The use of external controls: to what extent can it currently be recommended? Pharm. Stat. (2021). Published online ahead of print. 10.1002/pst.2120. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2