

Abstract
Congenital disorder of glycosylation type Ig (ALG12‐CDG) is a rare inherited metabolic disease caused by a defect in alpha‐mannosyltransferase 8, encoded by the ALG12 gene (22q13.33). To date, only 15 patients have been diagnosed with ALG12‐CDG globally. Due to a newborn Slovak patient's clinical and biochemical abnormalities, the isoelectric focusing of transferrin was performed with observed significant hypoglycosylation typical of CDG I. Furthermore, analysis of neutral serum N‐glycans by mass spectrometry revealed the accumulation of GlcNAc2Man5–7 and decreased levels of GlcNAc2Man8–9, which indicated impaired ALG12 enzymatic activity. Genetic analysis of the coding regions of the ALG12 gene of the patient revealed a novel homozygous substitution mutation c.1439T>C p.(Leu480Pro) within Exon 10. Furthermore, both of the patient's parents and his twin sister were asymptomatic heterozygous carriers of the variant. This comprehensive genomic and glycomic approach led to the confirmation of the ALG12 pathogenic variant responsible for the clinical manifestation of the disorder in the patient described.
Keywords: ALG12‐CDG, CDG Ig, N‐glycans
1. INTRODUCTION
Congenital disorders of glycosylation (CDGs) comprise a set of at least 150 genetically and clinically heterogeneous diseases caused by defects in various steps of glycan modification pathways (Verheijen et al., 2020; Brucker et al., 2020; Ng & Freeze, 2018). Most of the secreted molecules are glycosylated (Varki et al., 2009), all human organs/systems are equipped with glycoconjugates, and glycosylation process occurs in every cell. This explains why most CDG diseases involve multiple organs (Marques‐da‐Silva et al., 2017). Furthermore, CDGs are often challenging to diagnose because of the considerable variability of clinical features that characterize this set of diseases (Cylwik et al., 2013).
ALG12‐CDG (OMIM#607143) is a rare disease with autosomal recessive inheritance caused by either a homozygous or compound heterozygous mutation in the ALG12 (22q13.33) gene that encodes the dolichyl‐P‐mannose Man‐7‐GlcNAc‐2‐PP‐dolichyl‐alpha‐6‐mannosyltransferase. This enzyme transfers the eighth mannose residue from dolichyl‐P‐mannose to lipid‐linked oligosaccharides. To date, only 15 ALG12‐CDG patients have been reported worldwide (Chantret et al., 2002; de la Morena‐Barrio et al., 2020; Di Rocco et al., 2005; Eklund et al., 2005; Esfandiari et al., 2019; Grubenmann et al., 2002; Kranz et al., 2007; Murali et al., 2014; Sturiale et al., 2019; Tahata et al., 2019; Thiel et al., 2002; Zdebska et al., 2003). These patients are of different ethnic origins and share some common CDG features. However, significant individual variations in symptoms and severity of disease have been observed (Table S1).
Here, we describe a patient carrying a homozygous pathogenic variant in the ALG12 gene. Clinical and biochemical findings, complemented by N‐glycan analysis and genome sequencing, confirmed the first identified ALG12‐CDG patient of Slovak origin.
2. MATERIAL AND METHODS
2.1. Editorial policies and ethical considerations
Written informed consent was given by parents, and the experimental study protocol was reviewed and approved by the hospital ethics committee.
2.2. Samples
Blood samples from the suspected ALG12‐CDG patient, his asymptomatic twin sister, both of the parents, and two other healthy newborns were obtained from the National Institute of Children's Diseases, Bratislava, Slovakia, in accordance with standard operating procedures. The samples were processed under the same conditions. Serum and blood samples were stored at −20°C immediately after the collection.
2.3. Isoelectric focusing of serum transferrin
Screening for protein N‐glycosylation by transferrin (Tf) via isoelectric focusing (IEF) was performed at the National Institute of Children's Diseases, Bratislava, Slovakia using PhastSystem (GE Healthcare, Chicago, IL) equipment, as described previously (de Jong & van Eijk, 1988; Hackler et al., 1995). Briefly, iron‐saturated serum was applied to a rehydrated PhastGel matrix that ranged from pH 5 to 8. After the separation process, Tf isoforms were immunofixed using polyclonal anti‐Tf rabbit antibodies (Dako, Glostrup, Denmark). The remaining serum proteins were removed by washing, and Tf immunocomplexes were stained with PhastGel Blue R‐350. The resulting pattern was compared to that of the negative control.
2.4. Analysis of serum N‐glycans by matrix‐assisted laser desorption/ionization time‐of‐flight (MALDI TOF) mass spectrometry (MS)
Ten microliters of serum were premixed with 40 μl of 10 mM Tris buffer containing 0.1% sodium dodecyl sulfate, pH 7.5. Serum proteins were alkylated and reduced using 10 mM dithiothreitol and 25 mM iodoacetamide before the addition of 1 U of peptide‐N‐glycosidase (PNG‐ase F; Roche Diagnostics GmbH, Mannheim, Germany). After overnight incubation at 37°C, released N‐glycans were isolated using the Supelclean ENVI‐Carb SPE system (Supelco/Sigma Aldrich, Bellefonte, PA), and the neutral glycan fraction was eluted using 40% acetonitrile (ACN, v/v) (Hykollari et al., 2013). The fraction was lyophilized, and free N‐glycans were directly analyzed by MS in reflectron positive ion mode using the UltrafleXtreme MALDI mass spectrometer (Bruker Daltonics, Billerica, MA). A 20 mg/ml 2,5‐dihydroxybenzoic acid (DHB) solution in 30% ACN supplemented with 0.1% trifluoroacetic acid (TFA) and 1 mM NaOH was used as a matrix solution. MALDI TOF data were processed and interpreted by software programs FlexAnalysis and ProteinScape with GlycomeDB glycan database (Bruker Daltonics; http://www.glycome-db.org). Patient's sample was analyzed in three technical replicates, a serum of asymptomatic sister and two age‐matching healthy individuals served as negative controls.
To provide additional quantitative data, stable isotope labeling was performed. Briefly, 150 μl of a homogenous mixture of NaOH in DMSO was added to samples. Each reaction was initiated by adding 150 μl 12C‐ or 13C‐iodomethane to permethylate neutral N‐glycans from the patient or negative control. The reaction was terminated by the addition of ice‐cold water, and permethylated N‐glycans were extracted with chloroform, dried at room temperature, and dissolved in 50% methanol (Palmigiano et al., 2018). The two samples were premixed and analyzed from a single MALDI target plate spot as described above.
2.5. Sanger sequencing
Genomic DNA was extracted from the whole blood samples from the patient, his twin sister, and his parents. Primers were designed to amplify the entire coding region of the ALG12 gene (Exons 2–10). Standard polymerase chain reactions (PCR, 25 μl) used 60–100 ng of template DNA, conditions included 7 min at 95°C, followed by 40 cycles of 30 s at 95°C, 30 s at 58°C, and 40 s at 72°C. The reaction was completed by the final elongation step for 7 min at 72°C. PCR products of the expected size were sequenced using a 3500 Genetic Analyzer (Applied Biosystems, Foster City, CA). Primers used in these experiments are described in Table S2.
2.6. Clinical exome sequencing
Coding exons of 4.490 Online Mendelian Inheritance in Man (OMIM)‐listed genes were enriched from 200 ng of blood‐derived genomic DNA using a clinical exome solution (CES) sequencing panel (Sophia Genetics SA, Saint Sulpice, Switzerland) according to the manufacturer's instructions. Sequencing was performed using an Illumina MiSeq sequencer (Illumina, San Diego, CA). Reads were aligned to the reference genome (GRCh37/hg19), and the variant calling and clinical interpretation of data were performed using the SOPHIA DDM platform (Sophia Genetics SA).
3. RESULTS
3.1. Clinical phenotype
Our study reports a case of a dysmorphic male born prematurely, after a complicated pregnancy, as a second child from binovular twins. Comprehensive clinical phenotype was described separately (Lekka et al., 2021). Newborn screening for cystic fibrosis was repeatedly false positive. Bleeding, thrombocytopenia, and hypocoagulation led to the continuous substitution of plasma and antithrombin III. Hypoglycemia, mild unconjugated hyperbilirubinemia, and elevated hepatic enzyme levels with subsequent hepatomegaly occurred. Chronic hypoproteinemia and hypogammaglobulinemia required the regular parenteral substitution of albumin and human immunoglobulins. Cardiological findings indicated the progressively developed hypertrophic cardiomyopathy, and neurological findings revealed axial hypotonia and acral hypertonia. Magnetic resonance imaging of the brain confirmed the presence of an arteriovenous malformation of the vena Galeni magna, delayed white matter myelination, and hypoplasia of optic nerves. Hepatomegaly, edemas, and tachycardia persisted, while septic fevers, oliguria, and enteral uptake failure complicated the patient's condition. The patient died at the age of 164 days.
3.2. IEF of serum Tf
Described clinical symptoms resembled defects associated with CDG‐type disorders. Therefore, IEF of serum Tf was performed (Figure 1). Findings revealed a pattern typical for patients affected by CDG type I (CDG I), and consisted of increased disialo‐ and asialo‐Tf, along with mildly decreased levels of tetra‐ and penta‐sialo‐Tf. Although IEF suggested CDG I, the precise subtype remained unclear. To determine whether the N‐glycan synthesis disorder was general or Tf‐specific, an N‐glycoprofile assessment of the patient serum was performed.
FIGURE 1.
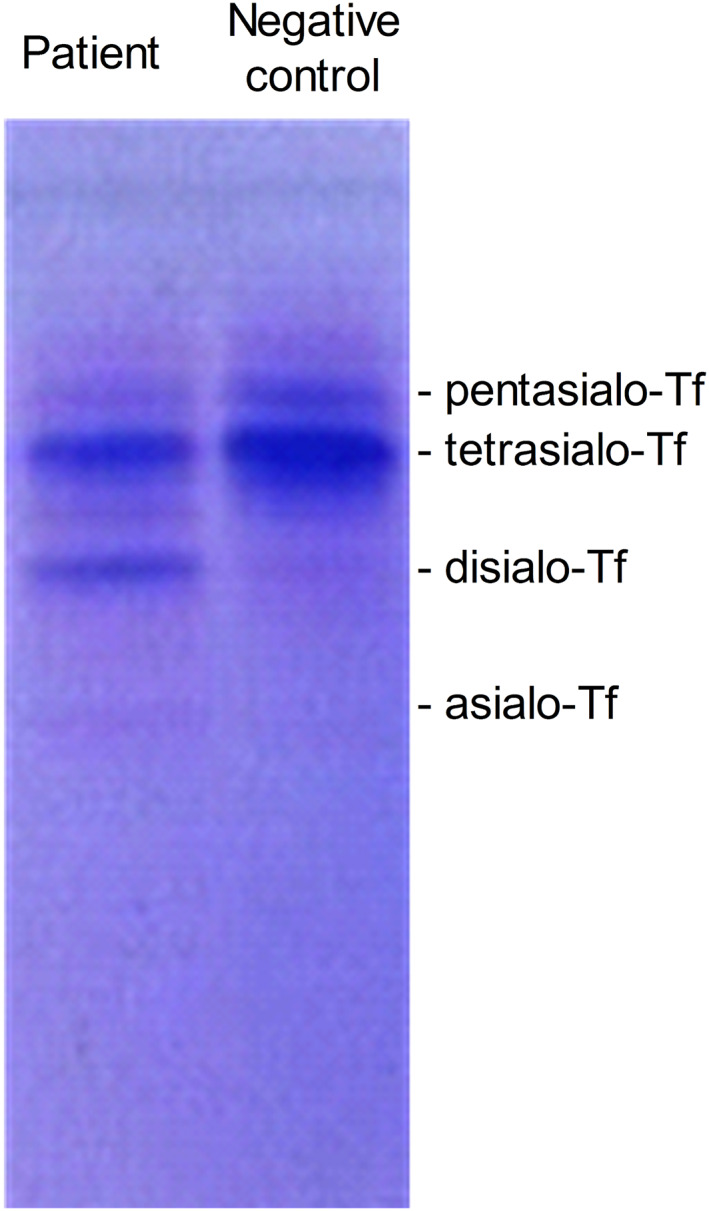
Modified serum transferrin glycosylation pattern determined by isoelectric focusing (IEF) suggested the diagnosis of congenital disorders of glycosylation type I (CDG I) in the patient. Isoelectric focusing of transferrin (Tf) from the patient and negative control with marked individual sialylated protein bands. In the patient's sample, increased disialo‐ and asialo‐Tf along with mildly decreased levels of tetra‐ and pentasialo‐Tf are consistent with CDG I profile
3.3. Qualitative and quantitative analyses of N‐glycans
Analysis of serum N‐glycans revealed significant aberrations, primarily observed in high‐mannose structures collected in neutral fraction of the N‐glycan pool. The N‐glycoprofile of the patient differed from the negative controls concerning the distribution of individual glycan structures. Increased levels of GlcNAc2Man5–7 and decreased levels of GlcNAc2Man8–9 and GlcNAc2Man9Glc1 were observed in the spectrum of free, underivatized N‐glycans released from the patient's serum (Figure 2a). Quantification of individual N‐glycan structures, based on the average values of relative intensities, calculated from three technical replicates of the patient's sample or three age‐matching negative controls, is shown in Figure 2b. In the patient's sample, the relative intensities corresponding to GlcNAc2Man5–7 were 1.3–2.2‐fold higher than in negative controls. Furthermore, the relative intensities of GlcNAc2Man8 and GlcNAc2Man9 were 3.4‐ and 22.4‐fold lower than negative controls, respectively. GlcNAc2Man9Glc1 appeared to be almost absent from the N‐glycoprofile of the patient.
FIGURE 2.
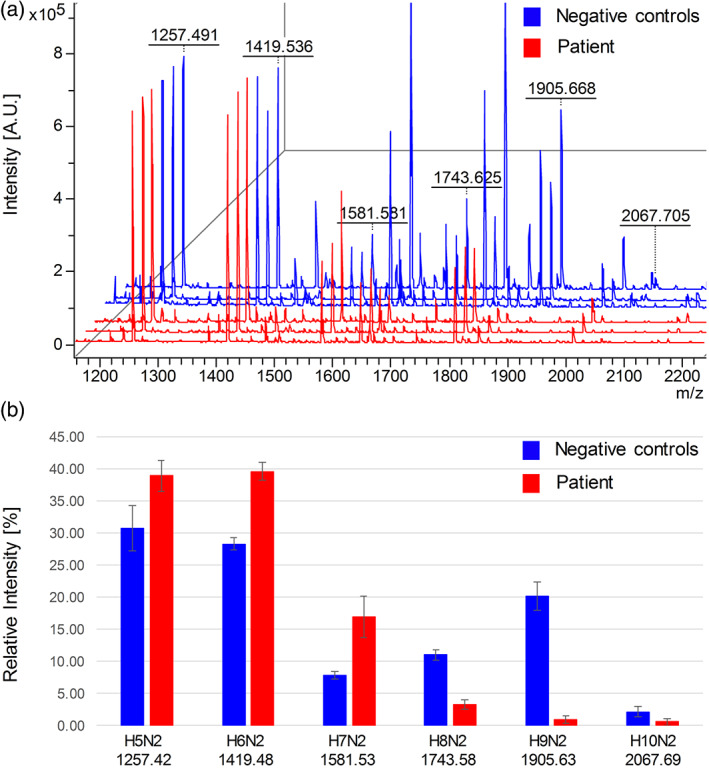
Decreased levels of serum GlcNAc2Man8–9 and GlcNAc2Man9Glc1 indicated ALG12 dysfunction. (a) MALDI TOF analysis of the neutral fraction of free, underivatized N‐glycans released from three technical replicates of patient's serum (red) and three age‐matching negative controls (blue) revealed decreased signals corresponding to GlcNAc2Man8–9 (marked as H8N2 and H9N2) and GlcNAc2Man9Glc1 (marked as H10N2) as well as increased signals corresponding to GlcNAc2Man5–7 (marked as H5N2–H7N2) in the patient's N‐glycome. (b) Average values and standard deviations of relative intensities of indicated high‐mannose N‐glycan structures from (a). Depicted m/z values in (a) and (b) are experimental and theoretical [M + Na]+, respectively
In the attempt to obtain structural information on accumulated GlcNAc2Man7 (m/z 1581.5), MS/MS fragmentation analysis was performed (Figure S1a). The oligosaccharide composition was unambiguously assigned as Hex7HexNAc2 (H7N2); however, a precise isomeric configuration could not be determined. Database search results of the relevant human glycan structures revealed two main isomeric glycans with similar score values. A slightly higher score corresponded to the core N‐glycan with linear A‐ and B‐arms (Figure S1b), and the second structure corresponded to the core N‐glycan with A‐, B‐, and C‐arms (Figure S1c).
To provide an additional quantitative comparison between individual high‐mannose structures, a stable‐isotope labeling strategy was applied. N‐glycans from the patient and negative control (asymptomatic sister) were permethylated with 12C‐ and 13C‐iodomethane, respectively. Labeled N‐glycans from both samples were premixed and analyzed directly from a single MALDI target plate spot. Absolute signal intensities corresponding to structures of interest confirmed the results obtained from the analysis of free N‐glycans (Figure S2).
3.4. Genomics analysis
Based on MS data, the N‐glycosylation pathway was impaired at the step of converting GlcNAc2Man7 to GlcNAc2Man8, catalyzed by alpha‐mannosyltransferase 8, and encoded by ALG12 gene. DNA analysis was performed to determine the sequence of the coding region of the ALG12 gene of the patient, his parents, and sister (asymptomatic twin). The patient's sample possessed a homozygous variant in Exon 10, which was heterozygous in both of his parents and sister (Figure 3). The variant c.1439T>C results in a p.(Leu480Pro) substitution within the C‐terminal part of the ALG12 protein. The missense mutation, recently reported within the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/), was predicted to be likely pathogenic (submission accession number: SCV000927994.1) and has a global minor allele frequency of 4 × 10−6. According to the Exome Aggregation Consortium (ExAC/gnomAD) database (https://gnomad.broadinstitute.org/), no known biallelic form of the variant exists to date. Based on the ACMG/AMP criteria (Nykamp et al., 2017; Rehm, 2017; Richards et al., 2015), results from various prediction software (Mutation taster, http://www.mutationtaster.org/; PolyPhen, http://genetics.bwh.harvard.edu/pph2/; Provean/SIFT, http://provean.jcvi.org/index.php; and UMD_Predictor, http://umd-predictor.eu/) and the occurrence of the mutated allele in the population (approx. 1:250,000), we concluded that the variant is responsible for the manifestation of disease (Table S3).
FIGURE 3.
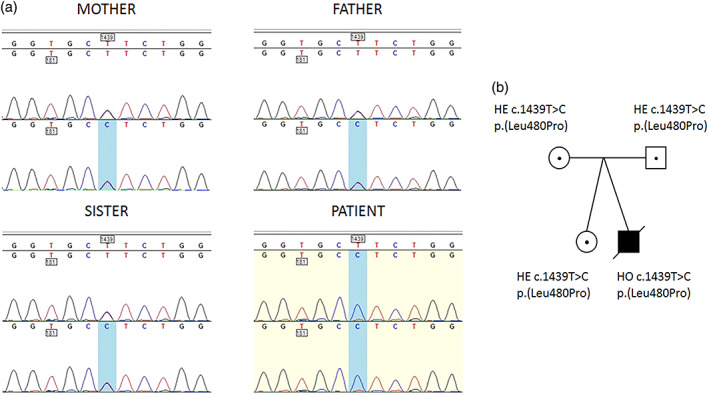
An autosomal recessive mutation in ALG12 gene of the patient. (a) Sequence alignment of PCR‐amplified genomic DNA fragments of the mother, father, asymptomatic twin sister, and patient. The mother, father, and sister are heterozygous, and the patient is homozygous for a T1439C transition, which leads to the substitution of leucine 480 with proline. (b) Pedigrees of family members based on the presence of T1439C
According to Clinvar, the patient was also homozygous in three other benign, or likely benign variants. The first was synonymous variant c.885A>G p.(Ala295=) in Exon 7 (rs8135963, benign). The other two variants, c.664+9G>C and c.664+12A>G (rs9616368, rs9616204), were located within the intron between Exons 5 and 6 were predicted to be benign and likely benign, respectively.
To provide an additional genetic evidence for a critical role of the discovered ALG12 mutation in pathogenesis, and to exclude any other possible genetic variants that could be responsible for aberant glycoprofile or phenotype, we performed the sequencing analysis of the clinical exome (CES by Sophia Genetics SA). For a more detailed analysis of CDG‐related genes, we created a virtual panel using Sophia DDM software. We focused on both homozygous and heterozygous variants of the gene, since inheritance of the majority of CDG diseases is autosomal recessive, and compound heterozygosity is frequently observed. From selected genes, no variants of clinical relevance were identified in the patient sample. Based on these results, we can link the identified homozygous variant within Exon 10 of ALG12 with the phenotypic manifestation of disease and biochemical changes in glycosylation in the patient.
4. DISCUSSION
This report describes the first Slovak patient with ALG12‐CDG, which brings the total number of published cases of this subtype to 16. Clinical symptoms, prenatal history, and cardiorespiratory, gastrointestinal, musculoskeletal findings, together with the biochemical assessment applied in each published ALG12‐CDG case, are summarized in Table S1. Most ALG12‐CDG patients were born preterm, and most pregnancies were complicated. Every ALG12‐CDG patient possessed characteristic dysmorphism, psychomotor retardation, hypotonia, and/or skeletal abnormalities. Most patients suffered from feeding difficulties, respiratory distress, and frequent infections. Furthermore, IgG levels of patients were low. Typical biochemical findings consisted of decreased coagulation factors and cholesterol levels and increased transaminase levels. Hypoglycemia is another finding associated with ALG12‐CDG diagnosis. According to the summarized data, clinical and biochemical findings of the patient described in this report corresponded to the previously described ALG12‐CDG patients.
This study presents ALG12‐CDG diagnostics based on the analysis of the neutral N‐glycans attached to serum proteins by using MALDI‐TOF. The spectrum acquired from the neutral fraction of serum N‐glycoprofile appeared to be characteristic of ALG12‐CDG. We observed significantly decreased GlcNAc2Man8–9 and GlcNAc2Man9Glc1 structures (Figure 2a,b). Accumulation of the GlcNAc2Man7 oligosaccharide was also observed in the spectra of serum N‐glycans, which is indicative of ALG12‐CDG. Due to the short lifespan of the presented patient, only two serum sample collections were performed. However, both revealed an unambiguously pathological IEF Tf profile indicating CDG I. Analysis of serum N‐glycoprofile by MS immediately confirmed the suspected diagnosis.
Although the N‐glycoprofile of serum glycoproteins of the patient was abnormal, some complex glycans were formed (data not shown). The presence of abnormal N‐glycan structures on secreted glycoproteins suggests that aberrantly glycosylated glycoproteins may escape ER‐associated quality control (ERQC) mechanisms (Davis et al., 2017). The discovery of increased levels of GlcNAc2Man7 attached to serum proteins, which presumably originated from incomplete lipid‐linked oligosaccharide synthesis, is in accordance with the existence of side pathways, which are used to add glycan intermediates to the proteins or indicates that the specificity of oligosaccharidyltransferase enzymes is broader than initially expected. Based on experiments conducted in fibroblasts from ALG12‐CDG patients, it is known that, in addition to the mature core Dol‐PP‐GlcNAc2Man9Glc3, two other oligosaccharide structures are transferred in approximately equal amounts from dolichol onto glycoproteins: GlcNAc2Man7Glc3 and GlcNAc2Man7 (Chantret et al., 2002; Sturiale et al., 2019).
The mechanism mentioned above and previously published studies of ALG12‐CDG explain the observation of both underoccupancy of glycosylation sites (CDG I) and glycan processing defects (CDG type II [CDG II]), thus ALG12‐CDG having the features of a dual/combined CDG (CDG I and II) (Sturiale et al., 2019). Our data support this presumption; however, N‐glycan analysis by MS was targeted to high‐mannose structures (neutral fraction of serum N‐glycans), and complex N‐glycans, forming in the Golgi apparatus during the processing pathway, were not quantified.
Immunodeficiency and low serum IgG levels are considered as a hallmark of this disease (Pascoal et al., 2020). The majority of the ALG12‐CDG patients experience severe infections associated with hypogammaglobinemia and B‐cell dysfunction. This may be due to deficient N‐glycosylation of immunoglobulins, which is necessary for their effector functions such as complement binding and antibody‐dependent cell cytotoxicity and pathogen phagocytosis (Anthony & Ravetch, 2010; Monticelli et al., 2016). Thus, parenteral IgG substitution administered to the presented patient posed a limitation of IgG N‐glycoprofile study, and no significant alterations in IgG N‐glycoprofile were observed (data not shown). In previously published ALG12‐CDG case, MALDI MS showed the presence of additional signals in IgG N‐glycoprofile, that is, the mono‐antennary sialylated species and the hybrid structures bearing one and two mannose units; an overall decrease of the agalactosylated structures, increase of the di‐galactosylated structure, and an increase of the mono‐ and di‐sialylated structures (Sturiale et al., 2019).
Eighteen different mutations in ALG12 have been previously reported to cause ALG12‐CDG, four introducing a premature stop codon, and 14 others leading to amino acid changes. The presented patient had a homozygous transition mutation (c.1439T>C), resulting in the substitution of leucine to proline at position 480. The same variant was first reported by Sturiale et al. and, along with another compound heterozygous variant identified in the patient, was assumed to be responsible for the clinical manifestation of ALG12‐CDG disorder (Sturiale et al., 2019). The function of the C‐terminal part of ALG12 has not yet been elucidated. However, its importance is highlighted by examination of the pathogenic variant p.Y414Stop* (Thiel et al., 2002) that causes a premature insertion of the stop codon and results in the loss of 75 C‐terminal amino acid residues. The C‐terminal domain may be involved in the enzyme's catalytic activity or protein–protein interactions with other glycosylation enzymes within the synthetic pathway.
Decreased signal corresponding to GlcNAc2Man8 is a reliable and specific biomarker of ALG12‐CDG. However, not all pathogenic variants have such a significant impact on the function of proteins, and therefore, some ALG12 cases produce GlcNAc2Man8 or even higher oligosaccharides in sufficient amounts. We hypothesize that the more impaired enzyme activity, the more severe phenotypic presentation of ALG12‐CDG, and thus the lower the signals corresponding to GlcNAc2Man8 in the serum glycoprofile of the patient will be observed.
The correlation between genotype and phenotype of ALG12‐CDG patients varies dramatically, as demonstrated by the summary of all previously published cases, including the one presented in this study (Table S1). In some patients, compound heterozygosity (Thiel et al.2002; Grubenmann et al., 2002; Eklund et al., 2005; Kranz et al., 2007; Murali et al., 2014; Tahata et al., 2019; Sturiale et al., 2019) of mutations in ALG12 gene was observed, and some of these patients are still living, however, with respective disabilities (growth retardation, developmental disability, etc.). Their therapy is mainly symptomatic, and with adequate attention from physicians, they can reach normal adulthood. On the contrary, one ALG12‐CDG patient with homozygous variant c.77T>A p.(Val26Asp) was reported as an unusual adult patient without facial dysmorphism or intellectual disability (de la Morena‐Barrio et al., 2020). There is thus an important heterogeneity in the degree of severity and the phenotypic spectrum of this CDG. This phenomenon is not exceptional in CDG. In PMM2‐CDG, the phenotypes range from neonatal mortality to near‐normal adulthood (de la Morena‐Barrio et al., 2020; Grunewald, 2009). As described in the presented study, homozygous pathogenic mutation possesses a serious and life‐threatening condition of the patient that suffered from an early death. However, further research is needed to enhance our understanding of the mechanism by which identified amino acid substitutions affect ALG12 enzymatic activity and produce pathologic phenotypes.
5. CONCLUSIONS
This study characterized the clinical, biochemical, genomic, and glycomic features of the first ALG12‐CDG patient identified in Slovakia. Characteristics of the patient, including psychomotor retardation, decreased levels of IgG, and coagulopathy, together with typical Tf IEF pattern, indicated that the patient was likely afflicted with CDG I. Subsequent analysis of serum N‐glycans revealed reduced α‐mannosyltransferase 8 (ALG12) activity. We have identified a homozygous variant c.1439T>C p.(Leu480Pro) in Exon 10 of the ALG12 gene that caused the clinical manifestation of ALG12‐CDG disorder in the patient. This study demonstrates the efficacy of the combined multi‐OMICs approach, where the synergy between glycan structure analysis and genomic data interpretation leads to comprehensive and personalized diagnostics.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
AUTHOR CONTRIBUTIONS
Jana Ziburová, Gabriela Hrčková, and Sergej Šesták performed DNA sequencing and genomic data analysis. Marek Nemčovič, Zuzana Pakanová and Ján Mucha performed analyses of N‐glycoprofiles by mass spectrometry. Jana Bellová and Barbara Siváková analyzed the mass spectrometric data. Anna Šalingová, Claudia Šebová, and Mária Ostrožlíková performed selective screening based on IEF. Clinicians Vladimír Bzdúch, Anna Hlavatá, Jana Brucknerová, Dimitra‐Evanthia Lekka, and Ingrid Brucknerová evaluated the clinical parameters and observed phenotype. Martina Skokňová and Alexandra McCullough initiated CDG diagnostics. Jana Ziburová drafted the first version of the manuscript and Peter Baráth and Zuzana Pakanová critically revised the data and edited the manuscript. All authors provided edits and approved the final version of the manuscript.
Supporting information
Table S1 Summary of described pathogenic ALG12 gene variants and their clinical and biochemical parameters
Table S2 List of PCR and sequencing primers used in analysis human ALG12 exomes
Table S3 Summary of the results from in silico analysis of the substitution mutation c.1439T>C p.(Leu480Pro) in human ALG12/ALG12 using various prediction software programs and databases with attached result files
Figure S1 MALDI TOF/TOF fragmentation analysis of H7N2 (m/z 1581.5) N‐glycan accumulated in the patient sample (a). Annotations corresponding to two isomeric human glycan structures with the highest scores using GlycomeDB are in (b) and (c). H, hexose; N, N‐acetylglucosamine
Figure S2 A quantitative comparison of absolute signal intensities produced by high‐mannose structures from patient (red) and control (blue) samples after the permethylation using 12C‐ and 13C‐iodomethane (stable isotope labeling). H, hexose; N, N‐acetylglucosamine
ACKNOWLEDGMENTS
We thank to the family of the patient for the collaboration and kindness. This work was supported by the Slovak Research and Development Agency under Contract nos. APVV‐17‐0300 and APVV‐18‐0336. Financial support was provided from VEGA 2/0060/21 and the Ministry of Health of the Slovak Republic with no. 2019/7‐CHÚ SAV‐4. This publication was created with the support of the Operational Program Integrated Infrastructure for the project study of structural changes of complex glycoconjugates in the process of inherited metabolic and civilization diseases, ITMS: 313021Y920, co‐financed by the European Regional Development Fund.
Ziburová, J. , Nemčovič, M. , Šesták, S. , Bellová, J. , Pakanová, Z. , Siváková, B. , Šalingová, A. , Šebová, C. , Ostrožlíková, M. , Lekka, D.‐E. , Brucknerová, J. , Brucknerová, I. , Skokňová, M. , Mc Cullough, A. , Hrčková, G. , Hlavatá, A. , Bzdúch, V. , Mucha, J. , & Baráth, P. (2021). A novel homozygous mutation in the human ALG12 gene results in an aberrant profile of oligomannose N‐glycans in patient's serum. American Journal of Medical Genetics Part A, 185A:3494–3501. 10.1002/ajmg.a.62474
Funding information Agentúra na Podporu Výskumu a Vývoja, Grant/Award Numbers: APVV‐17‐0300, APVV‐18‐0336; European Regional Development Fund, Grant/Award Number: ITMS: 313021Y920; Ministry of Health of the Slovak Republic, Grant/Award Number: 2019/7‐CHÚ SAV‐4; Vedecká Grantová Agentúra MŠVVaŠ SR a SAV, Grant/Award Number: 2/0060/21
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Anthony, R. M. , & Ravetch, J. V. (2010). A novel role for the IgG Fc glycan: The anti‐inflammatory activity of sialylated IgG Fcs. Journal of Clinical Immunology, 30(Suppl. 1), S9–S14. [DOI] [PubMed] [Google Scholar]
- Brucker, W. J. , Croteau, S. E. , Prensner, J. R. , Cullion, K. , Heeney, M. M. , Lo, J. , McAlvin, J. B. , Peeler, K. , Shah, N. , Yee, C. S. K. , Berry, G. T. , & Bodamer, O. (2020). An emerging role for endothelial barrier support therapy for congenital disorders of glycosylation. Journal of Inherited Metabolic Disease, 43, 880–890. 10.1002/jimd.12225 [DOI] [PubMed] [Google Scholar]
- Chantret, I. , Dupré, T. , Delenda, C. , Bucher, S. , Dancourt, J. , Barnier, A. , Charollais, A. , Heron, D. , Bader‐Meunier, B. , Danos, O. , Seta, N. , Durand, G. , Oriol, R. , Codogno, P. , & Moore, S. E. H. (2002). Congenital disorders of glycosylation type Ig is defined by a deficiency in dolichyl‐P‐mannose:Man7GlcNAc2‐PP‐dolichyl mannosyltransferase. The Journal of Biological Chemistry, 277, 25815–25822. 10.1074/jbc.M203285200 [DOI] [PubMed] [Google Scholar]
- Cylwik, B. , Naklicki, M. , Chrostek, L. , & Gruszewska, E. (2013). Congenital disorders of glycosylation. Part I. Defects of protein N‐glycosylation. Acta Biochimica Polonica, 60, 151–161. [PubMed] [Google Scholar]
- Davis, K. , Webster, D. , Smith, C. , Jackson, S. , Sinasac, D. , Seargeant, L. , Wei, X. C. , Ferreira, P. , Midgley, J. , Foster, Y. , Li, X. , He, M. , & al‐Hertani, W. (2017). ALG9‐CDG: New clinical case and review of the literature. Molecular Genetics and Metabolism Reports, 13, 55–63. 10.1016/j.ymgmr.2017.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong, G. , & van Eijk, H. G. (1988). Microheterogeneity of human serum transferrin: A biological phenomenon studied by isoelectric focusing in immobilized pH gradients. Electrophoresis, 9, 589–598. 10.1002/elps.1150090921 [DOI] [PubMed] [Google Scholar]
- de la Morena‐Barrio, M. E. , Sabater, M. , de la Morena‐Barrio, B. , Ruhaak, R. L. , Miñano, A. , Padilla, J. , Toderici, M. , Roldán, V. , Gimeno, J. R. , Vicente, V. , & Corral, J. (2020). ALG12‐CDG: An unusual patient without intellectual disability and facial dysmorphism, and with novel variant. Molecular Genetics & Genomic Medicine, 8, e1304. 10.1002/mgg3.1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rocco, M. , Hennet, T. , Grubenmann, C. E. , Pagliardini, S. , Allegri, A. E. M. , Frank, C. G. , Aebi, M. , Vignola, S. , & Jaeken, J. (2005). Congenital disorder of glycosylation (CDG) Ig: Report on a patient and review of the literature. Journal of Inherited Metabolic Disease, 28, 1162–1164. 10.1007/s10545-005-0137-3 [DOI] [PubMed] [Google Scholar]
- Eklund, E. A. , Newell, J. W. , Sun, L. , Seo, N. S. , Alper, G. , Willert, J. , & Freeze, H. H. (2005). Molecular and clinical description of the first US patients with congenital disorder of glycosylation Ig. Molecular Genetics and Metabolism, 84, 25–31. 10.1016/j.ymgme.2004.09.014 [DOI] [PubMed] [Google Scholar]
- Esfandiari, H. , Mets, M. B. , Kim, K. H. , & Kurup, S. P. (2019). Ocular abnormalities in a patient with congenital disorder of glycosylation type Ig. Ophthalmic Genetics, 40, 549–552. 10.1080/13816810.2019.1692361 [DOI] [PubMed] [Google Scholar]
- Grubenmann, C. E. , Frank, C. G. , Kjaergaard, S. , Berger, E. G. , Aebi, M. , & Hennet, T. (2002). ALG12 mannosyltransferase defect in congenital disorder of glycosylation type lg. Human Molecular Genetics, 11, 2331–2339. 10.1093/hmg/11.19.2331 [DOI] [PubMed] [Google Scholar]
- Grunewald, S. (2009). The clinical spectrum of phosphomannomutase 2 deficiency (CDG‐Ia). Biochimica et Biophysica Acta (BBA)—Molecular Basis of Disease, 1792(9), 827–834. 10.1016/j.bbadis.2009.01.003 [DOI] [PubMed] [Google Scholar]
- Hackler, R. , Arndt, T. , Kleine, T. O. , & Gressner, A. M. (1995). Effect of separation conditions on automated isoelectric focusing of carbohydrate‐deficient transferrin and other human isotransferrins using the PhastSystem. Analytical Biochemistry, 230, 281–289. 10.1006/abio.1995.1475 [DOI] [PubMed] [Google Scholar]
- Hykollari, A. , Balog, C. I. A. , Rendić, D. , Braulke, T. , Wilson, I. B. H. , & Paschinger, K. (2013). Mass spectrometric analysis of neutral and anionic N‐glycans from a dictyostelium discoideum model for human congenital disorder of glycosylation CDG IL. Journal of Proteome Research, 12, 1173–1187. 10.1021/pr300806b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranz, C. , Basinger, A. A. , Güçsavaş‐Calikoğlu, M. , Sun, L. , Powell, C. M. , Henderson, F. W. , Aylsworth, A. S. , & Freeze, H. H. (2007). Expanding spectrum of congenital disorder of glycosylation Ig (CDG‐Ig): Sibs with a unique skeletal dysplasia, hypogammaglobulinemia, cardiomyopathy, genital malformations, and early lethality. American Journal of Medical Genetics, 143A, 1371–1378. 10.1002/ajmg.a.31791 [DOI] [PubMed] [Google Scholar]
- Lekka, D. E. , Brucknerova, J. , Salingova, A. , Sebova, C. , Ostrozlikova, M. , Ziburova, J. , Nemcovic, M. , Sestak, S. , Bellova, J. , Pakanova, Z. , Sivakova, B. , Skoknova, M. , Bzduch, V. , Mucha, J. , Barath, P. , & Brucknerova, I. (2021). Congenital disorders of glycosylation—An umbrella term for rapidly expanding group of rare genetic metabolic disorders—Importance of physical investigation. Bratislava Medical Journal, 122(3), 190–195. 10.4149/BLL_2021_030 [DOI] [PubMed] [Google Scholar]
- Marques‐da‐Silva, D. , Dos Reis Ferreira, V. , Monticelli, M. , Janeiro, P. , Videira, P. A. , Witters, P. , Jaeken, J. , & Cassiman, D. (2017). Liver involvement in congenital disorders of glycosylation (CDG). A systematic review of the literature. Journal of Inherited Metabolic Disease, 40, 195–207. 10.1007/s10545-016-0012-4 [DOI] [PubMed] [Google Scholar]
- Monticelli, M. , Ferro, T. , Jaeken, J. , dos Reis Ferreira, V. , & Videira, P. A. (2016). Immunological aspects of congenital disorders of glycosylation (CDG): A review. Journal of Inherited Metabolic Disease, 39, 765–780. 10.1007/s10545-016-9954-9 [DOI] [PubMed] [Google Scholar]
- Murali, C. , Lu, J. T. , Jain, M. , Liu, D. S. , Lachman, R. , Gibbs, R. A. , Lee, B. H. , Cohn, D. , & Campeau, P. M. (2014). Diagnosis of ALG12‐CDG by exome sequencing in a case of severe skeletal dysplasia. Molecular Genetics and Metabolism Reports, 1, 213–219. 10.1016/j.ymgmr.2014.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, B. G. , & Freeze, H. H. (2018). Perspectives on glycosylation and its congenital disorders. Trends in Genetics, 34(6), 466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nykamp, K. , Anderson, M. , Powers, M. , Garcia, J. , Herrera, B. , Ho, Y. Y. , Kobayashi, Y. , Patil, N. , Thusberg, J. , Westbrook, M. , Invitae Clinical Genomics Group , & Topper, S. (2017). Sherloc: A comprehensive refinement of the ACMG‐AMP variant classification criteria. Genetics in Medicine, 19, 1105–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmigiano, A. , Messina, A. , Bua, R. O. , Barone, R. , Sturiale, L. , Zappia, M. , & Garozzo, D. (2018). CSF N‐Glycomics using MALDI MS techniques in Alzheimer's disease. Methods in Molecular Biology, 1750, 75–91. 10.1007/978-1-4939-7704-8_5 [DOI] [PubMed] [Google Scholar]
- Pascoal, C. , Francisco, R. , Ferro, T. , dos Reis Ferreira, V. , Jaeken, J. , & Videira, P. A. (2020). CDG and immune response: From bedside to bench and back. Journal of Inherited Metabolic Disease, 43, 90–124. 10.1002/jimd.12126 [DOI] [PubMed] [Google Scholar]
- Rehm, H. L. (2017). A new era in the interpretation of human genomic variation. Genetics in Medicine, 19, 1092–1095. 10.1038/gim.2017.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17, 405–423. 10.1016/j.jmoldx.2016.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturiale, L. , Bianca, S. , Garozzo, D. , Terracciano, A. , Agolini, E. , Messina, A. , Palmigiano, A. , Esposito, F. , Barone, C. , Novelli, A. , Fiumara, A. , Jaeken, J. , & Barone, R. (2019). ALG12‐CDG: Novel glycophenotype insights endorse the molecular defect. Glycoconjugate Journal, 36, 461–472. 10.1007/s10719-019-09890-2 [DOI] [PubMed] [Google Scholar]
- Tahata, S. , Gunderson, L. , Lanpher, B. , & Morava, E. (2019). Complex phenotypes in ALG12‐congenital disorder of glycosylation (ALG12‐CDG): Case series and review of the literature. Molecular Genetics and Metabolism, 128, 409–414. 10.1016/j.ymgme.2019.08.007 [DOI] [PubMed] [Google Scholar]
- Thiel, C. , Schwarz, M. , Hasilik, M. , Grieben, U. , Hanefeld, F. , Lehle, L. , von Figura, K. , & Körner, C. (2002). Deficiency of dolichyl‐P‐Man:Man7GlcNAc2‐PP‐dolichyl mannosyltransferase causes congenital disorder of glycosylation type Ig. The Biochemical Journal, 367, 195–201. 10.1042/BJ20020794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki, A. , Esko, J. D. , & Colley, K. J. (2009. (Chapter 3)). Cellular organization of glycosylation. In Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., & Etzler M. E. (Eds.), Essentials of Glycobiology (2nd ed.). Cold Spring Harbor Laboratory Press. https://www.ncbi.nlm.nih.gov/books/NBK1926/ [PubMed] [Google Scholar]
- Verheijen, J. , Tahata, S. , Kozicz, T. , Witters, P. , & Morava, E. (2020). Therapeutic approaches in congenital disorders of glycosylation (CDG) involving N‐linked glycosylation: An update. Genetics in Medicine, 22, 268–279. 10.1038/s41436-019-0647-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdebska, E. , Bader‐Meunier, B. , Schischmanoff, P. O. , Dupré, T. , Seta, N. , Tchernia, G. , Kościelak, J. , & Delaunay, J. (2003). Abnormal glycosylation of red cell membrane band 3 in the congenital disorder of glycosylation Ig. Pediatric Research, 54, 224–229. 10.1203/01.PDR.0000072327.55955.F7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Summary of described pathogenic ALG12 gene variants and their clinical and biochemical parameters
Table S2 List of PCR and sequencing primers used in analysis human ALG12 exomes
Table S3 Summary of the results from in silico analysis of the substitution mutation c.1439T>C p.(Leu480Pro) in human ALG12/ALG12 using various prediction software programs and databases with attached result files
Figure S1 MALDI TOF/TOF fragmentation analysis of H7N2 (m/z 1581.5) N‐glycan accumulated in the patient sample (a). Annotations corresponding to two isomeric human glycan structures with the highest scores using GlycomeDB are in (b) and (c). H, hexose; N, N‐acetylglucosamine
Figure S2 A quantitative comparison of absolute signal intensities produced by high‐mannose structures from patient (red) and control (blue) samples after the permethylation using 12C‐ and 13C‐iodomethane (stable isotope labeling). H, hexose; N, N‐acetylglucosamine
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.