

Abstract
In August 2020, the International Council on Harmonisation (ICH) released a new draft document, which for the first time combined nonclinical (S7B) and clinical (E14) Questions and Answers (Q&As) into 1 document. FDA describes the revision as a “value proposition”: if the human ether‐à‐go‐go assay and the in vivo study are performed in a standardized way, the number of dedicated thorough QT (TQT) studies can be reduced. In this article, we describe and discuss the Q&As that relate to clinical ECG evaluation. If supported by negative standardized nonclinical assays, Q&A 5.1 will obviate the need for a TQT study in the case that a >2‐fold exposure margin vs high clinical scenario cannot be obtained. Q&A 6.1 addresses drugs that are poorly tolerated in healthy subjects and cannot be studied at high doses or in placebo‐controlled studies; it therefore mainly applies to oncology drugs. It will enable sponsors to claim that a new drug has a “low likelihood of proarrhythmic effects” in the case that the mean corrected QT effect is <10 milliseconds at the time of market application. The E14 2015 revision allowed application of concentration–corrected QT analysis on data from routinely performed clinical pharmacology studies, for example, the first‐in‐human study and the proportion of dedicated TQT studies has since steadily decreased. It can be foreseen that the proposed new revision will further reduce the number of TQT studies. To achieve harmonization across regulatory regions, it seems important to reach consensus within the International Council on Harmonisation group on the new threshold proposed in 6.1. For this purpose, the Implementation Working Group has asked for public comments.
Keywords: QTc, ICH E14, ICH S7B, cardiology, arrhythmias, regulatory, QTc prolongation
In August 2020, the International Council on Harmonisation (ICH) released a new Question and Answer (Q&A) document, 1 which is meant to clarify the nonclinical (ICH S7B) 2 and clinical (ICH E14) 3 guidance on how to study new chemical entities (NCEs) for their potential to delay repolarization, that is, to prolong the corrected QT (QTc) interval and cause proarrhythmias. For the first time, the Q&A document addresses both nonclinical assays and the clinical evaluation, in an effort to describe how nonclinical assays can be standardized and thereby improve the integrated nonclinical/clinical risk assessment. The proposed Q&A document is now currently open for public review and comments, and in October 2020, ICH hosted a webinar on the topic, from which presentations are also available. 4 In this commentary, we outline the content with focus on proposed changes for the clinical evaluation of QT liability, place it into the context of the current cardiac safety paradigm and the ICH goal and mission, that is, to achieve efficient and harmonized processes for drug development across regulatory jurisdictions. We also share some thoughts and considerations in terms of what this may mean for drug developers.
Background
Since the release of the S7B and E14 guidances in May 2005, all NCEs with systemic exposure have to undergo an evaluation as to whether they cause an effect on the heart rate QTc. The feared consequence of pronounced QTc prolongation in susceptible patients is that it may trigger a potentially lethal ventricular arrhythmia called torsades de pointes (TdP). 5 , 6 , 7 , 8 The incidence of TdP is very low, often lower than 1 in 10 000 even for those nonantiarrhythmic drugs that were removed from the market due to pronounced QTc prolongation and reported events of TdP and sudden death. 9 Consequently, TdP is rarely observed in clinical trials during drug development. 10 , 11 The ICH E14 guidance established the use of the QTc interval as a biomarker for proarrhythmic risk and described a dedicated study, the thorough QT (TQT) study, designed and powered to exclude an effect on the QTc interval exceeding 10 milliseconds. The purpose of this study is to guide the extent of cardiac safety monitoring in phase III. The statistical analysis described in the original E14 document was based on excluding a QTc effect >10 milliseconds at serial postdose time points after a supratherapeutic dose of the drug, that is, a dose that resulted in clearly higher plasma concentrations of the drug and its metabolites as compared to a therapeutic dose in patients. An additional feature of the TQT study is the need to demonstrate assay sensitivity. This has usually been done by using a drug, moxifloxacin, which is known to cause a mild prolongation of QTc and to demonstrate that this effect can be detected. To achieve sufficient statistical power for the analysis using the so‐called intersection‐union test (IUT), 12 around 44 to 60 subjects need to be enrolled in a stand‐alone study if a crossover design is appropriate. In case a parallel group design is mandated, this number is 2 to 4 times higher. Since 2005 most NCEs have undergone a clinical evaluation of their effect on the QTc interval, and the risk assessment, from a regulatory perspective, has largely been based on clinical electrocardiographic data, whereas the role of nonclinical assays has been to ensure that the drug can be safely taken into the first‐in‐human (FIH) study. With improved performance and predictive value, nonclinical assays have also been extensively used by sponsors for internal decision making in screening and selection of NCEs.
Emerging Role of Concentration‐QTc Modeling
The 2015 revision of the E14 Q&A document (R3) allowed concentration‐QTc (C‐QTc) analysis to be applied to data from studies in healthy subjects to definitively demonstrate that a drug did not cause clinically relevant QTc prolongation. 13 This revision was based on extensive experience from 10 years of TQT studies 14 , 15 , 16 and on the Consortium for Innovation and Quality in Pharmaceutical Development (IQ)–Cardiac Safety Research Consortium (CSRC) study, designed in collaboration between regulators, industry, and the Cardiac Safety Research Consortium. 17 , 18 , 19 Application of C‐QTc analysis created an alternative path for the evaluation of a drug's QTc liability by collecting serial electrocardiograms (ECGs) paired with pharmacokinetic (PK) samples in a study in healthy subjects. In contrast to the “by time point” analysis based on IUT, data from all doses and time points can be used for the prediction of the effect on QTc at any given concentration in the covered range. This considerably increases the statistical power of C‐QTc analysis as compared to IUT. An important point in the 2015 (R3) revision was that a separate positive control would not be necessary if “there are data characterizing the response at a sufficiently high multiple of the clinically relevant exposure (see ICH E14 Section 2.2.2).” The 2015 (R3) revision thereby enabled C‐QTc analysis to be applied to data from, for example, the FIH study and, provided sufficiently high concentrations were obtained, waive the request for a later, designated TQT study. 14 , 19 If procedures are implemented in the FIH study to ensure that subjects are supinely resting at prespecified time points for PK sampling and continuous 12‐lead ECGs (Holters) are recorded, the ECG waveforms can be stored. At a later stage, an informed decision can then be made on whether to extract ECGs from the stored waveforms, based on observed concentrations of parent and metabolites and the development project's viability. This “collect and store” approach thereby represents a very efficient way of obtaining a sufficient amount of ECG data to exclude a small effect on the QTc interval and other ECG parameters and avoids the more resource intensive, stand‐alone TQT study.
Since the 2015 (R3) revision of E14, there has been a steady trend away from TQT studies with a positive control. These studies have been replaced by, for the most part, FIH studies with the added objective of excluding small effects on ECG parameters (herein called “5.1 studies”; see below). On average, the US Food and Drug Administration (FDA) has reviewed 55 QT studies per year since 2016 (personal communication, Dr Garnett, FDA). Between 2016 and August 2020, the proportion of TQT studies has decreased from 62% to 34%, and the proportion of 5.1 studies has gone from 10% to 42% (Figure 1A). The lemborexant development program can serve as an example on how C‐QTc analysis was successfully applied to standard clinical pharmacology studies, thereby waiving the request to perform a stand‐alone TQT study in all major regulatory regions. 20 Lemborexant, a novel dual orexin receptor antagonist, has been developed and was recently approved for the treatment of insomnia. Serial ECG monitoring paired with PK samples were implemented in the FIH multiple ascending dose (MAD) study and a MAD bridging study in Japanese and non‐Japanese healthy subjects. Both studies followed the same basic design with subjects were dosed in the evening for 14 days, and the same ECG technique was employed. In Figure 2, it can be seen that the prespecified linear C‐QTc model captured the observed data in an acceptable way and predicted that an effect of lemborexant on the QTc interval exceeding 10 milliseconds can be excluded (ie, the 90% confidence interval [CI] is <10 milliseconds) within the full range of observed plasma concentrations, up to ≈600 ng/mL. These levels exceed clinically relevant plasma levels by at least 5‐fold, and the result was therefore accepted as conclusive and a TQT study was not requested by FDA, Pharmaceuticals and Medical Devices Agency, or the European Medicines Agency. Lemborexant was approved by the FDA in December 2019 and by the Pharmaceuticals and Medical Devices Agency in January 2020. In the Japanese label, a potential QTc effect is not mentioned, whereas the US label describes the negative result.
Figure 1.
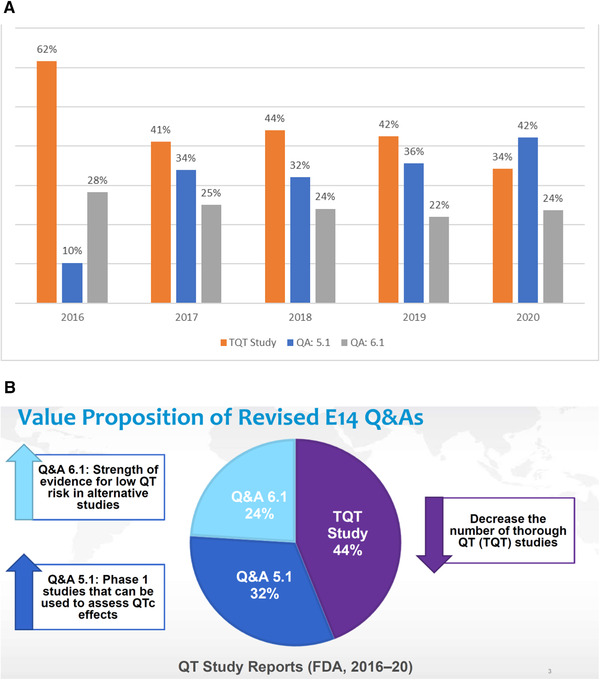
(A) Distribution of electrocardiographic studies intended to demonstrate a lack of a corrected QT (QTc) effect. Between 2016 and August 2020, the US Food and Drug Administration (FDA) on average reviewed 55 studies, which included the objective to evaluate whether a drug caused clinically relevant effect on electrocardiogram (ECG) parameters. The 2015 revision of the E14 Question and Answer (Q&A) document enabled conducting definitive ECG evaluation in standard clinical pharmacology studies, planned as part of the development program, for example, first‐in‐human studies, without inclusion of a positive control (here called 5.1 studies). It is clear that the 2015 revision has resulted in a steady decline of the number of TQT studies, which have been replaced by 5.1 studies. The proportion of studies without placebo and often with the therapeutic dose of the drug in the targeted patient population (6.1 studies) have been constant over the years. (Numbers and graph kindly provided by Drs Johannesen and Garnett, FDA.) (B) The FDA's “value proposition.” The proposed S7B/E14 revision is presented by the FDA representatives on the IWG as a value proposition. With standardized hERG and in vivo assays, as described in the S7B part of the proposed Q&A revision, (1) TQT studies with a positive control can in some cases by replaced by 5.1 studies, and (2) sponsors may claim low likelihood of proarrhythmic effects due to delayed repolarization if the mean effect on ΔQTc is <10 milliseconds in a study in, for example, patients with cancer without placebo and with the therapeutic dose. (Source: Slide 3 in Dr Garnett's presentation on the public webinar on the revised S7B/E14 Q&A document in October 2020.) https://www.fda.gov/drugs/news‐events‐human‐drugs/new‐approaches‐integrated‐nonclinical‐clinical‐qtproarrhythmic‐risk‐assessment‐10152020‐10162020.
Figure 2.
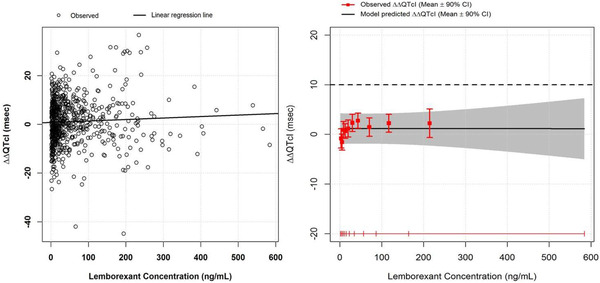
Lemborexant concentration‐QTc analysis. The left panel shows a scatter plot with observed plasma concentration/ΔΔQTcI pairs pooled from the 2 MAD studies. Placebo‐corrected ΔQTcI was derived by subtracting the mean ΔQTcI in the placebo group at the same nominal time as the observed ΔQTcI at each time for each subject. The right panel is a goodness‐of‐fit plot for observed and predicted relation between lemborexant plasma levels and ΔΔQTcI on the same data. Red squares with vertical bars denote the observed mean ΔΔQTcI with 90%CI displayed at the median plasma concentration within each decile. The solid black line with gray‐shaded area shows the predicted mean ΔΔQTcI with 90%CI based on the C‐QTc analysis. The horizontal red lines with notches show the range of plasma concentrations divided into deciles. CI, confidence interval; C‐QTc, concentration–corrected QT; Δ, change from baseline; ΔΔ, placebo‐corrected, change from baseline; MAD, multiple ascending dose; QTcI, individualized corrected QT interval. (Source: Figure 3 in Murphy et al. 20 Reproduced with permission from the authors and The Journal of Clinical Pharmacology.)
It is worth pointing out that the vast majority of TQT studies are today performed with C‐QTc analysis as the primary statistical method, thereby dramatically reducing the required sample size to exclude an effect on the QTc interval >10milliseconds. These studies can today be performed with 24 to 28 healthy subjects, as compared to more than twice these numbers when analyzed with IUT.
Current Paradigm for ECG Evaluation in Drug Development
The current paradigm for ECG evaluation is based on excluding clinically concerning ECG effects (ie, not only on the QTc interval but on ECG parameters in general, ie, heart rate [HR], PR and QRS interval) in appropriately powered studies in healthy subjects before initiation of late‐stage patient trials. When such effects can be excluded, routine safety monitoring in line with the standards in the therapeutic area can be applied in subsequent patient trials. When concerning effects cannot be excluded or a satisfactory ECG evaluation has not been performed, the regulatory expectation is to record high‐quality ECGs in all patients at a few time points, that is, before dosing and at maximum concentration (Cmax) at steady‐state concentrations. Based on this, the ECG effects will be described in the label and, depending on the effect size, will result in precautionary statements, as an example in terms of administration to subjects with cardiovascular disease and/or arrhythmias, with hypokalemia or bradycardia or on other drugs known to cause QTc prolongation. It must be emphasized that this safety paradigm with appropriately powered ECG evaluation in healthy subjects has been successful: No new drugs have been withdrawn from the market due to concerns related to delayed repolarization since the implementation of S7B and E14 in 2005. It has also meant that effects on not only the QTc interval, but on ECG parameters in general, that is, HR and PR and QRS interval, have been detected and are described in labels, as appropriate. 21
Proposed S7B/E14 Q&A Revision
The “value proposition,” as the FDA calls the proposed revision of S7B/E14 Q&A, is that the number of TQT studies can be further reduced by implementing standardized hERG and in vivo assays, as described in Q&A 5.1 and 6.1 (Figure 1B).
Question and Answer 5.1
Q&A 5.1 refers to studies in healthy subjects that use C‐QTc analysis to exclude that a drug has a >10‐millisecond effect on the QTc interval but without a positive control, for example, a FIH study. Current FDA practice, not strictly followed by other regulators, has been as follows: High clinical exposure has been defined as mean Cmax in patients with impaired clearance of the drug, based on intrinsic or extrinsic factors, that is, the worst‐case scenario in terms of concentrations. As an example, if a drug is a 3A4 substrate and mean Cmax is ≈200 ng/mL in patients on a therapeutic dose (ie, not reduced) administered concomitantly with a potent 3A4 inhibitor, this level corresponds to high clinical exposure. The FDA's position has been that if obtained concentrations do not exceed this level by 2‐fold—in this example, do not reach mean Cmax around 400 ng/mL—then a study with a positive control would be needed to demonstrate that the drug does not cause QTc prolongation of concern. In such case, results from a study with a positive control, that is, a TQT study, excluding a QTc effect >10 milliseconds would be needed to justify that the drug can be taken into late‐stage development without an expanded ECG safety evaluation. 3
In the new Q&A 5.1 revision, it is now stated under point 4) (bold added by authors):
A separate positive control would not be necessary if either of the following conditions is met:
There are data characterizing the response at a sufficiently high multiple of the clinically relevant exposure (see ICH E14 Section 2.2.2);or
If the maximum therapeutic exposure has been fully covered in the clinical ECG assessment (eg, concentrations representative of the maximum recommended dose at steady‐state in situations of intrinsic and/or extrinsic factors that increase bioavailability), but sufficiently high multiples cannot be obtained (eg, for reasons of safety, tolerability, saturating absorption), then a nonclinical integrated risk assessment that includes the hERG assay, an in vivo QT assay, and any follow up studies can be used as supplementary evidence. See ICH S7B Q&A 1.1 for details; in summary, the nonclinical studies should include (1) a hERG safety margin higher than the safety margins computed under the same experimental protocol for a series of drugs known to cause torsade de pointes (TdP) and (2) no QTc prolongation in an in vivo assay of sufficient sensitivity conducted at exposures of parent compound and human‐specific major metabolites that exceed clinical exposures.
The change proposed in Q&A 5.1 will therefore, to some extent, further reduce the number of TQT studies and enable acceptance of C‐QTc analysis of high‐quality ECG data in, for example, FIH trials and supportive nonclinical data to demonstrate that the drug does not cause clinically concerning QT prolongation, even in case ECG effects at supratherapeutic concentrations have not been studied. It is, however, important to note that according to the proposed text, this would apply to drugs for which sufficiently high concentrations cannot be obtained, for example, for reasons of safety, tolerability, and saturating absorption. In this context, it should be clarified that high drug concentrations drive the power the C‐QTc analysis to detect mild QTc prolongation. This point can be illustrated with C‐QTc analysis results from the development program of JNJ‐54861911, a beta‐site amyloid precursor protein cleaving enzyme‐1 inhibitor developed for treatment of Alzheimer disease by reducing the production of beta‐amyloid fragments. In the nonclinical workup, QTc prolongation and increased HR was noted in male telemetered conscious dogs following high oral doses (30 mg/kg). In the 1‐month repeat‐dose toxicity study in dogs, QTc prolongation was observed at 100 mg/kg/d. Concentrations at which no action potential/QTc prolongation was seen in dogs were ≈20‐fold higher than Cmax associated with the pharmacologic target of a 50% reduction in Aβ concentrations over 24 hours in dogs. In Figure 3, the model‐predicted effect on QTc is shown with data from the first part of a MAD study with doses up to 90 mg once daily and when a higher dose, 150 mg, was added. The slope of the C‐QTc relationship with doses up to 90 mg is shallow and an effect exceeding 10 milliseconds can be excluded within the observed range of concentrations. When the higher dose was added, the C‐QTc relationship is now clearly positive and demonstrates that the drug causes QTc prolongation, even at concentrations for which the previous model was negative. This QTc effect seen at high concentrations was also confirmed in a fully powered TQT study.
Figure 3.
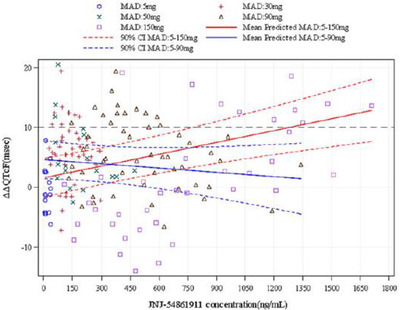
Importance of high concentrations in C‐QTc analysis. Scatter plot with observed plasma concentration/ΔΔQTcF pairs and predicted effect on ΔΔQTc using C‐QTc analysis from a MAD study with JNJ‐54861911. The blue lines demonstrate the predicted QTc effect (mean ± 90%CI) using C‐QTc analysis applied to data for doses between 5 and 90 mg. The predicted QT effect is very small and an effect exceeding 10 ms can be excluded. When a higher dose group with 150 mg was added, the model, shown in red lines, predicted that the drug has an effect on the QTc interval, also within concentrations ranges that were negative in the initial analysis. CI, confidence interval; C‐QTc, concentration–corrected QT; Δ, change from baseline; ΔΔ, placebo‐corrected, change from baseline; MAD, multiple ascending dose; QTcI, individualized corrected QT interval. (Source: Figure 5, Panel C in Timmers et al. 39 Reprinted with permission from the authors and The Journal of Clinical Pharmacology.)
We have made the same observations in other studies, and it is clear, in our view, that high concentrations determine the power of the concentration‐QTc analysis to detect small QT effect. In preparations for the IQ‐CSRC study, this was also demonstrated through simulations that the precision of the C‐QTc slope increased, that is, the width of the 90%CI became narrower, when a higher dose of a positive drug was added (Table 4 in Darpo et al 22 ).
Figure 4 is a slide from Dr Garnett's presentation at the public ICH webinar on the proposed Q&A revision and describes 2 paths by which the regulatory expectation for a study with a positive control can be waived. It should then be noticed that this represents the FDA's view on how to interpret Q&A 5.1, and the slide and a subsequent publication on the same topic 23 do not acknowledge that the text in 5.1 specifically says that it applies to drugs for which supratherapeutic concentrations cannot be obtained, “for reasons of safety, tolerability, and saturating absorption.” The figure does not provide insight as to whether doses were not pushed high enough or supratherapeutic concentrations could not be obtained for the reasons mentioned above. It is therefore not clear, in our view, to which extent the proposed Q&A 5.1 revision will further reduce the proportion of TQT studies. To date, the most frequently debated topic between regulators and sponsors is the selection of a supratherapeutic dose in the QT evaluation study, and it seems that this discussion will continue. It may be that the FDA will accept that sponsors now have 2 equal options to obviate the need for a positive control. In our view, however, it is advisable to strive for concentrations clearly higher than those observed in the high clinical scenario when possible; it also seems likely that some regulators will point to the text in Q&A 5.1 and request that the sponsor demonstrates on which basis concentrations 2‐fold above the highest clinical scenario cannot be achieved.
Figure 4.

Waiving positive controls in clinical QT studies. In the US Food and Drug Administration's presentation of the public webinar and in a subsequent publication, 23 2 equal options are described to waive the request for a positive control in case supratherapeutic concentrations have not been achieved in, for example, a first‐in‐human study with serial electrocardiograms paired with pharmacokinetic samples. This interpretation does not seem to be aligned with the proposed text in the revised Question and Answer (Q&A) 5.1 and is not shared by other regulators. The Q&A document would benefit from clarification on this point. The pie chart shows proportion of studies by observed exposure margin in “QT reports” as compared to mean maximum concentration levels in patients on a therapeutic dose of the drug. It is not clear how many studies did not achieve “2 × high clinical” exposure, based on the prerequisite in the revised 5.1 Q&A: “… but sufficiently high multiples cannot be obtained (eg, for reasons of safety, tolerability, saturating absorption).…” (Source: Slide 3 in Dr Garnett's presentation on the public webinar on the revised S7B/E14 Q&A document in October 2020. https://www.fda.gov/drugs/news‐events‐human‐drugs/new‐approaches‐integrated‐nonclinical‐clinical‐qtproarrhythmic‐risk‐assessment‐10152020‐10162020.)
Question and Answer 6.1
Q&A 6.1 addresses drugs that are poorly tolerated at high doses/concentrations or cannot be studied in a placebo‐controlled trial. The question therefore mainly refers to development programs for oncology drugs. The proposed revision of Q&A 6.1 reads:
An integrated nonclinical and clinical QT/QTc risk assessment can be particularly valuable under scenarios where a placebo‐controlled comparison is not possible; safety considerations preclude administering supratherapeutic doses to obtain high clinical exposures and/or safety or tolerability prohibit the use of the product in healthy participants. The design elements that include placebo and healthy participant dosing assist in decreasing variability, but their absence does not preclude interpretation.
The integrated non‐clinical and clinical QT/QTc risk assessment should include:
The hERG assay, an in vivo QT assay, and any follow‐up non‐clinical studies, especially those selected to overcome the challenges encountered in the clinical studies (see ICH S7B Q&As 1.1 and 1.2); and
- Alternative QT clinical study designs incorporating ECG assessments with as many of the usual “thorough QT/QTc” design features as possible (see ICH E14 section 2.2 and Q&A 5.1).In situations where it is not possible to evaluate the QT/QTc effects at higher exposures than are anticipated with the recommended therapeutic dose, it is particularly important that the non‐clinical in vivo studies are conducted at exposures exceeding the clinical therapeutic exposures.
An integrated QT/QTc risk assessment can also be particularly valuable for drugs with confounding heart rate effects (ie, >20 bpm) that could impact accurate determination of the QTc. Advanced methodologies for controlling or correcting for heart rate changes in the nonclinical in vivo studies and/or conducting QTc assessments in patients with the disease might be informative in this situation. If tolerance to the chronotropic effect develops with repeat dosing, upward titration regimens can sometimes be employed to avoid or minimize the confounding effects of drug‐induced heart rate changes on the QTc assessment.”
The current paradigm has been to apply as many components from the TQT study as possible into a clinical study with high‐quality ECGs. For oncology drugs, ECG monitoring implemented into the escalation or expansion part of the FIH study has therefore often been successfully used to replace the formal TQT study. 24 The FDA's standard for labeling over the past few years has been that in case an effect >10 milliseconds can be excluded in such study, that is, the upper bound of the 90%CI of ΔQTc <10 milliseconds, then the label will state that it seems unlikely that the drug will cause a QTc effect >20 milliseconds.
Under point 2, Decision Making, the following is now stated (bold added by authors):
A totality of evidence argument based on the results of an integrated non‐clinical and clinical QT/QTc assessment could be made at the time of marketing application. Evidence to support that drug has a low likelihood of proarrhythmic effects due to delayed repolarization should include the following:
The nonclinical studies, following best practice considerations for in vitro studies (see ICH S7B Q&A 2) and in vivo studies (see ICH S7B Q&A 3), show low risk, which includes (1) a hERG safety margin higher than the safety margins computed under the same experimental protocol for a series of drugs known to cause Torsade; and (2) no QTc prolongation in an in vivo assay of sufficient power to detect a QTc prolongation effect of a magnitude similar to a dedicated clinical QT studies and at exposures of parent compound and human‐specific major metabolites that exceed clinical exposures (see ICH S7B Q&A for details).
The high‐quality ECG data (see ICH E14 and ICH E14 Q&A [1]) collected in the alternative QT clinical assessment do not suggest QT prolongation, generally defined as ΔQTc greater than 10 msec as computed by the concentration‐response analysis (see E14 Q&A 5.1 for details) or the IUT. The strength of the clinical ECG data depends on the upper bound of the 2‐sided 90% confidence interval around the mean ΔQTc estimate. If applicable, there should be no notable imbalances between treatment/dose arms in the proportion of subjects exceeding outlier thresholds.
A cardiovascular safety database that does not suggest increased rate of adverse events that signal potential for proarrhythmic effects (ICH E14 Section 4).
Below this paragraph, the following question is asked:
The ICH E14/S7B Implementation Working Group is seeking input via public comment on how to define the lack of clinically relevant QT prolongation in the context of the specific #2 criteria above when #1 and #3 would also be met.
Then the current FDA labeling standard is brought into the document:
If non‐clinical studies do not show low risk (or are not performed), there is reluctance to draw conclusions of lack of an effect in an absence of a positive control; however, if the upper bound of the 2‐sided 90% confidence interval around the estimated maximal effect on ΔQTc is less than 10 msec, the treatment is unlikely to have an actual mean effect as large as 20 msec.”
Unfortunately, the text in #2 is not unambiguous. Seen from a statistical perspective, “generally defined as ΔQTc greater than 10 msec” means that the estimated effect including an appropriate confidence interval meets this criterion, so, this means no change. On the other hand, “do not suggest QT prolongation” is open to alternative interpretations, such as the estimate itself meeting this criterion. Based on Dr Garnett's presentation and subsequent publication, the FDA's view is that this refers to the estimated effect.
Again, it should be borne in mind that this text is relevant mainly to oncology drugs, for which the therapeutic dose is often the highest tolerated and the clinical study is conducted in cancer patients, and placebo is thereby often difficult to justify. The key point is that a new threshold is introduced, namely, that the mean effect on ΔQTc should be <10 milliseconds in a clinical study with high‐quality ECGs. The new criterion of the mean QTc effect being <10 milliseconds has a qualifier: the strength of the clinical ECG data depends on the upper bound of the 2‐sided 90%CI around the mean ΔQTc estimate. This new proposed criterion is clearly the most debated topic among regulators on the Implementation Working Group, and thus the request for public comments, specifically on this point. It is not clear how a low likelihood of proarrhythmic effects would translate into labeling language, perhaps intentionally since labeling is handled differently across regulatory jurisdictions. A pertinent question is obviously whether a drug with a low likelihood of proarrhythmic effects can be safely given to patients at high risk for arrhythmias, for example, patients with history of ventricular arrhythmias, with hypokalemia (often seen in cancer patients with vomiting or diarrhea), or to patients on other drugs that also prolong the QTc interval, that is, what this means in terms of precautionary labeling statements.
At the public webinar, Dr Garnett gave the following example (slide 20‐21 in Garnett 25 ), asking whether an integrated QT assessment would support the claim of low likelihood of proarrhythmic effects under the circumstances listed in Q&A 6.1:
The sponsor conducted an uncontrolled, dose‐escalation study with high‐quality ECGs for an oncology drug in 30 patients, and the highest dose was the labeled dose.
No evidence of QTc prolongation using concentration‐response relationship (ΔQTc at Cmax at steady state = 4 milliseconds with an upper bound of the CI of 12 milliseconds and no QTc outliers [QTc>480 milliseconds or ΔQTc > 60 milliseconds])
Double negative nonclinical assessments using best practices and in vivo study was powered to detect QT prolongation similar to a TQT study.
Dr Garnett's answer was yes, if the cardiac safety database at marketing application does not suggest proarrhythmic risk. In other words, Dr Garnett understands criterion #2 in the alternative way, that is, in the sense of its estimate, while other regulators argue that the threshold of concern should be the same for all drugs (even though the benefit/risk assessment will be different) and that the agreed‐upon concept paper 26 does not describe a revised threshold. 27 A more challenging example, still within the definition of the proposed revision, would be a drug with a mean effect on ΔQTc in a clinical study of 8 milliseconds, with the same width of the CI as above, that is, an upper bound of 16 milliseconds. It seems highly likely that there will be differences in opinion across regulators whether this means that the drug has “low likelihood of proarrhythmic effects” and therefore can be safely given to patients also on other drugs with a QT effect and to patients at risk for proarrhythmic events. 27 The reason for using a less stringent threshold of concern in patients with cancer is, in our view, not imminently apparent. It is certainly true that a higher risk of rare serious adverse events can be accepted for a drug that may prolong survival, but this is not the same as claiming that the drug is “safe” (“low likelihood of proarrhythmic effect”) and therefore can be given without informing prescribing physicians and patients about potential and sometimes avoidable risks. Instead of introducing a new, less stringently defined criterion without describing what this may mean in terms of labeling, it seems better to retain the threshold of concern and decide, as is the case today, on approvability and labeling case by case based on an integrated risk assessment, which then can give more weight to the newly proposed, standardized nonclinical assays. For an efficacious cancer drug, this integrated risk assessment would obviously be favorable in the overwhelming majority of cases, even with some precautionary statements in the label giving guidance to prescribers.
The example of drugs with a pronounced HR effect, defined as, for example, >20 bpm heart rate increase, is poorly chosen. It may seem correct to claim that a drug is unlikely to be proarrhythmic based on delayed repolarization if it increases HR, but it may well trigger coronary ischemia or worsen heart failure and thereby lead to life‐threatening arrhythmias. A drug that causes a reduction of HR at this level, that is, has a strong negative chronotropic effect, is likely to cause clinically significant bradycardia and may trigger sinus pauses and atrioventricular blocks. This example is therefore torsade‐centric, ignoring other mechanisms that clearly warrant caution and potentially ECG monitoring in susceptible patients. In this context, it is important to reiterate that the role of ECG evaluation in early studies is to identify drugs for which ECG monitoring in late‐stage clinical trials would be warranted, not to per se define which drugs are proarrhythmic. To claim that a drug with such substantial HR effect has low likelihood of proarrhythmic effects due to delayed repolarization is at best noninformative, in our view, and may lead prescribers in the wrong direction.
Looking Ahead: Phase 2 of the Q&A Process, the Comprehensive In Vitro Proarrhythmia Assay Project and the Role of Clinical ECG Evaluation
Once the current version of the S7B/E14 document is endorsed, stage 2 will commence, addressing additional ways of applying integrated nonclinical/clinical risk assessment for proarrhythmic risk. The topics will be addressed: “… pending sufficient data are available to support the revision of E14 Q&A additions. ” 26 If enough data do not exist at this stage: “… the implementation working group (IWG) will make recommendations for what additional data are required. ” In view of this, and to promote their positions, 2 recent publications from industry and from FDA representatives on the IWG are of interest. Vargas and coauthors 28 represent industry in all major regions and outline their expectations on stage 2. The position of this industry group can be summarized as follows: (1) nonclinical assays, especially the in‐vitro hERG assay and nonrodent in vivo studies, have been substantially improved over the past 10 years and must be given more weight in regulatory decision making in regard to the need for clinical QT assessment, need for ECG monitoring, and labeling of mildly QT prolonging drugs; (2) clinical “QT evaluation” is burdensome, costly, and can in many cases be replaced by nonclinical assays; and (3) by applying the principles of comprehensive in vitro Proarrhythmia Assay CiPA, drugs with mild QT prolongation can be identified as safe and thereby avoid “clinical QT” studies and “inappropriate labeling.” The overriding theme has essentially been the same over the past 10 years, the need to reduce the “burden” of clinical ECG assessment, even though it is at times difficult to follow or agree with the logic of the arguments. The authors claim that “… there are several scenarios where a robust ICH S7B data package provides a solid foundation for a fully integrated proarrhythmia risk assessment that obviates the need for an enhanced QTc assessment in the clinic,” without explaining how a “fully integrated” approach can be undertaken without confirmation of the lack of clinically relevant ECG effects in humans. The authors acknowledge that ECG evaluation can be performed more efficiently today, based on C‐QTc analysis on, for example, FIH trials, but state that “a potential consequence of the clinical concentration‐QTc response modeling in E14 is that it unintentionally diminished the regulatory impact and value of the ICH S7B core assays and mechanistic approaches inspired by the CIPA proposal.” Unless one assumes a priori that proarrhythmic liability is always better addressed through nonclinical assays, it is hard to understand how improving the efficiency of a clinical evaluation can be depicted as something negative, because it reduces the value of nonclinical assays. Vargas et al 28 also state that “there are no standard protocols, experimental conditions, or regulatory expectations to guide the execution of the nonclinical core assays,” which may, in our view, have contributed to the perceived small impact of these assays. The authors suggest that some drugs should be exempt from clinical testing based on preclinical findings and take the example of large therapeutic proteins, such as monoclonal antibodies, as an example of currently exempted drugs. In our view, the example seems awkward since such proteins cannot enter the hERG ion channel pore and therefore cannot biologically exert an effect on cardiac repolarization. A corresponding example from the class of small molecules could be NCEs without systemic exposure, such as those formulated for topical application on the skin. To claim that such a drug should not be required to undergo testing for potential effects on the QTc interval is fully logical and also compliant with the current E14 document. It should, however, be recognized that there is still a regulatory expectation to perform some level of ECG evaluation with large therapeutic proteins in the targeted patient population, since there are such molecules with indirect, cardiotoxic effects. 29 To use the example to justify that nonclinical assays can replace the need for a careful, that is, appropriately powered, ECG evaluation in humans seems to be a weak argument. The authors also claim that “… any preclinical or clinical signal, ie, hERG blockade or QTc prolongation may have had the unintended consequence of preventing the development of otherwise safe and efficacious drugs,” and yet state that there is “… little to no impact on approval and product labeling for drugs that demonstrate a positive TQT finding.” It seems unlikely that internal sponsor decisions would be based on such minimal impact on approvability and labeling rather than being well informed and made on an approach based on totality of evidence. It also seems highly unlikely that the development of promising NCEs would be prevented by the requirement to inform prescribers and patients of an electrocardiographic effect, which in certain circumstances warrants caution. The statement suggests that the authors underestimate the value of informing prescribers and patients that a drug may cause QT prolongation and potentially proarrhythmias, and should, in certain cases, be avoided or given with caution. To further promote reducing clinical ECG evaluation, the authors also argue that it is difficult to perform ECG evaluation in clinical studies in patients with cancer; this is in contrast to our experience, which also entails numerous studies from sponsors represented in the publication. In many cases, ECG evaluation with oncology drugs is today conducted in ≈30 to 40 patients as part of a study planned for other purposes, either in the FIH trial or in a substudy of pivotal patient trials. This obviously comes with some effort from the side of sponsors, investigators, and clinical sites, but with increasing experience cannot reasonably be described as very cumbersome, strenuous, or resource intensive as compared to many other study activities.
Strauss and colleagues, 23 all representing the FDA on the IWG, published a commentary and editorial soon after. In this paper, the predictive value of nonclinical assays, especially a “double negative” outcome of hERG and an in vivo study, is further discussed. It is also pointed out that the current version of E14 encourages sponsors to explore additional nonclinical assays and proarrhythmia models to better understand the proarrhythmic liability of a drug. The approach of CiPA is promoted, 30 , 31 including the use of the JTpeak interval to define a mildly QT prolonging drug as safe enough to undergo clinical testing in patients with reduced or eliminated ECG evaluation. For drugs with balanced, mixed‐ion‐channel inhibition (equipotent L‐type calcium or late sodium, in addition to hERG inhibition) with a QTc effect of not more than 20 milliseconds, the authors reiterate that the JTpeak interval can be used to differentiate potentially proarrhythmic drugs from those that are safe. 30 , 31 , 32 Under the CiPA paradigm, the role of clinical ECG evaluation would be to “check for missed or unanticipated effects.” Near‐term goals for stage 2 are also discussed, including small molecules with low systemic exposure and drugs for which the QT effect plateaus, which may indicate that the effect is indirect and not caused by hERG inhibition (for a discussion on this topic, see, eg, Darpo et al 33 ).
It is certainly correct that the current version of the E14 document discusses the role of additional nonclinical assays to improve the understanding the proarrhythmic liability of a drug, but, as Strauss and colleagues know better than most of us, this area is driven by regulatory precedence as evidenced, for example, by feedback from reviewing divisions, drug approvals, and labeling. As an example, JTpeak as a safety biomarker with the role as described above is controversial and not accepted by other regulators at this stage. We obviously do not have full insight but are not aware of drugs approved by the FDA for which JTpeak was accepted as a biomarker to signal safety, but we have encountered some examples that point toward nonacceptance in the regulatory decision making. Some published examples have demonstrated other ECG effects, for example, QRS widening and PR prolongation, for drugs with mixed channel inhibition and lack of JTpeak prolongation, suggesting that this biomarker is not very specific. 17
It would benefit the IWG discussions if FDA representatives demonstrated consensus within the agency by presenting cases for which nonclinical assays and novel clinical biomarkers (JTpeak), as described under the CiPA paradigm, contributed to positioning the drug as safe enough to undergo late‐phase patient trials with reduced or eliminated ECG monitoring and to label the drug without precautionary statements, in the presence of mild (<20 millisecond) QTc prolongation.
It seems highly unlikely that other regulators would accept a novel biomarker, proposed by regulatory scientists, for regulatory decision making unless this biomarker had been endorsed as evidenced in drug approvals, by the agency the scientists represent. This is, in particular, true if the innovative approach is intended to replace a safety paradigm that apparently works well. 34 To date, the validation of JTpeak as a biomarker to signal safety despite QT prolongation has been conducted internally by CiPA researchers. In our view, it would benefit the ICH process and enhance the acceptance of this new biomarker if also endorsed by an independent, highly credited, group of independent cardiologists with extensive experience from drug development, for example, the Cardiorenal Advisory Committee. In our view, the arguments favoring the use of JTpeak as a novel biomarker for safety are weak, and we foresee that other regulators will remain unconvinced without an independent validation process. A recent example that illustrates this point is ranolazine, which is described as a safe drug by CiPA scientists based on its balanced late sodium and hERG inhibition and lack of JT prolongation, despite pronounced QT prolongation 35 when carefully studied. Ranolazine was approved in Canada as late as January 2021, with a black box warning for QT prolongation and observations of TdP during postmarket surveillance. 36
CiPA has clearly added value in terms of highlighting the importance of standardized nonclinical assays. On the clinical side, the application of a well‐defined threshold (exclusion of a QTc effect >10 milliseconds) has served this purpose within a flexible framework. Several ECG methods and also innovations based on industry‐wide experience, such as C‐QTc analysis, have thereby been introduced without compromising patient safety. Both cited publications 23 , 28 focus on replacing the need for “QT‐centric” clinical ECG evaluation by nonclinical assays and defer, to a varying extent, the role of ECG evaluation to “checking for missed or unanticipated effects.” In our view, clinical ECG evaluation is only QT‐centric to the extent that it is powered to exclude a small increase of this interval. Since the QTc interval is the most variable parameter, such study is also able to detect small changes of other intervals, that is, HR and PR and QRS intervals.
Soon after the adoption of the E14 in 2005, there were cases of TQT studies in which only QT changes were statistically analyzed, but early on, regulators started to request an analysis of all ECG parameters, which today is standard practice in any type of study intended for ECG evaluation. Applying a well‐defined criterion (exclusion of a QTc effect >10 milliseconds) has thereby provided a framework that has enabled a careful characterization of a drug's electrophysiological effects, including effects on L‐type calcium and sodium ion channels. We agree that it makes sense to require a higher degree of standardization of nonclinical assays, which so far has been lacking, but it would dismiss 15 years’ experience if this comes at the price of replacing current strict criteria for clinical ECG evaluation by general statement such as “checking for missed or unanticipated effects.” After close to 8 years of CiPA activities, it would not be unreasonable to expect that such a proposal, coming from regulatory representatives in the IWG, should be better defined.
In our view, appropriately powered clinical ECG evaluation remains the cornerstone of cardiac safety assessment in drug development. Although centered on the QTc interval, this evaluation must include all quantitative ECG parameters. It has ensured that clinically relevant ECG effects are known and described when the drug enters the market.
TQT studies are sometimes conducted based on an informed choice by the sponsor, that is, a choice is made to spend resources on surviving projects rather than on all FIH studies. However, we often encounter study teams who have not realized that ECG effects have to be evaluated as part of the clinical safety package and therefore end up having to perform a dedicated study, in most cases a TQT study. Much of the burden of clinical ECG testing can be reduced by ECG evaluation in FIH trials and if a collect‐and‐store approach is taken, resources that represent a fraction of the cost of 1 single drug‐drug interaction study would be spent. If the program achieves proof of concept and the decision is then taken to analyze the stored ECGs, spent resources would still represent <1 single drug‐drug interaction study. It is certainly true that the cost of performing ECG evaluation in late‐stage clinical trials may be more costly and sometimes constrained by feasibility concerns from clinical sites, but with the right choices in early development, this would apply only to drugs for which concerning ECG effects have not been excluded. In our view, expanded ECG monitoring is then fully justified by the need to further characterize the ECG effects in the targeted patient population and inform prescribers how such effects can be handled.
To proceed with the ICH discussions without independent, critical assessment of whether sufficient data exist to allow revisions of the S7B/E14 document to enable further reductions of clinical ECG evaluation is likely to lead to a guidance document with very general, noncommitting statements. This may in turn result in discrepant interpretations across regulators and thereby contradict the objective of ICH guidance documents, to achieve harmonization in drug development across regions and, as a result, enable global development plans. As shown many times, this does not prevent individual regulators from proceeding with safety initiatives that they view as important, perhaps best illustrated by the FDA's draft guidance on blood pressure evaluation in clinical development 37 and the requirement to study antidiabetic drugs in cardiovascular outcome trials. 38
Summary and Concluding Comments
The 2015 revision of the ICH E14 document enabled ECG evaluation, including C‐QTc analysis applied to standard clinical pharmacology studies, for example, FIH studies (so‐called 5.1 studies), to replace a dedicated TQT study. For the purpose of excluding a clinically relevant effect on the QTc interval, the proportion of dedicated TQT studies has thereby decreased from 62% in 2016 to 34% in 2020, whereas the proportion of 5.1 has increased from 10% to 42%. The new proposed S7B/E14 Q&A document states that in case supratherapeutic concentrations of a drug cannot be obtained (eg, for reasons of safety, tolerability, or saturating absorption), a positive control may not be needed if a standardized hERG assay and in vivo study, as described in the revised S7B Q&As, are negative. Based on this proposed revision, a continued reduction of the proportion of TQT studies can to some extent be foreseen.
The S7B/E14 Q&A 6.1 addresses drugs for which the ECG evaluation cannot be undertaken in healthy subjects, at doses higher than the therapeutic or in placebo‐controlled studies; it therefore mainly applies to oncology drugs. The IWG seeks public comments on the change of the “threshold of concern” from excluding an effect on the QTc interval exceeding 10 milliseconds (ie, the upper bound of the 90%CI is below 10 millilseconds) to a mean effect of 10 milliseconds to allow that a drug is labeled as having “low likelihood of proarrhythmic effect.” The document does not describe what this may mean in terms of administration to high‐risk patients or with other drugs with an effect on the QTc interval, but it can be assumed that such a precautionary label statement will then not be applied. Based on public comments, it is clear that this new threshold is debated also among regulators.
ICH is driven by regulatory consensus, and guidance documents must balance the interest of different regions. To provide useful guidance that can be applied to global development programs and be accepted by all authorities at the time of market application, documents need to be specific and not written in a way that can result in widely different interpretations. In our view, the current proposed version of the S7B/E14 Q&A document provides useful guidance on how to standardize nonclinical assays, and we hope that the on‐going discussions in the IWG will result in further clarification of how to apply Q&A 5.1 and what 6.1 may mean in terms of instructions to prescribers and patients if the new threshold of concern is agreed upon.
Conflicts of Interest
B.D. is a consulting cardiologist and chief scientific officer, Cardiac Safety, for ERT, a central ECG laboratory. He owns stock and is eligible for stock options with ERT. He represented the European Federation of Pharmaceutical Industries and Associations on the ICH E14 Working Group from 2001 to the adoption of the document in 2005 and subsequently on the E14 IWG between 2005 and 2008. G.F. is statistical consultant. He owns stock from Novartis and was member of the Statistical Expert Team of PhARMA between 2002 and 2005.
Acknowledgment
The authors thank J. Rick Turner, PhD, DSc, DRT Strategies Inc, for valuable editorial assistance.
[The copyright line for this article was changed on 29 January 2022 after original online publication.]
References
- 1. ICH E14/S7B Implementation Working Group . Clinical and nonclinical evaluation of QT/QTc interval prolongation and proarrhythmic potential questions and answers draft version, August 2020. https://database.ich.org/sites/default/files/ICH_E14‐S7B_QAs_Step2_2020_0827_0.pdf. Accessed April 30, 2021.
- 2. ICH Harmonized Tripartite Guideline S7B . Safety pharmacology assessment of the potential for delayed ventricular repolarization (QT interval prolongation) by human pharmaceuticals. https://www.ich.org/page/safety-guidelines. Accessed April 30, 2021.
- 3. ICH E14 the clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non‐antiarrhythmic drugs. https://www.ich.org/page/efficacy-guidelines. Accessed April 30, 2021.
- 4. ICH webinar: new approaches for an integrated nonclinical‐clinical QT/proarrhythmic risk assessment. https://www.fda.gov/drugs/news-events-human-drugs/new-approaches-integrated-nonclinical-clinical-qtproarrhythmic-risk-assessment-10152020-10162020. Published October 2020. Accessed April 30, 2021.
- 5. Roden DM. Torsade de pointes. Clin Cardiol. 1993;16:683‐686. [DOI] [PubMed] [Google Scholar]
- 6. Roden DM. Taking the “idio” out of “idiosyncratic”: predicting torsades de pointes. Pacing Clin Electrophysiol. 1998;21:1029‐1034. [DOI] [PubMed] [Google Scholar]
- 7. Roden DM. Drug induced prolongation of the QT interval. N Eng J Med. 2004;350:1013‐1022. [DOI] [PubMed] [Google Scholar]
- 8. Turner JR, Rodriguez I, Mantovani E, et al. Drug‐induced proarrhythmia and torsade de pointes: a primer for students and practitioners of medicine and pharmacy. J Clin Pharmacol. 2018;58:997‐1012. [DOI] [PubMed] [Google Scholar]
- 9. Darpo B. Detection and reporting of drug‐induced proarrhythmias: room for improvement. Europace. 2007;9:iv23‐iv36. [DOI] [PubMed] [Google Scholar]
- 10. Hohnloser SH, Woosley RL. Sotalol. N Engl J Med. 1994;331:31‐38. [DOI] [PubMed] [Google Scholar]
- 11. Tan HH, Hsu LF, Kam RM, et al. A case series of sotalol‐induced torsade de pointes in patients with atrial fibrillation—a tale with a twist. Ann Acad Med Singapore. 2003;32:403‐407. [PubMed] [Google Scholar]
- 12. Zhang J, Machado SG. Statistical issues including design and sample size calculation in thorough QT/QTc studies. J Biopharm Stat. 2008;18:451‐467. [DOI] [PubMed] [Google Scholar]
- 13. ICH E14 Questions & Answers (R3) December 10, 2015. https://www.ich.org/page/efficacy-guidelines. Accessed April 30, 2021.
- 14. Darpo B, Garnett C. Early QT assessment—how can our confidence in the data be improved? Br J Clin Pharmacol. 2013;76:642‐648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Darpo B, Garnett C, Benson CT, et al. Cardiac safety research consortium: can the thorough QT/QTc study be replaced by early QT assessment in routine clinical pharmacology studies? Scientific update and a research proposal for a path forward. Am Heart J. 2014;168:262‐272. [DOI] [PubMed] [Google Scholar]
- 16. Garnett C, Beasley N, Bhattaram VA, et al. Concentration‐QT relationships play a key role in the evaluation of proarrhythmic risk during regulatory review. J Clin Pharmacol. 2008;48:13‐18. [DOI] [PubMed] [Google Scholar]
- 17. Darpo B, Benson C, Brown R, et al. Evaluation of the effect of 5 QT‐positive drugs on the JTpeak interval ‐ an analysis of ECGs from the IQ‐CSRC study. J Clin Pharmacol. 2020;60:125‐139. [DOI] [PubMed] [Google Scholar]
- 18. Darpo B, Benson C, Dota C, et al. Results from the IQ‐CSRC prospective study support replacement of the thorough QT study by QT assessment in the early clinical phase. Clin Pharmacol Ther. 2015;97:326‐335. [DOI] [PubMed] [Google Scholar]
- 19. Darpo B, Garnett C, Keirns J, et al. Implications of the IQ‐CSRC prospective study: time to revise ICH E14. Drug Saf. 2015;38:773‐780. [DOI] [PubMed] [Google Scholar]
- 20. Murphy PJ, Yasuda S, Nakai K, et al. Concentration‐response modeling of ECG data from early‐phase clinical studies as an alternative clinical and regulatory approach to assessing QT risk—experience from the development program of lemborexant. J Clin Pharmacol. 2017; 57: 96‐104. [DOI] [PubMed] [Google Scholar]
- 21. Mason JW, Bellibas SE, Huang NY, et al. Electrocardiographic effects of a supratherapeutic dose of oritavancin. Clin Pharmacol Drug Dev. 2016;5:502‐508. [DOI] [PubMed] [Google Scholar]
- 22. Darpo B, Sarapa N, Garnett C, et al. The IQ‐CSRC prospective clinical phase 1 study: “Can early QT assessment using exposure response analysis replace the thorough QT study?” Ann Noninvasive Electrocardiol. 2014;19:70‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Strauss DG, Wu WW, Li Z, et al. Translational models and tools to reduce clinical trials and improve regulatory decision making for QTc and proarrhythmia risk (ICH E14/S7B updates). Clin Pharmacol Ther. 2021;109:319‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ogasawara K, Xu C, Yin J, et al. Evaluation of the potential for QTc prolongation with repeated oral doses of fedratinib in patients with advanced solid tumors. Clin Pharmacol Drug Dev. 2021;10(4):366‐375. [DOI] [PubMed] [Google Scholar]
- 25. Garnett C. Revised E14 Q&As and presentation of examples to the impact of nonclinical data on clinical development and interpretation. https://www.fda.gov/drugs/news-events-human-drugs/new-approaches-integrated-nonclinical-clinical-qtproarrhythmic-risk-assessment-10152020-10162020. Published 2020. Accessed April 30, 2021.
- 26. ICH E14 Questions & Answers (R3) December 10: ICH S7B and E14 Q&A final concept paper. https://database.ich.org/sites/default/files/E14S7B_IWG_Concept_Paper.pdf. Published 2018. Accessed April 30, 2021.
- 27. Shinagawa K.Current status and future perspective of clinical proarrhythmic risk assessment from a regulatory point of view. https://www.diaglobal.org/course-listing/webinar/2021/01/8th-dia-cardiac-safety-workshop-in-japan. Published January 2021. Accessed April 30, 2021.
- 28. Vargas HM, Rolf MG, Wisialowski TA, et al. Time for a fully integrated nonclinical‐clinical risk assessment to streamline QT prolongation liability determinations: a pharma industry perspective. Clin Pharmacol Ther. 2021;109:310‐318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rodriguez I, Erdman A, Padhi D, et al. Electrocardiographic assessment for therapeutic proteins—scientific discussion. Am Heart J. 2010;160:627‐634. [DOI] [PubMed] [Google Scholar]
- 30. Vicente J, Hosseini M, Johannesen L, et al. Electrocardiographic biomarkers to confirm drug's electrophysiological effects used for proarrhythmic risk prediction under CiPA. J Electrocardiol. 2017;50:808‐813. [DOI] [PubMed] [Google Scholar]
- 31. Vicente J, Strauss DG, Upreti VV, et al. The potential role of the J‐Tpeak interval in proarrhythmic cardiac safety: current state of the science from the American college of clinical pharmacology and the cardiac safety research consortium. J Clin Pharmacol. 2019;59:909‐914. [DOI] [PubMed] [Google Scholar]
- 32. Johannesen L, Vicente J, Mason JW, et al. Late sodium current block for drug‐induced long QT syndrome: results from a prospective clinical trial. Clin Pharmacol Ther. 2016;99:214‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Darpo B, Zhou M, Bai SA, et al. Differentiating the effect of an opioid agonist on cardiac repolarization from micro‐receptor‐mediated, indirect effects on the QT interval: a randomized, 3‐way crossover study in healthy subjects. Clin Ther. 2015;38(2):315‐326 [DOI] [PubMed] [Google Scholar]
- 34. Mason JW. Cardiologist's point of view: novel ECG biomarkers and in silico models for proarrhythmic risk prediction: are we ready? J Electrocardiol. 2017;50:825‐827. [DOI] [PubMed] [Google Scholar]
- 35. Vicente J, Zusterzeel R, Johannesen L, et al. Assessment of multi‐ion channel block in a phase‐1 randomized study design: results of the cipa phase 1 ECG biomarker validation study. Clin Pharmacol Ther. 2018;105:943‐953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Health Canada Corzyna (ranolazine) product monograph. https://kyepharma.com/wp-content/uploads/2021/02/CORZYNA-PM-19JAN2021_EN_Clean.pdf. Published 2021. Accessed April 30, 2021.
- 37. Garnett C, Johannesen L, McDowell TY. Redefining blood pressure assessment—the role of the ambulatory blood pressure monitoring study for drug safety. Clin Pharmacol Ther. 2020;107:147‐153. [DOI] [PubMed] [Google Scholar]
- 38. US Food and Drug Administration .Guidance for industry: diabetes mellitus—evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. https://wayback.archive-it.org/7993/20191020084020/https://www.fda.gov/regulatory-information/search-fda-guidance-documents/diabetes-mellitus-evaluating-cardiovascular-risk-new-antidiabetic-therapies-treat-type-2-diabetes. Published December 2008. Accessed April 30, 2021.
- 39. Timmers M, Sinha V, Darpo B, et al. Evaluating potential QT effects of JNJ‐54861911, a BACE inhibitor in single‐ and multiple‐ascending dose studies, and a thorough QT trial with additional retrospective confirmation, using concentration‐QTc analysis. J Clin Pharmacol. 2018;58:952‐964. [DOI] [PubMed] [Google Scholar]