

Abstract
Introduction
Valoctocogene roxaparvovec is an investigational AAV5‐based factor VIII (FVIII) gene therapy that has demonstrated sustained clinical benefit in people with severe haemophilia A.
Aim
To report safety, tolerability, efficacy, and quality of life (QOL) among participants who received valoctocogene roxaparvovec in a phase 1/2 clinical study (NCT02576795).
Methods
Men ≥18 years of age with severe haemophilia A (FVIII ≤1 IU/dl) without history of FVIII inhibitors or anti‐AAV5 antibodies received a single infusion of valoctocogene roxaparvovec and were followed for 5 years (6 × 1013 vg/kg dose, n = 7) and 4 years (4 × 1013 vg/kg dose, n = 6).
Results
Over the past 2 years, few adverse events and no FVIII inhibitors were reported. Per chromogenic substrate (CSA) assay at years 5 and 4, four of seven and three of six participants in the 6 × 1013 and 4 × 1013 vg/kg cohorts, respectively, maintained median FVIII levels >5 IU/dl, corresponding to mild haemophilia. By regression analysis, rate of change in FVIII activity was ‐0.14 (95% confidence interval [CI]: ‐.32 to .03) IU/dl/wk in the 6 × 1013 vg/kg cohort in year 5 and ‐.06 (95% CI: ‐.14 to .01) IU/dl/wk in the 4 × 1013 vg/kg cohort in year 4. No participants resumed FVIII prophylaxis, and eight of 13 participants reported zero bleeds in the past 2 years. Improved QOL from baseline persisted in the 6 × 1013 vg/kg cohort; all six Haemo‐QOL‐A domain scores increased. For the 4 × 1013 vg/kg cohort, high baseline Haemo‐QOL‐A scores persisted.
Conclusion
These results demonstrate transgene expression and haemostatic response for up to 5 years in individuals with haemophilia A.
Keywords: haemophilia A, factor VIII, genetic therapy, haemostasis, quality of life
1. INTRODUCTION
Standard of care for people with severe haemophilia A is prophylactic administration of exogenous factor VIII (FVIII) or emicizumab to reduce frequency of bleeding. 1 , 2 However, some people with severe haemophilia A on prophylaxis still experience bleeding. 3 , 4 By eliminating peaks and troughs associated with exogenous FVIII, gene therapy may transform treatment options and yield improved clinical outcomes for severe haemophilia A.
Valoctocogene roxaparvovec (AAV5‐hFVIII‐SQ) is an adeno‐associated virus serotype 5 (AAV5) vector encoding a B‐domain–deleted human FVIII coding sequence (hFVIII‐SQ) with a hepatocyte‐selective promoter. 5 , 6 , 7 In an ongoing phase 1/2 study (NCT02576795), an infusion of AAV5‐hFVIII‐SQ in 13 adults with severe haemophilia A at 6 × 1013 or 4 × 1013 vg/kg substantially reduced bleeding and eliminated prophylactic exogenous FVIII use for up to 3 years. 5 , 6 The most common adverse event (AE) was transient, asymptomatic increases in alanine aminotransferase (ALT). 5 , 6 We present updated results from this phase 1/2 study, assessing safety, efficacy, and quality of life (QOL) from the 6 × 1013 vg/kg cohort (n = 7) through 5 years and the 4 × 1013 vg/kg cohort (n = 6) through 4 years.
2. MATERIALS AND METHODS
2.1. Study design and participants
Primary objectives of this phase 1/2 dose‐escalation study were to assess safety of valoctocogene roxaparvovec and determine the dose required to achieve FVIII activity ≥5 IU/dl at 16 weeks. Secondary objectives were to assess immune responses, exogenous FVIII utilisation, and annualised treated bleeding rate. Participant QOL was evaluated with the haemophilia‐specific health‐related QOL questionnaire for adults (Haemo‐QOL‐A). 8 , 9 , 10 , 11 , 12
As previously published, eligible participants were men ≥18 years of age with severe haemophilia A (FVIII ≤1 IU/dl) without history of FVIII inhibitors (and ≥150 days of exposure to FVIII concentrates or cryoprecipitate) and without anti‐AAV5 antibodies. 5 , 6 , 13 Key exclusion criteria included significant liver dysfunction, significant liver fibrosis, and liver cirrhosis. Participants received one infusion of valoctocogene roxaparvovec.
2.2. Vector construct
The recombinant, replication‐incompetent AAV5 vector contains a hepatocyte‐selective promoter, B‐domain‐deleted human FVIII coding sequence (SQ‐hFVIII), and synthetic polyadenylation sequence. 5 , 6 , 7 The vector is manufactured with a baculovirus—Spodoptera frugiperda (Sf9) insect‐cell production system, 14 as described previously. 5
2.3. Procedures
Participants received AAV5‐hFVIII‐SQ doses as follows: Participant one received 6 × 1012 vg/kg, participant two received 2 × 1013 vg/kg, participants three through nine received 6 × 1013 vg/kg, and participants 10–15 received 4 × 1013 vg/kg. The 4 × 1013 vg/kg dose was selected after some participants who received 6 × 1013 vg/kg expressed circulating FVIII above normal during year 1. As previously described, per‐protocol prophylactic glucocorticoids were administered to participants four through nine following ALT elevation and glucocorticoid initiation postinfusion in participant three; following a protocol amendment, participants 10–15 only received glucocorticoids in response to ALT elevation (Table S1). 5 , 6 After 1 year, participants could enrol in a liver biopsy substudy, which required a liver ultrasound and participants to be brought to FVIII activity >50 IU/dl.
2.4. Assessments
Safety was assessed with physical examination, laboratory assessments, and AEs graded with Common Terminology Criteria for Adverse Events v4.0.3. FVIII activity assays were performed at a central laboratory (Esoterix, Englewood, CO, USA) using both chromogenic substrate (CSA) and one‐stage clotting assays (OSA), as previously reported. 5 , 6 Annualised bleeding rate (ABR) and annualised exogenous FVIII infusion rate were calculated as previously described. 5 , 6 Baseline ABR and FVIII infusion rate were based on the 12 months prior to enrolment. QOL was evaluated with the Haemo‐QOL‐A questionnaire, comprising 41 items scored on a 6‐point Likert‐type scale grouped into six domains: Consequences of Bleeding (7 items), Emotional Impact (6 items), Physical Functioning (9 items), Role Functioning (11 items), Treatment Concern (3 items), and Worry (5 items). 8 , 9 , 10 , 11 , 12 Higher scores indicate better QOL.
2.5. Statistical analysis
Data were summarised using descriptive statistics. Regression analyses using all FVIII samples were used to determine rate of change in FVIII activity each year. Safety analyses included all infused participants. The 6 × 1013 vg/kg cohort participant receiving on‐demand FVIII at baseline was not included in summaries of ABR and FVIII infusion rate. Missing data were not imputed.
3. RESULTS
3.1. Participants
The study was initiated June 2015; the cutoff date was 29 March, 2021. Data include 5 years of follow‐up for the 6 × 1013 vg/kg cohort and 4 years of follow‐up for the 4 × 1013 vg/kg cohort. Baseline participant characteristics were previously presented (Table S2). 5 , 6 Prior to infusion, bleeds requiring exogenous FVIII use occurred despite 12 of 13 participants being on a prophylactic FVIII regimen; baseline mean and median ABR were 17.6 and 24.0, respectively, for the 6 × 1013 vg/kg cohort and 12.2 and 8.0, respectively, for the 4 × 1013 vg/kg cohort. At baseline, one participant in the 6 × 1013 cohort and two in the 4 × 1013 vg/kg cohort had at least three target joints.
3.2. Safety
Overall, the most common AEs associated with valoctocogene roxaparvovec were transient, asymptomatic, and mild‐to‐moderate ALT elevations. Table 1 summarises AEs per year. In the past 2 years, no unexpected safety events occurred (Table S3), and the overall safety profile of valoctocogene roxaparvovec remains unchanged from previous reports. 5 , 6 No participants experienced thrombotic events, developed FVIII inhibitors, or discontinued study participation.
TABLE 1.
Summary of incidence of adverse events in each year by cohort
| 6 × 1013 vg/kg cohort (n = 7) | 4 × 1013 vg/kg cohort (n = 6) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Y1 | Y2 | Y3 | Y4 | Y5 | Y1 | Y2 | Y3 | Y4 | |
| Any AEa | 7 (100.0) | 6 (85.7) | 7 (100.0) | 7 (100.0) | 6 (85.7) | 6 (100.0) | 5 (83.3) | 5 (83.3) | 4 (66.7) |
| Any SAEa | 0 | 1 (14.3) | 1 (14.3) | 1 (14.3) | 0 | 1 (16.7) | 0 | 1 (16.7) | 1 (16.7) |
| Any treatment‐related AEa,b | 6 (85.7) | 1 (14.3) | 1 (14.3) | 2 (28.6) | 0 | 6 (100.0) | 0 | 0 | 0 |
| Grade 1 treatment‐related AEa | 4 (57.1) | 1 (14.3) | 1 (14.3) | 2 (28.6) | 0 | 4 (66.7) | 0 | 0 | 0 |
| Grade 2 treatment‐related AEa | 2 (28.6) | 0 | 0 | 0 | 0 | 2 (33.3) | 0 | 0 | 0 |
| Grade 3 treatment‐related AEa | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Any treatment‐related SAEa,b | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 |
| Any AE of Grade ≥3a | 1 (14.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (16.7) |
| Fatal AEs | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| AEs of special interesta | |||||||||
| ALT elevation | 6 (85.7) | 0 | 0 | 1 (14.3)c | 1 (14.3)d | 4 (66.7) | 0 | 1 (16.7)e | 0 |
| AEs of liver dysfunction | 6 (85.7) | 1 (14.3) | 0 | 1 (14.3)c | 1 (14.3)d | 5 (83.3) | 0 | 1 (16.7)e,f | 0 |
| Potential Hy's law case | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Infusion‐related reactions | 3 (42.9) | 0 | 0 | 0 | 0 | 4 (66.7) | 0 | 0 | 0 |
| Systemic hypersensitivity | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Anaphylactic or anaphylactoid reactions | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Thromboembolic events | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Abbreviations: AE, adverse event; ALT, alanine aminotransferase; AST, aspartate aminotransferase; SAE, serious adverse event; ULN, upper limit of normal; Y, year.
Data are presented as n (%).
Percentages were calculated using the total number of participants (N) in each analysis population as the denominator. Participants with more than one AE of the same category were counted only once for that category.
Relationship to study drug was assessed by the investigator.
A Grade 1 AE of ALT increase (1.4 × ULN) occurred on study day 1389. The AE was deemed not related to the study drug by the investigator and resolved by the next study visit on day 1431 without treatment.
A Grade 1 AE of ALT increase (1.3 × ULN) occurred on study day 1626. The AE was deemed not related to the study drug by the investigator and resolved by study day 1631.
A Grade 1 AE of ALT increase (2.1 × ULN) occurred on study day 1100. The AE was deemed not related to the study drug by the investigator and resolved by the next study visit on day 1135 without treatment.
A Grade 1 AE of AST increase (1.7 × ULN) occurred on study day 1100. The AE was deemed not related to the study drug by the investigator and resolved by the next study visit on day 1135 without treatment.
No treatment‐related serious AEs (SAEs) occurred beyond the single event of Grade 2 pyrexia with myalgia and headache within 24 h of infusion reported previously. 5 , 6 In year 4, treatment‐related AEs of Grade 1 diarrhoea and Grade 1 hepatic steatosis were each reported in a single 6 × 1013 vg/kg cohort participant. Hepatic steatosis was an incidental liver ultrasound finding; this participant had a Grade 1 ALT elevation during year 1 and normal liver function test results in other years.
In the past 2 years, there were three SAEs unrelated to treatment. A manually ruptured furuncle bleed led to an overnight hospital stay for a participant in the 6 × 1013 vg/kg cohort; his FVIII activity level was 9.9 IU/dl per CSA at the time of the event. In the 4 × 1013 vg/kg cohort, one participant had pre‐existing severe chronic knee arthropathy necessitating elective total knee replacement surgery, and one participant reported a rectal haemorrhage due to an unrelated medical condition.
In the past 2 years, three additional participants reported asymptomatic ALT elevations; none were deemed treatment‐related by the investigator. In the 6 × 1013 cohort, a participant reported an ALT elevation 1.3‐times the upper limit of normal (ULN) in year 5 that resolved after 6 days and an ALT elevation 1.4‐times ULN in year 4 with no clear causality that resolved after 43 days (next study visit). No glucocorticoids were administered for either event. This participant had a history of hepatitis C, which had cleared at enrolment. In year 3, a 4 × 1013 cohort participant reported an ALT elevation 2.1‐times ULN associated with concomitant amoxicillin/clavulanate to treat an ear infection that resolved without glucocorticoid treatment after 36 days (next study visit); this participant had no history of liver disease. All events were mild, transient, and asymptomatic, and none were associated with a decrease in FVIII levels.
3.3. Efficacy
FVIII activity is shown in Figure 1. For participants in the 6 × 1013 cohort (n = 7), mean (standard deviation [SD]) and median FVIII activity levels at the end of year 5 were 11.6 (12.2) and 8.2 IU/dl, respectively, as measured by CSA and 18.7 (17.5) and 15.7 IU/dl, respectively, as measured by OSA. At the end of year 4, mean (SD) and median FVIII activity levels for participants in the 4 × 1013 cohort (n = 6) were 5.6 (5.6) and 4.8 IU/dl, respectively, per CSA and 9.5 (7.0) and 7.5 IU/dl, respectively, per OSA (Table S4). As previously reported, OSA results are approximately 1.6‐times higher than CSA results; this ratio has not changed over time. 5 , 15 Regression analysis of each year (Figure 2) shows that in both cohorts, FVIII activity rose to a peak in year 1 and declined in the second year in proportion to peak FVIII expression, with the lower peak in the 4 × 1013 vg/kg cohort associated with shallower year‐over‐year declines. Beyond year 2, the slopes have become more consistent and predictable with the 95% confidence interval (CI) for each slope becoming narrower each year (Figure 2). The rate of change in FVIII activity measured by CSA was ‐0.14 (95% CI, ‐.32 to .03) IU/dl/wk in the 6 × 1013 vg/kg cohort in year 5 and ‐.06 (95% CI, ‐.14 to .01) IU/dl/wk in the 4 × 1013 vg/kg cohort in year 4 (Figure 2). The pattern of increasing consistency in the rate of FVIII activity change over time is also apparent at the individual participant level (Figure S1).
FIGURE 1.
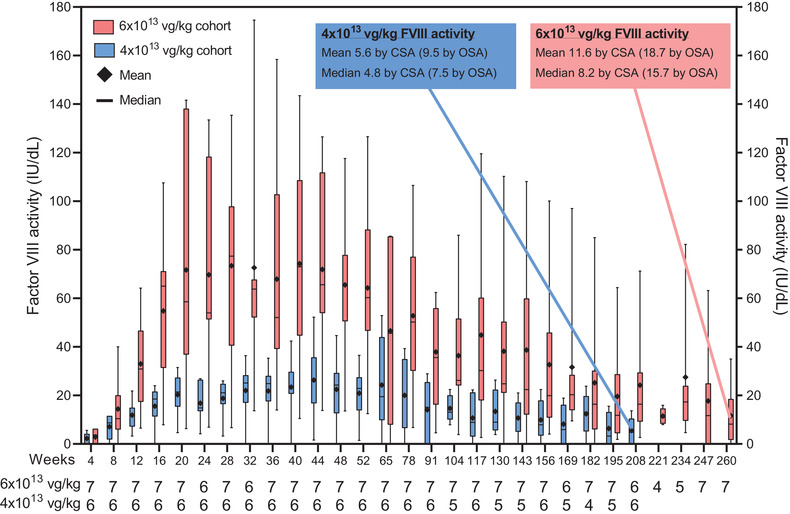
FVIII activity measured by the chromogenic substrate assay for participants in the 6 × 1013 and 4 × 1013 vg/kg cohorts over 5 and 4 years, respectively. FVIII activity levels taken within 72 h of exogenous FVIII administration were excluded. FVIII activity that fell below the lower limit of quantitation (<3.0 IU/dl) was imputed as 0 IU/dl. Whiskers represent the minimum and maximum values; boxes represent the 25th and 75th percentiles. Diamonds represent the mean, and horizontal bars represent the median. CSA, chromogenic substrate assay; FVIII, factor VIII; OSA, one‐stage assay
FIGURE 2.

FVIII activity measured by the chromogenic substrate assay and regression for all samples. (A) Participants in the 6 × 1013 vg/kg cohort over 5 years, and (B) participants in the 4 × 1013 vg/kg cohort over 4 years. FVIII activity levels taken within 72 h of exogenous FVIII administration were excluded. FVIII activity that fell below the lower limit of quantitation (<3 IU/dl) was imputed as 0 IU/dl. FVIII, factor VIII
Participants in the 6 × 1013 vg/kg cohort on prophylaxis at baseline (n = 6) had a mean (SD) ABR of 0.7 (1.6) treated bleeds/year during year 5, resulting in a cumulative mean (SD) ABR of 0.8 (2.0) treated bleeds/year from week 5 onward; this is a 95% reduction from baseline (Figure 3A; Table S5). Participants in the 4 × 1013 vg/kg cohort had a mean (SD) ABR of 1.7 (2.3) treated bleeds/year during year 4, resulting in a cumulative mean (SD) ABR of 1.0 (1.9) treated bleeds/year from week 5 onward, a 92% reduction from baseline.
FIGURE 3.

Pre‐ and postinfusion of valoctocogene roxaparvovec annualised rates of bleeding and FVIII infusion. (A) Annualised rates of bleeding and FVIII infusion for the 6 × 1013 vg/kg cohort at baseline and in years 1, 2, 3, 4, and 5. (B) Annualised rates of bleeding and FVIII infusion for the 4 × 1013 vg/kg cohort at baseline and in years 1, 2, 3, and 4. The participant in the 6 × 1013 cohort receiving on‐demand FVIII treatment at baseline was excluded. Year 1 included weeks 5–52 only; Year 2 included weeks 53–104; Year 3 included weeks 105–156; Year 4 included weeks 157–208; Year 5 included weeks 209–260. †In year 5, in addition to treatment for bleeding, two participants in the 6 × 1013 vg/kg cohort had 12 instances of exogenous FVIII use due to surgery. ‡In year 4, in addition to treatment for bleeding, two participants in the 6 × 1013 vg/kg cohort had two instances of exogenous FVIII use due to surgery. §In year 3, in addition to treatment for bleeding, one participant in the 6 × 1013 vg/kg cohort had 23 instances of exogenous FVIII use due to surgery, including knee replacement. ¶In year 2, in addition to treatment for bleeding, two participants in the 6 × 1013 vg/kg cohort had 48 instances of exogenous FVIII use due to surgery. ††In year 1, in addition to treatment for bleeding, no participants in the 6 × 1013 vg/kg cohort used exogenous FVIII as prophylaxis for surgery. ‡‡In year 4, in addition to treatment for bleeding, one participant in the 4 × 1013 vg/kg cohort had three instances of FVIII use due to surgery, including colonoscopy with biopsy. §§In year 3, in addition to treatment for bleeding, three participants in the 4 × 1013 vg/kg cohort had 46 instances of exogenous FVIII use due to surgery, including knee replacement and liver biopsy. ¶¶In year 2, in addition to treatment for bleeding, one participant in the 4 × 1013 vg/kg cohort had one instance of exogenous FVIII use due to sigmoidoscopy. †††In year 1, in addition to treatment for bleeding, one participant in the 4 × 1013 vg/kg cohort had one instance of exogenous FVIII use due to surgery. FVIII, factor VIII
For participants in the 6 × 1013 vg/kg cohort receiving prophylaxis at baseline (n = 6), the mean annualised rate of FVIII infusion was 5.5 infusions/year in year 5, a 96% cumulative reduction from baseline. For participants in the 4 × 1013 vg/kg cohort, year 4 annualised FVIII infusion rate was 7.8 infusions/year, a cumulative 95% reduction from baseline (Figure 3B). All participants in the 4 × 1013 and 6 × 1013 vg/kg cohorts continue to remain off FVIII prophylaxis.
Individual participant ABRs and annualised FVIII usage before and after infusion are shown in Figure 4, along with their FVIII range at the end of year 5 (6 × 1013 vg/kg cohort) and year 4 (4 × 1013 vg/kg cohort). At last analysis, four of seven and three of six participants in the 6 × 1013 and 4 × 1013 vg/kg cohorts, respectively, maintained median FVIII levels >5 IU/dl, corresponding to mild haemophilia.
FIGURE 4.
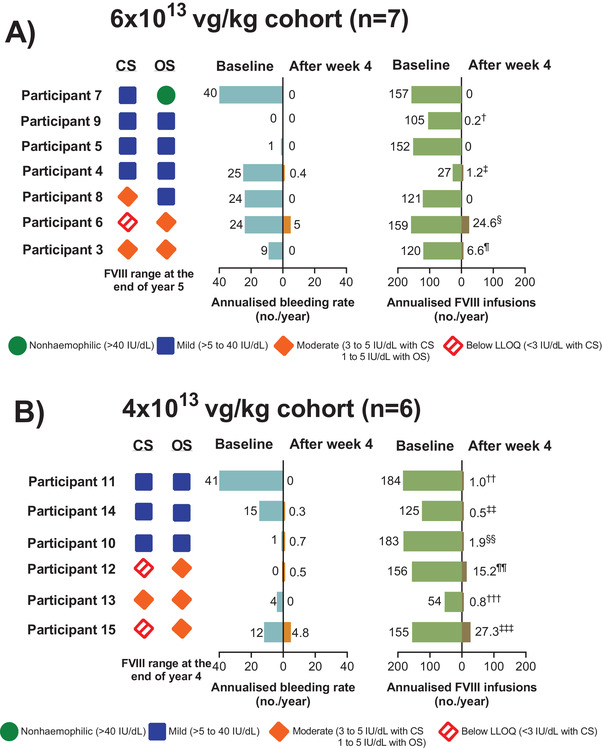
Individual participants’ annualised FVIII infusion rate and ABR at baseline and from week 5 and onward and FVIII activity at the end of year 5 or 4. (A) The 6 × 1013 vg/kg cohort. (B) The 4 × 1013 vg/kg cohort. Participants are ordered by descending current FVIII activity (week 260 for 6 × 1013 cohort; week 208 for 4 × 1013 cohort). Current FVIII activity ranges are based on median value in a ±4‐week window. †From week 5 onward, participant nine had one instance of exogenous FVIII use for surgery prophylaxis. ‡From week 5 onward, in addition to treatment for bleeding, participant four had four instances of exogenous FVIII use due to prophylaxis for surgery and liver biopsy. §From week 5 onward, in addition to treatment for bleeding, participant six had 47 instances of exogenous FVIII use due to surgery prophylaxis, including total knee replacement. ¶From week 5 onward, participant three had 34 instances of exogenous FVIII use due to prophylaxis for total knee replacement surgery and liver biopsy. ††From week 5 onward, in addition to treatment for bleeding, participant 11 had two instances of exogenous FVIII use due to liver biopsy. ‡‡From week 5 onward, participant 14 only used exogenous FVIII as treatment for bleeding. §§From week 5 onward, in addition to treatment for bleeding, participant 10 had one instance of exogenous FVIII use due to prophylaxis for a sigmoidoscopy. ¶¶From week 5 onward, in addition to treatment for bleeding, participant 12 had three instances of exogenous FVIII use due to prophylaxis for procedures, including colonoscopy with biopsies taken. †††From week 5 onward, in addition to treatment for bleeding, participant 13 had three instances of exogenous FVIII use due to prophylaxis for surgery, a tooth extraction, and steroid injection. ‡‡‡From week 5 onward, in addition to treatment for bleeding, participant 15 had 43 instances of exogenous FVIII use due to general surgery, total knee replacement, and liver biopsy. ABR, annualised bleeding rate; CS, chromogenic substrate assay; FVIII, factor VIII; LLOQ, lower limit of quantitation; OS, one‐stage assay; SAE, serious adverse event. [Correction added on 28 August 2021, after first online publication: In Figure 4, the values for Participants 3 and 13 were wrongly aligned and have been corrected in this version.]
In the 6 × 1013 cohort, five of seven participants reported no treated bleeds in either year 4 or 5. Participant six reported eight bleeds during year 4: five spontaneous bleeds in prior target joints and three traumatic non‐joint bleeds (two after falling down stairs, one finger bleed occurred at work); and four bleeds during year 5: one spontaneous bleed in prior target joint, and three traumatic non‐joint bleeds (one from pulled lower back muscle, one from stubbed toe, and one from knocking his hand). At week 260, participant six's FVIII activity was 3.3 IU/dl. Compared with his baseline ABR of 24 treated bleeds/year while on prophylaxis, his bleeding rate decreased by 79% following treatment with valoctocogene roxaparvovec. Participant four reported the SAE of traumatic bleeding due to manual rupture of a furuncle.
In the 4 × 1013 cohort, three of six participants reported no treated bleeds in either year 3 or 4. Participant 10 reported four traumatic non‐target joint bleeds (one occurred after jumping from a bridge into a boat, one after playing football, one at the gym, and one from punching a punching bag). In the year prior to enrolment, participant 10 experienced one treated bleed per year. At weeks 134 and 174, around the time of these bleeds, participant 10 had an FVIII activity level of 10.5 and 9.5 IU/dl, respectively. Participant 15 reported nine bleeds: three spontaneous bleeds in prior target joints, three traumatic bleeds from prolonged standing at work, and three of unknown causality. At week 208, participant 15 had FVIII activity levels <3 IU/dl (LLOQ of CSA). His bleeding rate was reduced by 63% from his baseline ABR of 12 treated bleeds/year while on prophylaxis. Participant 12 reported one bleed of unknown causality as well as the SAE of rectal haemorrhage related to Crohn's disease. At baseline, his ABR was zero treated bleeds/year, and from week 168 onward his FVIII activity was <3.0 IU/dl.
During the past 2 years, most exogenous FVIII use was one‐time prophylaxis for procedures, including dental surgery, intra‐articular glucocorticoid injections, gastroscopy, and biopsies. Participant 15 received prophylactic FVIII treatments following total knee replacement surgery. As noted previously, participants four, six, 10, 12, and 15 received FVIII treatment for their bleeding events. Over all 5 years of follow‐up, two participants in the 6 × 1013 vg/kg cohort (participants four and nine) had five procedures or surgeries, including fluoroscopy‐guided steroid injection and dental extraction, without prior FVIII prophylaxis. At the time of these procedures, FVIII levels were in the mild haemophilia range by CSA.
3.4. Quality of life
At baseline, participants in the 6 × 1013 vg/kg cohort had a total Haemo‐QOL‐A score of 71.9 (16.6), which increased by a mean (SD) of 10.3 (13.6) to 82.2 (18.1) at the end of year 5 (n = 7) (Figure 5 and Figure S2). Participants in the 4 × 1013 vg/kg cohort had a high baseline mean (SD) total Haemo‐QOL‐A score of 80.8 (9.1), which was maintained over time. At the end of year 4, mean (SD) total Haemo‐QOL‐A score was 80.3 (10.4) for the 4 × 1013 vg/kg cohort (n = 5). On average, all 6 domains of the Haemo‐QOL‐A improved relative to baseline in participants in the 6 × 1013 vg/kg cohort. In the 4 × 1013 vg/kg cohort, three domains (Role Functioning [mean change from baseline, 1.82], Treatment Concern [5.33], and Worry [8.8]) improved from baseline on average, while the others did not change (Consequences of Bleeding [0]) or decreased slightly (Emotional Impact [‐2.0] and Physical Functioning, [‐4.4]).
FIGURE 5.
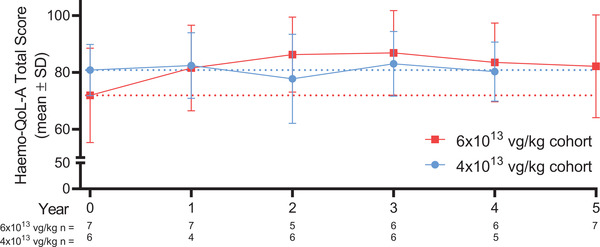
Haemo‐QOL‐A Total Score over 5 years. Dotted lines indicate baseline values for each cohort. Total score is based on six domains: Consequences of Bleeding (7 items), Emotional Impact (6 items), Physical Functioning (9 items), Role Functioning (11 items), Treatment Concern (3 items), and Worry (5 items). Haemo‐QOL‐A, haemophilia‐specific health‐related QOL questionnaire for adults; SD, standard deviation
4. DISCUSSION
These results represent the longest duration of follow‐up from any clinical gene therapy study for severe haemophilia A. Safety, efficacy, and QOL results for up to 5 years are presented for participants following a single dose of valoctocogene roxaparvovec. These data demonstrate continued clinical benefit and an acceptable safety profile, and maintenance of increased QOL from baseline or high baseline QOL.
Five years postinfusion, safety results were aligned with previous observations, showing no unexpected new safety signals. 5 , 6 No participants developed FVIII inhibitors. Previously observed liver enzyme elevations had no apparent sequelae, though mild liver damage cannot be ruled out without biopsy evidence. In the past 2 years of follow‐up, one participant in the 6 × 1013 cohort reported two ALT elevations, and one participant in the 4 × 1013 vg/kg cohort reported one ALT elevation. None impacted FVIII expression and all resolved without glucocorticoid treatment. The lengthy temporal separation between these ALT elevations and those in the first year post‐gene therapy suggests they were separate events.
Efficacy at 5 years postinfusion in the 6 × 1013 vg/kg cohort was similar to that observed at 3 years, as assessed by treated ABR and exogenous FVIII utilisation. 5 , 6 All 13 participants in both cohorts continued to experience substantial reductions in ABR compared to baseline. All participants, including those who had relatively low FVIII activity levels, remain off prophylaxis. After 5 years of follow‐up, observed clinical effects align with other gene therapy trials and the natural history of the disease in that even low levels of endogenously expressed clotting factors can ameliorate the haemophilic phenotype. 16 , 17 , 18
FVIII activity is reported based on both CSA and OSA. Recent work demonstrates that the CSA more closely estimates the specific activity of the transgene‐produced FVIII protein, due to kinetic differences in Factor Xa and thrombin formation by transgenic FVIII‐SQ 15 ; however, investigation in a larger dataset is required to determine the clinical haemostatic predictiveness of either assay. At most recent measurement, 10 out of 13 participants continued to have FVIII levels above the severe haemophilia range (<1 IU/dl) per CSA, while three participants had FVIII levels below the CSA LLOQ (<3 IU/dl). Of seven participants in the 6 × 1013 vg/kg cohort, two had FVIII activity levels corresponding to moderate haemophilia (3–5 IU/dl) and four were in the mild haemophilia range (>5–40 IU/dl) at year 5. Of six participants in the 4 × 1013 vg/kg cohort, one had FVIII activity levels corresponding to moderate haemophilia and three had FVIII activity corresponding to mild haemophilia at year 4. A regression analysis of the rate of FVIII activity change provides insight into the trajectory over time and reveals increasing consistency and predictability of that trajectory, both between and within participants, in the recent years of this study.
Our data also suggest clinical benefits of sustained FVIII levels, without frequent prophylactic intravenous infusions, may be associated with improved QOL. The existence of a disability paradox in haemophilia indicates general health questionnaires such as the EuroQol‐5D‐5L may underestimate haemophilia‐related QOL burdens. 19 The haemophilia‐specific Haemo‐QOL‐A has demonstrated robust validity among adults undergoing standard treatment for haemophilia, 9 , 10 , 20 , 21 and a change in Haemo‐QOL‐A Total Score of approximately seven is considered meaningful in that population. 22 , 23 QOL scores in the 6 × 1013 vg/kg cohort increased by >7 over 5 years. These QOL improvements are in addition to benefits derived from prophylaxis, as all but one participant were on prophylaxis at baseline. 10 , 24 In the 4 × 1013 vg/kg cohort, high baseline scores were maintained, perhaps reflecting a ceiling effect. However, the Haemo‐QOL‐A was not developed for use with gene therapy and may therefore not fully capture its impact on QOL. Work is ongoing to validate the Haemo‐QOL‐A in this context to ascertain the clinically important difference in total score. 25
Limitations of this study include the small sample size and open‐label trial design, which may weaken QOL data interpretability. Participants with the highest unmet need in terms of high baseline bleeding rates may also have been most motivated to enrol. High intra‐ and interparticipant variability, particularly notable during the first couple of years after treatment, are likely due to the large number of molecular events that must occur for AAV‐mediated gene therapy to result in predictable FVIII protein production. 26 However, the exact mechanisms underlying individual participant variation in FVIII and observed declines in FVIII over time are not yet known. Additional basic research is underway, and further clinical studies with larger sample sizes may elucidate contributors to variability and suggest minimisation or management strategies.
5. CONCLUSION
These results demonstrate that valoctocogene roxaparvovec gene therapy can lead to reductions from baseline ABR and FVIII utilisation in people with severe haemophilia A. Haemostatic efficacy was largely maintained in these two dose groups over 4 and 5 years, and the safety profile remained acceptable. Average participant QOL was improved or maintained from baseline. Gene therapy could represent a paradigm shift in the treatment of haemophilia A, enabling long‐lasting treatment and improving clinical and patient‐centred outcomes, including QOL. A phase 3 clinical trial (NCT03370913) of valoctocogene roxaparvovec is currently assessing these possibilities.
ACKNOWLEGEMENTS
Funding for this study was provided by BioMarin Pharmaceutical. Medical writing and editorial support were provided by Kathleen Pieper, PhD; and Atreju Lackey, PhD, of AlphaBioCom, LLC, and funded by BioMarin Pharmaceutical. We thank all the study participants and study site personnel: Stefan Tiefenbacher and Mary Robinson, of Esoterix, for factor VIII assays and consulting; and the following employees of BioMarin Pharmaceutical: Jennifer Quinn for Haemo‐QOL‐A data interpretation; Brian Long for immunogenicity assessments; Kala Jayaram for safety assessments; Mingjin Li for statistical analyses; Joshua Henshaw for peripheral blood analyses and interpretation; Steve Zoog and Christian Vettermann for assistance with bioanalytic strategy, assay design and validation, and data interpretation; and Kendra Bolt, Sara Hawley, and Micah Robinson for writing and project management support.
CONFLICTS OF INTEREST
K John Pasi is an employee for Roche and reports receiving consulting payments from Alnylam, APCintex, BioMarin, Bioverativ, Catalyst Bio, Catapult, Chugai, Roche, Novo Nordisk, Sanofi, and Sobi; participating as a clinical trial investigator for BioMarin, Sanofi, and UniQure; receiving speaker fees from Bayer, BioMarin, Biotest, Novo Nordisk, Sanofi, Shire, Sobi, Octapharma, Pfizer, and UniQure; and receiving travel support from Alnylam, Bayer, BioMarin, Bioverativ, Novo Nordisk, Octapharma, Pfizer, Sobi, Sanofi, and Shire. Michael Laffan reports receiving grants from BioMarin; personal fees from Bayer, LEO Pharma, LFB Biopharmaceuticals, Pfizer, Roche, Shire, and Sobi; and travel support from Bayer, LFB Biopharmaceuticals, and Sobi. Savita Rangarajan reports receiving grants from Roche and Sangamo; travel support from Reliance Life Sciences and Shire/Takeda; and consulting payments from Pfizer, Reliance Life Sciences, Sanofi, and Shire/Takeda. Will Lester reports receiving grants from BioMarin; personal fees from Bayer, LFB, Novo Nordisk, Sobi, and Takeda; and travel support from Takeda and CSL. Emily Symington reports receiving grants from BioMarin and travel support from CSL Behring and Novo Nordisk. Bella Madan reports nothing to disclose. Nina Mitchell is a former employee of BioMarin Pharmaceutical and may own stock. Tara M. Robinson, Xinqun Yang, Benjamin Kim, and Wing Yen Wong are employees and stockholders of BioMarin Pharmaceutical. Glenn F. Pierce is an employee of Voyager Therapeutics; reports receiving consulting payments from Ambys Medicines, BioMarin Pharmaceutical, BridgeBio, CRISPR Therapeutics, Decibel Therapeutics, Frontera, Generation Bio, Geneception, Novo Nordisk, Pfizer, Regeneron, Spark, and Third Rock Ventures; and is a board member of the World Federation of Haemophilia, Voyager Therapeutics, Global Blood Therapeutics, VarmX, and the Medical and Scientific Advisory Council of the US National Haemophilia Foundation.
AUTHOR CONTRIBUTIONS
K John Pasi, Glenn F Pierce, and Wing Yen Wong contributed to conception and design of the study. K John Pasi, Savita Rangarajan, Will Lester, Emily Symington, Bella Madan, and Michael Laffan carried out the clinical study. Benjamin Kim, Tara M. Robinson, Nina Mitchell, and Wing Yen Wong oversaw the conduct of the study; Tara M. Robinson and Nina Mitchell were the medical monitors. Xinqun Yang performed the statistical and data analyses. All authors critically reviewed the manuscript, provided input on data interpretation, and approved the final draft for submission.
DATA AVAILABIITY STATEMENT
De‐identified individual participant data underlying these results will be made available together with the clinical protocol and data dictionaries, for noncommercial, academic purposes. Additional supporting documents may be available upon request. Investigators will be able to request access to these data and supporting documents via the Publication Data Request page at www.BioMarin.com beginning 6 months and ending 2 years after publication. Data associated with any ongoing development program will be made available within 6 months after approval of the relevant product. Requests must include a research proposal clarifying how the data will be used, including proposed analysis methodology. Research proposals will be evaluated relative to publicly available criteria available at www.BioMarin.com to determine if access will be given, contingent upon execution of a data access agreement with BioMarin Pharmaceutical Inc.
Supporting information
Supplementary Information
Pasi KJ, Laffan M, Rangarajan S, et al. Persistence of haemostatic response following gene therapy with valoctocogene roxaparvovec in severe haemophilia A. Haemophilia. 2021;27:947–956. 10.1111/hae.14391
[Correction added on 28 August 2021, after first online publication: The copyright line was changed.]
REFERENCES
- 1. Srivastava A, Santagostino E, Dougall A, et al. WFH guidelines for the management of hemophilia, 3rd edition. Haemophilia. 2020;26(6):1‐158. [DOI] [PubMed] [Google Scholar]
- 2. Srivastava A, Brewer AK, Mauser‐Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19(1):e1‐47. [DOI] [PubMed] [Google Scholar]
- 3. Mazepa MA, Monahan PE, Baker JR, et al. Men with severe hemophilia in the United States: birth cohort analysis of a large national database. Blood. 2016;127(24):3073‐3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kruse‐Jarres R, Oldenburg J, Santagostino E, et al. Bleeding and safety outcomes in persons with haemophilia A without inhibitors: results from a prospective non‐interventional study in a real‐world setting. Haemophilia. 2019;25(2):213‐220. [DOI] [PubMed] [Google Scholar]
- 5. Rangarajan S, Walsh L, Lester W, et al. AAV5‐factor VIII gene transfer in severe hemophilia A. N Engl J Med. 2017;377(26):2519‐2530. [DOI] [PubMed] [Google Scholar]
- 6. Pasi KJ, Rangarajan S, Mitchell N, et al. Multiyear follow‐up of AAV5‐hFVIII‐SQ gene therapy for hemophilia A. N Engl J Med. 2020;382(1):29‐40. [DOI] [PubMed] [Google Scholar]
- 7. Bunting S, Zhang L, Xie L, et al. Gene therapy with BMN 270 results in therapeutic levels of FVIII in mice and primates and normalization of bleeding in hemophilic mice. Mol Ther. 2018;26(2):496‐509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rentz A, Flood E, Altisent C, et al. Cross‐cultural development and psychometric evaluation of a patient‐reported health‐related quality of life questionnaire for adults with haemophilia. Haemophilia. 2008;14(5):1023‐1034. [DOI] [PubMed] [Google Scholar]
- 9. Sun HL, Mcintosh KA, Squire SJ, et al. Patient powered prophylaxis: a 12‐month study of individualized prophylaxis in adults with severe haemophilia A. Haemophilia. 2017;23(6):877‐883. [DOI] [PubMed] [Google Scholar]
- 10. Manco‐Johnson MJ, Lundin B, et al. Effect of late prophylaxis in hemophilia on joint status: a randomized trial. J Thromb Haemost. 2017;15(11):2115‐2124. [DOI] [PubMed] [Google Scholar]
- 11. Stemberger M, Kallenbach F, Schmit E, et al. Impact of adopting population pharmacokinetics for tailoring prophylaxis in haemophilia A patients: a historically controlled observational study. Thromb Haemost. 2019;119(3):368‐376. [DOI] [PubMed] [Google Scholar]
- 12. St‐Louis J, Urajnik DJ, Menard F, et al. Generic and disease‐specific quality of life among youth and young men with Hemophilia in Canada. BMC Hematol. 2016;16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Protocol for : Rangarajan S, Walsh L, Lester W, et al. AAV5–factor VIII gene transfer in severe hemophilia A. N Engl J Med. 2017;377:2519‐2530. [DOI] [PubMed] [Google Scholar]
- 14. Kotin RM, Snyder RO. Manufacturing clinical grade recombinant adeno‐associated virus using invertebrate cell lines. Hum Gene Ther. 2017;28(4):350‐360. [DOI] [PubMed] [Google Scholar]
- 15. Rosen S, Tiefenbacher S, Robinson M, et al. Activity of transgene‐produced B‐domain‐deleted factor VIII in human plasma following AAV5 gene therapy. Blood. 2020;136(22):2524‐2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nathwani AC, Reiss UM, Tuddenham EGD, et al. Long‐term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. 2014;371(21):1994‐2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nathwani AC, Tuddenham EGD, Rangarajan S, et al. Adenovirus‐associated virus vector‐mediated gene transfer in hemophilia B. N Engl J Med. 2011;365(25):2357‐2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Den Uijl IEM, Fischer K, Van Der Bom JG, et al. Analysis of low frequency bleeding data: the association of joint bleeds according to baseline FVIII activity levels. Haemophilia. 2011;17(1):41‐44. [DOI] [PubMed] [Google Scholar]
- 19. O'hara J, Martin AP, Nugent D, et al. Evidence of a disability paradox in patient‐reported outcomes in haemophilia. Haemophilia. 2021;27(2):245‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Limperg PF, Terwee CB, Young NL, et al. Health‐related quality of life questionnaires in individuals with haemophilia: a systematic review of their measurement properties. Haemophilia. 2017;23(4):497‐510. [DOI] [PubMed] [Google Scholar]
- 21. Wharfe G, Buchner‐Daley L, Gibson T, et al. The Jamaican Haemophilia Registry: describing the burden of disease. Haemophilia. 2018;24(4):e179‐e186. [DOI] [PubMed] [Google Scholar]
- 22. Wyrwich KW, Krishnan S, Poon JL, et al. Interpreting important health‐related quality of life change using the Haem‐A‐QoL. Haemophilia. 2015;21(5):578‐584. [DOI] [PubMed] [Google Scholar]
- 23. Santagostino E, Lentz SR, Busk AK, et al. Assessment of the impact of treatment on quality of life of patients with haemophilia A at different ages: insights from two clinical trials on turoctocog alfa. Haemophilia. 2014;20(4):527‐534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Usuba K, Price VE, Blanchette V, et al. Impact of prophylaxis on health‐related quality of life of boys with hemophilia: an analysis of pooled data from 9 countries. Res Pract Thromb Haemost. 2019;3(3):397‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Quinn J, Mitchell N & Wong WY Clinical Validation of the Haemo‐QoL‐A Questionnaire in Adults with Severe Hemophilia A Treated with Valoctocogene Roxaparvovec: Results from 2 Studies. Abstract presented at Hemophilia Federation of America Symposium. 2020.
- 26. Pierce GF. Uncertainty in an era of transformative therapy for haemophilia: addressing the unknowns. Haemophilia. 2021;27(3):103‐113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information