

Abstract
In the last 15 years, increasing evidence linking epigenetics to various aspects of cancer biology has prompted the investigation of histone post‐translational modifications (PTMs) and histone variants in the context of clinical samples. The studies performed so far demonstrated the potential of this type of investigations for the discovery of both potential epigenetic biomarkers for patient stratification and novel epigenetic mechanisms potentially targetable for cancer therapy. Although traditionally the analysis of histones in clinical samples was performed through antibody‐based methods, mass spectrometry (MS) has emerged as a more powerful tool for the unbiased, comprehensive, and quantitative investigation of histone PTMs and variants. MS has been extensively used for the analysis of epigenetic marks in cell lines and animal tissue and, thanks to recent technological advances, is now ready to be applied also to clinical samples. In this review, we will provide an overview on the quantitative MS‐based analysis of histones, their PTMs and their variants in cancer clinical samples, highlighting current achievements and future perspectives for this novel field of research. Among the different MS‐based approaches currently available for histone PTM profiling, we will focus on the ‘bottom‐up’ strategy, namely the analysis of short proteolytic peptides, as it has been already successfully employed for the analysis of clinical samples.
Keywords: biomarker, cancer, chromatin, epigenetics, histone post‐translational modification, histone variant, histone‐modifying enzyme, mass spectrometry, proteomics
In the last decade, epigenetics has emerged as a crucial player in various aspects of cancer biology, prompting the investigation of epigenetic features in the context of clinical samples. In this review, we provide an overview on the application of quantitative mass spectrometry‐based approaches for the analysis of histone post‐translational modifications and histone variants in cancer clinical samples, highlighting current achievements, challenges, and future perspectives.

Abbreviations
- DDA
data‐dependent acquisition
- DIA
data‐independent acquisition
- FFPE
formalin‐fixed paraffin‐embedded
- HAT
histone acetyltransferases
- HDAC
histone deacetylase
- KDM
lysine demethylase
- KMT
lysine methyltransferase
- LMD
laser microdissection
- MALDI
matrix‐assisted laser desorption ionization
- MMD
manual macrodissection
- MS
mass spectrometry
- OCT
optimal cutting temperature
- PTM
post‐translational modification
- SILAC
stable isotope labeling by amino acids in cell culture
Introduction
In the nucleus of eukaryotes, the genomic DNA is packed into chromatin, a nucleoprotein structure that regulates many fundamental nuclear processes, including DNA replication, DNA repair, and transcription. The majority of the protein content of chromatin is constituted by histones, which comprise core and linker histones. Core histones (H2A, H2B, H3, and H4) build the histone octamer, around which approximately 146 bp of DNA wrap, forming the basic unit of chromatin, the nucleosome. Linker histone H1 binds the nucleosome and the free DNA in between nucleosomes, contributing to the formation of higher‐order chromatin structures [1]. The assembly of nucleosomes occurs mainly during DNA replication and is essential for chromatin replication and epigenetic inheritance. Chromatin remodelers contribute to nucleosome assembly by regulating nucleosome assembly/eviction and organization, chromatin access, and nucleosome positioning [2], while histone chaperones ensure the transport of newly synthetized histones from the cytoplasm to specific sites in the genome [3].
Crucial for the function of core histones are a number of reversible post‐translational modifications (PTMs) that are deposited mainly at their N‐terminal tails. Histone PTMs comprise methylation, acylation (including acetylation, propionylation, and butyrylation), phosphorylation, SUMOylation, ubiquitylation, ADP‐ribosylation, deamination, and crotonylation [4, 5, 6]. The type, location, and combination of histone modifications have been proposed to generate a ‘code’—known as the histone code—which determines the functional state of the underlying DNA [7]. Such functional regulation is achieved either by influencing chromatin accessibility or by generating binding sites for the recruitment of enzymes and protein complexes that mediate downstream events [8, 9]. Histone PTMs are deposited and removed by histone‐modifying enzymes known as ‘writers’ and ‘erasers’, respectively, and are recognized by effector proteins called ‘readers’, which are characterized by specialized domains that bind to modified residues [such as Tudor domain, chromodomain, PWWP domain, plant homeodomain, WD40, and bromodomain (BRD)] [10]. Methylation and acetylation on lysine residues are the most common and best‐studied histone PTMs. Acetylation of histone tails is associated with transcriptional activation, while methylation, depending on the location, residue, and the number of methyl groups involved, can cause transcriptional activation or repression [11]. To add another level of complexity, all histones exist in several variants, which can be locally enriched at distinct chromatin regions and contribute to the fine‐tuning of chromatin‐related processes [12].
In the last decade, the increasing evidence that epigenetics plays a crucial role in various aspects of cancer biology has prompted the investigation of epigenetic features in the context of clinical samples [13]. Differences detected in the levels of histone PTMs and histone variants in different types of tissues or patient cohorts can not only represent potential epigenetic biomarkers for patient stratification, but also suggest epigenetic mechanisms associated with cancer, and indicate potential novel epigenetic pathways targetable for cancer therapy (Fig. 1). The investigation of histones in the context of patient‐derived samples has been traditionally performed using antibody‐based methods, such as immunoblots and immunohistochemistry (IHC). However, mass spectrometry (MS) has emerged as a more suitable tool for the unbiased, comprehensive, and quantitative investigation of histone PTMs and histone variants. Although MS has been applied so far to the analysis of histone PTMs mostly in the context of cell lines, thanks to recent technological advances, it is now ready for the application to clinical samples.
Fig. 1.
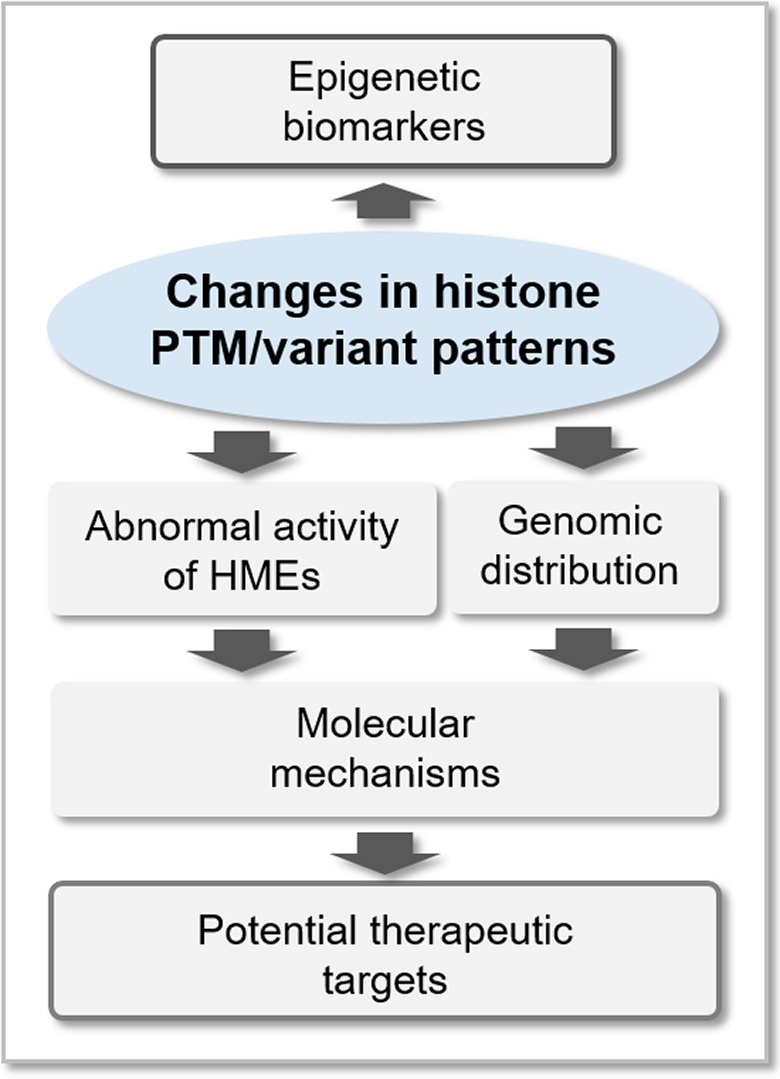
Potential contribution of histone analysis in cancer research. Histone PTM and variant profiling in clinical samples can generate both novel epigenetic biomarkers for patient stratification and epigenetic mechanisms targetable for cancer therapy. HME, histone‐modifying enzyme.
In this review, we will provide a state‐of‐the‐art overview on the quantitative MS‐based analysis of histones, their PTMs, and their variants in clinical samples. Our goal is providing to an audience of nonmass spectrometry experts the basic tools to understand how MS can be employed to investigate epigenetic features in cancer clinical specimens. Therefore, while giving a general overview on all the aspects concerning histone PTM and variant analysis by MS, we will focus in detail on those strategies that have already been applied to patient‐derived tissues, referring to other reviews for more general technical aspects (for instance, [14] provides a very comprehensive and detailed review on histone PTM analysis by MS). In particular, among the different MS‐based approaches currently available, we will focus on the ‘bottom‐up’ strategy, namely the analysis of short (5–20 amino acid long) proteolytic peptides, as it has been already successfully applied to the analysis of clinical samples. The review will articulate in three main parts: (a) a summary of what is currently known regarding the role of histone PTMs and histone variants in cancer, (b) the basics of MS‐based histone PTMs and variant analysis, and (c) the contribution of MS‐based analysis of histones in cancer research.
Histones and cancer
Histone post‐translational modifications as cancer biomarkers
In recent years, the functional implications of histone modifications in cancer have been the object of intensive investigations. Histone PTMs have been studied in tissue biopsies for their potential role as biomarkers for cancer development, progression, histological grading, and response to therapy (Fig. 2). A decrease in histone H4 acetylated at lysine 16 (H4K16ac) and trimethylated at lysine 20 (H4K20me3) occurs early during tumorigenesis and was observed across various cancer cell lines and tissue types, thus representing a general epigenetic hallmark of cancer [15, 16]. Loss of acetylation was correlated with reduced recruitment at repeated sequences of the acetyltransferases MOZ, MOF, and MORF [15, 17]. Correlation between H4K20me3 and its methyltransferases is more controversial: loss of H4K20me3 was correlated with lower levels of the enzymes SUV420H1 and/or SUV420H2 in different studies [15, 16], but no association between SUV420H2 expression and H4K20me3 was identified in other studies where both cell lines and clinical samples were analyzed [18, 19]. Loss of H4K16ac and H4K20me3 occurs at DNA repetitive regions that are hypomethylated in cancer cells. Because changes in histone PTMs influence DNA methylation and vice versa [20, 21, 22], one hypothesis is that DNA hypomethylation could drive the observed changes in histone PTMs, but no conclusive evidence on the mechanisms through which these changes occur exists
Fig. 2.
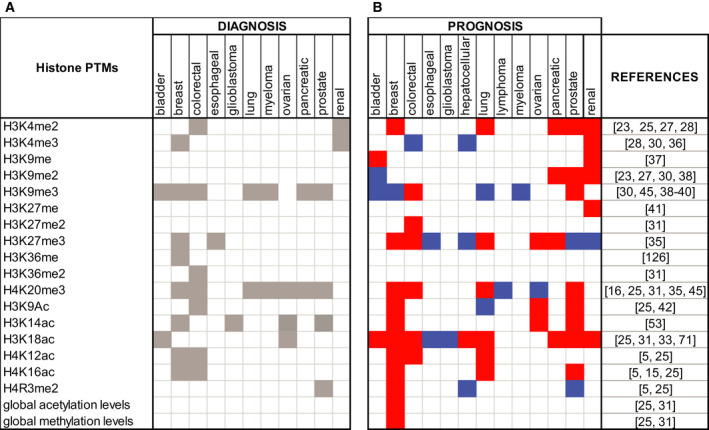
List of histone PTM cancer biomarkers. Histone PTMs as cancer biomarkers for diagnosis (including differences between tumor and normal tissues, among tumor subtypes, and tumors with different stages or grades) (A) and prognosis (B). The red color in (B) indicates a positive correlation between the level of the histone PTM and the prognosis, while the blue color indicates a negative correlation.
Following the landmark discovery of histone PTMs as hallmarks of cancer, other histone PTMs have been linked with tumor onset and development. Different studies showed that altered global levels of acetylation and methylation of histones H3 and H4 correlate with tumor progression and clinical outcome [6]. In general, hypoacetylation was identified as a marker of cancer progression and was found to be a potential prognostic factor in many types of solid tumors [23]. In particular, low levels of H4K12ac correlate with cancer progression in lung cancer [16] and increasing tumor grade in colon [24] and breast cancer [25]. On the contrary, an increase of H3K4ac levels in breast cancer cell lines is associated with cancer metastasis and is considered a predictor of an aggressive and metastatic phenotype [26].
Moderate to low levels, compared with healthy tissues, of H3K4me2 were reported in prostate, kidney, breast, pancreas, liver, and lung cancers with a correlation with worse patient outcome and poor prognosis [23, 25, 27, 28]. A decrease in H3K9me3 was observed in lung and prostate cancer [29, 30], while a global increase in the same PTM correlates with poor prognosis and tumor recurrence in other tumor types [31]. Several other examples of histone PTM changes having opposite effects depending on the tumor type exist. For instance, loss of H3K18ac is associated with a better prognosis in glioblastoma [32], but seems to correlate with poor survival and cancer recurrence in many solid tumors, including prostate, kidney, lung, breast, colon, and pancreatic cancer [23, 25, 33]. Reduced levels of H4R3me2 correlate with poor survival and tumor size in breast cancer and tumor grade in prostate cancer, while higher levels correlate with poor survival in hepatocellular carcinoma [25, 34]. Furthermore, a decrease in H3K27me3 was associated with breast, ovarian, pancreatic, colon, kidney, and lung cancer, while an increase in the same mark correlates with tumor progression in prostate, liver, and esophageal cancer [31, 35]. Additional potential histone PTM biomarkers for cancer diagnosis or prognosis have been identified and are shown in Figure 2 [36, 37, 38, 39, 40, 41, 42]. In addition to the best characterized PTMs, less‐studied histone modifications are also being evaluated for their role as tumor suppressors and oncogenes. For instance, phosphorylation, ADP‐ribosylation, and ubiquitination have been studied for their role in regulation of tumorigenesis, as they are involved in DNA damage detection, carcinogenesis, and modulation of transcription and chromatin remodeling [43].
Although histone levels and their associated PTMs in tumor tissues hold great promise as cancer biomarkers, the requirement for invasive biopsies of an established tumor for their analysis reduces their applicability, especially to early‐stage tumors or tumors in sites that are difficult to reach (e.g., brain). It has been shown that nucleosomes can be released into the blood from cancer tissue, as a consequence of cell death and apoptosis [44], and have been proposed as non‐invasive biomarkers for different types of cancer. Generally, circulating nucleosomes are present in higher amounts in sera from cancer patients compared with healthy samples [6, 45], due to higher cellular turnover, and are increased by cell death induced by chemotherapy. However, the amount of circulating nucleosomes on its own does not allow discriminating tumor patients from subject with benign inflammatory diseases [45]. A more promising avenue is represented by the quantification of histone PTMs and combinations thereof, as increasing evidence suggests that circulating nucleosomes may be representative of the nucleosomes found inside tumor cells in terms of DNA methylation [46], nucleosome fingerprinting [47], and also combinatorial histone marks [48]. For instance, lower levels of the tumor hallmarks H4K20me3 and H4K16ac were found in circulating nucleosomes isolated from tumor patients compared with normal subjects, together with differences in additional histone PTMs [48]. The potential held by circulating histone PTMs is confirmed by two studies where two panels of epigenetic modifications were identified as diagnostic biomarkers in colorectal and pancreatic cancers [49, 50].
Chromatin modifiers and cancer
Changes in the levels of histone PTMs are often the consequence of the aberrant expression, mutation, or mislocalization of histone‐modifying enzymes. The best‐studied histone‐modifying enzymes are those responsible for transferring or removing acetyl and methyl groups, which include acetyltransferases (HATs) and deacetylases (HDACs), and methyltransferases (KMTs) and demethylases (KDMs). Remarkably, chromatin modifiers have been reported to be one of the most heavily mutated protein classes in cancer [51]. Accordingly, a mutational analysis of the samples present in The Cancer Genome Atlas (TCGA) Program pan‐cancer Atlas study [52] showed that a remarkable portion of tumors (from 20% to 90%, depending on the tumor type) presents at least one mutation in one of the histone‐modifying enzymes involved in the deposition and removal of methylations or acetylations [53].
While HATs and HDACs show low substrate specificity and are able to acetylate and deacetylate multiple histone sites, KMTs and KDMs typically act in a more specific manner, on one or few histone residues [54, 55]. Overexpression, mutations, and amplification of HAT genes have been identified in many cancers, especially those of epithelial and hematological origin. HATs were reported as either tumor suppressor genes or oncogenes, depending on the specific type of cancer and the type of mutation [56, 57]. For example, p300/CBP are generally considered as tumor suppressors [58], but are associated with poor prognosis in lung cancer [59]. Aberrant expression of HDACs and/or mutation in the corresponding genes were also identified in many cancers [60]. Generally, HDACs are upregulated in cancer and their expression correlates with poor prognosis [61].
Lysine methyltransferases and KDMs have been also extensively evaluated for their role in cancer. In general, demethylases seem to be upregulated, while methyltransferase levels may change in a more variegate manner, depending on tumor type and grade, when compared to normal tissue [53]. In accordance with the increase in H3K9 methylation observed in many solid tumors, overexpression of the methyltransferases SUV39H1, SUV39H2, and SETDB1 was observed in several cancers and reported to correlate with poor prognosis and shorter survival time [62, 63, 64, 65, 66]. Also in accordance with H3K9 methylation increase, the H3K9 demethylase PHF2 was found to be downregulated in breast cancer [67]. One of the most studied histone‐modifying enzymes is EZH2, which mediates the deposition of H3K27me3. EZH2 is overexpressed in all the most common cancers [68], and high EZH2 expression is a prognostic indicator of poor survival and correlates with metastasis and tumor progression [69]. In prostate and esophageal cancers, increased EZH2 levels correlate with an increase in H3K27 methylation [70, 71]. However, in most of the cases no association has been observed between EZH2 and H3K27me3 [35], highlighting how other factors may concur to the level of histone PTMs, in addition to the expression levels of histone‐modifying enzymes. These include enzyme mutation and turnover rates, the altered function of the multi‐subunit complexes to which enzymes belong to, and differences in proliferation rates [53]. Many other methyltransferases acting on other important histone PTMs, such as H3K4, H3K36, H3K79, and H4K20, have also been linked with cancer prognosis or disease mechanisms (reviewed in [72]).
Given their involvement not only in cancer but also in several other diseases, histone‐modifying enzymes are considered as promising novel therapeutic targets, and intensive research has been devoted to the design, test, and development of novel drugs targeting them. There are currently more than 20 HDAC inhibitors in different phases of clinical trials, some of which have already been approved by the food and drug administration for the treatment of hematological malignancies [73]. Drugs targeting HATs were also investigated, but the majority of the inhibitors developed so far show modest potency and low selectivity [56, 74]. However, some highly selective and potent inhibitors were developed to target p300/CBP [75]. In the last decade, there has been also a remarkable progress in the development of inhibitors of methyltransferases and demethylases, as they show more specificity for their substrates [76, 77]. The first methyltransferase inhibitor, chaetocin, was discovered in 2005 [78], and in the following years, many other inhibitors were identified. While most of them are still in preclinical development, several inhibitors of EZH2 are now under investigation in clinical trials [79]. Among demethylase inhibitors, some of the most studied are the inhibitors of LSD1, for which various Phase I and II clinical trials are ongoing [80].
Chromatin remodelers, histone chaperones, and histone readers alterations have also been widely investigated in cancer. One of the most studied chromatin remodelers is SWI/SNF, which has been shown to be mutated in almost 20% of all tumor types [81, 82]. Among histone chaperones, HJURP is upregulated in cancer and described as a predictor of poor prognosis in prostate [83] and breast cancer [84]. Concerning histone readers, the most remarkable example of disregulation in cancer is represented by the BRD containing readers proteins of the BET family, which regulate gene transcription by binding to acetylated lysine residues on histones, causing the aberrant expression of genes involved in carcinogenesis. Mutation, misregulation, and oncogenic fusions of BRD‐containing proteins were investigated in different studies as reviewed in [85]. BET proteins promote MYC overexpression in both hematological and solid tumors [86], and several BET inhibitors are now in preclinical and clinical trials [87].
Histone variants in cancer and oncohistones
The so‐called ‘canonical’ histones are expressed during the S phase of the cell cycle and deposited on chromatin in a replication‐dependent manner. The histone pool also includes histone variants, which are encoded by a different set of genes, can be slightly or very different from the canonical counterparts, and are expressed throughout the cell cycle [12, 88]. Histone H3 has three major variants (H3.1, H3.2, and H3.3), a testis‐specific variant (H3T), and a variant that is mainly localized to centromeric chromatin (CENP‐A). Histones H2A and H2B have 20 and 17 variants, respectively, while H4, which was believed to exist as a single histone sequence, has been recently shown to have one variant (H4G) [89]. Histone variants can differ from their canonical counterpart in their expression patterns and PTMs, and be enriched at specific genomic regions, mediating different chromatin‐related processes. Aberrations in the deposition of histone variants have been linked to various diseases, including cancer (reviewed in [90]). Overexpression of H2A.Z is frequently observed in cancers and correlates with poor survival [12, 88]. In malignant melanoma, H2A.Z.2 is implicated in cell proliferation and drug sensitivity, suggesting inhibition of H2A.Z deposition as a potential therapeutic strategy [91]. Another variant of H2A, macroH2A, is generally considered a tumor suppressor in a wide range of cancers, although the effect of its deregulation might have opposite effects in specific contexts [88]. Consistent with its role in DNA damage repair, H2A.X also acts mainly as a tumor suppressor [88]. Aberrations in its coding gene have been observed in many types of tumors and are linked with oncogenic translocations and genomic instability [92, 93]. Furthermore, γ‐H2A.X, which has been extensively used as a specific and sensitive molecular marker of DNA damage, was described as a prognostic and predictive factor in cancer [88]. Because DNA double‐strand breaks may lead to cancer, but are also increased by cytotoxic cancer treatment, using γH2A.X detection to determine the extent of DNA damage could be useful to monitor the effectiveness of cancer therapies, as well as to detect precancerous cells and determine cancer stage, based on the levels of DNA damage [94]. γ‐H2A.X can also be detected in liquid biopsies [95], which makes it a particularly attractive biomarker.
Histone H3 variant H3.3 is continuously expressed independently of cell cycle, and its presence at promoters is associated with active transcription. In pediatric brain tumors, several studies identified driver mutations in histone H3, the majority of which were found exclusively or at higher frequency in the H3.3 variant compared with canonical H3.1/2 [96] (also see the last paragraph of this section). CENP‐A is one of the most studied H3 variants, it localizes at the centromere in all eukaryotes and plays an essential role in cell division [12]. Increased expression of CENP‐A was associated with cancer development and progression and was proposed as a prognostic and predictive cancer biomarker in several cancers, with correlation with poor prognosis [97]. Finally, histone H4 variant H4G appears to be preferentially expressed in breast cancer (both cell lines and human tissues) compared with normal cells, and its levels correlate with breast cancer stage progression [89].
Linker histone H1 also exists in 11 variants in human and mouse. Of the seven somatic variants, histones H1.1–H1.5 are replication‐dependent, while histones H1.0 and H1x are transcribed throughout the cell cycle [98]. By binding differently to the nucleosome, H1 variants can generate distinct higher‐order chromatin structures and contribute to the regulation of nuclear functions (reviewed in [99]). Alterations in the global levels of histone H1 and the levels of specific variants have been observed in cancer (reviewed in [100]). Normally, the levels of replication‐dependent variants are increased in tumor compared with normal tissues, because of increased cell proliferation. Instead, H1.0 is expressed in a heterogeneous manner in cancers and is overall reduced, particularly in undifferentiated and aggressive tumors [100]. These results suggest that H1 levels, as well as the levels of other histone variants, could be exploited as biomarkers to distinguish between normal and tumor tissue, benign and malignant lesions, and patient with different prognosis.
In addition to altered histone levels, recurrent mutations in both core and linker histones have been found in different types of cancer (reviewed in [96, 100]). Several mutations have been reported in histone H3 residues that are substrates or are close to substrates of histone‐modifying enzymes, such as H3K27 and H3K36. Because mutant histones contain missense oncogenic mutations, they are usually termed as ‘oncohistones’. The first oncohistones were reported in pediatric high‐grade gliomas and contain the H3.1/H3.3K27M and H3.3 G34R/V mutations [101, 102, 103, 104, 105]. The H3.3K27M mutation was later identified also in posterior fossa ependymoma [106], and additional histone mutations include H3.3G34W/L and H3.3 K36M in pediatric bone tumors, and H3.1/H3.3K36M in head and neck cancer [107, 108]. The K27 and K36 mutations impair the binding of methyltransferases and display a dominant‐negative effect, causing a general decrease of methylation levels, despite the fact that mutant histones typically represent a minor fraction of total histone H3 [109, 110]. Epigenetic therapies aimed at reverting the changes in chromatin caused by oncohistones are currently being investigated.
MS‐based analysis of histone PTMs and variants
Mass spectrometry is an analytical technique that allows determining with high accuracy the mass (or more precisely the mass/charge ratio, m/z) of different types of molecules, including peptides and proteins. In recent years, MS has become the method of choice for the identification and quantitation of histone and their PTMs. Prior to the advent of the MS analysis of protein and polypeptides, histone PTMs were traditionally analyzed through antibody‐based methods, such as immunoblotting, immunofluorescence, ELISA, and IHC, the latter having particular relevance for the analysis of clinical samples. All these methods shared several limitations, including the ability to measure only one or few known modifications at the time, the need for reliable and specific antibodies, poor signal linearity, cross‐reactivity and epitope masking (poor efficiency to detect a modification when another one is present nearby). PTMs are identified by MS and localized to specific residues by detecting a ‘delta‐mass’ between the theoretical and experimentally measured masses of peptide/proteins. As a consequence, theoretically any PTM or combination of PTMs can be profiled in a single run, without requiring a priori knowledge of the type or site of the modification, and in a highly quantitative manner. In addition, MS provides a unique tool to detect mutations in histone sequences, such as oncohistones, as well as to quantify histone variants, which can only differ in a few amino acids and may be difficult to distinguish using traditional methods.
Three main MS‐based approaches can be used to study histone PTMs in biological samples (reviewed in detail in [14] and schematized in Fig. 3A). In ‘top‐down’ approaches, intact histones are chromatographically separated and directly ionized and MS‐analyzed, providing information on the complete panel of histone isoforms present in a sample and their overall stoichiometry. In ‘middle‐down’ approaches, long histone peptides (> 5 kDa) are obtained through digestion with endoproteases that cleave at residues occurring with low frequency, such as Glu‐C and Asp‐N. Through this approach, intact N‐terminal tails can be obtained. For example, Asp‐N generates the H4 1–24 peptide, while Glu‐C generate the H3 1–50 peptide, which contain most of the known PTM sites in histones H4 and H3, respectively. Although top‐ and middle‐down MS approaches are ideal to detect long‐range PTM associations and extrapolate their stoichiometry, one of the major challenges related to these two approaches is the difficulty in distinguishing isobaric species (namely peptides carrying the same modifications, but on different positions). Other issues involve the lower sensitivity and computationally demanding data analysis. As a consequence, top‐ and middle‐down MS approaches are usually carried out in few, specialized laboratories, and no applications to clinical samples have been reported so far.
Fig. 3.
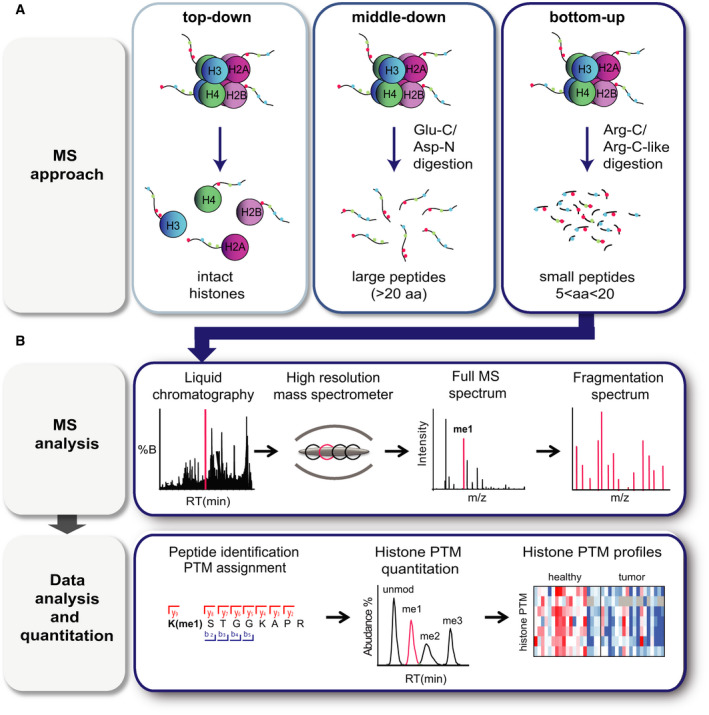
MS‐based analysis of histone PTMs. (A) Scheme summarizing the three MS‐based approaches that can be used for histone PTM analysis. In ‘top‐down’ approaches, intact histones are chromatographically separated and directly analyzed by MS; in ‘middle‐down’ approaches, long histone peptides (> 5 kDa) are obtained through digestion with Glu‐C or Asp‐N; in ‘bottom‐up’ approaches, histones are enzymatically digested into shorter (5–20 amino acid long) peptides, for instance using the Arg‐C protease. Panel B shows a general bottom‐up MS workflow, which involves liquid chromatography separation, acquisition of full MS, and fragmentation spectra in the mass spectrometer, followed by identification and quantification of the modified peptides.
In ‘bottom‐up’ MS approaches, which are by far the most employed for PTM analysis, histones are enzymatically digested into relatively short (5–20 amino acid long) peptides prior to MS analysis. Trypsin, which is the most commonly used protease for global proteomics studies and cuts at the carboxylic terminus of arginine and lysines, can be used to analyze histone H1 variants, but is not ideal for core histones. Due to the high number of basic amino acids present in core histone sequences, trypsin generates peptides that are often too short for MS analysis. In addition, trypsin does not cut efficiently next to modified residues, thus generating peptides of inconsistent length that cannot be quantified accurately (Fig. 4). To solve these issues and generate histone peptides of proper length, two main strategies can be employed: a digestion with the Arg‐C protease, which cleaves at the C‐terminal of arginine residues, or an ‘Arg‐C like’ digestion (Fig. 4). The latter is achieved by using the trypsin protease following chemical acylation of lysines with agents that impair trypsin digestion, such as deuterated acetic anhydride [111, 112], or, more commonly, propionic anhydride [113]. This approach is also useful to discriminate isobaric peptides [112], and—when combined with a second round of derivatization of the digested peptides with propionic anhydride or phenyl isocyanate (PIC)—significantly improves the chromatographic retention and detectability of short and hydrophilic peptides ([114] and unpublished results). Bottom‐up approaches provide limited information about co‐occurring modifications (up to four), especially in the case of distant marks, but represent a more flexible tool that has also been applied for the analysis of patient‐derived samples. Figure 3B shows a typical bottom‐up workflow for histone PTM analysis, which include chromatographic separation, MS acquisition, and data analysis and quantitation.
Fig. 4.
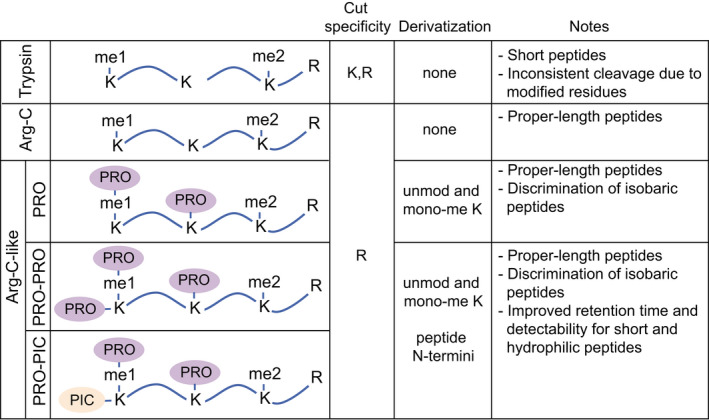
Digestion approaches for MS bottom‐up histone PTM analysis. Schematic representation and main features of the most common bottom‐up digestion methods for the MS‐based analysis of histone PTMs. Different ‘Arg‐C‐like’ digestions are obtained by derivatizing lysines with agents that impair protease cutting, in combination with a trypsin digestion. PRO: propionic anhydride (deuterated acetic anhydride can be employed as an alternative to propionic anhydride to acylate lysines).
Below, we will provide a general overview of the key aspects of the analysis of histones and their PTMs by MS, and discuss the different options available for the analysis of clinical samples, focusing mostly on bottom‐up approaches.
Types of clinical samples and their preparation for MS‐based histone PTM and variant analysis
Histones are particularly small and hydrophilic proteins, and unlike the majority of the proteins, they are soluble in strong acids, such as HCl or H2SO4 [115]. Therefore, most standard protocols to isolate histones from cells involve the purification of nuclei followed by acidic extraction. While these protocols generate highly pure histones, there is a considerable loss of material associated with each purification step, which may represent a problem when dealing with a low number of cells. This is often the case of patient‐derived primary cells, which cannot be expanded indefinitely and are available in limited amounts. For these cells, simplified protocols tailored to different ranges of starting cell amounts have been developed [116] (Fig. 5). In addition, during the last years, approaches suitable for the analysis of histone PTMs—which can also be used for the analysis of histone variants—from the most common sources of patient‐derived specimens have been implemented by our group and others [116, 117, 118, 119, 120, 121, 122] (Fig. 4). Typically, these methods must be followed by SDS/PAGE or other strategies (e.g., StageTip microcolumn enrichment [123]) to remove detergents and other MS contaminants, and to separate histones from other proteins present in the gel.
Fig. 5.
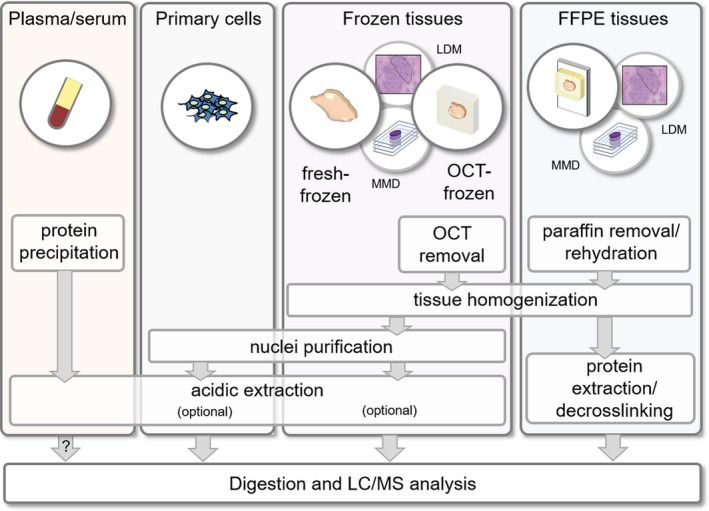
Overview of protocols for the isolation and enrichment of histones from patient‐derived samples prior to MS analysis. The protocols for FFPE and frozen tissues can also be applied to manually macrodissected (MMD) or LMD samples. The question mark indicates that more studies need to be performed to fully evaluate the feasibility of analyzing the circulating nucleosomes present in plasma/serum.
Patient‐derived samples are stored in hospital biobanks as either formalin‐fixed paraffin‐embedded (FFPE), fresh‐frozen, or optimal cutting temperature (OCT)‐frozen tissues. Fresh‐freezing is considered the ideal storage method for proteomics studies, since it avoids potential artifacts and do not contain MS contaminants. However, sections are difficult to obtain from this type of samples and often do not completely preserve the morphology of the original tissue. On the contrary, OCT generates a matrix around the samples that allows the morphological preservation of the tissue during section cutting. However, OCT is a strong contaminant for MS analysis, which must be carefully eliminated during sample preparation prior to LC‐MS [116, 118, 122, 124]. Histones can be enriched from frozen tissues though protocols similar to those used for cells, after tissue homogenization and removal of OCT, if present [116, 118, 119, 122] (Fig. 5). Purification steps, such as acidic extraction, can be avoided to maximize the yield of histones, when dealing with low amounts of starting material.
Despite providing several advantages, the collection of frozen samples is not routine in hospitals and their availability in biobanks is limited. A much more common preservation technique to archive patient specimens is formalin fixation followed by embedding in paraffin. FFPE samples represent a precious source of patient samples, in particular for retrospective studies, and are stable at room temperature, but their analysis requires reverting the extensive protein crosslinking generated by formaldehyde fixation, which is usually performed through heat‐induced antigen retrieval techniques similar to those used in IHC [125]. All the reported protocols for extraction of histones from FFPE tissues share few main steps: (a) paraffin removal; (b) tissue rehydration and homogenization; and (c) protein extraction and decrosslinking, which is achieved through incubation at high temperature in the presence of high concentrations of strong detergents [117, 120, 121] (Fig. 5). The comparison of matched FFPE and frozen breast cancer tissues through the PAThology Tissue analysis of Histones by MS (PAT‐H‐MS) protocol that we developed showed very similar histone PTM patterns for the two storage methods, allowing the definition of a list of 52 differentially modified histone H3 and H4 peptides that can be reliably identified and quantified in FFPE samples up to 7 years old [120, 123]. It must be noted that a few methylations and formylations were significantly and systematically increased in FFPE tissues (the most relevant changes were on H3K18me1 and K79me1/me2), suggesting that some artifacts due to FFPE storage are not eliminated by the decrosslinking step of the protocol, in accordance with previous reports [117].
A major limitation of both FFPE and frozen tissues is related to tissue heterogeneity, which may come from the presence of different cell types (e.g., normal or immune cells) or from differences among different areas within the same tumor. This problem can be overcome by selecting gross tissue areas or specific areas/cell populations by manual macrodissection or laser microdissection (LMD), respectively. After proving that MS‐based histone PTM analysis can be coupled with manual macrodissection and LMD, and showing that all the most common histone PTMs can be quantified from approximately 450 000 cells [126], more recently we have optimized the sample preparation step in order to scale down the starting amount needed. Our results show that at least 33 differentially modified histone peptides can be quantified from microdissected tissue areas corresponding to as low as 1000 cells (unpublished results).
Finally, histones can be isolated from blood, which represents a particularly attractive source of non‐invasive molecular biomarkers. As an alternative to ELISA methods, a dual acid extraction protocol for isolation of circulating histones from serum of patients with solid tumors was proposed [48]. Western blot analysis of few selected histone PTMs showed a comparable pattern in paired sera and cancer tissues, suggesting that analyzing circulating nucleosomes may be a viable option to study epigenetic changes in cancer patients. Although the presence of histones in the preparation was confirmed by MS, no systematic analysis of PTMs by MS was performed. Such an analysis would be important not only to expand the number of modifications that can be profiled in serum, but also to better understand to which extent histone PTMs in circulating nucleosomes reflect those found in tumors.
Instrument set‐ups for MS profiling of histones
The majority of the histone PTM studies conducted so far have been performed using a liquid chromatography (LC)‐MS setup, where a high‐resolution mass spectrometer is combined with reversed‐phase high‐performance LC (HPLC, as shown in Fig. 3B). The chromatographic step allows the separation of complex peptide mixtures obtained by digestion of histones, prior to MS analysis, and is particularly useful for the identification and quantitation of isobaric peptides. As an alternative, a recent study proposed the use of direct injection MS, which allows analyzing 200 histone PTMs with a 1 min of MS analysis, as part of a workflow that theoretically allows analyzing more than 1000 of samples per day [127]. Although this method would solve the robustness issues often linked with nano‐LC systems, and provide a throughput that would be extremely useful in the context of clinical studies, the applicability of this workflow to patient‐derived samples, where histones are often not present at high purity, has still to be verified.
A completely different set‐up of particular interest in the context of clinical samples is matrix‐assisted laser desorption ionization (MALDI) imaging [128]. This approach involves the analysis of tissue sections, allowing the definition of the in situ spatial distribution of m/z values for hundreds of molecules. The identification assignment of such m/z values typically requires LC‐MS to be used in parallel. Although MALDI imaging has been used in various studies to profile either intact or digested proteins [128], only few examples of histone analysis have been reported (reviewed in [129]). Most of them were not specifically tailored to histone PTMs, but were rather global proteomics profiling experiments aimed at identifying possible cancer biomarkers, among which histones were identified. For instance, histone H4K16ac and K20me2 were identified as potential biomarkers for microvascular invasion, a major risk factor for tumor recurrence and postoperative mortality in hepatocellular carcinoma [130]. Another study reported the development of a workflow based on intact protein detection, which allows characterizing the in situ distribution of histone PTMs and histone H1 variants in the mouse brain [131]. MALDI imaging was also used to follow histone acetylation changes during treatment with HDAC inhibitors in gastrointestinal cancer cells [132]. The spatial molecular profiles generated by MALDI imaging can be used to guide the selection of areas by LMD, representing an alternative to the evaluation of morphological features usually carried out by a pathologist. This type of approach has been employed to select heterogeneous breast cancers regions to be analyzed by microproteomics [133], where one tissue section was subjected to MALDI imaging analysis, and adjacent sections were used for LMD followed by LC/MS analysis. A similar approach could be imagined for histone PTM and variant analysis.
MS data acquisition
For MS analysis, the peptides obtained from digesting histones are ionized and separated based on their m/z, obtaining a ‘full MS spectrum’ (or MS1 spectrum, Fig. 3B, top panel). Ions with a specific m/z are then selected and fragmented, generating smaller fragments that are detected in a ‘fragmentation spectrum’ (or MS2 spectrum, Fig. 3B, top panel). The experimentally determined spectra are then searched against the theoretical spectra present in a database, to determine the mass and the sequence of the peptides, and the presence and location of PTMs (Fig. 3B, bottom panel). Most of the currently used data acquisition routines for histone PTMs are based on data‐dependent acquisition (DDA) methods, where the ion precursors with the highest relative abundance in the full MS scan are selected for subsequent fragmentation. While being a very successful strategy to analyze well‐known and abundant modifications, this type of analysis imposes an intensity bias that may limit the detection of less abundant PTMs in the absence of specific enrichment/fractionation steps preceding the MS analysis. To overcome this issue, targeted or data‐independent acquisition (DIA) approaches (reviewed in [134]) can represent a useful alternative to DDA. Targeted MS methods, such as single‐, multiple‐, and parallel‐reaction monitoring (SRM, MRM, and PRM, respectively) and single ion monitoring (SIM), allow the selection of specific ions to be analyzed with higher sensitivity and throughput, but require an a priori knowledge of the peptide of interest, and cannot be employed in a discovery experiment. Although targeted methods cannot be used to discover previously unknown modifications, they allow analyzing many differentially modified peptides in the same run. For instance, targeted methods were applied to follow up 20 modification sites in cells and human brain [131]. Differently from DDA and targeted methods, DIA involves the fragmentation of all precursor ions within a given m/z window, avoiding intensity biases. Because data analysis and quantitation can be performed from both MS1 and MS2 scans, DIA can be useful to distinguish between isobaric and coeluting peptides [127, 135]. DIA was successfully used to assess dynamic changes in methylations and acetylations in histones H3 and H4 in response to treatment with the HDAC inhibitor SAHA [136]. In addition, both DIA and SIM were employed to quantify 199 peptides from histones extracted from mouse brain and liver [127].
Histone PTM quantitation strategies
Because most histone PTMs are common to all sample types, but are present at different levels, an accurate quantitation of their abundance is essential to characterize a biological sample (Fig. 6). Different MS‐based strategies can be used to quantify histone PTMs (reviewed in [14]). Typically, they involve extracting from the chromatographic profile the peaks corresponding to the m/z value and chromatography retention time of the peptide of interest, known as eXtracted Ion Chromatograms (XICs; Fig. 7). Isobaric and coeluting peptides can be quantitated based on the abundance of fragment ions at the MS/MS level [137, 138].
Fig. 6.
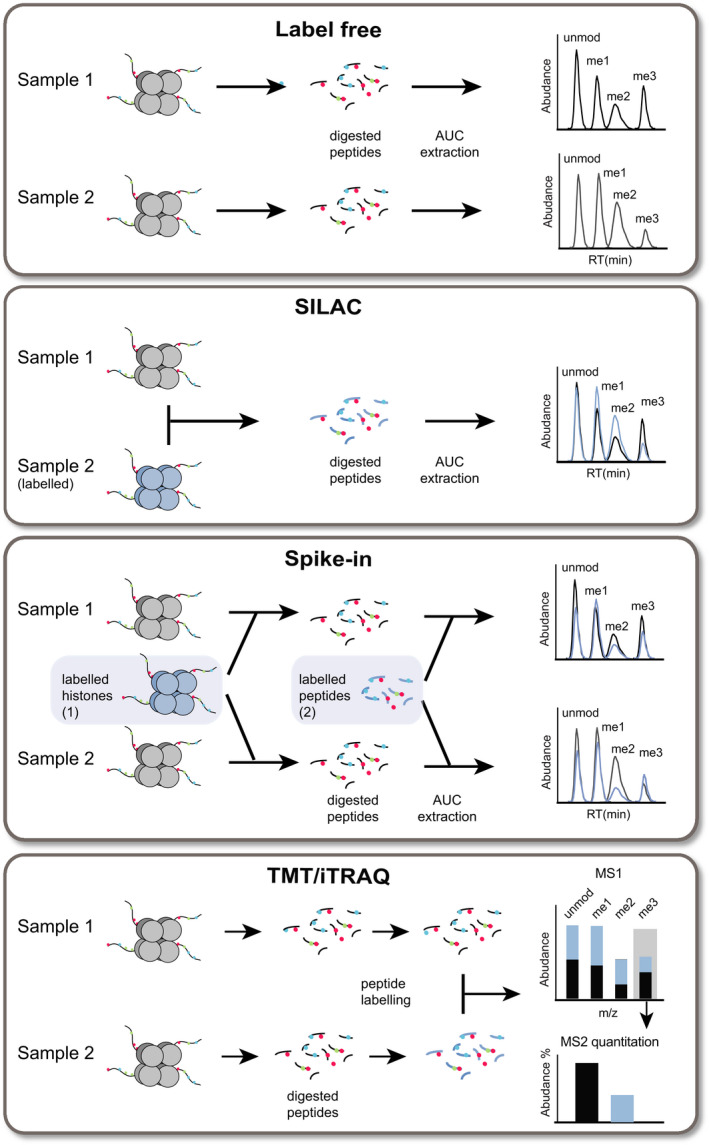
MS‐based histone PTM quantitation strategies. Simplified scheme depicting the most common histone PTM quantitation methods in bottom‐up MS approaches. In label‐free experiments, unlabeled samples are acquired in distinct runs and then compared. When using labeling strategies, different samples are labeled, combined, and acquired in a single run, from which the information regarding the abundance of distinct samples can be extracted. Labeling can be achieved by growing cells in media containing isotope‐encoded amino acids, a method known as SILAC. Unlabeled samples can also be compared with an internal standard, which can be SILAC‐labeled histones purified from cell lines, or synthetic isotope‐labeled peptides. Alternatively, peptides can be chemically modified after digestion, using TMT or iTRAQ. Up to 16 samples can be labeled using different tags, which have the same chemical structures and mass, but contain isotopes that allow them to be distinguished at the MS2 level.
Fig. 7.
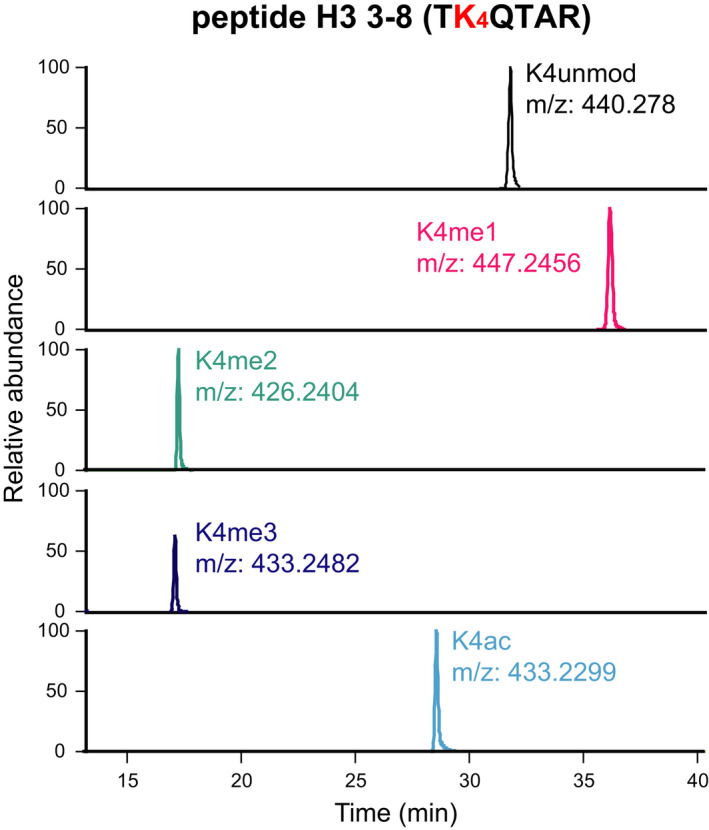
XICs. XICs corresponding to doubly charged ions for five differentially modified forms of the histone H3 3–8 peptide TKQTAR, containing H3K4. The peptides were digested with the PRO‐PIC method. The PTMs and m/z values of the peptides are indicated.
In a label‐free analysis, the XICs of different unlabeled samples, which are acquired in distinct runs, are compared. When using labeling strategies, different samples are labeled, combined, and acquired in a single run, from which the XICs of distinct samples can be extracted. Labeling can be achieved by growing cells in media containing isotope‐encoded amino acids, a method known as stable isotope labeling by amino acids in cell culture (SILAC) [139]. This method allows comparing up to three different cell populations, but requires actively diving cells to be applicable. In the case of clinical samples, spike‐in approaches are typically used, where unlabeled samples are compared with a labeled internal standard, which can be represented by SILAC‐labeled histones purified from one or more cell lines [140, 141], or by a library of synthetic isotope‐labeled peptides [142]. The use of an internal standard usually reduces experimental variability compared with label‐free strategies, and offers higher flexibility (e.g., the possibility to run large cohorts of samples in different batches, to be normalized to the standard) compared with standard SILAC approaches. In addition, using synthetic peptides allows the absolute quantitation of histones and their PTMs, as demonstrated by using a library of 93 synthetic Protein‐AQUA™ peptides carrying the most common histones H3, H4, and H2A PTMs [142]. Targeted MS represents a particularly suitable data acquisition strategy for relative and absolute quantitation of peptides and proteins in combination with internal standards.
Another labeling approach involves the chemical modification of histone peptides after digestion, using tandem mass tags (TMT) [143] or isobaric tags for relative and absolute quantification (iTRAQ) [144]. By using these strategies, up to 16 samples can be labeled using different tags, which have the same chemical structures and mass, but contain isotopes that allow them to be distinguished at the MS2 level. The possibility to combine up to 16 samples significantly reduces acquisition times and improves the multiplexing capabilities. Although this would be beneficial for profiling large cohorts of samples, these strategies have only been applied to cell lines so far [145, 146].
Although the above‐mentioned quantitative strategies can provide useful information when profiling clinical samples in a discovery stage, their use in the clinical routine in the near future is unlikely. Therefore, validation and routine analysis of potential epigenetic biomarkers could be performed using other more straightforward approaches, such as ELISA or IHC, which however suffer from the limitations already described in the introduction. Alternatively, one MS‐based method for the analysis of histones that could be currently implemented in the clinic is MRM, which requires instrumentation often already in use in hospitals for other applications. MRM‐based methods were developed for the analysis of a specific histone modification (H3K56ac [147]) or 42 differentially modified histone peptides [148], using stable isotopic labeled peptides as internal standards to measure the absolute concentrations. These studies provide the proof of concept for the applicability of MRM approaches for histone PTM analysis, although they have not been applied to clinical samples yet.
Histone variants and oncohistones
Because histone variants often show limited sequence differences, their identification through conventional antibody‐based methods is challenging. Although all three main proteomics strategies—bottom‐up, middle‐down, and top‐down—have been used to study core histone variants (thoroughly reviewed in [149]), bottom‐up proteomics has limitations in discriminating many of the variants. Indeed, because of the small size of the peptides resulting from this type of digestion, the peptides containing residues that are different among variants may be undetectable. For instance, H3.3 differs from H3.1 in five amino acids, of which only one (amino acid 31) falls in a peptide detectable by bottom‐up MS (peptide 27–40) [150]. However, there is no peptide derived from bottom‐up approaches that allows distinguishing all the H2B variants one from another. This problem can be overcome by using an offline chromatographic separation to isolate histone variants prior to bottom‐up analyses [149], or, alternatively, by using top‐ and middle‐down MS. These approaches have been employed to characterize H2A [151, 152] and H3 [153, 154] variants, which showed interesting differences in their PTM patterns. A combination of top‐down and middle‐down MS was also applied to the investigation of histone H3 and H2B clipping [155], namely the proteolytic cleavage of histone N‐terminal tails by different proteases, which has been implicated in various cellular processes [156]. MS can also be very useful to identify and quantify mutations on oncohistones, as shown in a study reporting the role of the H3K27M mutation in pediatric glioblastoma [110].
Linker histone H1 sequences contain a conserved globular domain, whereas N‐ and C‐terminal tails are much less conserved among the different variants [157], so that they can be distinguished more easily by MS. Differently from core histones, histone H1 variants can be reliably detected and quantified using a simple trypsin digestion. Because trypsin is commonly used in global proteomics studies, differences in histone H1 variant levels emerged from the global proteomics profiling of patient‐derived samples. For instance, differences in several linker histone variants were identified between patient‐derived epithelial cells from healthy and cancerous ovarian tissues [158]. In a recent study, we focused specifically on linker histone and implemented a label‐free quantification approach to analyze somatic histone H1 variants in clinical samples, which is applicable to laser‐microdissected tissue regions containing as low as 1000 cells. By applying this approach to breast cancer patient samples, we identified differences in histone H1 variants patters in triple negative breast tumors with and without relapse after chemotherapy [159]. PTMs on histone H1 variants can also be investigated by MS. Phosphorylation, which is the best characterized PTM on linker histone, as well as other PTMs, were characterized by both top‐down [160] and bottom‐up MS methods [161, 162]. Despite the potential of profiling histone variants in clinical samples through MS‐based approaches, the vast majority of the studies performed so far were done on cell lines, while only a few used mouse tissue [154, 163].
MS contribution to cancer epigenetics
The growing evidence regarding an involvement of epigenetic mechanisms in cancer, together with the technological advances in MS‐based histone PTM analysis witnessed in recent years, has fueled the application of MS‐based approaches to profile the epigenomes of cancer clinical samples, with the goal of identifying epigenetic biomarkers, and investigate epigenetic mechanisms underpinning cancer onset and development [164]. As already mentioned, many of the studies performed so far employed cancer cells lines as a proxy for tumor tissues. In some cases, the goal of the study was the identification of epigenetic biomarkers, for instance, associated with cigarette smoking [165], with an invasive phenotype in esophageal squamous cell carcinoma [166] or tumor grade in breast cancer [167]. Other studies investigated the levels of histone PTMs in the context of changes in histone‐modifying enzymes. For instance, a super‐SILAC‐based histone PTMs profiling of 115 cancer cell lines revealed an increase of H3K36me2 in cells containing mutations in the methyltransferase NSD2, which hence was suggested as a potential therapeutic target in pediatric acute lymphoblastic leukemia [168]. Top‐down MS identified the presence of hypermethylated proteoforms in human cell lines derived from multiple myeloma patients overexpressing the histone methyltransferase MMSET [169], while a targeted MS‐based assay showed that EZH2 can affect H3K9 methylation in drug‐tolerant cell precursors in the presence of the repressive mark H3S10P, which supported a role of this methyltransferase in the initiation of drug tolerance [170]. In another study, profiling of histone modifications and histone‐modifying enzyme gene expression levels in 24 cell lines, mostly cancerous, highlighted an enrichment of H3K27me3, along with its main methyltransferase (EZH2) in breast cancer cell lines compared with the cell lines of different origin [171]. This finding prompted the investigation of the effects of knocking down EZH2, which reduced tumor growth in a mouse mammary xenograft model.
Although using cancer cell lines that can be easily manipulated is the easiest method to study epigenetic mechanisms in cancer, caution must be given to the interpretation of the results that are obtained from such investigations. It is indeed known that cancer cell lines represent some but not all the features of primary tumors, also from the epigenetic point of view [119, 172]. By profiling histone PTMs in cancer cell lines, primary cells, and cancer tissues in different tumor models, we showed that cells undergo a rapid, extensive, and systematic rewiring after transition to culture conditions [119]. Such rewiring involved important changes in methylations/acetylations on a number of histone H3 residues, including K9, K14, K18, K27, K36, K79, and also several histone H4 residues (unpublished results). These changes are relevant to the point that differences between tissues and cell lines are more marked than those observed between tumor and normal samples [53]. Importantly, these changes revert when cells are returned in an in vivo situation, such as in tumor xenograft. Therefore, while cell lines and long‐term primary cells are a useful tool, it is advisable to validate the results obtained in patient tissues or in vivo animal models [119]. This was done in the already mentioned seminal study investigating histone PTM changes in tumor compared with normal samples [15], where the decrease in H4K16ac and H4K20me3 initially observed in tumor cell lines was then validated in patient tissues. Along the same line of investigation, more recently, we employed MS approaches to profile histone H3 modifications in a panel tumor and normal tissues for several cancer types, revealing various changes, some of which tumor‐ and subtype‐specific, and some more general [53, 123] (Fig. 2, left panel). Most notably, we detected in all the tumor types tested compared with their normal counterpart a widespread decrease of H3K14ac, which could represent a novel epigenetic hallmark of cancer. We also detected a decrease in H3K27me3 in several tumor types/subtypes, including breast cancer, compared with normal tissue [53, 123]. This result is in contrast with the increase in the methyltransferase EZH2 taking place in the majority of tumors, and suggests that a mechanism different from deregulation of this histone‐modifying enzyme levels is responsible for the change in the H3K27me3 mark. One such mechanism could be the increased proliferations rates in tumors, since H3K27me3 shows decreased levels in proliferating cells [53, 123]. In breast cancer tissues, we also detected significant differences in various histone PTMs, including H3K9me3, H3K36me1/me2 and H3K27me3 marks among different subtypes [120, 126]. Changes in proliferation rates may at least partially account for the difference in H3K27me3—as well as H3K36 methylation—also in this setting, while the increase in H3K9me3 in the triple negative and HER‐positive subtypes appears to be specific and cell cycle‐independent [53, 123].
The H3K27me3 mark was also investigated in malignant peripheral nerve sheath cancer, where the analysis of histone PTMs in FFPE tumors revealed a number of changes due to loss‐of‐function alterations in Polycomb‐repressive complex 2 (PRC2), which contains the EZH2 enzyme. In particular, an increase in PTMs associated with active transcription, including H3K27Ac and H3K36me2, and loss of H3K27me2/me3 were observed [121]. These changes suggested NSD2, the methyltransferase responsible for the deposition of H3K36me2, as a potential therapeutic target. NSD2 knockdown restored the expression of pathways linked to interferon signaling and antigen presentation, which were altered in PRC2‐deficient tumors.
Lastly, we found a significant decrease in the levels of all somatic histone variants in breast cancer belonging to the triple negative subtype relapsing tumors when comparing patients with and without relapse 3 years after chemotherapy [159], suggesting histone H1 variant levels as potential biomarkers useful for prediction of response to therapy.
Conclusion and perspectives
In the last 15 years, MS‐based proteomics has proved its value for the analysis of histone PTMs and histone variants, outperforming antibody‐based approaches for the analysis of bulk histone PTM levels in terms of comprehensiveness and quantitative accuracy of the data that it can provide. In the context of cancer research, where epigenetic components are being increasingly recognized as important players, histone analysis by MS holds great promise for the investigation of biomarkers and identification of therapeutic targets. Nevertheless, its application to clinical samples has been limited so far. From the technical point of view, there have been great advances, which now allow using all the most common clinical specimens. Indeed, established protocols exist for histone PTM and variant analysis of FFPE, OCT‐frozen, and fresh‐frozen tissues, as well as for patient‐derived primary cells. One important improvement to be achieved in the near future would be the ability to accurately quantify histone PTMs from circulating nucleosomes, which would dramatically expand the possibilities to discover cancer biomarkers, also in the presence of an early‐stage disease. Another technical point that has already been addressed is represented by the possibility to analyze a good portion of the most common histone PTMs, as well as histone H1 variants, from very small starting amounts of material. These results open the way for a more extensive and easy application of LMD, which can be used not only to obtain more homogeneous cell populations, but also to investigate tumor heterogeneity.
This review focuses on bottom‐up MS approaches for histone PTM analysis, because they have been already applied successfully to patient‐derived tissues. However, while bottom‐up strategies offer efficient amino acid sequencing and higher throughput for complex samples, they involve loss of information about combinatorial PTM patterns, including crosstalk and interplay between distant modifications, as well as variant information. This type of information is maintained when using by top‐down and middle‐down approaches, which have witnessed encouraging progress from the technical point of view in recent years, particularly in the case of middle‐down approaches (reviewed in [173]). For instance, an optimized middle‐down setup allowing increased automation of LC‐MS acquisition and data analysis has been proposed [174]. Despite these advances, an issue of top‐ and middle‐down approaches remains the lower sensitivity and the requirement of substantial amounts of starting material, which is particularly relevant in the case of clinical samples. Furthermore, the development of bioinformatics tools suitable for the analysis of top‐ and middle‐down data still represents a challenge that will have to be solved for routine application to patient‐derived samples.
Although histone PTM analyses are usually carried out from bulk histones, focusing on specific genomic locations could provide valuable information regarding the histone modification patterns at the level of disease‐associated genes. Various proteomics methods for the analysis of histone PTMs at specific genomic loci have been developed (reviewed in [175]) and could benefit from the recent improvements in the quantitation of modifications from low abundance samples. Another important layer of information is represented by the genomic distribution of histone PTMs. Even if it is highly accurate, MS usually provides information on the total levels of histone PTMs, without localizing the modification to specific genome regions. The combination of MS‐based approaches with ChIP‐seq analysis of histone PTMs/variants would lead to a more comprehensive view on different types of epigenetic alterations linked with cancer and could highlight aberrant pathways that could be targeted for therapy (Fig. 1). In this scenario, the integration of the data obtained through proteomics and genomics, as well as transcriptomics and global proteomics, will be fundamental to fully exploit the information obtained using these powerful analytical methods, and to elucidate how epigenetic aberrations can affect the tumor phenotype.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgments
TB is supported by the Italian Association for Cancer Research, grant number IG‐2018‐21834 (to TB) and by EPIC‐XS, project number 823839, funded by the Horizon 2020 Programme of the European Union. RN is supported by the Italian Ministry of Health, grant number GR‐2016‐02361522 (to RN).
[Correction added on 28 February 2022, after first online publication: the title was updated to remove the plural from progress.]
Contributor Information
Roberta Noberini, Email: roberta.noberini@ieo.it.
Tiziana Bonaldi, Email: tiziana.bonaldi@ieo.it.
References
- 1. Cutter AR & Hayes JJ (2015) A brief review of nucleosome structure. FEBS Lett 589, 2914–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Clapier CR, Iwasa J, Cairns BR & Peterson CL (2017) Mechanisms of action and regulation of ATP‐dependent chromatin‐remodelling complexes. Nat Rev Mol Cell Biol 18, 407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hammond CM, Stromme CB, Huang H, Patel DJ & Groth A (2017) Histone chaperone networks shaping chromatin function. Nat Rev Mol Cell Biol 18, 141–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Audia JE & Campbell RM (2016) Histone modifications and cancer. Cold Spring Harb Perspect Biol 8, a019521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chervona Y & Costa M (2012) Histone modifications and cancer: biomarkers of prognosis? Am J Cancer Res 2, 589–597. [PMC free article] [PubMed] [Google Scholar]
- 6. Khan SA, Reddy D & Gupta S (2015) Global histone post‐translational modifications and cancer: biomarkers for diagnosis, prognosis and treatment? World J Biol Chem 6, 333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Strahl BD & Allis CD (2000) The language of covalent histone modifications. Nature 403, 41–45. [DOI] [PubMed] [Google Scholar]
- 8. Jenuwein T & Allis CD (2001) Translating the histone code. Science 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- 9. Bannister AJ & Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21, 381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Biswas S & Rao CM (2018) Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur J Pharmacol 837, 8–24. [DOI] [PubMed] [Google Scholar]
- 11. Kouzarides T (2007) Chromatin modifications and their function. Cell 128, 693–705. [DOI] [PubMed] [Google Scholar]
- 12. Vardabasso C, Hasson D, Ratnakumar K, Chung CY, Duarte LF & Bernstein E (2014) Histone variants: emerging players in cancer biology. Cell Mol Life Sci 71, 379–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Portela A & Esteller M (2010) Epigenetic modifications and human disease. Nat Biotechnol 28, 1057–1068. [DOI] [PubMed] [Google Scholar]
- 14. Huang H, Lin S, Garcia BA & Zhao Y (2015) Quantitative proteomic analysis of histone modifications. Chem Rev 115, 2376–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fraga MF, Ballestar E, Villar‐Garea A, Boix‐Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K et al. (2005) Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 37, 391–400. [DOI] [PubMed] [Google Scholar]
- 16. Van Den Broeck A, Brambilla E, Moro‐Sibilot D, Lantuejoul S, Brambilla C, Eymin B & Gazzeri S (2008) Loss of histone H4K20 trimethylation occurs in preneoplasia and influences prognosis of non‐small cell lung cancer. Clin Cancer Res 14, 7237–7245. [DOI] [PubMed] [Google Scholar]
- 17. Zhu L, Yang J, Zhao L, Yu X, Wang L, Wang F, Cai Y & Jin J (2015) Expression of hMOF, but not HDAC4, is responsible for the global histone H4K16 acetylation in gastric carcinoma. Int J Oncol 46, 2535–2545. [DOI] [PubMed] [Google Scholar]
- 18. Isin H, Ozgur E, Talu CK, Trabulus DC, Karacetin D & Gezer U (2020) Impact of histone methyltransferase SUV420H2 in breast cancer. Biomed Rep 13, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang Y, Huang J, Li Q, Chen K, Liang Y, Zhan Z, Ye F, Ni W, Chen L & Ding Y (2018) Histone methyltransferase SETDB1 promotes cells proliferation and migration by interacting withTiam1 in hepatocellular carcinoma. BMC Cancer 18, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bachman KE, Park BH, Rhee I, Rajagopalan H, Herman JG, Baylin SB, Kinzler KW & Vogelstein B (2003) Histone modifications and silencing prior to DNA methylation of a tumor suppressor gene. Cancer Cell 3, 89–95. [DOI] [PubMed] [Google Scholar]
- 21. Espada J, Ballestar E, Fraga MF, Villar‐Garea A, Juarranz A, Stockert JC, Robertson KD, Fuks F & Esteller M (2004) Human DNA methyltransferase 1 is required for maintenance of the histone H3 modification pattern. J Biol Chem 279, 37175–37184. [DOI] [PubMed] [Google Scholar]
- 22. Tamaru H & Selker EU (2001) A histone H3 methyltransferase controls DNA methylation in Neurospora crassa . Nature 414, 277–283. [DOI] [PubMed] [Google Scholar]
- 23. Seligson DB, Horvath S, McBrian MA, Mah V, Yu H, Tze S, Wang Q, Chia D, Goodglick L & Kurdistani SK (2009) Global levels of histone modifications predict prognosis in different cancers. Am J Pathol 174, 1619–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ashktorab H, Belgrave K, Hosseinkhah F, Brim H, Nouraie M, Takkikto M, Hewitt S, Lee EL, Dashwood RH & Smoot D (2009) Global histone H4 acetylation and HDAC2 expression in colon adenoma and carcinoma. Dig Dis Sci 54, 2109–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Elsheikh SE, Green AR, Rakha EA, Powe DG, Ahmed RA, Collins HM, Soria D, Garibaldi JM, Paish CE, Ammar AA et al. (2009) Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res 69, 3802–3809. [DOI] [PubMed] [Google Scholar]
- 26. Messier TL, Gordon JA, Boyd JR, Tye CE, Browne G, Stein JL, Lian JB & Stein GS (2016) Histone H3 lysine 4 acetylation and methylation dynamics define breast cancer subtypes. Oncotarget 7, 5094–5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Manuyakorn A, Paulus R, Farrell J, Dawson NA, Tze S, Cheung‐Lau G, Hines OJ, Reber H, Seligson DB, Horvath S et al. (2010) Cellular histone modification patterns predict prognosis and treatment response in resectable pancreatic adenocarcinoma: results from RTOG 9704. J Clin Oncol 28, 1358–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ellinger J, Kahl P, Mertens C, Rogenhofer S, Hauser S, Hartmann W, Bastian PJ, Buttner R, Muller SC & von Ruecker A (2010) Prognostic relevance of global histone H3 lysine 4 (H3K4) methylation in renal cell carcinoma. Int J Cancer 127, 2360–2366. [DOI] [PubMed] [Google Scholar]
- 29. Song JS, Kim YS, Kim DK, Park SI & Jang SJ (2012) Global histone modification pattern associated with recurrence and disease‐free survival in non‐small cell lung cancer patients. Pathol Int 62, 182–190. [DOI] [PubMed] [Google Scholar]
- 30. Ellinger J, Kahl P, von der Gathen J, Rogenhofer S, Heukamp LC, Gutgemann I, Walter B, Hofstadter F, Buttner R, Muller SC et al. (2010) Global levels of histone modifications predict prostate cancer recurrence. Prostate 70, 61–69. [DOI] [PubMed] [Google Scholar]
- 31. McAnena P, Brown JA & Kerin MJ (2017) Circulating nucleosomes and nucleosome modifications as biomarkers in cancer. Cancers (Basel) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu BL, Cheng JX, Zhang X, Wang R, Zhang W, Lin H, Xiao X, Cai S, Chen XY & Cheng H (2010) Global histone modification patterns as prognostic markers to classify glioma patients. Cancer Epidemiol Biomarkers Prev 19, 2888–2896. [DOI] [PubMed] [Google Scholar]
- 33. Halasa M, Wawruszak A, Przybyszewska A, Jaruga A, Guz M, Kalafut J, Stepulak A & Cybulski M (2019) H3K18Ac as a marker of cancer progression and potential target of anti‐cancer therapy. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M & Kurdistani SK (2005) Global histone modification patterns predict risk of prostate cancer recurrence. Nature 435, 1262–1266. [DOI] [PubMed] [Google Scholar]
- 35. Wei Y, Xia W, Zhang Z, Liu J, Wang H, Adsay NV, Albarracin C, Yu D, Abbruzzese JL, Mills GB et al. (2008) Loss of trimethylation at lysine 27 of histone H3 is a predictor of poor outcome in breast, ovarian, and pancreatic cancers. Mol Carcinog 47, 701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. He C, Xu J, Zhang J, Xie D, Ye H, Xiao Z, Cai M, Xu K, Zeng Y, Li H et al. (2012) High expression of trimethylated histone H3 lysine 4 is associated with poor prognosis in hepatocellular carcinoma. Hum Pathol 43, 1425–1435. [DOI] [PubMed] [Google Scholar]
- 37. Rogenhofer S, Kahl P, Holzapfel S, VON Ruecker A, Mueller SC & Ellinger J (2012) Decreased levels of histone H3K9me1 indicate poor prognosis in patients with renal cell carcinoma. Anticancer Res 32, 879–886. [PubMed] [Google Scholar]
- 38. Ellinger J, Bachmann A, Goke F, Behbahani TE, Baumann C, Heukamp LC, Rogenhofer S & Muller SC (2014) Alterations of global histone H3K9 and H3K27 methylation levels in bladder cancer. Urol Int 93, 113–118. [DOI] [PubMed] [Google Scholar]
- 39. Gezer U, Ustek D, Yoruker EE, Cakiris A, Abaci N, Leszinski G, Dalay N & Holdenrieder S (2013) Characterization of H3K9me3‐ and H4K20me3‐associated circulating nucleosomal DNA by high‐throughput sequencing in colorectal cancer. Tumour Biol 34, 329–336. [DOI] [PubMed] [Google Scholar]
- 40. Leszinski G, Gezer U, Siegele B, Stoetzer O & Holdenrieder S (2012) Relevance of histone marks H3K9me3 and H4K20me3 in cancer. Anticancer Res 32, 2199–2205. [PubMed] [Google Scholar]
- 41. Rogenhofer S, Kahl P, Mertens C, Hauser S, Hartmann W, Buttner R, Muller SC, von Ruecker A & Ellinger J (2012) Global histone H3 lysine 27 (H3K27) methylation levels and their prognostic relevance in renal cell carcinoma. BJU Int 109, 459–465. [DOI] [PubMed] [Google Scholar]
- 42. Ellinger J, Schneider AC, Bachmann A, Kristiansen G, Muller SC & Rogenhofer S (2016) Evaluation of global histone acetylation levels in bladder cancer patients. Anticancer Res 36, 3961–3964. [PubMed] [Google Scholar]
- 43. Shanmugam MK, Arfuso F, Arumugam S, Chinnathambi A, Jinsong B, Warrier S, Wang LZ, Kumar AP, Ahn KS, Sethi G et al. (2018) Role of novel histone modifications in cancer. Oncotarget 9, 11414–11426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lichtenstein AV, Melkonyan HS, Tomei LD & Umansky SR (2001) Circulating nucleic acids and apoptosis. Ann N Y Acad Sci 945, 239–249. [DOI] [PubMed] [Google Scholar]
- 45. Holdenrieder S, Stieber P, Bodenmuller H, Busch M, Fertig G, Furst H, Schalhorn A, Schmeller N, Untch M & Seidel D (2001) Nucleosomes in serum of patients with benign and malignant diseases. Int J Cancer 95, 114–120. [DOI] [PubMed] [Google Scholar]
- 46. Duforestel M, Briand J, Bougras‐Cartron G, Heymann D, Frenel JS, Vallette FM & Cartron PF (2020) Cell‐free circulating epimarks in cancer monitoring. Epigenomics 12, 145–155. [DOI] [PubMed] [Google Scholar]
- 47. Snyder MW, Kircher M, Hill AJ, Daza RM & Shendure J (2016) Cell‐free DNA comprises an in vivo nucleosome footprint that informs its tissues‐of‐origin. Cell 164, 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Reddy D, Khade B, Pandya R & Gupta S (2017) A novel method for isolation of histones from serum and its implications in therapeutics and prognosis of solid tumours. Clin Epigenetics 9, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bauden M, Pamart D, Ansari D, Herzog M, Eccleston M, Micallef J, Andersson B & Andersson R (2015) Circulating nucleosomes as epigenetic biomarkers in pancreatic cancer. Clin Epigenetics 7, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rahier JF, Druez A, Faugeras L, Martinet JP, Gehenot M, Josseaux E, Herzog M, Micallef J, George F, Delos M et al. (2017) Circulating nucleosomes as new blood‐based biomarkers for detection of colorectal cancer. Clin Epigenetics 9, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Morgan MA & Shilatifard A (2015) Chromatin signatures of cancer. Genes Dev 29, 238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, Shen R, Taylor AM, Cherniack AD, Thorsson V et al. (2018) Cell‐of‐origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173, 291–304.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Noberini R, Restellini C, Savoia EO, Raimondi F, Ghiani L, Jodice MG, Bertalot G, Bonizzi G, Capra M, Maffini FA et al. (2019) Profiling of epigenetic features in clinical samples reveals novel widespread changes in cancer. Cancers (Basel) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Albert M & Helin K (2010) Histone methyltransferases in cancer. Semin Cell Dev Biol 21, 209–220. [DOI] [PubMed] [Google Scholar]
- 55. Nottke A, Colaiacovo MP & Shi Y (2009) Developmental roles of the histone lysine demethylases. Development 136, 879–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sheikh BN & Akhtar A (2019) The many lives of KATs ‐ detectors, integrators and modulators of the cellular environment. Nat Rev Genet 20, 7–23. [DOI] [PubMed] [Google Scholar]
- 57. Zhang K & Dent SY (2005) Histone modifying enzymes and cancer: going beyond histones. J Cell Biochem 96, 1137–1148. [DOI] [PubMed] [Google Scholar]
- 58. Iyer NG, Ozdag H & Caldas C (2004) p300/CBP and cancer. Oncogene 23, 4225–4231. [DOI] [PubMed] [Google Scholar]
- 59. Gao Y, Geng J, Hong X, Qi J, Teng Y, Yang Y, Qu D & Chen G (2014) Expression of p300 and CBP is associated with poor prognosis in small cell lung cancer. Int J Clin Exp Pathol 7, 760–767. [PMC free article] [PubMed] [Google Scholar]
- 60. Ropero S & Esteller M (2007) The role of histone deacetylases (HDACs) in human cancer. Mol Oncol 1, 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Glozak MA & Seto E (2007) Histone deacetylases and cancer. Oncogene 26, 5420–5432. [DOI] [PubMed] [Google Scholar]
- 62. Chiba T, Saito T, Yuki K, Zen Y, Koide S, Kanogawa N, Motoyama T, Ogasawara S, Suzuki E, Ooka Y et al. (2015) Histone lysine methyltransferase SUV39H1 is a potent target for epigenetic therapy of hepatocellular carcinoma. Int J Cancer 136, 289–298. [DOI] [PubMed] [Google Scholar]
- 63. Rodriguez‐Paredes M, Martinez de Paz A, Simo‐Riudalbas L, Sayols S, Moutinho C, Moran S, Villanueva A, Vazquez‐Cedeira M, Lazo PA, Carneiro F et al. (2014) Gene amplification of the histone methyltransferase SETDB1 contributes to human lung tumorigenesis. Oncogene 33, 2807–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sun Y, Wei M, Ren SC, Chen R, Xu WD, Wang FB, Lu J, Shen J, Yu YW, Hou JG et al. (2014) Histone methyltransferase SETDB1 is required for prostate cancer cell proliferation, migration and invasion. Asian J Androl 16, 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vougiouklakis T, Saloura V, Park JH, Takamatsu N, Miyamoto T, Nakamura Y & Matsuo Y (2018) Development of novel SUV39H2 inhibitors that exhibit growth suppressive effects in mouse xenograft models and regulate the phosphorylation of H2AX. Oncotarget 9, 31820–31831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li B, Zheng Y & Yang L (2019) The oncogenic potential of SUV39H2: a comprehensive and perspective view. J Cancer 10, 721–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Park JH, Jung M & Moon KC (2018) The prognostic significance of nuclear expression of PHF2 and C/EBPalpha in clear cell renal cell carcinoma with consideration of adipogenic metabolic evolution. Oncotarget 9, 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gan L, Yang Y, Li Q, Feng Y, Liu T & Guo W (2018) Epigenetic regulation of cancer progression by EZH2: from biological insights to therapeutic potential. Biomark Res 6, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jiang T, Wang Y, Zhou F, Gao G, Ren S & Zhou C (2016) Prognostic value of high EZH2 expression in patients with different types of cancer: a systematic review with meta‐analysis. Oncotarget 7, 4584–4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ellinger J, Kahl P, von der Gathen J, Heukamp LC, Gutgemann I, Walter B, Hofstadter F, Bastian PJ, von Ruecker A, Muller SC et al. (2012) Global histone H3K27 methylation levels are different in localized and metastatic prostate cancer. Cancer Invest 30, 92–97. [DOI] [PubMed] [Google Scholar]
- 71. Tzao C, Tung HJ, Jin JS, Sun GH, Hsu HS, Chen BH, Yu CP & Lee SC (2009) Prognostic significance of global histone modifications in resected squamous cell carcinoma of the esophagus. Mod Pathol 22, 252–260. [DOI] [PubMed] [Google Scholar]
- 72. Li J, Zhu S, Ke XX & Cui H (2016) Role of several histone lysine methyltransferases in tumor development. Biomed Rep 4, 293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Li Y & Seto E (2016) HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med 6, a026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dahlin JL, Nelson KM, Strasser JM, Barsyte‐Lovejoy D, Szewczyk MM, Organ S, Cuellar M, Singh G, Shrimp JH, Nguyen N et al. (2017) Assay interference and off‐target liabilities of reported histone acetyltransferase inhibitors. Nat Commun 8, 1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, Digiammarino EL, Hansen TM, Risi RM, Frey R, Manaves V et al. (2017) Discovery of a selective catalytic p300/CBP inhibitor that targets lineage‐specific tumours. Nature 550, 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kaniskan HU, Konze KD & Jin J (2015) Selective inhibitors of protein methyltransferases. J Med Chem 58, 1596–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kaniskan HU, Martini ML & Jin J (2018) Inhibitors of protein methyltransferases and demethylases. Chem Rev 118, 989–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Greiner D, Bonaldi T, Eskeland R, Roemer E & Imhof A (2005) Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3–9. Nat Chem Biol 1, 143–145. [DOI] [PubMed] [Google Scholar]
- 79. Gulati N, Beguelin W & Giulino‐Roth L (2018) Enhancer of zeste homolog 2 (EZH2) inhibitors. Leuk Lymphoma 59, 1574–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Fang Y, Liao G & Yu B (2019) LSD1/KDM1A inhibitors in clinical trials: advances and prospects. J Hematol Oncol 12, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J & Crabtree GR (2013) Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 45, 592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Shain AH & Pollack JR (2013) The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS One 8, e55119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chen YF, Liang YX, Yang JA, Yuan DZ, Li J, Zheng SS, Wan YP, Wang B, Han ZD & Zhong WD (2019) Upregulation of Holliday junction recognition protein predicts poor prognosis and biochemical recurrence in patients with prostate cancer. Oncol Lett 18, 6697–6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Montes de Oca R, Gurard‐Levin ZA, Berger F, Rehman H, Martel E, Corpet A, de Koning L, Vassias I, Wilson LO, Meseure D et al. (2015) The histone chaperone HJURP is a new independent prognostic marker for luminal A breast carcinoma. Mol Oncol 9, 657–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Fujisawa T & Filippakopoulos P (2017) Functions of bromodomain‐containing proteins and their roles in homeostasis and cancer. Nat Rev Mol Cell Biol 18, 246–262. [DOI] [PubMed] [Google Scholar]
- 86. Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L & Sims RJ 3rd (2011) Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci USA 108, 16669–16674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Alqahtani A, Choucair K, Ashraf M, Hammouda DM, Alloghbi A, Khan T, Senzer N & Nemunaitis J (2019) Bromodomain and extra‐terminal motif inhibitors: a review of preclinical and clinical advances in cancer therapy. Future Sci OA 5, FSO372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Corujo D & Buschbeck M (2018) Post‐translational modifications of H2A histone variants and their role in cancer. Cancers (Basel) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Long M, Sun X, Shi W, Yanru A, Leung STC, Ding D, Cheema MS, MacPherson N, Nelson CJ, Ausio J et al. (2019) A novel histone H4 variant H4G regulates rDNA transcription in breast cancer. Nucleic Acids Res 47, 8399–8409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Martire S & Banaszynski LA (2020) The roles of histone variants in fine‐tuning chromatin organization and function. Nat Rev Mol Cell Biol 21, 522–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Vardabasso C, Gaspar‐Maia A, Hasson D, Punzeler S, Valle‐Garcia D, Straub T, Keilhauer EC, Strub T, Dong J, Panda T et al. (2015) Histone variant H2A.Z.2 mediates proliferation and drug sensitivity of malignant melanoma. Mol Cell 59, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bassing CH, Suh H, Ferguson DO, Chua KF, Manis J, Eckersdorff M, Gleason M, Bronson R, Lee C & Alt FW (2003) Histone H2AX: a dosage‐dependent suppressor of oncogenic translocations and tumors. Cell 114, 359–370. [DOI] [PubMed] [Google Scholar]
- 93. Celeste A, Petersen S, Romanienko PJ, Fernandez‐Capetillo O, Chen HT, Sedelnikova OA, Reina‐San‐Martin B, Coppola V, Meffre E, Difilippantonio MJ et al. (2002) Genomic instability in mice lacking histone H2AX. Science 296, 922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S & Pommier Y (2008) GammaH2AX and cancer. Nat Rev Cancer 8, 957–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Garcia‐Villa A, Balasubramanian P, Miller BL, Lustberg MB, Ramaswamy B & Chalmers JJ (2012) Assessment of gamma‐H2AX levels in circulating tumor cells from patients receiving chemotherapy. Front Oncol 2, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Mohammad F & Helin K (2017) Oncohistones: drivers of pediatric cancers. Genes Dev 31, 2313–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Shrestha RL, Ahn GS, Staples MI, Sathyan KM, Karpova TS, Foltz DR & Basrai MA (2017) Mislocalization of centromeric histone H3 variant CENP‐A contributes to chromosomal instability (CIN) in human cells. Oncotarget 8, 46781–46800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zlatanova J & Doenecke D (1994) Histone H1 zero: a major player in cell differentiation? FASEB J 8, 1260–1268. [DOI] [PubMed] [Google Scholar]
- 99. Fyodorov DV, Zhou BR, Skoultchi AI & Bai Y (2018) Emerging roles of linker histones in regulating chromatin structure and function. Nat Rev Mol Cell Biol 19, 192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Scaffidi P (2016) Histone H1 alterations in cancer. Biochim Biophys Acta 1859, 533–539. [DOI] [PubMed] [Google Scholar]
- 101. Fontebasso AM, Papillon‐Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset PO, Bechet D, Faury D, De Jay N, Ramkissoon LA et al. (2014) Recurrent somatic mutations in ACVR1 in pediatric midline high‐grade astrocytoma. Nat Genet 46, 462–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Khuong‐Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L et al. (2012) K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 124, 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M et al. (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231. [DOI] [PubMed] [Google Scholar]
- 104. Taylor KR, Mackay A, Truffaux N, Butterfield Y, Morozova O, Philippe C, Castel D, Grasso CS, Vinci M, Carvalho D et al. (2014) Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet 46, 457–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M et al. (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non‐brainstem glioblastomas. Nat Genet 44, 251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Pajtler KW, Wen J, Sill M, Lin T, Orisme W, Tang B, Hubner JM, Ramaswamy V, Jia S, Dalton JD et al. (2018) Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol 136, 211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, Wedge DC, Cooke SL, Gundem G, Davies H et al. (2013) Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet 45, 1479–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Papillon‐Cavanagh S, Lu C, Gayden T, Mikael LG, Bechet D, Karamboulas C, Ailles L, Karamchandani J, Marchione DM, Garcia BA et al. (2017) Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat Genet 49, 180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Fang D, Gan H, Lee JH, Han J, Wang Z, Riester SM, Jin L, Chen J, Zhou H, Wang J et al. (2016) The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science 352, 1344–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ & Allis CD (2013) Inhibition of PRC2 activity by a gain‐of‐function H3 mutation found in pediatric glioblastoma. Science 340, 857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Smith CM, Haimberger ZW, Johnson CO, Wolf AJ, Gafken PR, Zhang Z, Parthun MR & Gottschling DE (2002) Heritable chromatin structure: mapping "memory" in histones H3 and H4. Proc Natl Acad Sci USA 99 (Suppl 4), 16454–16461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Soldi M, Cuomo A & Bonaldi T (2014) Improved bottom‐up strategy to efficiently separate hypermodified histone peptides through ultra‐HPLC separation on a bench top Orbitrap instrument. Proteomics 14, 2212–2225. [DOI] [PubMed] [Google Scholar]
- 113. Sidoli S, Bhanu NV, Karch KR, Wang X & Garcia BA (2016) Complete workflow for analysis of histone post‐translational modifications using bottom‐up mass spectrometry: from histone extraction to data analysis. J Vis Exp 111, 54112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Maile TM, Izrael‐Tomasevic A, Cheung T, Guler GD, Tindell C, Masselot A, Liang J, Zhao F, Trojer P, Classon M et al. (2015) Mass spectrometric quantification of histone post‐translational modifications by a hybrid chemical labeling method. Mol Cell Proteomics 14, 1148–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Shechter D, Dormann HL, Allis CD & Hake SB (2007) Extraction, purification and analysis of histones. Nat Protoc 2, 1445–1457. [DOI] [PubMed] [Google Scholar]
- 116. Noberini R, Restellini C, Savoia EO & Bonaldi T (2019) Enrichment of histones from patient samples for mass spectrometry‐based analysis of post‐translational modifications. Methods 184, 19–28. [DOI] [PubMed] [Google Scholar]
- 117. Bauden M, Kristl T, Andersson R, Marko‐Varga G & Ansari D (2017) Characterization of histone‐related chemical modifications in formalin‐fixed paraffin‐embedded and fresh‐frozen human pancreatic cancer xenografts using LC‐MS/MS. Lab Invest 97, 279–288. [DOI] [PubMed] [Google Scholar]
- 118. Farrelly LA, Dill BD, Molina H, Birtwistle MR & Maze I (2016) Current proteomic methods to investigate the dynamics of histone turnover in the central nervous system. Methods Enzymol 574, 331–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Noberini R, Osti D, Miccolo C, Richichi C, Lupia M, Corleone G, Hong SP, Colombo P, Pollo B, Fornasari L et al. (2018) Extensive and systematic rewiring of histone post‐translational modifications in cancer model systems. Nucleic Acids Res 46, 3817–3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Noberini R, Uggetti A, Pruneri G, Minucci S & Bonaldi T (2016) Pathology tissue‐quantitative mass spectrometry analysis to profile histone post‐translational modification patterns in patient samples. Mol Cell Proteomics 15, 866–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Wojcik JB, Marchione DM, Sidoli S, Djedid A, Lisby A, Majewski J & Garcia BA (2019) Epigenomic reordering induced by polycomb loss drives oncogenesis but leads to therapeutic vulnerabilities in malignant peripheral nerve sheath tumors. Cancer Res 79, 3205–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Zhang Q, Xue P, Li HL, Bao YH, Wu LH, Chang SY, Niu B, Yang FQ & Zhang T (2013) Histone modification mapping in human brain reveals aberrant expression of histone H3 lysine 79 dimethylation in neural tube defects. Neurobiol Dis 54, 404–413. [DOI] [PubMed] [Google Scholar]
- 123. Restellini C, Cuomo A, Lupia M, Giordano M, Bonaldi T & Noberini R (2019) Alternative digestion approaches improve histone modification mapping by mass spectrometry in clinical samples. Proteomics Clin Appl 13, e1700166. [DOI] [PubMed] [Google Scholar]
- 124. Rachdaoui N, Li L, Willard B, Kasumov T, Previs S & Sarkar D (2017) Turnover of histones and histone variants in postnatal rat brain: effects of alcohol exposure. Clin Epigenetics 9, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Fowler CB, O'Leary TJ & Mason JT (2011) Protein mass spectrometry applications on FFPE tissue sections. Methods Mol Biol 724, 281–295. [DOI] [PubMed] [Google Scholar]
- 126. Noberini R, Longuespee R, Richichi C, Pruneri G, Kriegsmann M, Pelicci G & Bonaldi T (2017) PAT‐H‐MS coupled with laser microdissection to study histone post‐translational modifications in selected cell populations from pathology samples. Clin Epigenetics 9, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Sidoli S, Kori Y, Lopes M, Yuan ZF, Kim HJ, Kulej K, Janssen KA, Agosto LM, Cunha J, Andrews AJ et al. (2019) One minute analysis of 200 histone posttranslational modifications by direct injection mass spectrometry. Genome Res 29, 978–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Kriegsmann J, Kriegsmann M & Casadonte R (2015) MALDI TOF imaging mass spectrometry in clinical pathology: a valuable tool for cancer diagnostics (review). Int J Oncol 46, 893–906. [DOI] [PubMed] [Google Scholar]
- 129. Lahiri S, Sun N, Buck A, Imhof A & Walch A (2016) MALDI imaging mass spectrometry as a novel tool for detecting histone modifications in clinical tissue samples. Expert Rev Proteomics 13, 275–284. [DOI] [PubMed] [Google Scholar]
- 130. Pote N, Alexandrov T, Le Faouder J, Laouirem S, Leger T, Mebarki M, Belghiti J, Camadro JM, Bedossa P & Paradis V (2013) Imaging mass spectrometry reveals modified forms of histone H4 as new biomarkers of microvascular invasion in hepatocellular carcinomas. Hepatology 58, 983–994. [DOI] [PubMed] [Google Scholar]
- 131. Lahiri S, Sun N, Solis‐Mezarino V, Fedisch A, Ninkovic J, Feuchtinger A, Gotz M, Walch A & Imhof A (2016) In situ detection of histone variants and modifications in mouse brain using imaging mass spectrometry. Proteomics 16, 437–447. [DOI] [PubMed] [Google Scholar]
- 132. Munteanu B, Meyer B, von Reitzenstein C, Burgermeister E, Bog S, Pahl A, Ebert MP & Hopf C (2014) Label‐free in situ monitoring of histone deacetylase drug target engagement by matrix‐assisted laser desorption ionization‐mass spectrometry biotyping and imaging. Anal Chem 86, 4642–4647. [DOI] [PubMed] [Google Scholar]
- 133. Alberts D, Pottier C, Smargiasso N, Baiwir D, Mazzucchelli G, Delvenne P, Kriegsmann M, Kazdal D, Warth A, De Pauw E et al. (2018) MALDI imaging‐guided microproteomic analyses of heterogeneous breast tumors‐a pilot study. Proteomics Clin Appl 12. [DOI] [PubMed] [Google Scholar]
- 134. Vidova V & Spacil Z (2017) A review on mass spectrometry‐based quantitative proteomics: targeted and data independent acquisition. Anal Chim Acta 964, 7–23. [DOI] [PubMed] [Google Scholar]
- 135. Sidoli S, Lin S, Xiong L, Bhanu NV, Karch KR, Johansen E, Hunter C, Mollah S & Garcia BA (2015) Sequential window acquisition of all theoretical mass spectra (SWATH) analysis for characterization and quantification of histone post‐translational modifications. Mol Cell Proteomics 14, 2420–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Krautkramer KA, Reiter L, Denu JM & Dowell JA (2015) Quantification of SAHA‐dependent changes in histone modifications using data‐independent acquisition mass spectrometry. J Proteome Res 14, 3252–3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Pesavento JJ, Mizzen CA & Kelleher NL (2006) Quantitative analysis of modified proteins and their positional isomers by tandem mass spectrometry: human histone H4. Anal Chem 78, 4271–4280. [DOI] [PubMed] [Google Scholar]
- 138. Garcia BA, Pesavento JJ, Mizzen CA & Kelleher NL (2007) Pervasive combinatorial modification of histone H3 in human cells. Nat Methods 4, 487–489. [DOI] [PubMed] [Google Scholar]
- 139. Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A & Mann M (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 1, 376–386. [DOI] [PubMed] [Google Scholar]
- 140. Cuomo A, Moretti S, Minucci S & Bonaldi T (2011) SILAC‐based proteomic analysis to dissect the "histone modification signature" of human breast cancer cells. Amino Acids 41, 387–399. [DOI] [PubMed] [Google Scholar]
- 141. Noberini R & Bonaldi T (2017) A super‐SILAC strategy for the accurate and multiplexed profiling of histone posttranslational modifications. Methods Enzymol 586, 311–332. [DOI] [PubMed] [Google Scholar]
- 142. Lin S, Wein S, Gonzales‐Cope M, Otte GL, Yuan ZF, Afjehi‐Sadat L, Maile T, Berger SL, Rush J, Lill JR et al. (2014) Stable‐isotope‐labeled histone peptide library for histone post‐translational modification and variant quantification by mass spectrometry. Mol Cell Proteomics 13, 2450–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK & Hamon C (2003) Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem 75, 1895–1904. [DOI] [PubMed] [Google Scholar]
- 144. Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S et al. (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine‐reactive isobaric tagging reagents. Mol Cell Proteomics 3, 1154–1169. [DOI] [PubMed] [Google Scholar]
- 145. Glen A, Evans CA, Gan CS, Cross SS, Hamdy FC, Gibbins J, Lippitt J, Eaton CL, Noirel J, Wright PC et al. (2010) Eight‐plex iTRAQ analysis of variant metastatic human prostate cancer cells identifies candidate biomarkers of progression: an exploratory study. Prostate 70, 1313–1332. [DOI] [PubMed] [Google Scholar]
- 146. Zhang K, Schrag M, Crofton A, Trivedi R, Vinters H & Kirsch W (2012) Targeted proteomics for quantification of histone acetylation in Alzheimer's disease. Proteomics 12, 1261–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Drogaris P, Villeneuve V, Pomies C, Lee EH, Bourdeau V, Bonneil E, Ferbeyre G, Verreault A & Thibault P (2012) Histone deacetylase inhibitors globally enhance h3/h4 tail acetylation without affecting h3 lysine 56 acetylation. Sci Rep 2, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Gao J, Liao R, Yu Y, Zhai H, Wang Y, Sack R, Peters AH, Chen J, Wu H, Huang Z et al. (2014) Absolute quantification of histone PTM marks by MRM‐based LC‐MS/MS. Anal Chem 86, 9679–9686. [DOI] [PubMed] [Google Scholar]
- 149. Arnaudo AM, Molden RC & Garcia BA (2011) Revealing histone variant induced changes via quantitative proteomics. Crit Rev Biochem Mol Biol 46, 284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Loyola A, Bonaldi T, Roche D, Imhof A & Almouzni G (2006) PTMs on H3 variants before chromatin assembly potentiate their final epigenetic state. Mol Cell 24, 309–316. [DOI] [PubMed] [Google Scholar]
- 151. Siuti N, Roth MJ, Mizzen CA, Kelleher NL & Pesavento JJ (2006) Gene‐specific characterization of human histone H2B by electron capture dissociation. J Proteome Res 5, 233–239. [DOI] [PubMed] [Google Scholar]
- 152. Boyne MT 2nd, Pesavento JJ, Mizzen CA & Kelleher NL (2006) Precise characterization of human histones in the H2A gene family by top down mass spectrometry. J Proteome Res 5, 248–253. [DOI] [PubMed] [Google Scholar]
- 153. Thomas CE, Kelleher NL & Mizzen CA (2006) Mass spectrometric characterization of human histone H3: a bird's eye view. J Proteome Res 5, 240–247. [DOI] [PubMed] [Google Scholar]
- 154. Tvardovskiy A, Schwammle V, Kempf SJ, Rogowska‐Wrzesinska A & Jensen ON (2017) Accumulation of histone variant H3.3 with age is associated with profound changes in the histone methylation landscape. Nucleic Acids Res 45, 9272–9289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Tvardovskiy A, Wrzesinski K, Sidoli S, Fey SJ, Rogowska‐Wrzesinska A & Jensen ON (2015) Top‐down and middle‐down protein analysis reveals that intact and clipped human histones differ in post‐translational modification patterns. Mol Cell Proteomics 14, 3142–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Yi SJ & Kim K (2018) Histone tail cleavage as a novel epigenetic regulatory mechanism for gene expression. BMB Rep 51, 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Zhou BR, Jiang J, Feng H, Ghirlando R, Xiao TS & Bai Y (2015) Structural mechanisms of nucleosome recognition by linker histones. Mol Cell 59, 628–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Francavilla C, Lupia M, Tsafou K, Villa A, Kowalczyk K, Rakownikow Jersie‐Christensen R, Bertalot G, Confalonieri S, Brunak S, Jensen LJ et al. (2017) Phosphoproteomics of primary cells reveals druggable kinase signatures in ovarian cancer. Cell Rep 18, 3242–3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Noberini R, Morales Torres C, Savoia EO, Brandini S, Jodice MG, Bertalot G, Bonizzi G, Capra M, Diaferia G, Scaffidi P et al. (2020) Label‐free mass spectrometry‐based quantification of linker histone H1 variants in clinical samples. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Chen Y, Hoover ME, Dang X, Shomo AA, Guan X, Marshall AG, Freitas MA & Young NL (2016) Quantitative mass spectrometry reveals that intact histone H1 phosphorylations are variant specific and exhibit single molecule hierarchical dependence. Mol Cell Proteomics. 15, 818–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Deterding LJ, Bunger MK, Banks GC, Tomer KB & Archer TK (2008) Global changes in and characterization of specific sites of phosphorylation in mouse and human histone H1 Isoforms upon CDK inhibitor treatment using mass spectrometry. J Proteome Res 7, 2368–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Wisniewski JR, Zougman A, Kruger S & Mann M (2007) Mass spectrometric mapping of linker histone H1 variants reveals multiple acetylations, methylations, and phosphorylation as well as differences between cell culture and tissue. Mol Cell Proteomics 6, 72–87. [DOI] [PubMed] [Google Scholar]
- 163. El Kennani S, Adrait A, Permiakova O, Hesse AM, Ialy‐Radio C, Ferro M, Brun V, Cocquet J, Govin J & Pflieger D (2018) Systematic quantitative analysis of H2A and H2B variants by targeted proteomics. Epigenetics Chromatin 11, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Onder O, Sidoli S, Carroll M & Garcia BA (2015) Progress in epigenetic histone modification analysis by mass spectrometry for clinical investigations. Expert Rev Proteomics 12, 499–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Sundar IK, Nevid MZ, Friedman AE & Rahman I (2014) Cigarette smoke induces distinct histone modifications in lung cells: implications for the pathogenesis of COPD and lung cancer. J Proteome Res 13, 982–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Zhang K, Li L, Zhu M, Wang G, Xie J, Zhao Y, Fan E, Xu L & Li E (2015) Comparative analysis of histone H3 and H4 post‐translational modifications of esophageal squamous cell carcinoma with different invasive capabilities. J Proteomics 112, 180–189. [DOI] [PubMed] [Google Scholar]
- 167. Harshman SW, Hoover ME, Huang C, Branson OE, Chaney SB, Cheney CM, Rosol TJ, Shapiro CL, Wysocki VH, Huebner K et al. (2014) Histone H1 phosphorylation in breast cancer. J Proteome Res 13, 2453–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Jaffe JD, Wang Y, Chan HM, Zhang J, Huether R, Kryukov GV, Bhang HE, Taylor JE, Hu M, Englund NP et al. (2013) Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat Genet 45, 1386–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Zheng Y, Fornelli L, Compton PD, Sharma S, Canterbury J, Mullen C, Zabrouskov V, Fellers RT, Thomas PM, Licht JD et al. (2015) Unabridged analysis of human histone H3 by differential top‐down mass spectrometry reveals hypermethylated proteoforms from MMSET/NSD2 overexpression. Mol Cell Proteomics 15, 776–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Pham V, Pitti R, Tindell CA, Cheung TK, Masselot A, Stephan JP, Guler GD, Wilson C, Lill J, Arnott D et al. (2020) Proteomic analyses identify a novel role for EZH2 in the initiation of cancer cell drug tolerance. J Proteome Res 19, 1533–1547. [DOI] [PubMed] [Google Scholar]
- 171. Leroy G, Dimaggio PA, Chan EY, Zee BM, Blanco MA, Bryant B, Flaniken IZ, Liu S, Kang Y, Trojer P et al. (2013) A quantitative atlas of histone modification signatures from human cancer cells. Epigenetics Chromatin 6, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Nestor CE, Ottaviano R, Reinhardt D, Cruickshanks HA, Mjoseng HK, McPherson RC, Lentini A, Thomson JP, Dunican DS, Pennings S et al. (2015) Rapid reprogramming of epigenetic and transcriptional profiles in mammalian culture systems. Genome Biol 16, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Sidoli S & Garcia BA (2017) Middle‐down proteomics: a still unexploited resource for chromatin biology. Expert Rev Proteomics 14, 617–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Sidoli S, Schwammle V, Ruminowicz C, Hansen TA, Wu X, Helin K & Jensen ON (2014) Middle‐down hybrid chromatography/tandem mass spectrometry workflow for characterization of combinatorial post‐translational modifications in histones. Proteomics 14, 2200–2211. [DOI] [PubMed] [Google Scholar]
- 175. Noberini R, Sigismondo G & Bonaldi T (2016) The contribution of mass spectrometry‐based proteomics to understanding epigenetics. Epigenomics 8, 429–445. [DOI] [PubMed] [Google Scholar]