

Abstract
Although atopic dermatitis (AD) has been reported to be a typical type 2 immune response disease, it is also an inflammatory skin disease that involves cytokines, such as Th1, Th17 and Th22. However, little is known about the mechanism by which the candidate cytokines, alone or in combination, are involved in AD pathology. Differences in cytokine balance, which contribute to the complexity of AD pathology, may influence the stratum corneum barrier function through tight junction (TJ) functional stability and contribute to disease severity. To confirm the regulatory mechanism of TJ protein expression in AD, we investigated the Th1 and Th17 pathways, which are the initiation factors of chronic AD pathology. We examined the effects of these cytokines on TJ protein expression in normal human epidermal keratinocytes in vitro, and also examined their function in a human skin equivalent model. We observed a time‐ and dose‐dependent inhibitory effect of IFN‐γ on claudin‐1 expression via the IFN‐γ receptor/JAK/STAT signalling pathway. IFN‐γ impaired TJ function in a human skin equivalent model. Moreover, we investigated co‐stimulation with IL‐17A, which is highly expressed in AD skin lesions and found that IL‐17A restores IFN‐γ‐induced TJ dysfunction. This restoration of TJ function was mediated by atypical protein kinase C zeta activation without recovery of TJ protein expression. These results are informative for personalized AD treatment via systemic therapies using anti‐cytokine antibodies and/or JAK inhibitors.
Keywords: atopic dermatitis, atypical protein kinase zeta, cytokines, Janus kinase, keratinocytes
Abbreviations
- AD
atopic dermatitis
- aPKC‐ζ
atypical protein kinase C zeta
- Ca
calcium
- CLDN1
claudin‐1
- GUSB
glucuronidase beta
- IFN
interferon
- IFNGR
interferon‐gamma receptor
- IgE
immunoglobulin E
- IL
interleukin
- JAK
Janus kinase
- NHEK
normal human epidermal keratinocytes
- qRT‐PCR
quantitative real‐time PCR
- SC
stratum corneum
- SD
standard deviation
- SE
standard error
- TEER
transepithelial electrical resistance
- TEWL
trans‐epidermal water loss
- Th
helper T cell
- TJ
tight junction
- TNF
tumor necrosis factor
- ZO
zona occludens
1. INTRODUCTION
Atopic dermatitis (AD) is characterized by diverse clinical symptoms with heterogeneous pathophysiology. AD has been considered a type 2 helper T‐cell (Th2) immune response disease; however, Th1 and Th17 cells are also involved in its pathogenesis. Acute AD is associated with increased expression of serum immunoglobulin E (IgE), and epidermal eosinophil counts are associated with elevated Th2 cytokines, such as interleukin (IL)‐4, IL‐5 and IL‐13. Th1 (tumor necrosis factor (TNF)‐α and interferon (IFN)‐γ)/Th17 (IL‐17 and IL‐22) activation occurs in the chronic phase of AD. 1 , 2 , 3 , 4 AD can be categorized into extrinsic and intrinsic types. Most cases of extrinsic AD occur in early childhood, and a shift in cytokine profiles during ageing changes the clinical presentation in younger patients. In older patients, Th2 activity is uniquely decreased, which contradicts the common ageing trends; conversely, the Th1 and Th17 axes are increased, combined with a decrease in serum IgE level and improvement of epithelial abnormalities. 5 , 6 In contrast, intrinsic AD exhibits different cytokine profiles, such as Th1 predominance. 7 , 8 Subtyping patients with AD showing different serum cytokine profiles is important for the effective use of novel molecular therapies, such as antibodies against IL‐4 receptor α and IL‐13 and Janus kinase (JAK) inhibitors that target specific immune pathways. 9 However, a detailed characterization of the effects of each cytokine (alone or in combination) on skin conditions is required for planning effective treatments using anti‐cytokine antibodies and JAK inhibitors.
One of the most important epidermal alterations in AD is the disruption of the stratum corneum (SC) barrier. 10 , 11 The outer epidermal SC barrier includes intercellular SC lipids and filaggrin, which defend against water loss and allergen penetration. High calcium (Ca2+) level or acidic pH is essential for differential protein and lipid expression in the SC and for controlling the intracellular and extracellular environment. 12
Tight junctions (TJs) are formed by transmembrane proteins, including occludin, claudins, junctional adhesion molecules and cytoplasmic plaque proteins, such as the zona occludens (ZO; ZO‐1, ZO‐2 and ZO‐3). 13 , 14 , 15 TJs are localized in the stratum granulosum and play an essential role in skin barrier function. 16 , 17 Mice with defective claudin‐1 (CLDN1), a TJ protein essential for barrier function, present with wrinkled skin as well as excessive trans‐epidermal water loss (TEWL) and die within 1 day of birth. 17 Moreover, SC lipid composition and filaggrin processing are damaged in the SC of these mice, 18 indicating that TJs are important for complete SC formation, including the skin water barrier.
We previously investigated the function of TJs in normal human epidermal keratinocytes (NHEKs) using the Transwell system and revealed that TJ dysfunction causes SC barrier damage through elevated pH and induces metabolic abnormalities in SC lipids and filaggrin. 19 Indeed, TJ dysfunction has been reported in AD. 20 , 21 Further, we previously reported that external UVB stimulation disrupts TJs, whereas pathogen‐associated molecular patterns, such as bacterial components that trigger pattern recognition receptor‐mediated immunity, enhance TJ barrier via a Toll‐like receptor. 22 , 23 Furthermore, endogenous cytokines, such as IL‐17, IL‐33 and a mixture of IL‐4, IL‐13 and IL‐31, have been reported to downregulate the expression of CLDN1, 21 , 24 , 25 which has also been shown to be downregulated in the lesional skin of AD patients. 21 Therefore, changes in TJ function caused by external and epidermal‐derived endogenous cytokine stimulation may affect the SC barrier. Thus, it is worth exploring the action and interaction of individual cytokines on TJs in vitro.
Here, we focussed on the effects of cytokines on epidermal TJ because endogenous cytokines first act on epidermal cells to affect TJ function and are thus likely to be involved in SC barrier functions. To this end, we performed quantitative analysis of TJ proteins to identify the effects of cytokines in patients with AD and their influence on TJ function using a skin equivalent model. Furthermore, we targeted IL‐17A, which is increased in several AD subtypes, to clarify the regulation mechanism of TJ function under complex cytokine situations associated with skin lesions in patients with AD.
2. METHODS
2.1. Cell cultures and reagents
Normal human epidermal keratinocytes (Lifeline Cell Technology, Frederick, MD, USA) were maintained on a type I collagen‐coated plate (Corning, Corning, NY, USA) in MCDB 153 HAA medium (Peptide Institute Inc., Yamagata, Japan) supplemented with 0.07 mM (low) Ca2+, 5 mg L−1 insulin, 100 ng L−1 epidermal growth factor, 180 μg L−1 hydrocortisone, 6.1 mg L−1 monoethanolamine, 14.1 mg L−1 O‐phosphorylethanolamine and 0.4% (v/v) bovine pituitary extract, as described previously. 26 Human recombinant cytokines (IL‐4, IL‐17A, IFN‐γ and TNF‐α) were purchased from FUJIFILM Wako Pure Chemical (Osaka, Japan). One day after the medium was replaced with a high‐Ca2+ differentiation medium (1.8 mM Ca2+), NHEKs were stimulated with cytokines. Ruxolitinib, a specific inhibitor of JAK1/2, was purchased from ChemScene (Monmouth Junction, NJ, USA). For RNA interference, cells were cultured with 5 nM negative control siRNA or STAT1 siRNA (Hs‐STAT1‐6, Qiagen, Hilden, Germany) using the HiPerFect transfection reagent (Qiagen), as previously reported. 27
For immunostaining of mature TJ formation, we plated cells on Transwell membranes with 0.4‐μm pore size (EMD Millipore, Burlington, MA, USA) and cultured them in a differentiation medium. After 2 days of incubation, the differentiation medium was replaced with fresh medium, and the TJ barrier was evaluated. 26 At this point, the cells were treated with cytokines.
The reconstructed human‐cultured epidermal model, LabCyte EPI‐MODEL24, is available commercially (Japan Tissue Engineering Co. Ltd., Aichi, Japan). Reconstruction of the human‐cultured epidermis was achieved by cultivating and proliferating NHEKs on an inert filter substrate at the air‐liquid interface. The process generated a multilayer structure comprising the fully differentiated epithelium with the SC. LabCyte EPI‐MODEL24 was maintained following the manufacturer's instructions. Culture medium was replaced daily with fresh medium (with or without IFN‐γ).
2.2. Immunostaining analysis
Frozen sections of LabCyte EPI‐MODEL and NHEKs cultured on Transwell filters were fixed with ice‐cold 90% (v/v) ethanol, incubated with 0.1% Triton X‐100 in phosphate‐buffered saline (PBS) for 5 min and blocked with 1% (v/v) bovine serum albumin in PBS for 1 h. The samples were incubated overnight at 4°C with rat anti‐occludin (MOC37), mouse anti‐ZO‐1 (T8‐754) (kind gifts from Prof. Furuse, Physiological Sciences) or rabbit anti‐ZO‐1 (Cell Signaling Technology, Danvers, MA, USA) antibody. After washing with PBS, Alexa Fluor 488‐ or 546‐conjugated secondary antibody (Life Technologies Japan Ltd., Tokyo, Japan) or Texas Red conjugated streptavidin (Merck, Darmstadt, Germany) was added, and the samples were incubated for 1 h. The fluorescence images were captured using a BZ‐9000 fluorescence microscope (KEYENCE Co., Osaka, Japan).
2.3. Quantitative real‐time PCR (qRT‐PCR)
Total RNA was isolated using a Nucleo Spin RNA kit (TaKaRa Bio Inc., Shiga, Japan), and cDNA was synthesized using a PrimeScript RT reagent kit (TaKaRa Bio Inc.). qRT‐PCR was performed on a Thermal Cycler Dice TP800 (TaKaRa Bio Inc.) using a TB green Premix EX Taq II (TaKaRa Bio Inc.) with 45 cycles of denaturation for 5 s at 95℃ and annealing and extension for 30 s at 60℃. Relative quantification was performed by normalization against the level of glucuronidase beta (GUSB) mRNA as a housekeeping gene.
The specific PCR primer sequences were as follows: CLDN1: 5′‐GGGCAGATCCAGTGCAAAG‐3′ and 5′‐GGATGCCAACCACCATCAAG‐3′; STAT1: 5′‐GCTGGCACCAGAACGAATGA‐3′ and 5′‐TACCAAACCAGGCTGGCACA‐3′; GUSB: 5′‐CATTATTCAGAGCGAGTATGGAGCA‐3′ and 5′‐TCTTCAGTGAACATCAGAGGTGGA‐3′.
2.4. TJ permeability assay
Transepithelial electrical resistance (TEER) reflects the water‐soluble ion permeability of the keratinocyte sheet. A higher TEER value indicates lower ionic permeability. TEER was measured using a Millicell ERS‐2 voltohmmeter (EMD Millipore).
The paracellular trace flux was measured using 4‐kDa FITC‐dextran (Sigma‐Aldrich Japan, Tokyo, Japan). FITC‐dextran (0.5 mg ml−1) was added to the culture medium with 1 mM CaCl2 from the apical side. Cells were incubated for 1 h. The medium outside the cup was collected, and fluorescence was measured using the GloMax®‐Mult+Detection System with Instinct Software (Promega, Madison, WI, USA).
Additionally, TJ permeability of the skin model was determined using a biotinylation technique. This was evaluated using 1 mg ml−1 EZ‐Link Sulfo‐NHS‐LC‐Biotin (Life Technologies Japan Ltd) in PBS with 1 mM CaCl2 added to the basolateral side of LabCyte EPI‐MODEL. After 30 min of incubation, the skin model was removed and frozen sections were prepared.
2.5. Western blotting analysis
Total protein was extracted in RIPA buffer (Nacalai Tesque, Kyoto, Japan) containing a phosphate inhibitor cocktail (Nacalai Tesque) and separated by electrophoresis on 12.5% SuperSep Ace gels (FUJIFILM Wako Pure Chemical). The samples were transferred to Immobilon‐P membranes (Merck) and reacted with the following primary antibodies: rabbit anti‐CLDN1, rabbit anti‐ZO‐1, rabbit anti‐occludin, rabbit anti‐atypical protein kinase C zeta (aPKC‐ζ), rabbit anti‐phospho‐aPKC‐ζ (T410), rabbit anti‐STAT1, rabbit anti‐phospho‐STAT1 (p‐STAT1; Y701) (Cell Signaling Technology Inc) and mouse anti‐GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The primary antibodies were detected using HRP‐conjugated secondary antibodies (GE Healthcare, Chicago, IL, USA). Bands were detected with ImmunoStar LD (FUJIFILM Wako Pure Chemical) using an Amersham Imager 680 (GE Healthcare).
2.6. Statistical analysis
Data are means ± standard deviation (SD) for ≥3 independent experiments. For standard error (SE) calculations, n ≥ 4. Data were analysed using SPSS Statistics, version 24 (SPSS Inc., Chicago, IL, USA). The Dunnett's t test was used to identify the statistically significant differences between pairs of treatment group means. The Tukey's multiple comparisons test was used to identify statistically significant differences among ≥3 different treatment group means. Differences were considered statistically significant at p < 0.05.
3. RESULTS
3.1. IFN‐γ downregulates CLDN1 via the IFN‐γ receptor (IFNGR) signalling pathway in NHEKs
To investigate the effects of inflammatory cytokines on TJ function, we examined TJ protein expression, including Th1/Th2/Th17 and other inflammatory cytokines involved in all stages of AD using NHEKs. After NHEKs were transferred to a high‐Ca2+ medium, cytokines were added for 48 h. Using Western blotting, we found that IFN‐γ (30 ng ml−1) significantly reduced CLDN1 expression (86.7% inhibition, **p < 0.01) more remarkably than other cytokines (Figure 1A and Figure S1A–C). We then analysed the dose‐ and time‐dependent regulation of CLDN1 expression in NHEKs. We detected dose‐dependent CLDN1 protein downregulation by IFN‐γ (10‒100 ng ml−1, Figure 1B and Figure S1F). IFN‐γ showed time‐dependent CLDN1 downregulation, especially after 48 h (Figure 1C and Figure S1G). We then analysed the quick response of NHEKs by IFN‐γ treatment using qRT‐PCR. We found that CLDN1 mRNA expression was downregulated by ~36% after 6 h (30 ng ml−1, *p < 0.05, Figure 1D) in a dose‐dependent manner (10–50 ng ml−1, **p < 0.01, Figure 1E).
FIGURE 1.
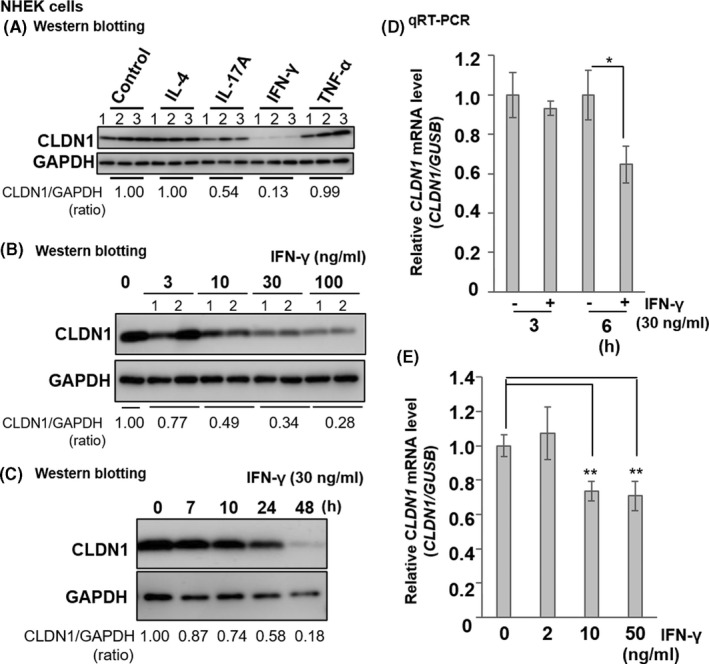
Dose‐ and time‐dependent inhibition of CLDN1 expression by IFN‐γ in normal human epidermal keratinocytes (NHEKs). (A) Effects of IL‐4, IL‐17A, IFN‐γ and TNF‐α (30 ng ml−1) on CLDN1 expression in NHEK cultures incubated with the cytokines in a 24‐well plate for 48 h. Samples were prepared from another three wells (n = 3). (B) Dose‐dependent inhibition of CLDN1 expression by IFN‐γ (3–100 ng ml−1) in NHEKs cultured with or without IFN‐γ for 48 h (n = 2). (C) Time‐dependent inhibition of CLDN1 expression in NHEKs treated with 30 ng ml−1 IFN‐γ for 0–48 h (n = 1). Western blotting performed with anti‐CLDN1 and anti‐GAPDH antibodies. Examples of typical blots in duplicate are illustrated. The expression level of CLDN1 was quantified by densitometry and normalized to GAPDH; the ratio is shown directly under each blot. Non‐treated control (0) is 1.00. The statistical analysis is shown in Figure S1. The results are representative of more than three independent experiments. (D) Changes in the relative CLDN1 mRNA expression in NHEKs cultured in a 24‐well plate with or without IFN‐γ (30 ng ml−1) for 3 or 6 h. (E) Changes in the relative CLDN1 mRNA expression in NHEKs treated with 2–50 ng ml−1 IFN‐γ for 6 h. mRNA expression was measured using qRT‐PCR. Data are presented as the mean ± SD (n = 4). Data were analysed using the Dunnett's test (*p < 0.05; **p < 0.01 vs. control (0 ng ml−1))
To identify the signals regulating IFN‐γ‐mediated CLDN1 downregulation, we used the JAK1/2 inhibitor ruxolitinib. NHEKs were incubated in high‐Ca2+ medium with various ruxolitinib concentrations (0.01–1.0 μM) for 1 h, with or without IFN‐γ for 48 h. IFN‐γ upregulated STAT1 and phosphorylated STAT1 (p‐STAT1) and downregulated CLDN1 protein (control; Figure 2A). However, ruxolitinib inhibited STAT1 phosphorylation in a dose‐dependent manner and rescued CLDN1 expression, especially at 1.0 μM (right line; Figure 2A and Figure S2).
FIGURE 2.
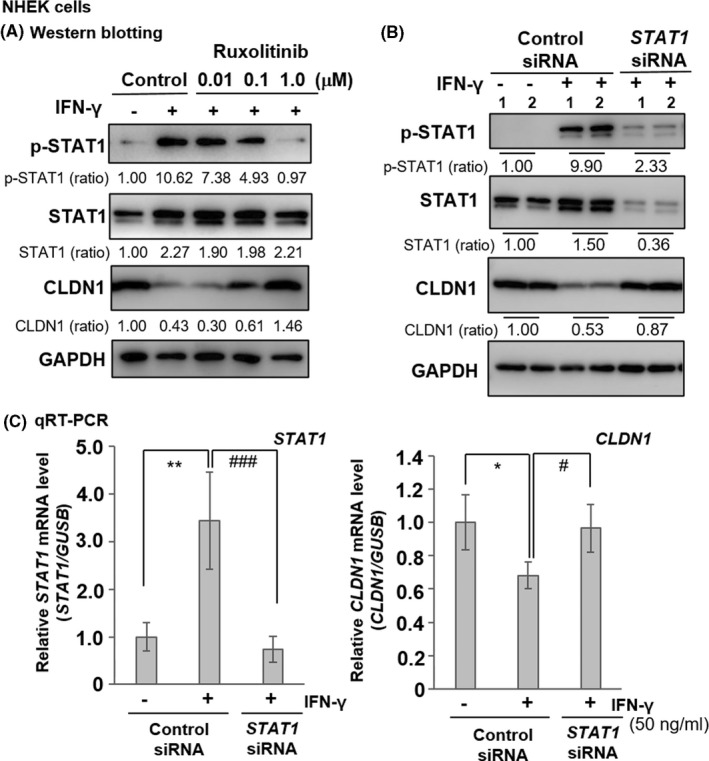
IFN‐γ receptor/JAK/STAT signalling pathway involvement in CLDN1 downregulation in NHEKs. (A) Ruxolitinib, a specific JAK1/2 inhibitor, reverses changes in p‐STAT1 and CLDN1 expression levels induced by IFN‐γ. NHEKs were pretreated for 1 h with 0.01–1.0 μM ruxolitinib and cultured with or without 30 ng ml−1 IFN‐γ for 48 h (n = 1). (B) Effects of STAT1 siRNA treatment on p‐STAT1, total STAT1 and CLDN1 expression levels in NHEKs stimulated with 50 ng ml−1 IFN‐γ for 48 h (n = 2). Western blotting was performed using anti‐p‐STAT1 (Y701), anti‐STAT1, anti‐CLDN1 and anti‐GAPDH antibodies. Protein expression levels were quantified by densitometry and normalized to the level of GAPDH; the ratio is shown directly under each blot. The non‐treated control is set at 1.00. The results are representative of three independent experiments. (C) Relative STAT1 and CLDN1 mRNA expression levels in NHEKs stimulated with 50 ng ml−1 IFN‐γ for 6 h in the presence of negative control siRNA or STAT1 siRNA. mRNA expression was measured by qRT‐PCR. Data are presented as the mean ± SD (n = 4). Data were analysed using Tukey's HSD test (*p < 0.05, **p < 0.01 vs. left group. #p < 0.05, ###p < 0.001 vs. middle group)
We then used siRNA‐mediated knockdown to analyse the effects of STAT1 signalling on CLDN1 protein and mRNA expression after 48 h and 6 h of IFN‐γ treatment, respectively. STAT1 siRNA recovered IFN‐γ‐induced CLDN1 downregulation, inhibited STAT1 expression and phosphorylation (Figure 2B), and restored CLDN1 mRNA expression (Figure 2C). Thus, in NHEKs, IFN‐γ downregulates CLDN1 via the IFNGR/JAK/STAT pathway.
3.2. IFN‐γ downregulates TJ function in a human skin model
Next, we measured TEER to establish whether IFN‐γ impairs skin barrier function. Low TEER indicates high ionic permeability and weak TJ construction. We used a skin equivalent model to measure TEER and assess the effects of IFN‐γ on the TJ barrier. The model was cultured with various IFN‐γ concentrations for 48 h; 1 ng ml−1 IFN‐γ was found to lower the TEER and 10–100 ng ml−1 IFN‐γ significantly decreased the TEER compared with the control (0 ng ml−1 IFN‐γ) (**p < 0.01, Figure 3A). We also used a fluorescent trace flux assay to determine whether IFN‐γ weakens the skin barrier function. IFN‐γ (50 ng ml−1) increased the 4‐kDa FITC‐dextran flux relative to the control (**p < 0.01, Figure 3B).
FIGURE 3.
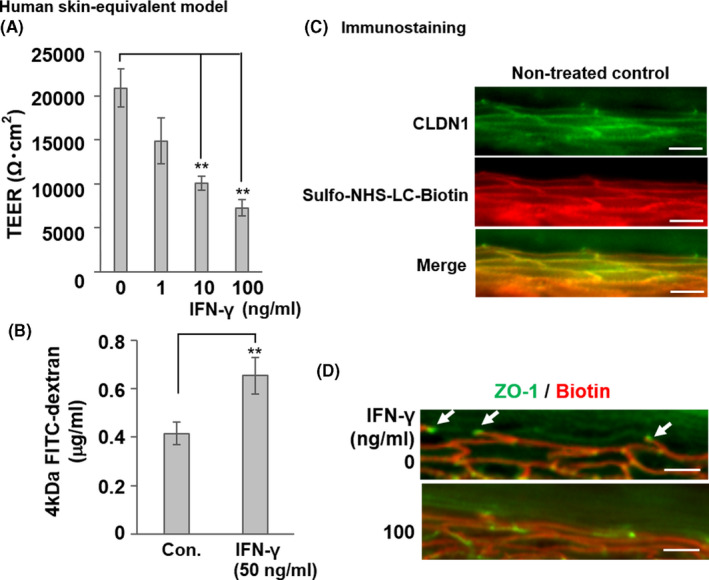
Impairment of TJ barrier by IFN‐γ in a human skin equivalent model. (a) Dose‐dependent suppression of TEER by IFN‐γ in the human skin equivalent model incubated with various IFN‐γ concentrations (1‒100 ng ml−1) for 48 h. Data are presented as the mean ± SE (n = 4). **p < 0.01 vs. control (0 ng ml−1). (B) Increased FITC‐dextran flux in the human skin equivalent model treated with 50 ng ml−1 IFN‐γ for 3 days (56.5% inhibition of TEER vs. control, *p < 0.05). Last 1 h, FITC‐dextran was added to the apical side of the skin equivalent model, and the flux to the basolateral side was measured. Data are presented as the mean ± SE (n = 4). **p < 0.01 vs. control (0 ng ml−1). (C) The normal human skin model was incubated with sulfo‐NHS‐LC‐biotin from the dermal side. After 30 min, the skin model was removed and frozen. Frozen sections were subjected to immunostaining with sulfo‐NHS‐LC‐biotin (red) to show TJ permeability and stained CLDN1 protein (green). The merged red and green image is shown in yellow. (D) Human skin equivalent models were incubated with or without IFN‐γ (100 ng ml−1) for 48 h and subjected to the TJ permeability assay with sulfo‐NHS‐LC‐biotin (red). Localization of TJs was detected using anti‐ZO‐1 (green), which was highly concentrated at the TJs (white arrows) in the skin equivalent models. Scale bars = 40 μm
The skin model was then subjected to a TJ tracer assay to evaluate TJ barrier function. The skin models were incubated with sulfo‐NHS‐LC‐biotin from the basal layer. In a healthy control skin model (non‐treated control), localization of CLDN1 protein was observed from the basal to granular layers, as reported previously (0 ng ml−1 IFN‐γ; Figure 3C, upper‐panel, green). 22 However, IFN‐γ significantly reduced the CLDN1 signal in the human skin model (Figure S3, right panel). ZO‐1, the general marker for TJs, was localized at the TJ sites of the granular layer (0 ng ml−1 IFN‐γ; Figure 3D, upper‐panel, green, white arrow). The intercellular tracer biotin (red) diffused through the basal layer, but its diffusion was blocked at the TJ site where ZO‐1 (green) had accumulated (0 ng ml−1 IFN‐γ; Figure 3D, upper‐panel, white arrow). In the skin model incubated with IFN‐γ (100 ng ml−1), the intercellular tracer biotin was found to pass through the site where ZO‐1 was localized (Figure 3D, lower panel). Thus, IFN‐γ damaged the TJ barrier in the skin equivalent model.
3.3. IL‐17A reversed IFN‐γ‐induced TJ barrier damage
Circulating cytokine levels in AD indicate Th1/Th17 activation, a transition from the acute to the chronic state, and disease severity. Thus, IFN‐γ and IL‐17A interact in terms of regulating TJ formation and barrier permeability. Here, we examined this association in a human skin equivalent model. We used TEER as a TJ barrier index. Two days after induction, 30 ng ml−1 IFN‐γ significantly lowered the TEER relative to the control (0 ng ml−1 IFN‐γ) (*p < 0.05, Figure 4A). IL‐17A (100 ng ml−1) slightly but non‐significantly decreased TEER compared with the control (Figure 4A). Cells co‐induced with IFN‐γ (30 ng ml−1) and IL‐17A (100 ng ml−1) showed higher TEER than those treated with IFN‐γ alone (*p < 0.05, Figure 4A).
FIGURE 4.
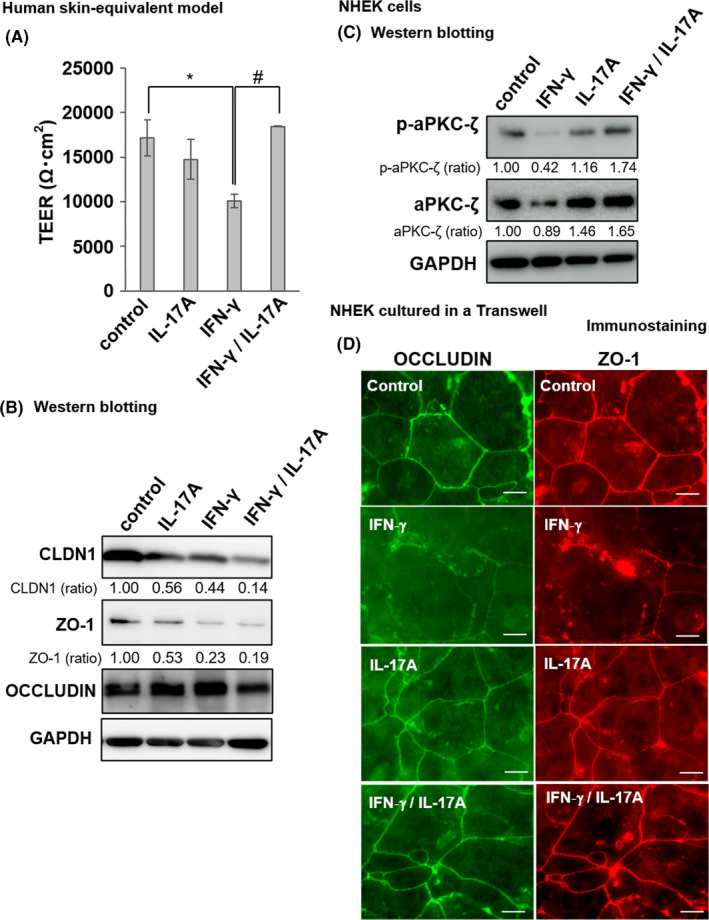
Recovery of IFN‐γ‐induced TJ barrier damage by IL‐17A through phosphorylated atypical protein kinase C (aPKC) reduction. (A) TEER of skin equivalent models incubated with or without IFN‐γ (30 ng ml−1) and/or IL‐17A (100 ng ml−1) for 48 h. Data are presented as the mean ± SE (n = 3). Data were analysed using Tukey's HSD test (*p < 0.05 vs. control; #p < 0.05 vs. IFN‐γ treatment group). (B) Changes in the relative TJ protein expression levels revealed by Western blotting of models incubated with/without IFN‐γ (30 ng ml−1) and/or IL‐17A (100 ng ml−1) for 48 h. Western blotting was performed using anti‐CLDN1, anti‐ZO‐1, anti‐occludin and anti‐GAPDH antibodies (n = 1). Protein expression levels were quantified by densitometry and normalized to GAPDH level; the ratio is shown directly under each blot. The non‐treated control was set at 1.00. The statistical analysis is shown in Figure S4. (C) NHEK cultures were incubated with or without IFN‐γ (30 ng ml−1) and/or IL‐17A (100 ng ml−1) for 12 h. Western blotting was performed using anti‐phospho‐aPKC‐ζ, anti‐aPKC‐ζ and anti‐GAPDH antibodies. The expression levels of phospho‐aPKC‐ζ and aPKC‐ζ were quantified by densitometry and normalized to GAPDH level; the ratio is shown directly under each blot. The non‐treated control is set at 1.00. The statistical analysis is shown in Figure S4. (D) Immunofluorescence staining for occludin and ZO‐1 was conducted. NHEKs were cultured in a Transwell for 4 days in the high‐Ca2+ differentiation medium. IFN‐γ (50 ng ml−1) and/or IL‐17A (100 ng ml−1) were added for the last 48 h. Occludin is visualized in green and ZO‐1 is visualized in red. Scale bars = 20 μm
We evaluated CLDN1 protein expression to establish the role of IL‐17A in recovery from IFN‐γ‐induced TJ damage. IL‐17A injection induced a slight decrease in CLDN1 expression and downregulation of ZO‐1 in NHEKs (Figure S1B–D). Using the skin equivalent model of Figure 4A, Western blotting showed that IL‐17A (100 ng ml−1) or IFN‐γ (30 ng ml−1) downregulated CLDN1 and ZO‐1 protein expression, which was not recovered by co‐treatment with IL‐17A (100 ng ml−1) and IFN‐γ (30 ng ml−1) (Figure 4B and Figure S4A,B). Occludin protein expression was unaffected. Thus, IL‐17A rescued the TJ damage caused by IFN‐γ in a manner independent of the recovery of damaged TJ protein expression.
To further examine the mechanism of the IL‐17A‐mediated TJ recovery, we focussed on atypical protein kinase C zeta (aPKC‐ζ), which is essential for TJ maturation. We examined the expression level of aPKC‐ζ and phosphorylated aPKC‐ζ in NHEKs subjected to treatment with IFN‐γ (30 ng ml−1) and/or IL‐17A (100 ng ml−1) for 12 h. Western blotting revealed that 30 ng ml−1 IFN‐γ downregulated phosphorylated aPKC‐ζ, whereas co‐treatment with 100 ng ml−1 IL‐17A reversed the effect of IFN‐γ (Figure 4C and Figure S4C).
We then explored the influences of IFN‐γ and IL‐17A on TJ maturation. Continuous TJs were detected in NHEK Transwell culture after 48‒96 h in a high‐Ca2+ medium. We thus measured TJ distribution in keratinocytes via occludin immunolabeling. 26 Occludin was concentrated in the TJ of cell‐cell adhesion sites. To investigate whether IL‐17A recovered TJ maturation, we cultured NHEKs in a Transwell and exposed them to 30 ng ml−1 IFN‐γ and/or 100 ng ml−1 IL‐17A (Figure 4D). We performed immunofluorescence staining for occludin and ZO‐1 at 48 h after induction and examined TJ protein arrangement. We observed a continuous occludin network in the cell‐cell borders of the control. The expression of occludin was detected as a distinct line, consistent with the expression of ZO‐1. Keratinocytes subjected to IFN‐γ exhibited discontinuous membranous pattern. Thus, IFN‐γ‐treated keratinocytes could not form mature TJ. However, keratinocytes co‐induced with IL‐17A recovered their localization of TJ proteins and TJ maturation.
4. DISCUSSION
In this study, we found that IFN‐γ, which is a Th1 cytokine with increased expression in skin lesions of patients with chronic AD, suppressed CLDN1 protein expression in a dose‐ and time‐dependent manner through IFNGR/JAK/STAT signalling (Figures 1 and 2). Furthermore, TEER and TJ permeability assays using a skin model indicated that IFN‐γ impaired TJ function (Figure 3). These results are consistent with previous reports that elevated TEWL and decreased TJ function are CLDN1 expression‐dependent in a mouse skin model with stepwise regulated CLDN1 protein expression. 28 Collectively, these data suggested that IFN‐γ‐mediated disruption of TJ function occurs due to CLDN1 expression downregulation.
In AD development, IFN‐γ is thought to suppress Th2 activity. 29 Moreover, circulating Th1 (IFN‐γ) responses in patients with early AD negatively correlate with clinical severity, and increased Th2/Th1 ratios have been linked to clinical disease severity. 30 Thus, IFN‐γ is believed to have a protective role, whereas Th2 responses likely promote atopic inflammation. However, the roles of IFN‐γ in AD skin lesions have not been sufficiently elucidated. We have shown, for the first time, that IFN‐γ can disrupt the TJ barrier by reducing CLDN1 expression via JAK/STAT signalling using a human skin equivalent model. In another study that used NHEKs, IFN‐γ was found to upregulate IL‐33 protein and mRNA expression in a dose‐ and time‐dependent manner. 31 IL‐33 acts as an alarmin to enhance Th2 activity and reportedly activates group 2 innate lymphoid cells and elicits AD‐like inflammation in mice. 32 Moreover, IFN‐γ decreases ceramides with long‐chain fatty acids in three‐dimensional cornified epidermal sheets 33 and reduces epidermal barrier function in a mouse AD model by affecting the fatty acid composition of ceramides. 34 These findings suggest that IFN‐γ both exacerbates and ameliorates pathological conditions and may influence AD pathophysiology, depending on the balance of T‐cell subtypes that produce Th17. IFN‐γ is also implicated in vitiligo pathogenesis as IFN‐γ is released in the vitiligo epidermis when type 1 innate lymphoid cells are increased. 35 TEWL recovery after tape‐strip stimulation was significantly slower in vitiligo than in the healthy epidermis, 36 suggesting that IFN‐γ‐mediated TJ barrier disruption is likely involved in vitiligo.
Recently, the novel JAK kinase inhibitor delgocitinib, which exhibits IC50 values in the nM range for JAK1/JAK2/JAK3/Tyk2, was approved as an ointment for AD treatment and was reportedly effective in Japanese adult patients. 37 According to our results, delgocitinib may exhibit its effect by improving IFN‐γ‐induced TJ dysfunction.
In this study, the Th17 cytokine IL‐17A, which is increased in atopic lesions, counteracted the strong inhibitory effect of IFN‐γ on TJ function (Figure 4). However, this effect was only observed when IFN‐γ and IL‐17A were co‐administered because IL‐17 alone had weak inhibitory effects on TJ function. IL‐17A could not recover the IFN‐γ‐mediated reduction in TJ protein expression, including CLDN1; however, we showed that its effect on TJ function was due to the re‐activation of aPKC‐ζ (Figure 4), which is essential in the early stage of TJ formation and in the maintenance of TJ function. 22
Previous studies have reported contrasting effects of IL‐17 on TJ function. Gutowska‐Owsiak et al. indicated that IL‐17 (200 ng ml−1) downregulated the expression of keratinocyte filaggrin and that of genes important for cellular adhesion, which could affect epidermal barrier formation. 38 Yuki et al. demonstrated that IL‐17 (1‒100 ng ml−1) impaired the TJ barrier and filaggrin monomer degradation (100 ng ml−1) in a skin model. 21 In contrast, Brewer et al. reported that IL‐17A (10 ng ml−1) increased the TEER and decreased paracellular flux (10 ng ml−1) 39 ; furthermore, the same study showed that IL‐4 inhibited the IL‐17A‐mediated enhancement of TJ function via blocking STAT3 phosphorylation. As described before, we observed weak inhibition of TJ function by treatment with IL‐17A alone (100 ng ml−1) (Figure 4A), which is consistent with the findings of Yuki et al. 21 On the contrary, we have shown that IL‐17A rescued IFN‐γ‐induced disruption of TJ function via the re‐activation of aPKC‐ζ (Figure 4C). In addition, Floudas et al. proposed a novel role for IL‐17 receptor A signalling in the maintenance of barrier integrity and immune system homeostasis in the skin because they showed that IL‐17 receptor A deficiency resulted in severe exacerbation of skin inflammation in the filaggrin mutant mouse model of spontaneous AD. 40 The IL‐17A‐mediated rescue of the IFN‐γ‐induced disruption of TJ function (Figure 4) in addition to the IFN‐γ‐induced suppression of Th2 proliferation and differentiation 29 may be related to improved serum IgE level and epithelial abnormalities in patients with chronic AD or in older patients with an increase in the Th1 (IFN‐γ)/Th17 (IL‐17) axis, 5 , 6 although further studies are required.
Anti‐IL‐17 and anti‐IL‐17 receptor A antibodies are effective in the treatment of psoriasis, 41 , 42 a well‐known IL‐17A‐dominated autoimmune disease. In contrast, the efficacy of targeted AD treatment with the IL‐17A antibody secukinumab remains obscure. In a recent randomized, double‐blind study in which IL‐17A was targeted with secukinumab in AD, Ungar et al. concluded that IL‐17A is not the only pathogenic contributor to AD. 43 This finding suggests that specific targeting approaches against other cytokines, such as IL‐17A, IL‐22, and IFN‐γ, in combination with a Th2‐targeting strategy may be needed for the complete resolution of AD in the majority of patients. Therefore, further studies are required to clarify the mechanism by which IL‐17A rescues TJ function and to elucidate the effects of cytokines other than IFN‐γ on epidermal barrier function to find new therapeutic targets.
The limitation of this study is that we used only an in vitro system, and not a real clinical case. However, our study provides a new perspective on clinical findings in patients with Th1‐ and Th17‐dominant AD. Our results may aid in the planning of theoretical treatments using anti‐cytokine antibodies and JAK inhibitors to improve the clinical presentation of heterogeneous AD.
CONFLICT OF INTEREST
Department of Cosmetic Health Science of the Gifu Pharmaceutical University was financially supported by a grant from Gifu City Hall, which, in turn, was financially supported by donations from Ichimaru Pharcos Co. Ltd., Gifu, Japan. The authors state no conflict of interest.
AUTHOR CONTRIBUTIONS
Y.M. and S.I. designed the experiments. Y.M., N.T. and H.N. performed the experiments. Y.M., N.T. and H.N. analysed the data. Y.M., N.T. and S.I. wrote the manuscript.
Supporting information
Fig S1. Effects of cytokines on TJ protein expression levels in NHEK.
Fig S2. Ruxolitinib, a specific JAK1/2 inhibitor, reverses changes in CLDN1 expression levels induced by IFN‐γ.
Fig S3. Downregulation of CLDN1 expression by IFN‐γ in a human skin‐equivalent model.
Fig S4. Relative levels of CLDN1, ZO‐1, phospho‐aPKC‐ζ, and total aPKC‐ζ in human skin‐equivalent models and NHEKs incubated with IFN‐γ, IL‐17A, or their combination.
Tab S1. Details of the cultured cell types and IFN‐γ lot numbers used in each figure.
ACKNOWLEDGMENTS
The authors thank Prof. Mikio Furuse of the Division of Cerebral Structure of the National Institute for Physiological Sciences (Okazaki, Japan) for the generous gift of occludin and ZO‐1 antibodies, Dr. Takuo Yuki of Kao Corp., Japan for providing helpful advice, and all of our laboratory assistants. We would like to thank Editage (www.editage.com) for English language editing.
Mizutani Y, Takagi N, Nagata H, Inoue S. Interferon‐γ downregulates tight junction function, which is rescued by interleukin‐17A. Exp Dermatol. 2021;30:1754–1763. 10.1111/exd.14425
DATA AVAILABILITY STATEMENT
Research data are not shared.
REFERENCES
- 1. Gittler JK, Shemer A, Suárez‐Fariñas M, et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol. 2012;130:1344‐1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Grewe M, Walther S, Gyufko K, Czech W, Schöpf E, Krutmann J. Analysis of the cytokine pattern expressed in situ in inhalant allergen patch test reactions of atopic dermatitis patients. J Invest Dermatol. 1995;105:407‐410. [DOI] [PubMed] [Google Scholar]
- 3. Grewe M, Bruijnzeel‐Koomen CAFM, Schöpf E, et al. A role for Th1 and Th2 cells in the immunopathogenesis of atopic dermatitis. Immunol Today. 1998;19:359‐361. [DOI] [PubMed] [Google Scholar]
- 4. Thepen T, Langeveld‐Wildschut EG, Bihari IC, et al. Biphasic response against aeroallergen in atopic dermatitis showing a switch from an initial TH2 response to a TH1 response in situ: an immunocytochemical study. J Allergy Clin Immunol. 1996;97:828‐837. [DOI] [PubMed] [Google Scholar]
- 5. Bakker DS, Nierkens S, Knol EF, et al. Confirmation of multiple endotypes in atopic dermatitis based on serum biomarkers. J Allergy Clin Immunol. 2021;147:189‐198. [DOI] [PubMed] [Google Scholar]
- 6. Zhou L, Leonard A, Pavel AB, et al. Age‐specific changes in the molecular phenotype of patients with moderate‐to‐severe atopic dermatitis. J Allergy Clin Immunol. 2019;144:144‐156. [DOI] [PubMed] [Google Scholar]
- 7. Tokura Y. Extrinsic and intrinsic types of atopic dermatitis. J Dermatol Sci. 2010;58:1‐7. [DOI] [PubMed] [Google Scholar]
- 8. Czarnowicki T, Krueger JG, Guttman‐Yassky E. Skin barrier and immune dysregulation in atopic dermatitis: an evolving story with important clinical implications. J Allergy Clin Immunol Pract. 2014;2:371‐379. [DOI] [PubMed] [Google Scholar]
- 9. Suga H, Sato S. Novel topical and systemic therapies in atopic dermatitis. Immunol Med. 2019;42:84‐93. [DOI] [PubMed] [Google Scholar]
- 10. Proksch E, Brandner JM, Jensen JM. The skin: an indispensable barrier. Exp Dermatol. 2008;17:1063‐1072. [DOI] [PubMed] [Google Scholar]
- 11. van Smeden J, Bouwstra JA. Stratum corneum lipids: their role for the skin barrier function in healthy subjects and atopic dermatitis patients. Curr Probl Dermatol. 2016;49:8‐26. [DOI] [PubMed] [Google Scholar]
- 12. Redoules D, Tarroux R, Périé J. Epidermal enzymes: their role in homeostasis and their relationships with dermatoses. Skin Pharmacol Appl Skin Physiol. 1998;11:183‐192. [DOI] [PubMed] [Google Scholar]
- 13. Fanning AS, Anderson JM. Zonula occludens‐1 and ‐2 are cytosolic scaffolds that regulate the assembly of cellular junctions. Ann N Y Acad Sci. 2009;1165:113‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Furuse M, Tsukita S. Claudins in occluding junctions of humans and flies. Trends Cell Biol. 2006;16:181‐188. [DOI] [PubMed] [Google Scholar]
- 15. Otani T, Furuse M. Tight junction structure and function revisited. Trends Cell Biol. 2020;30:805‐817. [DOI] [PubMed] [Google Scholar]
- 16. Brandner JM, Kief S, Grund C, et al. Organization and formation of the tight junction system in human epidermis and cultured keratinocytes. Eur J Cell Biol. 2002;81:253‐263. [DOI] [PubMed] [Google Scholar]
- 17. Furuse M, Hata M, Furuse K, et al. Claudin‐based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin‐1‐deficient mice. J Cell Biol. 2002;156:1099‐1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sugawara T, Iwamoto N, Akashi M, et al. Tight junction dysfunction in the stratum granulosum leads to aberrant stratum corneum barrier function in claudin‐1‐deficient mice. J Dermatol Sci. 2013;70:12‐18. [DOI] [PubMed] [Google Scholar]
- 19. Yuki T, Komiya A, Kusaka A, Kuze T, Sugiyama Y, Inoue S. Impaired tight junctions obstruct stratum corneum formation by altering polar lipid and profilaggrin processing. J Dermatol Sci. 2013;69:148‐158. [DOI] [PubMed] [Google Scholar]
- 20. Bergmann S, von Buenau B, Vidal‐y‐Sy S, et al. Claudin‐1 decrease impacts epidermal barrier function in atopic dermatitis lesions dose‐dependently. Sci Rep. 2020;10:2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yuki T, Tobiishi M, Kusaka‐Kikushima A, Ota Y, Tokura Y. Impaired tight junctions in atopic dermatitis skin and in a skin equivalent model treated with interleukin‐17. PLoS ONE. 2016;11:e0161759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yuki T, Hachiya A, Kusaka A, et al. Characterization of tight junctions and their disruption by UVB in human epidermis and cultured keratinocytes. J Invest Dermatol. 2011;131:744‐752. [DOI] [PubMed] [Google Scholar]
- 23. Yuki T, Yoshida H, Akazawa Y, Komiya A, Sugiyama Y, Inoue S. Activation of TLR2 enhances tight junction barrier in epidermal keratinocytes. J Immunol. 2011;187:3230‐3237. [DOI] [PubMed] [Google Scholar]
- 24. Gruber R, Börnchen C, Rose K, et al. Diverse regulation of claudin‐1 and claudin‐4 in atopic dermatitis. Am J Pathol. 2015;185:2777‐2789. [DOI] [PubMed] [Google Scholar]
- 25. Ryu WI, Lee H, Bae HC, et al. IL‐33 down‐regulates CLDN1 expression through the ERK/STAT3 pathway in keratinocytes. J Dermatol Sci. 2018;90:313‐322. [DOI] [PubMed] [Google Scholar]
- 26. Yuki T, Haratake A, Koishikawa H, Morita K, Miyachi Y, Inoue S. Tight junction proteins in keratinocytes: localization and contribution to barrier function. Exp Dermatol. 2007;16:324‐330. [DOI] [PubMed] [Google Scholar]
- 27. Biswas KB, Takahashi A, Mizutani Y, et al. GPNMB is expressed in human epidermal keratinocytes but disappears in the vitiligo lesional skin. Sci Rep. 2020;10:4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tokumasu R, Yamaga K, Yamazaki Y, et al. Dose‐dependent role of claudin‐1 in vivo in orchestrating features of atopic dermatitis. Proc Natl Acad Sci USA. 2016;113:E4061‐E4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Oriss TB, McCarthy SA, Morel BF, Campana MA, Morel PA. Crossregulation between T helper cell (Th)1 and Th2: inhibition of Th2 proliferation by IFN‐gamma involves interference with IL‐1. J Immunol. 1997;158:3666‐3672. [PubMed] [Google Scholar]
- 30. Brunner PM. Early immunologic changes during the onset of atopic dermatitis. Ann Allergy Asthma Immunol. 2019;123:152‐157. [DOI] [PubMed] [Google Scholar]
- 31. Meephansan J, Tsuda H, Komine M, Tominaga S, Ohtsuki M. Regulation of IL‐33 expression by IFN‐gamma and tumor necrosis factor‐alpha in normal human epidermal keratinocytes. J Invest Dermatol. 2012;132:2593‐2600. [DOI] [PubMed] [Google Scholar]
- 32. Imai Y, Yasuda K, Sakaguchi Y, et al. Skin‐specific expression of IL‐33 activates group 2 innate lymphoid cells and elicits atopic dermatitis‐like inflammation in mice. Proc Natl Acad Sci USA. 2013;110:13921‐13926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tawada C, Kanoh H, Nakamura M, et al. Interferon‐gamma decreases ceramides with long‐chain fatty acids: possible involvement in atopic dermatitis and psoriasis. J Invest Dermatol. 2014;134:712‐718. [DOI] [PubMed] [Google Scholar]
- 34. Kanoh H, Ishitsuka A, Fujine E, et al. IFN‐gamma reduces epidermal barrier function by affecting fatty acid composition of ceramide in a mouse atopic dermatitis model. J Immunol Res. 2019;2019:3030268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tulic MK, Cavazza E, Cheli Y, et al. Innate lymphocyte‐induced CXCR3B‐mediated melanocyte apoptosis is a potential initiator of T‐cell autoreactivity in vitiligo. Nat Commun. 2019;10:2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu J, Man WY, Lv CZ, et al. Epidermal permeability barrier recovery is delayed in vitiligo‐involved sites. Skin Pharmacol Physiol. 2010;23:193‐200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nakagawa H, Nemoto O, Igarashi A, Saeki H, Kaino H, Nagata T. Delgocitinib ointment, a topical Janus kinase inhibitor, in adult patients with moderate to severe atopic dermatitis: a phase 3, randomized, double‐blind, vehicle‐controlled study and an open‐label, long‐term extension study. J Am Acad Dermatol. 2020;82:823‐831. [DOI] [PubMed] [Google Scholar]
- 38. Gutowska‐Owsiak D, Schaupp AL, Salimi M, et al. IL‐17 downregulates filaggrin and affects keratinocyte expression of genes associated with cellular adhesion. Exp Dermatol. 2012;21:104‐110. [DOI] [PubMed] [Google Scholar]
- 39. Brewer MG, Yoshida T, Kuo FI, Fridy S, Beck LA, De Benedetto A. Antagonistic effects of IL‐4 on IL‐17A‐mediated enhancement of epidermal tight junction function. Int J Mol Sci. 2019;20:E4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Floudas A, Saunders SP, Moran T, et al. IL‐17 receptor a maintains and protects the skin barrier to prevent allergic skin inflammation. J Immunol. 2017;199:707‐717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hawkes JE, Chan TC, Krueger JG. Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol. 2017;140:645‐653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Papp KA, Leonardi C, Menter A, et al. Brodalumab, an anti‐interleukin‐17‐receptor antibody for psoriasis. N Engl J Med. 2012;366:1181‐1189. [DOI] [PubMed] [Google Scholar]
- 43. Ungar B, Pavel AB, Li R, et al. Phase 2 randomized, double‐blind study of IL‐17 targeting with secukinumab in atopic dermatitis. J Allergy Clin Immunol. 2021;147:394‐397. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. Effects of cytokines on TJ protein expression levels in NHEK.
Fig S2. Ruxolitinib, a specific JAK1/2 inhibitor, reverses changes in CLDN1 expression levels induced by IFN‐γ.
Fig S3. Downregulation of CLDN1 expression by IFN‐γ in a human skin‐equivalent model.
Fig S4. Relative levels of CLDN1, ZO‐1, phospho‐aPKC‐ζ, and total aPKC‐ζ in human skin‐equivalent models and NHEKs incubated with IFN‐γ, IL‐17A, or their combination.
Tab S1. Details of the cultured cell types and IFN‐γ lot numbers used in each figure.
Data Availability Statement
Research data are not shared.