

Abstract
Preterm birth (PTB), or birth that occurs earlier than 37 weeks of gestational age, is a major contributor to infant mortality and neonatal hospitalization. Mutations in the mitochondrial genome (mtDNA) have been linked to various rare mitochondrial disorders and may be a contributing factor in PTB given that maternal genetic factors have been strongly linked to PTB. However, to date, no study has found a conclusive connection between a particular mtDNA variant and PTB. Given the high mtDNA copy number per cell, an automated pipeline was developed for detecting mtDNA variants using low‐coverage whole‐genome sequencing (lcWGS) data. The pipeline was first validated against samples of known heteroplasmy, and then applied to 929 samples from a PTB cohort from diverse ethnic backgrounds with an average gestational age of 27.18 weeks (range: 21–30). Our new pipeline successfully identified haplogroups and a large number of mtDNA variants in this large PTB cohort, including 8 samples carrying known pathogenic variants and 47 samples carrying rare mtDNA variants. These results confirm that lcWGS can be utilized to reliably identify mtDNA variants. These mtDNA variants may make a contribution toward preterm birth in a small proportion of live births.
Keywords: human genetics, low‐coverage whole‐genome sequencing, mitochondrial disease, mitochondrial genome, preterm birth
Maternal genetic factors have been strongly linked to preterm birth (PTB). While mitochondrial DNA (mtDNA) has been proposed as a possible maternal factor that may contribute to preterm birth, no study has conclusively linked an mtDNA variant to PTB. As shown here, we have developed a new automated pipeline for mining mtDNA variants using low‐pass whole genome sequencing (lcWGS) data, and applied it to 929 preterm lcWGS samples to elucidate the contribution of mtDNA to preterm birth.
1. INTRODUCTION
The mitochondrial organelle plays a central role in the energy metabolism of the cell. The various functions of the mitochondrion are performed by approximately 1500 genes encoded in the nuclear genome of human cells, as well as a small number of genes carried by a separate genome carried within the mitochondrion itself, commonly referred to as “mitochondrial DNA,” or “mtDNA,” which is inherited virtually exclusively from the mother. Mitochondrial diseases can be caused by mutations in both the mtDNA as well as nuclear genomes. Variants in the human mtDNA have been linked to a variety of rare “primary” mitochondrial disorders, but may also influence more common forms of the disease, such as cancer and diabetes (H. Li et al., 2019, 2020; Thompson et al., 2020). In general, the diagnosis of mitochondrial diseases is difficult since symptoms and severity can vary widely among affected individuals.
Preterm birth (PTB), or birth that occurs earlier than 37 weeks of gestational age, affects nearly 10% of newborns and is a major contributor to infant mortality and neonatal hospitalization (Tucker & McGuire, 2004). Environmental factors (infection/inflammation, psychosocial stress, racism, or the age of the mother), as well as genetic factors, are known to play a role in the occurrence of PTB (Chang et al., 2020; Hallman et al., 2019). The occurrence of PTB appears to vary significantly based on ethnicity (Anum et al., 2009; Soltani et al., 2019; Tucker & McGuire, 2004). Regarding the role of genetic factors in PTB, several genetic loci have been previously shown to be associated with its occurrence (C. Zhang, Montooth, et al., 2017; G. Zhang, Feenstra, et al., 2017). However, there are still unknown genetic factors that remain to be uncovered. Mutations in the mitochondrial genome (mtDNA) have been linked to various rare mitochondrial disorders, and may also be a contributing factor in PTB given that maternal genetic factors have been consistently linked to the occurrence of PTB (Boyd et al., 2009; Hallman et al., 2019; Wu et al., 2015; York et al., 2013). However, to date, no study has found a decisive link between a particular mtDNA mutation and PTB, which may be due, in part, to the unusual genetic properties of the mitochondrial genome.
To properly examine whether or not mtDNA variants can contribute to PTB, we must first ensure that we have a validated methodology for detecting mtDNA sequence variants. Because there are hundreds of mtDNA copies per cell, mtDNA variants can exist at a continuum of frequencies of heteroplasmy. The severity of the disease associated with these pathogenic variants is often determined by their heteroplasmy level, with the severity increasing as the heteroplasmy level rises above a certain threshold. Due to the diversity of mitochondrial functions and physiological needs, mtDNA copy number and mutation frequencies can exist in varying levels across different tissues and cell types (M. Li et al., 2015; Robin & Wong, 1988). For these reasons, confirmation of the diagnosis in the clinical setting requires molecular genetic testing to determine both the presence as well as the heteroplasmy level of the pathogenic variant, given the critical role that the heteroplasmy level plays in the severity of the disease. This is traditionally accomplished by polymerase chain reaction (PCR) amplification of the mtDNA, followed by Sanger sequencing or next‐generation sequencing (Huang, 2011; Ma et al., 2015; Tang & Huang, 2010). However, there has been increasing interest in mining whole‐genome sequencing (WGS) data sets to help identify patients with previously unidentified, pathogenic mtDNA variants. For instance, given the fact that mtDNA data forms a subset of the sequence data provided by WGS, we have recently demonstrated that mtDNA heteroplasmy levels and mtDNA copy number can be calculated using whole‐genome DNA sequence (WGS) data (Brockhage et al., 2018; Husami et al., 2020).
Due to improving sequencing technology, improved genotyping data quality, and decreasing costs, low‐coverage whole‐genome sequencing (lcWGS) has started replacing genotyping arrays in genome‐wide association studies, particularly as it avoids the bias inherent in the choice of variants to include in the genotyping array and can also be utilized to detect structural and copy‐number variants (Homburger et al., 2019). While lcWGS data possesses a very low nuclear DNA coverage (usually around 1X to 2X), it still possesses a relatively high mtDNA coverage due to the much higher copy number of mtDNA, which is present at hundreds or thousands of copies per cell, depending on the cell type (Wallace & Chalkia, 2013). Thus, while standard variant calling on the low‐coverage nuclear DNA is challenging and prone to errors, reliable variant calling in the mitochondrial genome is feasible as the mtDNA read depth in the same sample is likely to be hundreds to thousands of times higher. In fact, lcWGS has been previously used in some cases to analyze mtDNA as part of the 1000 Genomes Project (Abecasis et al., 2010), as well as phylogenetic analysis of mitogenomes in other species (Baeza, 2020). However, until now, the mtDNA data generated by GWAS have been largely ignored in the clinical diagnostic context despite having a high enough read depth to meet standard variant calling thresholds.
Therefore, in this study, we have developed an automated pipeline for detecting mtDNA variants using lcWGS data, which can be used for the high‐throughput analysis of a large number of samples. This tool was then utilized for the task of analyzing mtDNA sequence variants that may contribute to PTB. This pipeline was first validated against previously analyzed samples of known heteroplasmy, and then applied to a set of 960 samples, including 929 samples from a PTB cohort and 31 non‐preterm samples as technical controls. This pipeline was able to accurately identify haplogroups and mtDNA variants for 957 out of the 960 lcWGS samples. Furthermore, we were able to identify the presence and heteroplasmy level of known pathogenic variants in eight of the preterm patient samples, which is overwhelmingly higher than in the general population. In addition, we also identified 47 preterm samples carrying variants of uncertain clinical significance (VUCS). The presence of these variants was confirmed using our established next‐generation DNA sequencing (NGS) techniques. These results suggest some new insights into the relationship between mtDNA genotypes and PTB and demonstrate that lcWGS is robust and efficient for identifying mtDNA variants and their heteroplasmy levels in patients.
2. MATERIALS AND METHODS
2.1. Editorial policies and ethical considerations
This study was approved by the Institutional Review Board (IRB) of Cincinnati Children's Hospital Medical Center (CCHMC) (Approval Study ID: 2013‐7868). All patients provided written informed consent to participate in this study. For the lcWGS data, patient tissues were originally collected with consent for research by the Discover Together Biobank, as approved by the IRB of CCHMC. The biobank samples were deidentified for this project. Additional information about the Discover Together Biobank can be found at https://www.cincinnatichildrens.org/service/c/clinical-trials/biobank.
2.2. lcWGS
The sequencing data were obtained from lcWGS of 929 patient samples from a PTB cohort within the Discover Together Biobank at CCHMC. The selection criteria for the samples were only limited to gestational age using the CCHMC i2b2 system (https://i2b2.cchmc.org/). Thirty‐one randomly chosen technical control samples, with no known reports of PTB, were also included to test the basic capabilities of our analytics pipeline. The sequencing data were first aligned against GRCH38, using Burrows–Wheeler alignment. The portion of the sequences aligned to the mitochondrial genome was then extracted as BAM files, which were subsequently used for the downstream analysis.
Multiplexed sequencing libraries were prepared using RipTide High Throughput Rapid DNA Library Prep (HT‐RLP) Kit from iGenomX, strictly following the manufacturer's protocol. The main steps of the HT‐RLP protocol are (1) barcode extension, (2) DNA capture, (3) extension, (4) amplification, and (5) size selection. In the first step of the protocol, a polymerase‐mediated primer/barcode extension reaction is performed in a 96‐well plate. The reaction products are then combined into one pool and all subsequent steps are performed with the single pool. An index barcode is added to the library during the PCR step. It acts as a plate identifier and permits the pooling of multiple 96‐reaction library preps on a single Illumina flow cell. We pooled ten 96‐plex libraries and generated a final pool of 960 libraries, which was then sequenced on NovaSeq 6000 sequencer using two S4 flow cells, generating a total of ~20 billion paired‐end 2 × 150 bp sequencing reads. The demux tool from iGenomX was used to demultiplex sequencing reads into individual sample reads.
The sequencing reads were first aligned against the GRCH38 human reference genome, using the Burrows–Wheeler alignment algorithm with options “bwa mem ‐B 4 ‐O 6 ‐E 1 ‐M.” The portion of the sequences aligned to the mitochondrial genome was then extracted as BAM files, which were subsequently used for the downstream analysis.
2.3. Automated pipeline for mtDNA variant calling
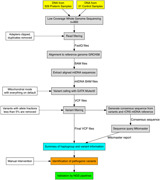
Variant calling on the aligned mtDNA sequences of each sample was performed using GATK4 Mutect2 (Cibulskis et al., 2013). Mutect2 was set to mitochondrial mode, with all other settings set to default. Variants with allele fractions below 5% were then removed from the VCF file with bcftools from samtools (the 5% cut‐off is based on previous experience with mtDNA variation and sequencing error rates associated with NGS) (H. Li et al., 2009). The final VCF file was used to create a consensus sequence using bcftools consensus with the rCRS mitochondrial genome reference (NC_012920.1). The consensus sequence was uploaded to Mitomaster using the Mitomaster API and a report containing haplogroup and variant information was obtained (Lott et al., 2013). The process of Mutect2 variant calling and upload of consensus sequence to Mitomaster was automated with a Python script, which can be provided upon request. Read depth at each base in the mtDNA portion of the aligned sequences was generated using samtools depth (H. Li et al., 2009). Read coverage of the mtDNA was generated by calculating the average read depth across all 16,569 positions in the rCRS. All variant positions were also numbered based on the corresponding positions in the CRS. A flowchart summarizing our analysis pipeline is included in Figure 1c.
Figure 1.
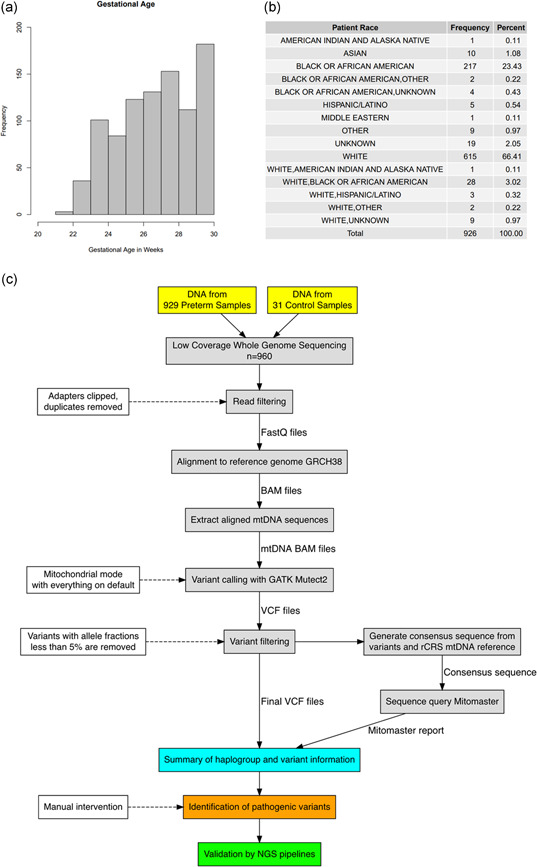
Summary of the preterm patient cohort and low‐coverage whole‐genome sequencing (lcWGS) analysis pipeline. (a) Histogram of gestational age in the 926 preterm samples, not including the three discarded samples and 31 technical controls. (b) Summary of racial demographics for the 926 preterm samples. (c) Flowchart for Mutect2 pipeline and visual summary of the lcWGS data analysis. The solid lines represent the flow of information while the dashed lines indicate the criteria and settings for the data filtration steps. Manual inspection was required for the identification of pathogenic variants (see Section 2)
2.4. Analysis of automated pipeline results
Each sample was manually checked for known pathogenic variants and likely pathogenic variants (Table S1). Mitochondrial variants were classified based on ACMG criteria (Richards et al., 2015), combined with additional information from MitoMap (http://mitomap.org/bin/view.pl/MITOMAP/WebHome).
2.5. PCR‐NGS validation
The samples with pathogenic variants were checked with PCR‐NGS and clinically validated techniques, according to our previously described approach for detecting mtDNA variants (Huang, 2011; Ma et al., 2015; Tang & Huang, 2010). For the PCR‐NGS analysis, whole mtDNA molecules were first amplified by long‐range PCR, using the following primers: mt16426F (CCGCACAAGAGTGCTACTCTCCTC) and mt16425R (GATATTGATTTCACGGAGGATGGTG). PCR amplification was performed with TaKaRa LA Taq Hot Start polymerase (TaKaRa Biotechnology) under conditions of 94°C for 1 min; 30 cycles of 98°C for 10 s and 68°C for 16 min; 72°C for 10 min; and a final hold at 4°C. The resulting PCR product was barcoded according to the Nextera XT library preparation protocol (Illumina). Sequencing was performed on the Illumina MiSeq platform (DNA Core Facility, CCHMC) and data were analyzed with NextGENe software (SoftGenetics). Briefly, sequence reads ranging from 100 to 200 bp in length were quality filtered and processed with NextGENe software and an algorithm similar to BLAT. The sequence error correction feature (condensation) was performed to reduce false‐positive variants and to produce a sample consensus sequence and variant calls. Alignment without sequence condensation was performed to calculate the percentage of the mitochondrial genome with a depth of coverage of 1000X. Quality FASTQ reads were quality filtered and converted to a FASTA format. Filtered reads were then aligned to the human mitochondrial sequence reference, NC_012920.1, followed by variant calling. Variant heteroplasmy was calculated with NextGENe software as follows: base heteroplasmy (mutant allele frequency %) = mutant allele (forward + reverse)/total coverage of all alleles C, G, T, A (forward + reverse) × 100. Clinical significance of the variants identified and the predicted haplogroups were analyzed with MitoMaster software (http://www.mitomap.org/MITOMASTER/WebHome).
3. RESULTS
3.1. Clinical summary
To test the ability of the automated pipeline to screen lcWGS data set for mtDNA variants, lcWGS data were obtained for 929 patient samples from a PTB cohort at CCHMC. Three of the 929 samples were eventually excluded from analysis due to low sequence quality and failure to pass the haplogroup‐identification step of our pipeline (see below), leaving 926 samples for further analysis. For those 926 preterm samples, the mean gestational age was 27.18 weeks with a standard deviation of 2.15 and a range of 21–30 weeks. The distribution of gestational age and patient race are provided in Figure 1a,b. For each preterm patient sample, the sex, race, ethnicity, gestational age, current age, nuclear genome and mtDNA coverage, and haplogroup were recorded. The haplogroups detected are shown in Table S1. As a technical control to verify the basic capabilities of our analytics pipeline, lcWGS data from 31 non‐preterm samples were also included in the analysis, resulting in a total of 957 samples for analysis.
3.2. Mutect2 pipeline validation
Before applying our automated Mutect2 pipeline to mitochondrial mutation detection on sequencing data, the pipeline was first validated using nine samples previously analyzed for mtDNA mutations (Husami et al., 2020). In this previous publication, these samples were analyzed using a traditional “PCR‐NGS” approach: That is, the DNA samples were first enriched for mtDNA by long‐range PCR amplification, and then sequenced with NGS and analyzed with clinically validated methods. In comparison to the PCR‐NGS results, the new, automated pipeline performed remarkably well, identifying the same nine pathogenic mutations at essentially the same heteroplasmy percentages, with a maximum difference of 2% for any given sample (average difference, 0.67%; standard deviation, 0.67%) (Table S2). A flowchart summarizing our analysis pipeline is included in Figure 1c, and a complete description can be found in Section 2.
3.3. Results of the automated Mutect2 pipeline for the 960 sample data set
To test whether or not particular mtDNA variants and haplotypes may be associated with PTB, lcWGS data were obtained for the 929 patient samples from our PTB cohort. Three out of the 960 samples were excluded from the analysis due to low sequencing quality and the inability of the automated pipeline to properly identify the haplogroup when it queried Mitomaster (see Section 2). This was likely due to the low coverage of the mitochondrial genome in these samples, which ranged from 39.8X to 336.1X.
After excluding the three low‐quality samples, this left 957 samples for further analysis. The mean mtDNA coverage for these samples was 1389X with a standard deviation of 797.6X and a range of coverage from 135.1X to 6169X (Figure 2a). In contrast, the mean nuclear genome coverage was very low (as would be expected for lcWGS data), averaging 1.9X with a standard deviation of 1.2X and a range of coverage from 0.12X to 12.5X (Figure 2b). In comparing the nuclear genome coverage to the mtDNA coverage for each sample, there was a moderate positive correlation between nuclear genome coverage and mtDNA coverage, with an R 2 value of .5959 (Figure 2c). Given the fact that mtDNA copy number can vary widely between individuals based on age or health status (Brockhage et al., 2018; Cheau‐Feng Lin et al., 2014; Xia et al., 2017; Zhou et al., 2014), while the nuclear genome remains relatively static in copy number, it is noteworthy that the two genomes retain this moderate level of correlation.
Figure 2.
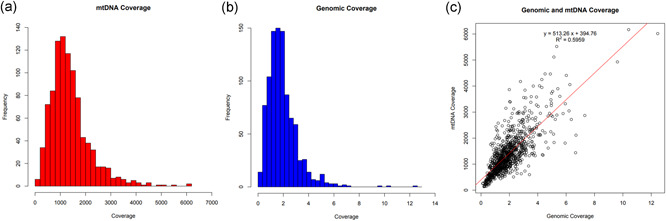
Comparison of sequencing coverage between the mitochondrial and nuclear genomes. (a) Mitochondrial genome (mtDNA) coverage for the low‐coverage whole‐genome sequencing (lcWGS) samples. The mean mtDNA read depth for the 957 samples that passed the haplogroup identification step was 1389X, with a standard deviation of 797.6X. The range of coverage was from 135.1X to 6169X. (b) Nuclear genome coverage for the lcWGS samples. The mean nuclear genome read depth for the 957 samples that passed the haplogroup identification step was 1.938X, with a standard deviation of 1.200. The range of coverage was from 0.1205X to 12.46X. (c) Comparison of the nuclear genome coverage to the mtDNA coverage across all samples. There was a moderate positive correlation between nuclear genome coverage and mtDNA coverage, with an R 2 value of .5959. Please note that the scale for the y axis (mtDNA coverage) is 500 times that of the x axis (nuclear genome coverage) to aid in the visualization of the line of best fit
3.4. Haplogroup composition of the preterm patient cohort
Mitochondrial DNA haplogroups are generally well correlated with ancestry and ethnic group, although there are notable exceptions, particularly in populations with a high degree of admixture (Cardena et al., 2013). Regarding the haplogroup composition of the PTB samples described in this study, the top five haplogroups were L2a (13.7%), J1c (6.4%), L3e (5.0%), K1a (4.8%), and L3 (4.6%) (Figure S1), representing some of the most prevalent mtDNA haplogroups in individuals of European or African ancestry (Johnson et al., 2015; Torroni et al., 1996). This is broadly in agreement with the overall demographic composition of the preterm patient cohort (Figure 1b and Table S1), further confirming the accuracy of the output derived from the lcWGS data set. Haplogroup composition of the 926 preterm samples that passed the haplogroup analysis and a heatmap of reported race and haplogroup are also provided in Figures 3 and S2, and further confirm the general concordance between the reported ethnicity of the patients and the observed mtDNA haplogroups. One striking discordance in the results, however, is the high frequency of haplogroups L2a (13.7%) and L3 (4.6%) in the patient samples reported as White, as both the L2 and L3 haplogroups are generally present in <1% of individuals in the United States who self‐identify as non‐Hispanic White (Mitchell et al., 2014). The heightened frequency of these haplogroups in our non‐Hispanic white PTB samples may suggest a possible link with PTB (see Section 4).
Figure 3.
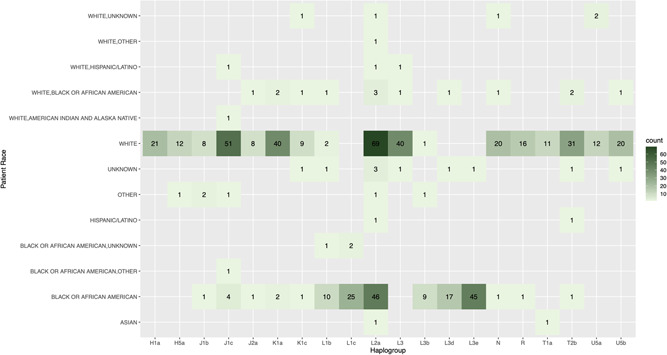
Distribution of the top 20 haplogroups in the preterm patient cohort by race. Heatmap shows the haplogroup count by race for the 20 most frequently observed haplogroups in the preterm patient cohort. The labels on the left axis represent the various self‐identified ethnic or ancestry groupings, while the labels along the bottom axis represent the 20 most frequently observed mitochondrial genome (mtDNA) haplogroups in the preterm patient cohort. The numbers in black within each square indicate the number of samples where a particular mtDNA haplogroup was detected for a particular racial or ethnic category. Each square is also color‐coded based on the number of samples with that particular combination of mtDNA haplogroup and self‐identified racial category, based on the key on the right side of the heatmap
3.5. Analysis of mtDNA variants found in preterm patient samples
The mtDNA variants uncovered in the 926 preterm patient samples were manually examined and classified based on ACMG criteria (Richards et al., 2015), combined with additional information from the human mitochondrial genome database “MITOMAP” (http://mitomap.org/bin/view.pl/MITOMAP/WebHome). MITOMAP contains a continually updated collection of information compiled by the entire mitochondrial field, including population frequency information curated from gnomAD, the Helix population database, GenBank, and so forth, as well as pathogenicity predictions and other relevant information about mtDNA sequence variation. By using these databases, we can effectively leverage hundreds of thousands of samples across the entire human population as our control set.
Using this approach, we identified eight samples (0.86%) containing known pathogenic variants (Table 1), which is a noticeably higher frequency than what is observed in the general population. For instance, 3 out of the 929 samples in our PTB cohort (or about 0.32%) showed the pathogenic m.3243A>G variant that is known to cause mitochondrial encephalopathy, lactic acidosis, and stroke‐like episodes (MELAS). However, as of April 2020, only 6 out of the 56,383 individuals with sequencing information at this position in gnomAD (https://gnomad.broadinstitute.org/variant/M-3243-A-G?dataset=gnomad_r3), or about 0.011% in total, possess the m.3243A>G variant as either a heteroplasmy or homoplasmy. Similarly, only 51 of the 195,983 individuals in the Helix database (https://www.mitomap.org/cgi-bin/helix?id=2654), or about ~0.026%, were found to have the m.3243A>G variant. This represents more than an order of magnitude difference between our PTB cohort and either of these databases, and even a simple Fisher's exact test will demonstrate that this is a significant difference in both cases (p = .0003 for gnomAD and p = .022 for the Helix database). Although the difference between our results and the frequency observed in gnomAD could be attributed to their higher detection threshold (gnomAD uses a cut‐off of 10%, while our cut‐off is 5%), the Helix database reports heteroplasmies down to a 1% cut‐off, which would make it more likely to detect and report m.3243>G variants than our own analysis. Yet, despite the fact that our cut‐off is more conservative than the one used by Helix, we still observe a much higher frequency for m.3243A>G in our preterm cohort than Helix reports for their population database. Furthermore, most of our known pathogenic variants are present at high heteroplasmy levels that are far above any of these cut‐offs (only one of the eight patients with known pathogenic variants would fall below the 10% cut‐off, and only two of the patients have heteroplasmies below 50%). Thus, it would appear that our PTB cohort shows fairly strong evidence for a heightened frequency of at least one well‐known pathogenic mtDNA variant.
Table 1.
Patient samples from the 960 sample data set that contain known pathogenic variants
| Sample name | Mutation | Heteroplasmy (%) | Phenotype associated with variant | Igenomix coverage | mtDNA coverage | Haplogroup | Current age (years) | Gestational age (months) | Gender | Patient race | Ethnicity |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient_116 | m.1494C>T | 85.1 | DEAF | 1.47553 | 559.1176293 | L1c | 19 | 24 | Male | Black or African‐American | Non‐Hispanic |
| Patient_152 | m.7471dup | 16.4 | PEM/AMDF/motor neuron disease‐like | 1.38653 | 738.1543847 | L2b | 16 | 25 | Male | Black or African‐American | Non‐Hispanic |
| Patient_203 | m.3243A>G | 7.1 | MELAS/LS/DMDF/MIDD/SNHL/CPEO/MM/FSGS/ASD/cardiac + multi‐organ dysfunction | 1.31811 | 809.5704629 | K2b | 16 | 24 | Female | White | Non‐Hispanic |
| Patient_242 | m.3243A>G | 53 | MELAS/LS/DMDF/MIDD/SNHL/CPEO/MM/FSGS/ASD/cardiac + multi‐organ dysfunction | 3.09628 | 1383.373529 | H | 16 | 30 | Female | White | Non‐Hispanic |
| Patient_727 | m.1555A>G | 68.6 | DEAF; autism spectrum intellectual disability; possibly antiatherosclerotic | 1.70454 | 1026.711509 | L3 | 2 | 24 | Female | Unknown | Non‐Hispanic |
| Patient_823 | m.1555A>G | 70.8 | DEAF; autism spectrum intellectual disability; possibly antiatherosclerotic | 2.26769 | 1117.326332 | T2b | 2 | 27 | Female | White | Non‐Hispanic |
| Patient_875 | m.11778G>A | 91.7 | LHON/progressive dystonia | 1.5207 | 1546.552296 | H27 | 3 | 28 | Male | White | Non‐Hispanic |
| Patient_877 | m.3243A>G | 85.8 | MELAS/LS/DMDF/MIDD/SNHL/CPEO/MM/FSGS/ASD/cardiac + multi‐organ dysfunction | 1.96847 | 1757.391514 | L1c | 11 | 29 | Female | Black or African‐American | Non‐Hispanic |
Note: All 957 samples that successfully completed the automated pipeline were manually examined for the presence of known pathogenic variants in their mtDNA. Eight samples were identified as containing known pathogenic variants, as shown in the table.
Abbreviations: AMDF, ataxia, myoclonus, and deafness; ASD, autism spectrum disorder; CPEO, chronic progressive external ophthalmoplegia; DEAF, deafness; DMDF, diabetes mellitus and deafness; FSGS, focal segmental glomerulosclerosis; LHON, Leber hereditary optic neuropathy; LS, Leigh syndrome; MELAS, mitochondrial encephalopathy, lactic acidosis, and stroke‐like episodes; MIDD, maternally inherited diabetes and deafness; MM, mitochondrial myopathy; mtDNA, mitochondrial genome; PEM, progressive encephalomyopathy; SNHL, sensorineural hearing loss.
Besides known pathogenic mtDNA variants, 47 of the samples (~5.1%) were also found to carry VUCS. Of the VUCS that affect either protein‐coding or RNA‐coding regions, five variants were observed in three or more individuals (m.7158A>G, m.15498G>A, m.3421G>A, m.3736G>A, and m.7080T>C). At the gene level, the largest number of variants were located in the MT‐ND1 gene (13 samples in total), and the second‐largest were located in the MT‐COI gene (8 samples in total). None of the VUCS were observed in more than four samples.
The eight samples containing known pathogenic mutations were confirmed using a clinically validated method based on PCR amplification of mtDNA and deep sequencing of the resulting amplicons (see Section 2). PCR‐NGS validation on the eight samples found the same variants as were observed in the lcWGS data using the automated pipeline, and at similar heteroplasmy levels (Table S3). Overall, there is a strong correlation between the heteroplasmy rate from lcWGS analysis and NGS results, with an R 2 value of .9228 (Figure 4). Some small deviations were observed for several of the samples with high heteroplasmy levels in the lcWGS results (ranging from 68.6% to 91.7%), which were found to be nearly homoplasmic according to the NGS results (93.6%–99.62%) (Table S3). Repeating analysis on the lcWGS data from the eight samples with more varied and more stringent quality filters did not result in heteroplasmy values closer to the NGS values (data not shown). The reasons for this discrepancy at high heteroplasmy levels are unclear. There is reason to believe that the PCR‐NGS values are likely to be closer to the true heteroplasmy levels due to the higher coverage (>1000X) generally observed in the PCR‐NGS data and the usage of sample preparation and analysis methods optimized for mtDNA sequencing. However, there is also a possibility that the geometric expansion inherent to the PCR component of PCR‐NGS may distort the detected frequency of the variant more than the lcWGS approach, which incorporates far fewer cycles of PCR during library prep. However, even in these cases, the corresponding lcWGS heteroplasmy values would still pass the threshold to flag these variants as likely contributing to pathogenesis in the clinical setting. Thus, the automated pipeline described here is confirmed as a reliable means of screening lcWGS data set for pathogenic mtDNA variants.
Figure 4.
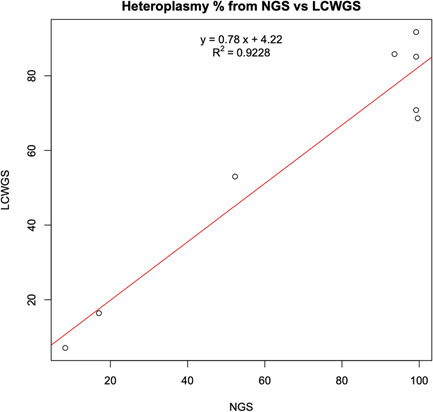
Correlation of estimated heteroplasmy between traditional PCR‐NGS and lcWGS. The heteroplasmy level for the eight pathogenic variants detected in the 960‐sample data set was calculated based on the novel lcWGS pipeline, versus a traditional PCR‐NGS methodology. The two methods showed a high degree of correlation for these variants, with an R 2 value of .9228. lcWGS, low‐coverage whole‐genome sequencing; NGS, next‐generation sequencing; PCR, polymerase chain reaction
4. DISCUSSION
4.1. Proof of concept
In this project, we have explored the effectiveness of using mtDNA data from lcWGS to screen for pathogenic mitochondrial variants. We found that the majority of the 960 lcWGS samples had at least 1000X mtDNA coverage, exceeding the necessary coverage threshold for mtDNA variant calling. In addition, we were able to reliably extract accurate haplogroup and variant heteroplasmy estimates from the lcWGS data, with the pathogenic variants fully confirmed by PCR‐NGS. Thus, these results have fully validated the process of mitochondrial variant calling from lcWGS data. More generally, since lcWGS data are readily available from existing projects that have used lcWGS for genotyping, we envision a straightforward process to extract the mitochondrial portions of these data and reveal any mtDNA variants in these patient samples without the need for any additional sequencing.
4.2. Possible relationship between mitochondrial genotypes and PTB
Nearly two‐thirds of PTBs are idiopathic spontaneous cases (Ferrero et al., 2016), and in the vast majority of these cases, genetic analyses are not performed. However, mothers experiencing spontaneous PTB or other pregnancy complications, such as pre‐eclampsia with medically indicated delivery are at increased risk for spontaneous PTB in subsequent pregnancies, suggesting that they share common pathways (Ananth et al., 2006). This makes their inclusion in our analysis potentially quite relevant. If labor is being induced for medical reasons and the presence of potentially pathogenic mitochondrial variants is unknown, then these variants could be very important factors in creating the medical need premature induced labor in the first place. The specific mitochondrial defects uncovered here could provide new knowledge that might suggest alternative therapy or management for such cases, thereby avoiding all the problems that come with medically induced premature delivery.
Previous attempts to uncover a direct link between mtDNA variation and PTB have shown inconclusive or mixed results. Most notably, a meta‐analysis of two large‐scale GWAS studies from Denmark and Norway showed no significant association between any mtDNA variants and preterm delivery (Alleman et al., 2012). However, this analysis was restricted to 135 preselected “common” SNPs with a minor allele frequency above 1% in Caucasian populations, which essentially excludes all known pathogenic mtDNA variants. In contrast, our analysis was based on an NGS approach that took an unbiased look at all possible mtDNA variants. These results show a strikingly high prevalence of pathogenic mtDNA variants in this PTB cohort. We found that approximately 1% of our 926 patient samples with identifiable haplogroups (8 out of 926) contained disease‐causing mutations and confirmed these results using clinically validated methods. This percentage is significantly higher than the occurrence of disease caused by pathogenic mtDNA variants in the general population, which traditional estimates place at approximately 1 in 5000 individuals (Gorman et al., 2015; Schaefer et al., 2008), suggesting that mitochondrial dysfunction may play at least some role in PTB. The existing literature provides some support for this hypothesis. Recent evidence suggests that the altered expression of proteins involved in the regulation of mitochondrial calcium levels may play a role in triggering PTB (Vishnyakova et al., 2019). More strikingly, a case‐comparison study comparing 67 women carrying the pathogenic mtDNA mutation m.3243A>G compared to 69 unaffected women found a significantly higher risk of pregnancy complications as well as PTB in the woman carrying the m.3243A>G variant (Feeney et al., 2019). A separate study from the Netherlands also found that mothers carrying the m.3243A>G mutation showed a wide range of obstetric complications, including premature delivery (~25%), pre‐eclampsia (~12%), and gestational diabetes (11%) (de Laat et al., 2015). In this light, we note that three of the eight samples with pathogenic variants among our preterm cohort possessed the m.3243A>G variant (Table 1). On the contrary, at least one study has found that pathogenic mtDNA mutations may be more common than the traditional “1 in 5000” estimate mentioned above, finding that ~1 in 200 neonates from a series of 3168 sequential live births harbored a known pathogenic variant (Elliott et al., 2008). In fact, when you expand the analysis beyond neonates to the general population, nearly 20% of the population appears to carry at least one mtDNA variant implicated in human disease (Ye et al., 2014), although nearly all are observed at an extremely low level of heteroplasmy. All of this would be more in line with the ~1 in 100 prevalence observed in our preterm cohort, which might suggest that the prevalence observed here is more of a general phenomenon and thus unrelated to PTB. However, we note that most of our samples with pathogenic variants (six out of eight) showed mtDNA variants at a high heteroplasmy level (>50%) that would traditionally be associated with an increased risk of disease presentation. In contrast, most of the cases from the report of a 1 in 200 prevalence in the general population had a heteroplasmy level below 50%, a level that is much less likely to be associated with overt mitochondrial disease.
It should also be noted that in this study, we are only reporting heteroplasmy from peripheral blood and not the placenta or other tissues. On the contrary, the energetic demands of the fetus appear to be the major trigger for parturition (Dunsworth et al., 2012). Thus, tissues where dysfunctional mtDNA variants would impinge upon the fetus, such as the placenta or the uterine lining of the mother, would be strong candidates for future investigation in these or other preterm patient populations.
Although less obviously associated with pathogenicity, the VUCS uncovered in over 5% of our PTB cohort also represent a source of new candidate loci for involvement in PTB. At least one previous study has linked mtDNA polymorphisms (such as m.4917A>G and m.4216T>C) to smoking‐induced spontaneous PTB (Velez et al., 2008), demonstrating that even seemingly common mtDNA variants may play an unexpected role in PTB under certain conditions. In particular, the high abundance of samples in our PTB cohort with variants in the MT‐ND1 and MT‐COI genes may warrant further investigation, although additional confirmation and contextualization will clearly be necessary before any clinical management changes can be recommended.
The unusually higher frequency of the L2a and L3 haplogroups in our patient samples reported with a European ancestry also bears further discussion in light of previous observations in the field. For one thing, these haplogroups are extremely common in African‐American populations, and it has been widely observed that the African‐American population experiences a higher rate of spontaneous PTB than other racial groups within the United States (Mohamed et al., 2014). Although the contributing factors to this difference are not fully understood, it is conceivable that these two haplogroups are major contributors to that increased frequency of PTB. Furthermore, although the L2 and L3 haplogroups are observed in <1% of individuals in the United States who self‐identify as non‐Hispanic White, they are observed much more frequently (4.8% and 7.8%, respectively) in other Hispanic populations with a greater degree of genetic admixture, many of whom self‐identify as White (Mitchell et al., 2014). This is particularly relevant in the context of PTB, as previous studies have indicated that individuals with more divergence between their mitochondrial and nuclear genome ancestry (as would more often occur with genetic admixture) have a higher risk of PTB (Crawford et al., 2018). Because the nuclear and mitochondrial genomes exhibit a great deal of interdependence and also tend to adapt to each other over evolutionary time, incompatibility between nuclear and mitochondrial genomes derived from genetically distinct populations has been observed across a variety of animal species (Healy & Burton, 2020; Ma et al., 2016; C. Zhang, Montooth, et al., 2017; G. Zhang, Feenstra, et al., 2017). Conclusive evidence for a clear health effect of mitochondrial nuclear mismatch in humans has remained elusive, although a recent analysis of admixed populations in humans provided evidence for the selection of nuclear‐encoded mitochondrial genes toward the source population of the mtDNA haplogroup (Zaidi & Makova, 2019). The results we have described here may provide further support for the idea that the interplay between particular mtDNA variants and haplogroups and the nuclear genome could play a role in triggering PTB.
5. CONCLUSION
Overall, these results demonstrate a practical procedure for evaluating mtDNA from lcDNA sequence data, as well as the first report of particular mitochondrial variants that may play a role in PTB. The results from our preterm cohort, combined with the existing literature on the subject, provide new insights into the relationship between mtDNA mutations/variants and PTB that likely warrant further investigation. Our automated Mutect2 analysis pipeline has also been shown to be effective at analyzing both PCR amplified NGS mtDNA sequences as well as lcWGS data, suggesting that this pipeline may be broadly applicable at extracting mtDNA variant data from a wide range of sequencing data types. These results suggest that, in the future, more attention should be paid to the feasibility of mining mtDNA data from existing genomic data sets that have not traditionally been utilized for this purpose.
CONFLICT OF INTERESTS
The authors declare that there is no conflict of interests.
WEB RESOURCES
CCHMC i2b2 framework: https://i2b2.cchmc.org
Discover Together Biobank: https://www.cincinnatichildrens.org/service/c/clinical-trials/biobank
gnomAD: https://gnomad.broadinstitute.org/variant/M-3243-A-G?dataset=gnomad_r3
Helix Database: https://www.mitomap.org/cgi-bin/helix?id=2654
MitoMap: http://mitomap.org/bin/view.pl/MITOMAP/WebHome
Mitomaster API: https://www.mitomap.org/mitomaster/websrvc.cgi
Supporting information
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
We wish to thank our collaborators and the patient participants for their contributions to this study. This study used samples, data, and/or services from the Discover Together Biobank at Cincinnati Children's Research Foundation. We thank the Discover Together Biobank for support of this study, as well as participants and their families, whose help and participation made this work possible. The study was supported in part by the Cincinnati Children's Hospital Research Foundation (TH).
Yang, Z. , Slone, J. , Wang, X. , Zhan, J. , Huang, Y. , Namjou, B. , Kaufman, K. M. , Pauciulo, M. , Harley, J. B. , Muglia, L. J. , Chepelev, I. , & Huang, T. (2021). Validation of low‐coverage whole‐genome sequencing for mitochondrial DNA variants suggests mitochondrial DNA as a genetic cause of preterm birth. Human Mutation, 42, 1602–1614. 10.1002/humu.24279
Zeyu Yang and Jesse Slone contributed equally to this study.
Contributor Information
John B. Harley, Email: Johnbharley@yahoo.com.
Louis J. Muglia, Email: LMuglia@BWFund.org.
Taosheng Huang, Email: thuang29@buffalo.edu.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon request.
REFERENCES
- Abecasis, G. R. , Altshuler, D. , Auton, A. , Brooks, L. D. , Durbin, R. M. , Gibbs, R. A. , Hurles, M. E. , & McVean, G. A. (2010). A map of human genome variation from population‐scale sequencing. Nature, 467(7319), 1061–1073. 10.1038/nature09534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alleman, B. W. , Myking, S. , Ryckman, K. K. , Myhre, R. , Feingold, E. , Feenstra, B. , Geller, F. , Boyd, H. A. , Shaffer, J. R. , Zhang, Q. , Begum, F. , Crosslin, D. , Doheny, K. , Pugh, E. , Pay, A. S. D. , Ostensen, I. H. G. , Morken, N.‐H. , Magnus, P. , Marazita, M. L. , & Murray, J. C. (2012). No observed association for mitochondrial SNPs with preterm delivery and related outcomes. Pediatric Research, 72(5), 539–544. 10.1038/pr.2012.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananth, C. V. , Getahun, D. , Peltier, M. R. , Salihu, H. M. , & Vintzileos, A. M. (2006). Recurrence of spontaneous versus medically indicated preterm birth. American Journal of Obstetrics and Gynecology, 195(3), 643–650. 10.1016/j.ajog.2006.05.022 [DOI] [PubMed] [Google Scholar]
- Anum, E. A. , Springel, E. H. , Shriver, M. D. , & Strauss, J. F., 3rd. (2009). Genetic contributions to disparities in preterm birth. Pediatric Research, 65(1), 1–9. 10.1203/PDR.0b013e31818912e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeza, J. A. (2020). Yes, we can use it: A formal test on the accuracy of low‐pass nanopore long‐read sequencing for mitophylogenomics and barcoding research using the Caribbean spiny lobster Panulirus argus. BMC Genomics, 21(1), 882. 10.1186/s12864-020-07292-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd, H. A. , Poulsen, G. , Wohlfahrt, J. , Murray, J. C. , Feenstra, B. , & Melbye, M. (2009). Maternal contributions to preterm delivery. American Journal of Epidemiology, 170(11), 1358–1364. 10.1093/aje/kwp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockhage, R. , Slone, J. , Ma, Z. , Hegde, M. R. , Valencia, C. A. , & Huang, T. (2018). Validation of the diagnostic potential of mtDNA copy number derived from whole genome sequencing. Journal of Genetics and Genomics, 45(18), 333–335. 10.1016/j.jgg.2018.06.001 [DOI] [PubMed] [Google Scholar]
- Cardena, M. M. , Ribeiro‐Dos‐Santos, A. , Santos, S. , Mansur, A. J. , Pereira, A. C. , & Fridman, C. (2013). Assessment of the relationship between self‐declared ethnicity, mitochondrial haplogroups and genomic ancestry in Brazilian individuals. PLOS One, 8(4), e62005. 10.1371/journal.pone.0062005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, Y. K. , Tseng, Y. T. , & Chen, K. T. (2020). The epidemiologic characteristics and associated risk factors of preterm birth from 2004 to 2013 in Taiwan. BMC Pregnancy and Childbirth, 20(1), 201. 10.1186/s12884-020-02903-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheau‐Feng Lin, F. , Jeng, Y. C. , Huang, T. Y. , Chi, C. S. , Chou, M. C. , & Chin‐Shaw Tsai, S. (2014). Mitochondrial DNA copy number is associated with diagnosis and prognosis of head and neck cancer. Biomarkers, 19(4), 269–274. 10.3109/1354750X.2014.902101 [DOI] [PubMed] [Google Scholar]
- Cibulskis, K. , Lawrence, M. S. , Carter, S. L. , Sivachenko, A. , Jaffe, D. , Sougnez, C. , Gabriel, S. , Meyerson, M. , Lander, E. S. , & Getz, G. (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature Biotechnology, 31(3), 213–219. 10.1038/nbt.2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford, N. , Prendergast, D. , Oehlert, J. W. , Shaw, G. M. , Stevenson, D. K. , Rappaport, N. , Sirota, M. , Tishkoff, S. A. , & Sondheimer, N. (2018). Divergent patterns of mitochondrial and nuclear ancestry are associated with the risk for preterm birth. The Journal of Pediatrics, 194, 40–46. 10.1016/j.jpeds.2017.10.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Laat, P. , Fleuren, L. H. J. , Bekker, M. N. , Smeitink, J. A. M. , & Janssen, M. C. H. (2015). Obstetric complications in carriers of the m.3243A>G mutation, a retrospective cohort study on maternal and fetal outcome. Mitochondrion, 25, 98–103. 10.1016/j.mito.2015.10.005 [DOI] [PubMed] [Google Scholar]
- Dunsworth, H. M. , Warrener, A. G. , Deacon, T. , Ellison, P. T. , & Pontzer, H. (2012). Metabolic hypothesis for human altriciality. Proceedings of the National Academy of Sciences of the United States of America, 109(38), 15212–15216. 10.1073/pnas.1205282109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott, H. R. , Samuels, D. C. , Eden, J. A. , Relton, C. L. , & Chinnery, P. F. (2008). Pathogenic mitochondrial DNA mutations are common in the general population. American Journal of Human Genetics, 83(2), 254–260. 10.1016/j.ajhg.2008.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feeney, C. L. , Lim, A. Z. , Fagan, E. , Blain, A. , Bright, A. , Maddison, J. , Devine, H. , Stewart, J. , Taylor, R. W. , Gorman, G. S. , Turnbull, D. M. , Nesbitt, V. , & McFarland, R. (2019). A case‐comparison study of pregnant women with mitochondrial disease—What to expect? BJOG, 126(11), 1380–1389. 10.1111/1471-0528.15667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero, D. M. , Larson, J. , Jacobsson, B. , Di Renzo, G. C. , Norman, J. E. , Martin, J. N., Jr. , D'Alton, M. , Castelazo, E. , Howson, C. P. , Sengpiel, V. , Bottai, M. , Mayo, J. A. , Shaw, G. M. , Verdenik, I. , Tul, N. , Velebil, P. , Cairns‐Smith, S. , Rushwan, H. , Arulkumaran, S. , … Simpson, J. L. (2016). Cross‐country individual participant analysis of 4.1 million singleton births in 5 countries with very high human development index confirms known associations but provides no biologic explanation for 2/3 of all preterm births. PLOS One, 11(9), e0162506. 10.1371/journal.pone.0162506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman, G. S. , Schaefer, A. M. , Ng, Y. , Gomez, N. , Blakely, E. L. , Alston, C. L. , Feeney, C. , Horvath, R. , Yu‐Wai‐Man, P. , Chinnery, P. F. , Taylor, R. W. , Turnbull, D. M. , & McFarland, R. (2015). Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Annals of Neurology, 77(5), 753–759. 10.1002/ana.24362s [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallman, M. , Haapalainen, A. , Huusko, J. M. , Karjalainen, M. K. , Zhang, G. , Muglia, L. J. , & Rämet, M. (2019). Spontaneous premature birth as a target of genomic research. Pediatric Research, 85(4), 422–431. 10.1038/s41390-018-0180-z [DOI] [PubMed] [Google Scholar]
- Healy, T. M. , & Burton, R. S. (2020). Strong selective effects of mitochondrial DNA on the nuclear genome. Proceedings of the National Academy of Sciences of the United States of America, 117(12), 6616–6621. 10.1073/pnas.1910141117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homburger, J. R. , Neben, C. L. , Mishne, G. , Zhou, A. Y. , Kathiresan, S. , & Khera, A. V. (2019). Low coverage whole genome sequencing enables accurate assessment of common variants and calculation of genome‐wide polygenic scores. Genome Medicine, 11(1), 74. 10.1186/s13073-019-0682-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, T. (2011). Next generation sequencing to characterize mitochondrial genomic DNA heteroplasmy. Current Protocols in Human Genetics, 71, 19.8.1–19.8.12. 10.1002/0471142905.hg1908s71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husami, A. , Slone, J. , Brown, J. , Bromwell, M. , Valencia, C. A. , & Huang, T. (2020). Clinical utility of whole genome sequencing for the detection of mitochondrial genome mutations. Journal of Genetics and Genomics, 47(3), 167–169. 10.1016/j.jgg.2020.03.001 [DOI] [PubMed] [Google Scholar]
- Johnson, D. C. , Shrestha, S. , Wiener, H. W. , Makowsky, R. , Kurundkar, A. , Wilson, C. M. , & Aissani, B. (2015). Mitochondrial DNA diversity in the African American population. Mitochondrial DNA, 26(3), 445–451. 10.3109/19401736.2013.840591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. , Durbin, R. , & 1000 Genome Project Data Processing Subgroup . (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England), 25(16), 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Slone, J. , Fei, L. , & Huang, T. (2019). Mitochondrial DNA variants and common diseases: A mathematical model for the diversity of age‐related mtDNA mutations. Cells, 8(6), 608. 10.3390/cells8060608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Slone, J. , & Huang, T. (2020). The role of mitochondrial‐related nuclear genes in age‐related common disease. Mitochondrion, 53, 38–47. 10.1016/j.mito.2020.04.012 [DOI] [PubMed] [Google Scholar]
- Li, M. , Schröder, R. , Ni, S. , Madea, B. , & Stoneking, M. (2015). Extensive tissue‐related and allele‐related mtDNA heteroplasmy suggests positive selection for somatic mutations. Proceedings of the National Academy of Sciences of the United States of America, 112(8), 2491–2496. 10.1073/pnas.1419651112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lott, M. T. , Leipzig, J. N. , Derbeneva, O. , Xie, H. M. , Chalkia, D. , Sarmady, M. , Procaccio, V. , & Wallace, D. C. (2013). mtDNA variation and analysis using Mitomap and Mitomaster. Current Protocols in Bioinformatics, 44(123), 1.23.1–1.23.26. 10.1002/0471250953.bi0123s44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, H. , Folmes, C. D. , Wu, J. , Morey, R. , Mora‐Castilla, S. , Ocampo, A. , Ma, L. , Poulton, J. , Wang, X. , Ahmed, R. , Kang, E. , Lee, Y. , Hayama, T. , Li, Y. , Van Dyken, C. , Gutierrez, N. M. , Tippner‐Hedges, R. , Koski, A. , Mitalipov, N. , … Mitalipov, S. (2015). Metabolic rescue in pluripotent cells from patients with mtDNA disease. Nature, 524(7564), 234–238. 10.1038/nature14546 [DOI] [PubMed] [Google Scholar]
- Ma, H. , Marti Gutierrez, N. , Morey, R. , Van Dyken, C. , Kang, E. , Hayama, T. , Lee, Y. , Li, Y. , Tippner‐Hedges, R. , Wolf, D. P. , Laurent, L. C. , & Mitalipov, S. (2016). Incompatibility between nuclear and mitochondrial genomes contributes to an interspecies reproductive barrier. Cell Metabolism, 24(2), 283–294. 10.1016/j.cmet.2016.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, S. L. , Goodloe, R. , Brown‐Gentry, K. , Pendergrass, S. A. , Murdock, D. G. , & Crawford, D. C. (2014). Characterization of mitochondrial haplogroups in a large population‐based sample from the United States. Human Genetics, 133(7), 861–868. 10.1007/s00439-014-1421-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed, S. A. , Thota, C. , Browne, P. C. , Diamond, M. P. , & Al‐Hendy, A. (2014). Why is preterm birth stubbornly higher in African‐Americans? Obstetrics & Gynecology International Journal, 1(3), 00019. 10.15406/ogij.2014.01.00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin, E. D. , & Wong, R. (1988). Mitochondrial DNA molecules and virtual number of mitochondria per cell in mammalian cells. Journal of Cellular Physiology, 136(3), 507–513. 10.1002/jcp.1041360316 [DOI] [PubMed] [Google Scholar]
- Schaefer, A. M. , McFarland, R. , Blakely, E. L. , He, L. , Whittaker, R. G. , Taylor, R. W. , Chinnery, P. F. , & Turnbull, D. M. (2008). Prevalence of mitochondrial DNA disease in adults. Annals of Neurology, 63(1), 35–39. 10.1002/ana.21217 [DOI] [PubMed] [Google Scholar]
- Soltani, M. , Tabatabaee, H. R. , Saeidinejat, S. , Eslahi, M. , Yaghoobi, H. , Mazloumi, E. , Rajabi, A. , Ghasemi, A. , Keyghobadi, N. , Enayatrad, M. , Noori, A. , Hashemi, S. A. , Zolfizadeh, F. , Mahdavi, S. , Valadbeigi, T. , Etemad, K. , Taghipour, A. , Salehnasab, C. , & Hajipour, M. (2019). Assessing the risk factors before pregnancy of preterm births in Iran: A population‐based case‐control study. BMC Pregnancy and Childbirth, 19(1), 57. 10.1186/s12884-019-2183-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, S. , & Huang, T. (2010). Characterization of mitochondrial DNA heteroplasmy using a parallel sequencing system. Biotechniques, 48(4), 287–296. 10.2144/000113389 [DOI] [PubMed] [Google Scholar]
- Thompson, K. , Collier, J. J. , Glasgow, R. , Robertson, F. M. , Pyle, A. , Blakely, E. L. , Alston, C. L. , Oláhová, M. , McFarland, R. , & Taylor, R. W. (2020). Recent advances in understanding the molecular genetic basis of mitochondrial disease. Journal of Inherited Metabolic Disease, 43(1), 36–50. 10.1002/jimd.12104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torroni, A. , Huoponen, K. , Francalacci, P. , Petrozzi, M. , Morelli, L. , Scozzari, R. , Obinu, D. , Savontaus, M. L. , & Wallace, D. C. (1996). Classification of European mtDNAs from an analysis of three European populations. Genetics, 144(4), 1835–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker, J. , & McGuire, W. (2004). Epidemiology of preterm birth. BMJ, 329(7467), 675–678. 10.1136/bmj.329.7467.675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velez, D. R. , Menon, R. , Simhan, H. , Fortunato, S. , Canter, J. A. , & Williams, S. M. (2008). Mitochondrial DNA variant A4917G, smoking and spontaneous preterm birth. Mitochondrion, 8(2), 130–135. 10.1016/j.mito.2007.10.007 [DOI] [PubMed] [Google Scholar]
- Vishnyakova, P. A. , Tarasova, N. V. , Volodina, M. A. , Tsvirkun, D. V. , Sukhanova, I. A. , Kurchakova, T. A. , Kan, N. E. , Medzidova, M. K. , Sukhikh, G. T. , & Vysokikh, M. Y. (2019). Gestation age‐associated dynamics of mitochondrial calcium uniporter subunits expression in feto‐maternal complex at term and preterm delivery. Scientific Reports, 9(1), 5501. 10.1038/s41598-019-41996-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace, D. C. , & Chalkia, D. (2013). Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harbor Perspectives in Biology, 5(11), a021220. 10.1101/cshperspect.a021220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, W. , Witherspoon, D. J. , Fraser, A. , Clark, E. A. , Rogers, A. , Stoddard, G. J. , Manuck, T. A. , Chen, K. , Esplin, M. S. , Smith, K. R. , Varner, M. W. , & Jorde, L. B. (2015). The heritability of gestational age in a two‐million member cohort: Implications for spontaneous preterm birth. Human Genetics, 134(7), 803–808. 10.1007/s00439-015-1558-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, C. Y. , Liu, Y. , Yang, H. R. , Yang, H. Y. , Liu, J. X. , Ma, Y. N. , & Qi, Y. (2017). Reference intervals of mitochondrial DNA copy number in peripheral blood for Chinese minors and adults. Chinese Medical Journal, 130(20), 2435–2440. 10.4103/0366-6999.216395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, K. , Lu, J. , Ma, F. , Keinan, A. , & Gu, Z. (2014). Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proceedings of the National Academy of Sciences of the United States of America, 111(29), 10654–10659. 10.1073/pnas.1403521111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- York, T. P. , Eaves, L. J. , Lichtenstein, P. , Neale, M. C. , Svensson, A. , Latendresse, S. , Långström, N. , & Strauss, J. F., 3rd. (2013). Fetal and maternal genes' influence on gestational age in a quantitative genetic analysis of 244,000 Swedish births. American Journal of Epidemiology, 178(4), 543–550. 10.1093/aje/kwt005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi, A. A. , & Makova, K. D. (2019). Investigating mitonuclear interactions in human admixed populations. Nature Ecology & Evolution, 3(2), 213–222. 10.1038/s41559-018-0766-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, C. , Montooth, K. L. , & Calvi, B. R. (2017). Incompatibility between mitochondrial and nuclear genomes during oogenesis results in ovarian failure and embryonic lethality. Development (Cambridge, England), 144(13), 2490–2503. 10.1242/dev.151951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, G. , Feenstra, B. , Bacelis, J. , Liu, X. , Muglia, L. M. , Juodakis, J. , Miller, D. E. , Litterman, N. , Jiang, P. P. , Russell, L. , Hinds, D. A. , Hu, Y. , Weirauch, M. T. , Chen, X. , Chavan, A. R. , Wagner, G. P. , Pavličev, M. , Nnamani, M. C. , Maziarz, J. , … Muglia, L. J. (2017). Genetic associations with gestational duration and spontaneous preterm birth. The New England Journal of Medicine, 377(12), 1156–1167. 10.1056/NEJMoa1612665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, W. , Zhu, M. , Gui, M. , Huang, L. , Long, Z. , Wang, L. , Chen, H. , Yin, Y. , Jiang, X. , Dai, Y. , Tang, Y. , He, L. , & Zhong, K. (2014). Peripheral blood mitochondrial DNA copy number is associated with prostate cancer risk and tumor burden. PLOS One, 9(10), e109470. 10.1371/journal.pone.0109470 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.