

Abstract
Introduction
The phase 2/3 PROTECT VIII main study demonstrated efficacy and safety of BAY 94–9027 (damoctocog alfa pegol; Jivi®), a B‐domain‐deleted recombinant factor VIII (FVIII), site‐specifically PEGylated to extend its half‐life.
Aim
To report the final efficacy and safety data for BAY 94–9027 from the PROTECT VIII extension.
Methods
Previously treated males aged 12–65 years with severe haemophilia A (FVIII <1%) who completed the multicentre, open‐label PROTECT VIII main study were eligible for the extension. Patients received either on demand or prophylaxis treatments (30‒40 IU/kg twice weekly [2 × W], 45‒60 IU/kg every 5 days [E5D], or 60 IU/kg every 7 days [E7D]) and could switch to any prophylaxis regimen (variable frequency) as needed. Annualised bleeding rates (ABR), zero bleeds and safety outcomes were included in this final analysis.
Results
At extension completion, patients (n = 121) received BAY 94–9027 for a median (range) total time of 3.9 (0.8–7.0) years. Median (Q1; Q3) total ABR was 1.49 (0.36; 4.80) for prophylaxis patients (n = 107), compared with 34.09 (20.3; 36.6) for on‐demand patients (n = 14). Median total ABRs for 2 × W (n = 23), E5D (n = 33), E7D (n = 23) and variable frequency (n = 28) groups were 1.57, 1.17, 0.65 and 3.10, respectively. Of prophylaxis patients, 20.6% were bleed‐free during the entire extension (median time, 3.2 years) and 50.0% were bleed‐free during the last 6 months. No patient developed FVIII inhibitors. No deaths or thrombotic events were reported.
Conclusions
Efficacy and safety of BAY 94–9027 was confirmed, with extension data supporting its use as a long‐term treatment option for patients with haemophilia A.
Keywords: Extended half‐life, haemophilia A, PEGylated, prophylaxis, recombinant proteins
1. INTRODUCTION
Routine prophylaxis with factor VIII (FVIII) replacement is the gold standard in haemophilia A management. 1 , 2 To improve adherence to prophylactic regimens, it is important to review current strategies and tailor treatment to patients’ specific needs. Such efforts should include individualising dosing regimens based on a patient's unique bleeding phenotype, pharmacokinetic profile and lifestyle. 3 Utilisation of FVIII products with improved pharmacokinetic properties may allow for less frequent dosing and/or increased FVIII activity levels, to potentially improve adherence among patients and provide better bleed protection and quality of life. 4
BAY 94–9027 (damoctocog alfa pegol; Jivi®) is a recombinant factor VIII (rFVIII) product that is site‐specifically conjugated with polyethylene glycol (PEG) to reduce clearance and extend time in circulation. 5 , 6 BAY 94–9027 is approved in several countries including the United States, EU, Canada and Japan for the treatment of previously treated adolescents and adults with haemophilia A. 7 , 8 , 9 , 10 PROTECT VIII demonstrated efficacy and safety of BAY 94–9027 for the prevention and treatment of bleeding episodes in adults and adolescents with severe haemophilia A. 11 Following completion of PROTECT VIII, patients could continue to receive BAY 94–9027 in an optional extension study that evaluated safety and efficacy for ≥100 cumulative exposure days (EDs) and examined long‐term treatment with BAY 94–9027.
A previously published interim analysis of the extension study reported that BAY 94–9027 prophylaxis was efficacious and well tolerated for up to 5 years. 12 Here, we report final data from the PROTECT VIII extension study and describe the long‐term safety and efficacy of BAY 94–9027 prophylaxis with a patient follow‐up of up to seven years.
2. MATERIALS AND METHODS
2.1. Patients
Males aged 12–65 years with severe haemophilia A (FVIII <1%) previously treated with any FVIII product for ≥150 EDs were eligible for inclusion in the main study. Key exclusion criteria included the presence or history of a FVIII inhibitor (≥0.6 BU/mL), diagnosis of any bleeding disorder other than haemophilia A, platelet count <100,000/mm3, creatinine >2 times the upper limit of normal (ULN), or aspartate aminotransferase (AST) or alanine aminotransferase (ALT) >5 times the ULN. 11
2.2. Study design
The PROTECT VIII extension study was a multicentre, open‐label, uncontrolled, optional study for patients who completed the 36‐week PROTECT VIII trial 11 (ClinicalTrials.gov identifier: NCT01580293), where they continued to receive BAY 94–9027 for ≥100 EDs (Figure S1). The extension study was conducted at 53 study centres in 18 countries (Table S1). Patients who decided to be treated on demand during the main study period had the option to either continue on‐demand treatment in the extension or switch to one of the three prophylaxis groups used during the main study (30–40 IU/kg twice weekly [2 × W], 45–60 IU/kg every 5 days [E5D] or 60 IU/kg every 7 days [E7D]). Patients who received prophylaxis in the main study could either continue their regimen or switch to another PROTECT VIII prophylaxis regimen at the start of the extension study, and any time during the extension study. The frequency and dose of BAY 94–9027 infusions were adapted to individual needs, but within the pre‐specified frequencies and doses used in PROTECT VIII. Patients who switched regimens at least once after the first 7 days of the extension were analysed in the variable frequency group (VAR). The dose and frequency of BAY 94–9027 infusions required to treat bleeding events were decided by the treating physician, up to a maximum recommended dose of 60 IU/kg/infusion or 6000 IU/infusion.
The study was approved by the institutional review board at each site and was carried out in compliance with the protocol, the principles of the Declaration of Helsinki and Good Clinical Practice guidelines. All patients or their guardians gave written informed consent before initiation of any study‐related procedures, before enrolment in the main study and at the end of the main study for continuation into the extension study.
2.3. Efficacy and safety assessments
Bleeding events and all administered infusions were recorded by patients using an electronic patient diary. Annualised bleeding rates (ABRs) were calculated for each dosing group and for the VAR group. Throughout the extension study, patients were closely monitored at visits every 6 months for the incidence of adverse events (AEs), which were documented in terms of type, severity and relationship to study drug, and also immunogenicity, including inhibitor development and anti‐PEG antibodies, and quantitative PEG levels in plasma. Inhibitor development was defined as a Nijmegen‐modified Bethesda assay measured titre of ≥0.6 BU, confirmed in a second independent sample (ideally collected within 2 weeks of the first inhibitor detection). Anti‐PEG antibodies were determined by enzyme‐linked immunosorbent assay. Quantitative PEG levels in plasma were measured using a combination of size exclusion chromatography and tandem mass spectrometry detection with a lower limit of quantification (LLOQ) of 0.1 mg/L. No routine pharmacokinetic sample collection was included in the extension study; however, blood samples were collected every 6 months during the extension and at the final extension visit for determining trough levels. Pre‐injection FVIII trough level measurements were based on chromogenic assay. For the sub‐analysis on FVIII trough levels, patients were grouped according to current prophylaxis regimen in order to assess pre‐injection geometric mean FVIII levels during the extension. Time since last injection prior to sampling was calculated based on electronic patient diary records of prophylaxis injections.
Patients were also assessed for hepatic and renal function. Serum and urine samples were collected during extension visits every 6 months and at the final extension visit. Hepatic biomarkers including ALT and AST were used to assess hepatic function. Renal biomarkers including serum creatinine level and calculated clearance of creatinine were used to assess renal function. Additional biomarkers of renal function were assessed, including cystatin C and lipocalin 2 in serum and, albumin and β2 microglobulin in urine.
2.4. Statistical analyses
Statistical analysis was performed using SAS® software 9.2 (SAS Institute Inc.). Summary statistics were calculated for continuous data, and frequencies were calculated for categorical data. The intent‐to‐treat (ITT) population (all patients who received ≥1 infusion of BAY 94–9027 for whom infusion and bleeding data were available) was used in the primary efficacy analysis. All patients who received ≥1 dose of study drug were included in the safety analysis. No formal statistics were performed to determine the sample size. The analysis was descriptive and based on extension data. Exposure data including total time and total EDs were based on total time in study (main and extension periods). Treatment groups were based on the treatment at the start of extension except for the VAR group, which included all patients who switched regimen at least once.
3. RESULTS
3.1. Patients
Of 134 patients treated with BAY 94–9027 in PROTECT VIII, 126 patients completed the main study, of which 121 patients enrolled into the extension study and are included in this analysis. Table 1 reports disease characteristics and patient demographics at baseline and at the end of extension. The median age of patients at enrolment was 36.0 years and at the end of extension was 40.0 years (Table 1). Regimen switching from extension enrolment to completion is shown in Figure 1. During the total time in extension, 83 out of 107 patients continued with the prophylaxis regimen they first received at the start of the extension. The VAR group (n = 28) included 20 patients who switched from a lower to a higher frequency regimen (E7D to E5D, n = 8; E7D to 2 × W, n = 5; E5D to 2 × W, n = 7), 4 patients who switched from a higher to a lower frequency regimen (2 × W to E7D, n = 1; E5D to E7D, n = 3) and 4 patients who switched more than once and were receiving the same treatment they started the extension with. At the last visit of the extension study, 72 patients (67.3%) were treated with regimens with extended intervals (E5D or E7D) and 35 patients (32.7%) were treated 2 × W. Twelve patients discontinued (AE, n = 2; withdrawn consent, n = 3; lack of follow‐up, n = 1; dropped‐out, n = 5; other, n = 1).
TABLE 1.
Disease characteristics and patient demographics at baseline and at the end of extension of PROTECT VIII
| Patient demographics | On‐demand (n = 14) | Prophylaxis | Total (n = 121) | ||||
|---|---|---|---|---|---|---|---|
| Twice a week (n = 23) | Every 5 days a (n = 33) | Every 7 days (n = 23) | Variable frequency b (n = 28) | Total prophylaxis (n = 107) | |||
| Age, y | |||||||
| Median (range) | |||||||
| Baseline | 43.5 (22–61) | 35.0 (12–58) | 33.0 (14–61) | 31.0 (13–53) | 39.5 (13–62) | 33.0 (12–62) | 36.0 (12–62) |
| End of extension | 47.5 (24–66) | 41.0 (17–61) | 38.0 (20–66) | 37.0 (17–60) | 43.0 (15–67) | 39.0 (15–67) | 40.0 (15–67) |
| Body mass index, kg/m2 | |||||||
| Median (range) | |||||||
| Baseline | 26.0 (19–31) | 22.6 (15–30) | 24.6 (17–34) | 23.4 (19–34) | 24.6 (18–42) | 23.9 (15–42) | 24.0 (15–42) |
| End of extension | 25.1 (19–33) | 22.4 (16–29) | 25.4 (17–33) | 25.5 (18–33) | 25.3 (20–36) | 24.3 (16–36) | 24.4 (16–36) |
| n (%) | 14 (100.0) | 22 (95.7) | 29 (87.9) | 19 (82.6) | 25 (89.3) | 95 (88.8) | 109 (90.1) |
| Patient demographics at baseline | |||||||
| Race, n (%) | |||||||
| White | 7 (50.0) | 16 (69.6) | 22 (66.7) | 12 (52.2) | 21 (75.0) | 71 (66.4) | 78 (64.5) |
| Black or African American | 1 (7.1) | 0 (0.0) | 1 (3.0) | 2 (8.7) | 1 (3.6) | 4 (3.7) | 5 (4.1) |
| Asian | 5 (35.7) | 6 (26.1) | 9 (27.3) | 6 (26.1) | 4 (14.3) | 25 (23.4) | 30 (24.8) |
| Not reported | 1 (7.1) | 1 (4.3) | 1 (3.0) | 3 (13.0) | 2 (7.1) | 7 (6.5) | 8 (6.6) |
| Previous FVIII treatment, n (%) | |||||||
| On demand | 14 (100.0) | 10 (43.5) | 5 (15.2) | 6 (26.1) | 5 (17.9) | 26 (24.3) | 40 (33.1) |
| Prophylaxis | 0 (0.0) | 13 (56.5) | 29 (87.9) | 17 (73.9) | 23 (82.1) | 82 (76.6) | 82 (67.8) |
| Baseline disease characteristics | |||||||
| Target joint present, n (%) | |||||||
| No | 3 (21.4) | 3 (13.0) | 11 (33.3) | 9 (39.1) | 7 (25.0) | 30 (28.0) | 33 (27.3) |
| Yes | 11 (78.6%) | 20 (87.0%) | 22 (66.7%) | 14 (60.9%) | 21 (75.0%) | 77 (72.0%) | 88 (72.7%) |
| Number of target joints per patient | |||||||
| Median (Q1;Q3) | 2.5 (1.0, 4.0) | 2.0 (1.0;2.0) | 1.0 (0.0;2.0) | 1.0 (0.0;2.0) | 1.5 (0.5;3.5) | 1.0 (0.0;2.0) | 1.0 (0.0;2.0) |
| Number of bleeds in the previous 12 months before screening | |||||||
| n (%) | 14 (100.0) | 23 (100.0) | 32 (96.9) | 23 (100.0) | 27 (96.4) | 105 (98.1) | 119 (98.3) |
| Median (Q1;Q3) | 25.5 (12.0;47.0) | 15.0 (9.0;25.0) | 3.5 (2.0;12.0) | 3.0 (0.0;12.0) | 6.0 (1.0;15.0) | 8.0 (2.0;16.0) | 9.0 (2.0;21.0) |
| Number of joint bleeds in the previous 12 months before screening | |||||||
| n (%) | 14 (100.0) | 23 (100.0) | 32 (96.9) | 23 (100.0) | 27 (96.4) | 105 (98.1) | 119 (98.3) |
| Median (Q1;Q3) | 19.5 (10.0;47.0) | 9.0 (5.0;23.0) | 2.0 (0.0;10.5) | 1.0 (0.0;11.0) | 5.0 (1.0;12.0) | 5.0 (1.0;12.0) | 5.0 (1.0;15.0) |
Two patients in the ‘Every 5 days’ regimen at data cut‐off had an unknown prophylaxis treatment frequency pre‐study. One of these patients with unknown prophylaxis treatment frequency also received treatment as ‘on demand when having a bleed’.
Patients who switched regimens after a week of first infusion in the extension period. FVIII, factor VIII; Q, quartile; y, years.
FIGURE 1.
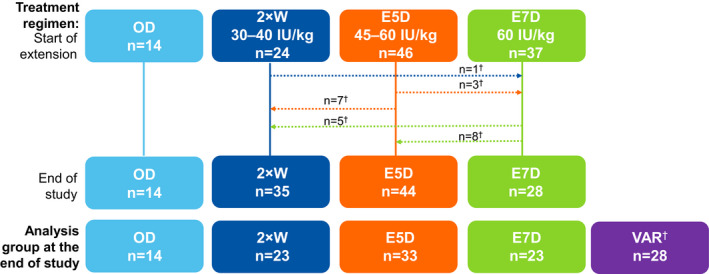
Patient disposition from the start to completion of extension period. †Dotted lines represent the overall shift in weekly prophylaxis regimens between the start and end of the extension study. Patients who switched regimens after a week of first infusion in the extension period were separately analysed in the variable (VAR) group, and other analysis groups represent patients who stayed on the regimen during total time in extension. 2 × W, twice weekly; ABR, annualised bleeding rate; E5D, every 5 days; E7D, every 7 days; IU, international units; OD, on‐ demand; VAR, variable frequency
3.2. Treatment exposure
At extension completion, median (range) total time in the full study (main and extension studies) was 3.9 (0.8–7.0) years with 223 (23–698) EDs. Seventy‐one patients were treated for ≥3 years, 53 for ≥4 years and 36 for ≥5 years. In the extension, prophylaxis patients (n = 107) accumulated a median (range) time in study of 3.2 (0.1–6.3) years with 211 (9–621) EDs (Table 2). Median (range) total number of infusions per year for the total prophylaxis group was 75.0 (48.7–143.9). Corresponding values for patients treated on 2 × W, E5D and E7D prophylaxis regimens were 106.0 (94.9–143.9), 74.0 (70.3–115.6) and 53.0 (48.7–67.6), respectively (Table 2). For those who completed ≥5 years of BAY 94–9027 treatment, median (Q1; Q3) FVIII utilisation and dose per infusion for the total prophylaxis group were 3332 (3144; 3991) IU/kg/year and 51 (44; 58) IU/kg/infusion, respectively.
TABLE 2.
FVIII utilisation during the PROTECT VIII extension study
| FVIII utilisation | On‐ demand (n = 14) | Prophylaxis | ||||
|---|---|---|---|---|---|---|
| Twice weekly n = 23 |
Every 5 days n = 33 |
Every 7 days n = 23 |
Variable frequency a n = 28 |
Total Prophylaxis (n = 107) | ||
| Total dose per year, IU/kg, median (range) | 1394.2 (736.5–3998.2) | 3916.9 (2776.8–5424.3) | 3503.5 (2995.2–4702.9) | 3129.7 (2623.5–5232.8) | 3826.2 (2789.6 (5328.0) | 3539.1 (2623.5–5424.3) |
| Total dose per infusion, IU/kg, median (range) | 32.8 (22.2–60.9) | 36.7 (26.8–42.8) | 44.8 (40.7–59.6) | 59.0 (51.2–107.5) | 50.68 (29.1–63.8) | 46.9 (26.8–107.5) |
| Number of infusions/year median (range) | 38.0 (18.6–108.0) | 106.0 (94.9–143.9) | 74 (70.3–115.6) | 53.0 (48.7–67.6) | 75.0 (54.2–125.2) | 75.0 (48.7–143.9) |
| Total EDs in extension, median (range) | 101.5 (13–176) | 168 (11–621) | 129 (23–450) | 163 (9–328) | 348.5 (46–584) | 211.0 (9–621) |
| Time in extension, years median (range) | 3.2 (0.6–4.1) | 1.3 (0.1–6.0) | 1.7 (0.3–6.2) | 3.1 (0.3–6.3) | 4.2 (0.5–6.2) | 3.2 (0.1–6.3) |
Abbreviations: ED, exposure day; FVIII, factor VIII.
Patients who switched regimens after a week of first infusion in the extension period.
3.3. Annualised bleeding rates
Median (Q1; Q3) total ABR during the extension study was 1.49 (0.36; 4.80) for all prophylaxis patients (n = 107), compared with 34.09 (20.3; 36.6) for on‐demand patients (n = 14). Among prophylaxis patients who did not switch dosing regimen, median total ABR was <2.0 (Figure 2). Spontaneous bleeds were also reduced with all prophylaxis regimens compared with on‐demand treatment: 0.75 (0.0; 2.9) vs 20.74 (12.9; 28.9) for total prophylaxis and on‐demand, respectively (Figure 2). Joint ABR for prophylaxis patients was 0.88 (0.0; 3.37), compared with a median joint ABR of 20.81 (12.3; 33.7) for patients treated on‐demand.
FIGURE 2.
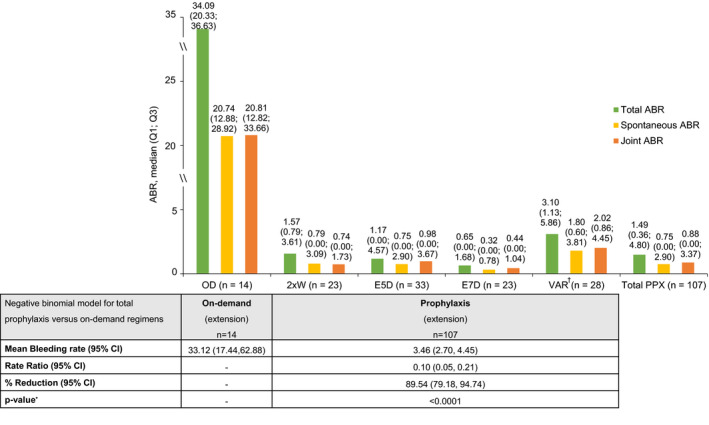
ABR by treatment regimen in PROTECT VIII extension study. 2 × W, twice weekly; ABR, annualised bleeding rate; CI, confidence interval; E5D, every 5 days; E7D, every 7 days; OD, on‐demand; PPX, prophylaxis; Q, quartile; VAR, variable. †Patients who switched regimens after a week of first infusion in the extension period. * p‐values were nominally derived from the negative binomial model, with no adjustments made for multiple comparisons
3.3.1. Zero bleeds
Throughout the extension, 20.6% of prophylaxis patients had 0 total bleeds and 29.9% had 0 joint bleeds during their participation in the extension study (Figure 3). In the last 12 months of the extension for patients who participated at least 12 months in the extension study (n = 85), 38.8% had 0 total bleeds and 45.9% had 0 joint bleeds. In the last six months of participation in the extension (n = 96), 50.0% and 58.3% had 0 total and joint bleeds, respectively.
FIGURE 3.
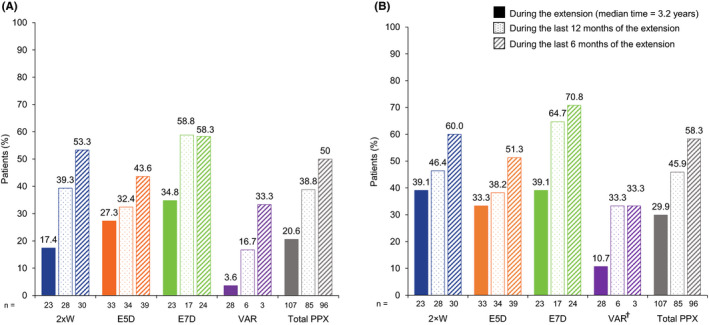
Patients with zero total (A) or zero joint bleeds (B) in the PROTECT VIII extension study. †Patients who switched regimens after a week of first infusion in the extension period. 2 × W, twice weekly; E5D, every 5 days; E7D, every 7 days; PPX, prophylaxis; VAR, variable
3.3.2. Bleeding patterns throughout pre‐study, main study and extension periods of PROTECT VIII
Patients who switched their regimen from on demand (pre‐study) to prophylaxis for PROTECT VIII (n = 22) had marked improvements in median ABRs, irrespective of which regimen they received, while median ABRs increased for patients who continued on‐demand treatment into the main study and extension (n = 14) (Figure 4). During the extension, median total ABRs (Q1; Q3) were 1.3 (0.0; 3.4) and 34.1 (20.3; 36.6) for on demand (pre‐study) to prophylaxis (PROTECT VIII) group and for those who continued their on‐demand regimen, respectively (Figure 4A). The respective median joint ABRs (Q1; Q3) were 0.6 (0.0; 3.1) and 20.8 (12.8; 33.7) (Figure 4B). At the start of the extension, 3 patients switched to prophylaxis from on‐demand regimen in the pre‐ and main study (2 × W, n = 2; VAR, n = 1).
FIGURE 4.
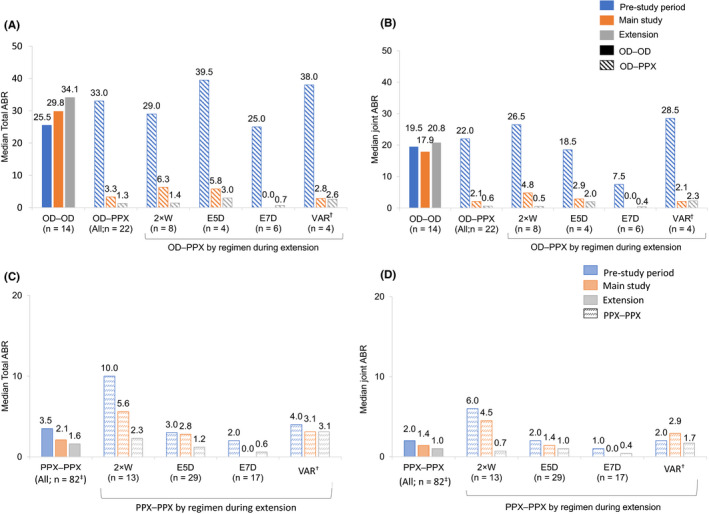
Median total (A, C) and joint (B, D) ABRs by pre‐study treatment regimen throughout PROTECT VIII. †Patients who switched regimens after a week of first infusion in the extension period. ‡This includes 3 patients who switched to prophylaxis at the start of the extension from OD regimen in the pre‐ and main study. 2 × W, twice weekly; ABR, annualised bleeding rate; E5D, every 5 days; E7D, every 7 days; OD, on‐demand; PPX, prophylaxis; VAR, variable
In prophylaxis patients who were also treated with prophylaxis prior to study entry (n = 82), median total ABR reduced from 3.5 (1.0; 10.0) pre‐study to 2.1 (0.0; 6.9) in the main study and further reduced to 1.6 (0.5; 4.9) during the extension; similarly, joint ABR reduced from 2.0 (0.0; 8.0) in the pre‐study to 1.4 (0.0; 4.5) in the main study, and to 1.0 (0.0; 3.7) during the extension (Figure 4).
3.3.3. Bleeding outcomes in patients with ≥5 years of BAY 94–9027
Of 134 patients who enrolled in the main study, 36 who continued into the extension had ≥5 years of prophylaxis treatment with BAY 94–9027: 2 × W (n = 4); E5D (n = 10); E7D (n = 8); VAR (n = 14). In total, these patients had a median (Q1; Q3) ABR of 1.14 (0.43; 2.10). Spontaneous and joint ABRs were 0.53 (0.09; 1.41) and 0.87 (0.36; 1.70), respectively, in these patients. In the last 6 and 12 months of the extension (n = 36), up to 63.9% and 50.0% of patients had 0 bleeds, respectively (Figure S2).
3.3.4. FVIII trough levels
Pre‐injection geometric mean (95% CI) FVIII trough levels in plasma during the extension period were 3.3% (1.4, 7.9) and 1.8% (1.1, 2.9) in patients who were treated with prophylaxis 2 × W with 3 days since last injection, and 4 days since last injection, respectively (Table 3). The corresponding values in patients treated with prophylaxis E5D, and E7D were 1.5% (1.0, 2.1) and 1.1% (0.8, 1.5), respectively.
TABLE 3.
Pre‐injection FVIII trough levels during the PROTECT VIII extension study
| Prophylaxis regimen (n) | Time since last injection | FVIII (%) trough level in plasma a | ||
|---|---|---|---|---|
| Arithmetic mean (SD) | Geometric mean (95% CI) | Geometric CV (%) | ||
| Twice weekly (9) |
3 days (61–84 h) |
5.0 (3.6) | 3.3 (1.4; 7.9) | 163.1 |
| Twice weekly (16) |
4 days (85–108 h) |
2.6 (2.4) | 1.8 (1.1; 2.9) | 106.5 |
| Every 5 days (22) |
5 days (109–132 h) |
2.1 (2.1) | 1.5 (0.9; 2.1) | 103.5 |
| Every 7 days (17) |
7 days (157–192 h) |
1.4 (1.1) | 1.1 (0.8; 1.5) | 66.4 |
Abbreviations: CI, confidence interval; CV, coefficient of variation; SD, standard deviation.
Trough levels were considered invalid if FVIII (%) >37.5%, >23.1%, >21.5% and >8.1% after 3, 4, 5 and 7 days, respectively.
3.4. Safety
During the extension, 10 (71.4%) on‐demand and 86 (80.4%) prophylaxis patients reported treatment‐emergent AEs (TEAEs) (Table 4). Ten (8.3%) patients reported study drug related TEAEs (most common types were musculoskeletal disorders [n = 4] and laboratory abnormal results [n = 3]). Study drug related serious AEs were reported in two (1.7%) patients (elevated liver function tests and migratory back pain, respectively), both leading to study drug discontinuation. Among patients who completed ≥5 years of BAY 94–9027 prophylaxis, 5 patients reported non‐serious drug‐related TEAEs: bone marrow oedema (moderate, n = 1), alanine aminotransferase increase (mild, n = 1), β2 microglobulin urine increase (mild, n = 1), arthralgia (mild, n = 1), meniscal degeneration (moderate, n = 1) and osteoarthritis (moderate, n = 1). None experienced serious drug‐related TEAEs. No patient had confirmed FVIII inhibitors during the extension. No allergic reactions, deaths or thrombotic events were reported.
TABLE 4.
TEAEs during the PROTECT VIII extension study
| TEAEs |
On‐demand (N = 14) |
Prophylaxis (N = 107) |
Total (N = 121) |
|---|---|---|---|
| Any AE, n (%) | 10 (71.4) | 86 (80.4) | 96 (79.3) |
| Mild | 1 (7.1) | 26 (24.3) | 27 (22.3) |
| Moderate | 5 (35.7) | 36 (33.6) | 41 (33.9) |
| Severe | 4 (28.6) | 24 (22.4) | 28 (23.1) |
| Any study drug related AE | 0 (0.0) | 10 (9.3) | 10 (8.3) |
| Mild | 0 (0.0) | 5 (4.7) | 5 (4.1) |
| Moderate | 0 (0.0) | 4 (3.7) | 4 (3.3) |
| Severe | 0 (0.0) | 1 (0.9) | 1 (0.8) |
| Any AE related to procedures as per protocol | 0 (0.0) | 3 (2.8) | 3 (2.5) |
| AE‐related deaths, n (%) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Any SAE, n (%) | 2 (14.3) | 34 (31.8) | 36 (29.8) |
| Any study drug related SAE | 0 (0.0) | 2 (1.9) a | 2 (1.7) |
| Discontinuation due to AE, n (%) | 0 (0.0) | 2 (1.9) | 2 (1.7) |
| Discontinuation due to SAE, n (%) | 0 (0.0) | 2 (1.9) | 2 (1.7) |
Abbreviations: AE, adverse events; SAE, serious adverse events; TEAE, treatment emergent adverse events.
Elevated liver function tests in one patient and severe migratory back pain (two cases) in another patient, both treated with the twice weekly prophylaxis regimen, resulting in discontinuation of the study drug.
At the final visit of the extension, patients had similar levels of markers of renal and hepatic function compared with baseline (Table S1). Overall, of the patients with available data, median levels of serum creatinine (n = 112), creatinine clearance (n = 112), ALT level (n = 109) and AST level (n = 108) remained in the normal range (Table S2).
During the extension, 8 (6.6%) patients were positive for low titre anti‐PEG antibodies. Of these, 7 (5.8%) patients were transiently positive for anti‐PEG antibodies at single visits during the study and negative at the last visit. One patient (n = 1, 0.8%) tested positive for anti‐PEG antibodies at the final visit only; by protocol, further testing was not allowed. None of the 8 patients had any associated clinical symptoms.
During the entire extension, 4 patients had very low levels of PEG in plasma just above the LLOQ at single visits without any clinical symptoms. In three of these patients, PEG was not detectable during the rest of the study. In one patient, low levels of PEG were detected in plasma at the final extension visit only; by protocol, further testing was not allowed.
4. DISCUSSION
Data from PROTECT VIII and its extension show that BAY 94–9027 prophylaxis is efficacious in adults and adolescents with severe haemophilia A at dose intervals of up to E7D. BAY 94–9027 prophylaxis reduced total and joint ABRs in the main and extension studies compared with pre‐study ABRs. Most prophylaxis patients in the main study continued their respective treatment regimen during the extension period. The trial design allowed patients to switch regimen to reflect real clinical practice, and most patients who switched chose to for improved protection. Overall, BAY 94– 9027 consumption remained stable over the years of treatment. Of note, the number of infusions received by patients treated with E7D prophylaxis during the extension was close to those who received treatment on‐demand. Nevertheless, E7D prophylaxis resulted in marked improvement in ABRs in these patients compared with the on‐demand group. These long‐term data corroborate those reported in the main study. 11
Total ABRs from pre‐study to the end of extension indicated a gradual decrease in bleeding events over time with BAY 94–9027 prophylaxis. These improvements in ABR were observed for all prophylaxis patients, irrespective of prior treatment regimen, and for all prophylaxis treatment regimens. For patients who were previously treated on demand, despite entering PROTECT VIII with higher ABRs than those who were previously on prophylaxis, the improvements in ABRs observed were such that the ABRs achieved in the extension were similar to those of patients previously on prophylaxis. Prophylaxis with BAY 94–9027 also resulted in a low incidence of joint bleeds with long‐term use for several years. Long‐term treatment with BAY 94–9027 thus resulted in continued improvement in bleed protection.
The ASPIRE and pathfinder2 extensions evaluated the long‐term efficacy and safety in patients with severe haemophilia who completed their treatment with rFVIII Fc fusion protein (rFVIIIFc, efmoroctocog alfa, Elocta®) and N8‐GP (turoctocog alfa pegol, Esperoct®) in the phase 3 main studies, respectively. 13 , 14 In the ASPIRE extension study, patients had a median cumulative treatment duration (range) of approximately 4.5 (0.7–5.9) years. 13 In the entire pathfinder2 study, patients were treated with N8‐GP for up to 6.6 years with a total median treatment duration of 5.4 (0.01–6.6) years. 15 Of note, while all these extension studies had the option for an E7D prophylaxis regimen, the study designs for the E7D treatment arms in all the three extension studies differ, including a higher dose (65 IU/kg rFVIIIFc; 75 IU/kg N8‐GP) compared with PROTECT VIII (60 IU/kg BAY 94–9027). Hence, due to differences in study design, a direct comparison of efficacy results from these studies is not possible.
BAY 94–9027 was well tolerated during the PROTECT VIII extension: no patients developed FVIII inhibitors; most patients had no detectable PEG in plasma. A few patients had transiently detectable PEG just above the LLOQ. One patient had detectable PEG in plasma only at the last visit of the study, and in accordance with the study protocol, further follow‐up was not allowed. There were few study drug related TEAEs, and markers of renal and hepatic function remained within normal limits for up to 7 years of observation of BAY 94–9027 treatment. These data support the favourable risk‐benefit profile of BAY 94–9027 and its use as a long‐term treatment option for patients with haemophilia A.
PROTECT VIII extension has the longest individual patient follow‐up of any other extension studies in adolescents and adults with severe haemophilia A. 13 , 15 ABR remained low in all prophylaxis groups and was maintained over ≥5 years’ using infusion schedules best fitting individual patients’ needs and low annual FVIII consumption, providing important long‐term efficacy and safety profiles.
FVIII replacement is the standard of care and has improved the quality of life and life expectancy of patients with haemophilia A. 1 , 16 , 17 Unlike non‐replacement therapies, FVIII replacement provides FVIII peaks and can be used for the treatment of bleeds and perioperative management. 18 In PROTECT VIII, the individualised approach of adjusting prophylactic regimen provided the necessary peak and trough levels for periods of high activity—an effective strategy as evidenced by the low overall bleeding rate in PROTECT VIII. With an established safety profile, long‐acting FVIII replacement therapies, including BAY 94–9027, provide comprehensive management of haemophilia A, allowing patients to meet their lifestyle goals. 19 , 20
CONFLICT OF INTERESTS
M. T. Reding has received honoraria for participation on advisory boards and/or speakers bureaus from Bayer, CSL Behring, Novo Nordisk, Sanofi Genzyme, Takeda, and grant funding from Bayer and Biomarin. I. Pabinger has received honoraria for lectures and advisory board meetings from Bayer, Biotest, CSL Behring, Octapharma, Pfizer, Shire, Novo Nordisk, Sobi, Roche, and has received unrestricted research grants from CSL Behring and Novo Nordisk. P. A. Holme has received speaker honoraria from Bayer, Novo Nordisk, Octapharma, Pfizer, and Shire and Sobi, and has received grant/research support from Bayer, Octapharma, Pfizer and Shire L. Poulsen has received honoraria for congresses from Bayer, Novo Nordisk, Sobi, Pfizer and Octapharma and has received grant/research support from Pfizer C. Negrier has received grant/research support, honoraria or consultation fees from Alnylam, Baxalta/Shire, Bayer, CSL Behring, LFB, Novo Nordisk, Octapharma, Roche, Pfizer and Sobi. P. Chalasani has received research support from Pfizer, not related to any haematologic products. M. Maas Enriquez and M. Wang and are employees of Bayer. K. Meijer has received honoraria from UniQure, and has received grant/research support from Bayer, Sanquin and Pfizer M. Elisa Mancuso has received honoraria for participation on advisory boards from Bayer, Bioverativ, Catalyst, CSL Behring, Grifols, Kedrion, Novo Nordisk, Octapharma, Pfizer, Roche, Shire and Sobi, and has received honoraria on speakers bureaus from Bayer, Biotest, CSL Behring, Grifols, Kedrion, Novo Nordisk, Octapharma, Pfizer, Roche, Shire and Sobi. S. Lalezari has received honoraria/consultation fees from Bayer, Pi Healthcare, Pfizer, Roche, Takeda and Teva, and for symposia/congresses from Alnylam, Bayer, Baxter, Biogen, BioMarin, Grifols, Novo Nordisk, Pfizer, Roche, Daiichi Sankyo and Janssen.
AUTHOR CONTRIBUTIONS
M. T. Reding, I. Pabinger, P. A. Holme, L. Poulsen, C. Negrier, P. Chalasani, K. Meijer and S. Lalezari are principal investigators, treated patients with study drug, and contributed to data acquisition and interpretation. M. Maas Enriquez was involved in analysis and interpretation of data. M. Wang was involved in design, analysis and interpretation of data. M. Elisa Mancuso was involved in interpretation of data. All authors contributed to the development of the manuscript, reviewed and commented on each draft and approved the final draft.
Supporting information
Figure S1
Figure S2
Table S1‐S2
ACKNOWLEDGEMENTS
This study and the PROTECT VIII main study were funded by Bayer. Medical writing assistance was provided by Sreerekha S. Pillai, PhD, of Darwin Healthcare Communications (Oxford, England) and was fully funded by Bayer.
DATA AVAILABILITY STATEMENT
Availability of the data that support the findings of this study will be determined according to Bayer's commitment to the EFPIA/PhRMA ‘Principles for responsible clinical trial data sharing’ and can be requested via www.clinicalstudydatarequest.com.
REFERENCES
- 1. Manco‐Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535‐544. [DOI] [PubMed] [Google Scholar]
- 2. Gringeri A, Lundin B, Von Mackensen S, Mantovani L, Mannucci PM. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J Thromb Haemost. 2011;9(4):700‐710. [DOI] [PubMed] [Google Scholar]
- 3. Ar MC, Vaide I, Fau ‐ Berntorp E, Björkman S. Methods for individualising factor VIII dosing in prophylaxis. Eur J Haematol Suppl. 2014;76:16‐20. [DOI] [PubMed] [Google Scholar]
- 4. Lambert T, Benson G, Dolan G, et al. Practical aspects of extended half‐life products for the treatment of haemophilia. Ther Adv Hematol. 2018;9(9):295‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mei B, Pan C, Jiang H, et al. Rational design of a fully active, long‐acting PEGylated factor VIII for hemophilia A treatment. Blood. 2010;116(2):270‐279. [DOI] [PubMed] [Google Scholar]
- 6. Coyle TE, Reding MT, Lin JC, Michaels LA, Shah A, Powell J. Phase I study of BAY 94–9027, a PEGylated B‐domain‐deleted recombinant factor VIII with an extended half‐life, in subjects with hemophilia A. J Thromb Haemost. 2014;12(4):488‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. European Medicines Agency . Jivi (damoctocog alfa pegol) summary of positive opinion. 2018; https://www.ema.europa.eu/en/documents/smop‐initial/chmp‐summary‐positive‐opinion‐jivi_en.pdf.
- 8. Pharmaceuticals and Medical Devices Agency . Review report (Jivi, damoctocog alfa pegol). 2018; https://www.pmda.go.jp/files/000229786.pdf.
- 9. US Food and Drug Administration . Jivi prescribing information. 2018; https://labeling.bayerhealthcare.com/html/products/pi/Jivi_PI.pdf.
- 10. Government of Canada . Regulatory decision summary ‐ Jivi ‐ Health Canada. 2018; https://hpr‐rps.hres.ca/reg‐content/regulatory‐decision‐summary‐detail.php?lang=en&linkID=RDS00474.
- 11. Reding MT, Ng HJ, Poulsen LH, et al. Safety and efficacy of BAY 94–9027, a prolonged‐half‐life factor VIII. J Thromb Haemost. 2017;15(3):411‐419. [DOI] [PubMed] [Google Scholar]
- 12. Lalezari S, Reding MT, Pabinger I, et al. BAY 94–9027 prophylaxis is efficacious and well tolerated for up to >5 years with extended dosing intervals: PROTECT VIII extension interim results. Haemophilia. 2019;25(6):1011‐1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nolan B, Mahlangu J, Pabinger I, et al. Recombinant factor VIII Fc fusion protein for the treatment of severe haemophilia A: Final results from the ASPIRE extension study. Haemophilia. 2020;26(3):494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Curry N, Albayrak C, Escobar M, et al. Once‐weekly prophylaxis with glycoPEGylated recombinant factor VIII (N8‐GP) in severe haemophilia A: Safety and efficacy results from pathfinder 2 (randomized phase III trial). Haemophilia. 2019;25(3):373‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Giangrande P, Abdul Karim F, Nemes L, et al. Long‐term safety and efficacy of N8‐GP in previously treated adults and adolescents with hemophilia A: Final results from pathfinder2. J Thromb Haemost. 2020;18(Suppl 1):5‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Darby SC, Kan SW, Spooner RJ, et al. Mortality rates, life expectancy, and causes of death in people with hemophilia A or B in the United Kingdom who were not infected with HIV. Blood. 2007;110(3):815‐825. [DOI] [PubMed] [Google Scholar]
- 17. Lofqvist T, Nilsson IM, Berntorp E, Pettersson H. Haemophilia prophylaxis in young patients–a long‐term follow‐up. J Intern Med. 1997;241(5):395‐400. [DOI] [PubMed] [Google Scholar]
- 18. Collins PW, Liesner R, Makris M, et al. Treatment of bleeding episodes in haemophilia A complicated by a factor VIII inhibitor in patients receiving emicizumab. interim guidance from UKHCDO inhibitor working party and executive committee. Haemophilia. 2018;24(3):344‐347. [DOI] [PubMed] [Google Scholar]
- 19. Aledort L, Mannucci PM, Schramm W, Tarantino M. Factor VIII replacement is still the standard of care in haemophilia A. Blood Transfus. 2019;17(6):479‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ar MC, Balkan C, Kavaklı K. Extended half‐life (EHL) coagulation factors: a new era in the management of haemophilia patients. Turk J Haematol. 2019;36(3):141‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Table S1‐S2
Data Availability Statement
Availability of the data that support the findings of this study will be determined according to Bayer's commitment to the EFPIA/PhRMA ‘Principles for responsible clinical trial data sharing’ and can be requested via www.clinicalstudydatarequest.com.