

Abstract
First‐degree relatives (FDRs) of familial pancreatic cancer (FPC) patients have increased risk of developing pancreatic ductal adenocarcinoma (PDAC). Investigating and understanding the genetic basis for PDAC susceptibility in FPC predisposed families may contribute toward future risk‐assessment and management of high‐risk individuals. Using a Danish cohort of 27 FPC families, we performed whole‐genome sequencing of 61 FDRs of FPC patients focusing on rare genetic variants that may contribute to familial aggregation of PDAC. Statistical analysis was performed using the gnomAD database as external controls. Through analysis of heterozygous premature truncating variants (PTV), we identified cancer‐related genes and cancer‐driver genes harboring multiple germline mutations. Association analysis detected 20 significant genes with false discovery rate, q < 0.05 including: PALD1, LRP1B, COL4A2, CYLC2, ZFYVE9, BRD3, AHDC1, etc. Functional annotation showed that the significant genes were enriched by gene clusters encoding for extracellular matrix and associated proteins. PTV genes were over‐represented by functions related to transport of small molecules, innate immune system, ion channel transport, and stimuli‐sensing channels. In conclusion, FDRs of FPC patients carry rare germline variants related to cancer pathogenesis that may contribute to increased susceptibility to PDAC. The identified variants may potentially be useful for risk prediction of high‐risk individuals in predisposed families.
Keywords: cancer genes, familial pancreatic cancer, first‐degree relatives, rare germline variants, whole genome sequencing
Whole‐genome sequencing (WGS) on first‐degree relatives (FDRs) of familial pancreatic cancer patients identified multiple genes enriched by rare germline premature truncating variants (PTVs) as well as 20 significant genes detected by gene‐based association test using the Proxy External Controls Association Test (ProxECAT) based on the Alternative Allele Frequencies (AAF) of functional and synonymous single‐nucleotide variants (SNVs). The Genome Aggregation Database (gnomAD) served as external controls. Enriched biological pathways were tested using Gene‐Set Enrichment Analysis (GSEA).

1. INTRODUCTION
The global incidence of pancreatic cancer is 5.5 per 100 000 for men and 4.0 per 100 000 for women. 1 Among the cases, 5%–10% are estimated to be familial pancreatic cancer (FPC). 2 FPC is an inherited malignancy with familial clustering, defined by presentation of at least one pair of first‐degree relatives (FDRs) with pancreatic ductal adenocarcinoma (PDAC), in the absence of a known hereditary cancer syndrome. 3 Familial aggregation of FPC confers increased risk of PDAC among relatives. 4 It is estimated that in FPC predisposed families, individuals with 2 FDRs with PDAC have a 6.4‐fold higher risk of developing PDAC, while individuals with at least 3 FDRs with PDAC have a 32‐fold higher risk of disease. 5 These reports characterize predisposed FDRs as high‐risk individuals (HRIs). International consensus guidelines recommend yearly screening of HRIs including FDRs in FPC predisposed families – with genetic testing for PDAC predisposition genes being a potential tool for future risk assessment and stratification of HRIs. 6 , 7
We have recently analyzed familial correlation of FPC in a Danish nationwide family cohort and estimated a high heritability of 51% in FDRs to FPC patients. 8 The high genetic predisposition for FPC calls for efforts to identify genetic variations underlying the pathogenesis of the disease. In the literature, multiple genome‐wide association studies (GWASs) have been performed and addressed common single nucleotide polymorphisms (SNPs) associated with sporadic pancreatic cancer (SPC). 9 , 10 , 11 However, only a few studies have focused on FPC 12 – perhaps due to limited sample availability, as FPC represents a rare sub‐group of pancreatic cancer, estimated to be accountable for around 10% of all PDAC cases.
Despite great efforts, previous genetic association studies have only detected a limited number of susceptibility loci for PDAC. 13 GWAS is based on a “common disease, common variant” hypothesis with common variants referring to allelic variants present in more than 1%–5% of the population. The common variants confer relatively small increments in the risk of disease. 14 Instead of focusing on common SNPs, a better choice is to analyze rare single nucleotide variants (SNVs) using next generation sequencing (NGS) and statistical models for joint analysis of rare variants.
Studies using NGS techniques have identified prominent gene mutations in BRCA1, BRCA2, CDKN2A, PALB2, and ATM to be associated with FPC. 15 , 16 Nonetheless, only about 12% of all FPC cases carry any of these mutations – meaning that the germline‐component of >80% of all FPC cases remains unknown. 7 , 17 A recent whole genome sequencing (WGS) analysis has demonstrated that the genetic architecture of FPC is highly heterogeneous, and the currently identified genetic variants account for a limited genetic component underlying the disease susceptibility. 16 The genetic heterogeneity of FPC means that susceptibility variants could be private to certain individuals or families – a situation that imposes challenges in identifying the relevant genetic variants.
The high degree of genetic similarity between FPC patients and their FDRs, 50% on average, suggests that the latter are valuable samples for conducting genetic association studies. We have performed the first Nordic WGS study on FDRs of FPC patients from a nation‐wide cohort in the Danish population. Analysis and characterization of rare germline variants in FDRs of FPC patients could help reveal the molecular basis underlying the high genetic susceptibility of FPC.
2. MATERIALS AND METHODS
2.1. Sample collection
A nationwide cohort of 27 Danish families with susceptibility to FPC is currently included in a screening program for PDAC at the Department of Medical Gastroenterology, Odense University Hospital, Denmark. Each family was diagnosed with genetic predisposition to FPC at the department of clinical genetics in their home region, prior to inclusion in the screening program. 18 In concordance with previous definitions at our institution and international consensus criteria, 19 familial predisposition for FPC was defined as presence of either: (1) Two FDRs with PDAC, with at least one of the cases debuting at age < 50 year; or (2) at least three FDRs with PDAC.
FDRs to the FPC cases in each family are offered inclusion in the screening program after reaching a certain age (i.e. 5 years younger than the earliest age of onset of PDAC in the family); but no later than at 50 years of age. The screening program for PDAC of FDRs includes yearly imaging of the pancreas (with endoscopic ultrasonography, and fine needle biopsy if relevant), along with PDAC blood markers (i.e. Cancer antigen 19–9, CA19‐9) – with the possibility to individualize the program for each individual.
Individuals in the screening program, comprising of FDRs to FPC patients currently without presentation of PDAC, received informed consent to participate in the WGS study. Sixty‐one FDRs were included in the study, and a sample of 10 mL full blood was collected from each individual for sequencing analysis. The cohort profiles of the FPC predisposed families included in the screening program are described in detail in a previous study. 8
2.2. Ethics
Data and sample collection from relevant individuals were conducted with the approvals from the Danish National Committee on Health Research Ethics (NVK) (project number: 1604008) and the Danish Data Protection Agency (project number: 18/54160).
2.3. Sequencing analysis
A total of 61 FDRs from FPC patients were whole genome sequenced using DNA extracted from peripheral blood. In brief, 20 μg of genomic DNA per sample was sequenced using the TruSeq DNA PCR free kit (Illumina, Inc). Sequencing was performed on a NovaSeq 6000 (Illumina, Inc). Sequence reads were analyzed and aligned to the human reference genome (hg19) using Illumina DRAGEN software. Variants were annotated using VarSeq (Golden Helix, Inc.) with (i) functional consequence in RefSeq gene transcripts, (ii) zygosity, (iii) minor allele frequency (MAF) determined using publicly available variant databases (gnomAD) and (iv) presence in ClinVar.
2.4. Filtering and interpretation of variants
We applied filtering using VarSeq (https://www.goldenhelix.com/products/VarSeq/), version 2.2.1 (Golden Helix, Inc.) for downstream filtering. All variants were first filtered with a minimum of 10× coverage, nonsynonymous, and presented in the exome region or splice sites, which represented a range of 60.4%–95.6% of targeted bases. Filtered variants were then processed twice, one for each parameter. The first parameter which covers the possibility of a compound heterozygous, autosomal recessive, multifactorial inheritance, or de novo, was set to a population frequency of ≤0.01 (genomAD and ExAC). The second parameter, which covers the dominant inheritance of single nucleotide polymorphisms (SNPs) and small insertions and deletions (INDELs), was set to a frequency of ≤0.0001. We removed sequence variants belonging to (1) pseudogenes using annotations provided by EnsDb.Hsapiens.v86 package in Bioconductor (DOI: 10.18129/B9.bioc.EnsDb.Hsapiens.v86); and (2) segmental duplication (humanparalogy.gs.washington.edu). Multi‐mapped reads and artefacts were also removed from subsequent analysis.
Variants were then classified into (1) a group of functional variants including frameshift variants, inframe deletion, inframe insertion, initiator codon variants, splice acceptor variants, splice donor variants, stop gained variants and missense variants; and (2) a group of synonymous variants including splice region variants, stop retained variants, and 5' UTR premature start codon gain variants.
VarSeq (https://www.goldenhelix.com/products/VarSeq/) was used for functional prediction of nonsynonymous variants. Clinical significance (benign, likely benign, pathogenic, likely pathogenic, uncertain significance, etc.) of variants was assessed based on ClinVar submitted records as recommended by ACMG/AMP, and on evaluation by local clinicians and biologists using an inhouse assessment catalog. Variants assessed as benign or likely benign were filtered out from the nonsynonymous group. Likewise, variants assessed as pathogenic or likely pathogenic were removed from the synonymous group.
Functional interpretation of SNVs was provided by dbNSFP (database for nonsynonymous SNPs' functional predictions), a database developed for functional prediction and annotation of all potential non‐synonymous SNVs in the human genome. 20 The dbNSFP via VarSeq contains variant effect classifications from six functional prediction algorithms. Pathogenicity prediction was provided by the PHRED‐like score, a scaled score based on CADD (Combined Annotation‐Dependent Depletion) scores 1.4. 21 CADD is a tool for scoring the deleteriousness of SNVs as well as insertion/deletions variants in the human genome, with CADD score (C‐Score) for a given variant assessed based on diverse genomic features derived from surrounding sequence context, gene model annotations, evolutionary constraint, epigenetic measurements and functional predictions. The PHRED‐like C‐Score is defined as −10*log10(rank/total), by ranking C‐Score of a variant relative to all possible 8.6 billion substitutions in the human reference genome.
2.5. The genome aggregation database
The Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org) is an open source database developed for aggregating and harmonizing both exome and genome sequencing data. It is the world's largest public collection of human genetic variations and a popular resource for basic research and clinical variant interpretation. The version 2 dataset (GRCh37/hg19) spans 125 748 exome sequences and 15 708 whole‐genome sequences from unrelated individuals sequenced by various genetic studies. We make use of the WGS data of gnomAD as an external control for statistical analysis.
2.6. Statistical analysis
Proxy external controls association test (ProxECAT): ProxECAT 22 is a statistical method specifically developed for analysis of WGS data using existing large databases as external controls (here gnomAD). Different from the conventional case–control design that focuses on genetic variants predicted as functional and compares their frequencies between groups, ProxECAT makes use of non‐functional variants as a proxy for how well variants within a genetic region are sequenced and called within a sample. It compares the ratio between variant frequency and proxy frequency (λg*, λproxy) in cases with that in the external controls to adjust for group differences in sequencing technology, in processing (i.e. processing of DNA samples), and in read depth for creating the internal and external datasets, with the null hypothesis:
Where g* represents the gene of interest and λ is the rate of variants per N cases or controls. As the maximum likelihood estimates have a closed form under Poisson distribution, statistical significance of estimates can be inferred by a likelihood ratio test. 22 In summary, ProxECAT is a gene‐based burden test that includes non‐functional variants to enable the use of existing databases as external controls in statistical testing. The model has been integrated in a R package, ProxECAT, to assist implementation of the method (https://github.com/hendriau/ProxECAT).
2.7. Over‐representation analysis
Over‐representation analysis (ORA) is used to assess if the overlap of identified significant genes with genes from a functional cluster (biological pathway, a compiled list of cancer related genes) is significantly different from being random by calculating the probability from a hypergeometric distribution:
where N is the number of all genes in the genome, m is the number of genes in a functional cluster, n is the number of genes identified as significant, k is the number of overlapping genes under testing. The R function phyper() was used for calculating the hypergeometric probability.
ORA has been implemented in a web tool for biological pathway analysis, the gene‐set enrichment analysis (GSEA), to test if genes in one biological pathway is over‐represented in a list of identified significant genes. GSEA was performed on canonical pathways at https://www.gsea-msigdb.org/gsea/index.jsp.
The analytical pipeline from sequencing analysis, filtering, statistical testing, functional annotations to the final report is illustrated in Figure 1.
FIGURE 1.
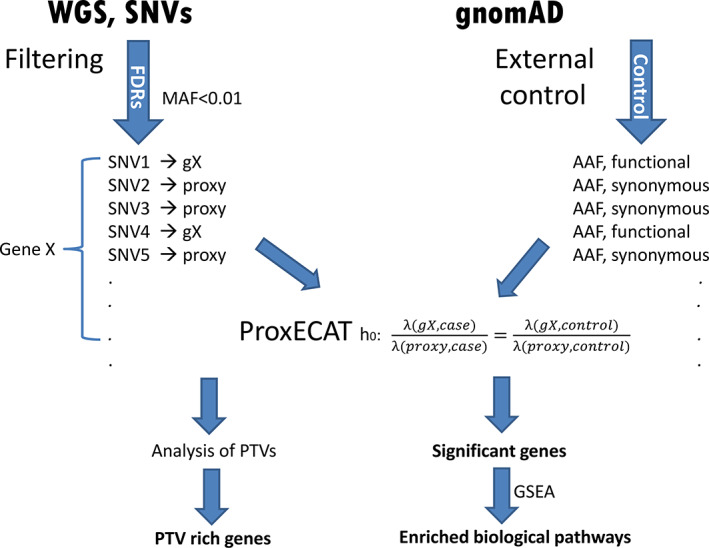
Workflow of whole genome sequencing analysis [Colour figure can be viewed at wileyonlinelibrary.com]
3. RESULTS
3.1. Sequencing analysis
We sequenced a total of 61 unaffected FDRs of FPC patients (sex: 25 female/36 male; median age: 59 years [37–84]) (Table S1), with an average output (median) of 15 161 SNVs per samples (range: 14678‐15 630), after applying in‐house filtering pipelines using VarSeq. In total, 60 778 SNVs were detected. Among them, 2397 SNVs were detected in all samples; and 16 533 SNVs were detected only once in 61 samples (referred to as private mutations, accounting for 27.2% of all detected SNVs). In Figure S1, we show the frequency of number of SNVs by number of times of detection in the 61 samples. As described in the Methods section, we further filtered all variants to remove SNVs from pseudogenes, pseudogene homology, segmental duplicates, and multi‐mapped variants, leaving 46 033 unique SNVs for subsequent analysis.
3.2. Analysis of premature truncating variants
PTVs represent a type of variants within a gene that create an early stop codon, leading to a shortened or truncated protein and resulting in serious functional consequences. Following Roberts et al. (2016), 16 we filtered SNVs using the following criteria (i) nonsense variants, splice‐site variants (splice donor variant, splice acceptor variant), and frameshift INDELs (frameshift variants); (ii) heterozygous in the germline; (iii) minor allele frequency (MAF) < 0.01 from gnomAD and (iv) present in only one individual, i.e. “private” or shared by FDRs in a family, i.e. “familial”, obtaining a total of 492 heterozygous PTVs in 448 genes. We then counted the number of PTVs in each gene.
A full list of genes with at least one PTV is shown in Table S2. Figure 2(A) is a histogram for the genes distributed by the number of PTVs they carry. The majority of genes have only 1 PTV. There are 22 genes with 2 PTVs and 8 with ≥3 PTVs (Table 1). Figure 2(B) plots the 448 genes ordered by the number of hosted PTVs (red labelled dots for genes with more than 4 PTVs). In Table 1 and Table S2, it is clear that the number of heterogenous PTVs harboured by the top genes are contributed mainly by private mutations, although there are also genes solely with PTVs shared by FDRs in a family.
FIGURE 2.
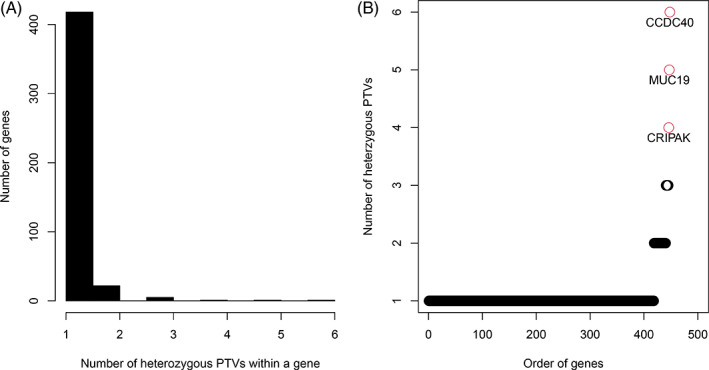
Premature truncating variant (PTV) analysis results illustrated with a histogram for the genes distributed by the number of PTVs they carry (a) and with a scatter plot for genes ordered by the number of hosting PTVs (b). The red colored dots are genes with 4 or more PTVs [Colour figure can be viewed at wileyonlinelibrary.com]
TABLE 1.
Thirty genes with two or more private heterozygous PTVs in FDRs of patients with FPC
| Gene names | Chr | Position 1st PTV | PTVs | Private PTVs | Familial PTVs |
|---|---|---|---|---|---|
| CCDC40 | 17 | 78 013 765 | 6 | 6 | 0 |
| MUC19 | 12 | 40 791 701 | 5 | 3 | 2 |
| CRIPAK | 4 | 1 388 414 | 4 | 3 | 1 |
| ATXN2 | 12 | 112 036 779 | 3 | 2 | 1 |
| HMCN2 | 9 | 133 061 568 | 3 | 3 | 0 |
| KLHL35 | 11 | 75 141 449 | 3 | 3 | 0 |
| SAMD1 | 19 | 14 200 862 | 3 | 2 | 1 |
| ZNF667 | 19 | 56 952 566 | 3 | 2 | 1 |
| ANO7 | 2 | 242 128 114 | 2 | 2 | 0 |
| ATP8B3 | 19 | 1 795 984 | 2 | 1 | 1 |
| BTN2A1 | 6 | 26 466 202 | 2 | 1 | 1 |
| C8orf44 | 8 | 67 590 023 | 2 | 2 | 0 |
| CCDC7 | 10 | 32 856 799 | 2 | 0 | 2 |
| CELSR1 | 22 | 46 832 075 | 2 | 0 | 2 |
| COL6A5 | 3 | 130 159 330 | 2 | 1 | 1 |
| COL6A6 | 3 | 130 284 459 | 2 | 2 | 0 |
| DNAH14 | 1 | 225 231 637 | 2 | 2 | 0 |
| DRD4 | 11 | 640 071 | 2 | 2 | 0 |
| GFY | 19 | 49 931 883 | 2 | 0 | 2 |
| HOMEZ | 14 | 23 744 823 | 2 | 0 | 2 |
| HSD3B1 | 1 | 120 056 818 | 2 | 0 | 2 |
| IQGAP3 | 1 | 156 509 221 | 2 | 2 | 0 |
| KRT77 | 12 | 53 086 340 | 2 | 2 | 0 |
| PKM | 15 | 72 523 315 | 2 | 0 | 2 |
| PRB3 | 12 | 11 420 755 | 2 | 2 | 0 |
| RBM33 | 7 | 155 532 530 | 2 | 2 | 0 |
| RETSAT | 2 | 85 576 627 | 2 | 1 | 1 |
| TEX15 | 8 | 30 703 647 | 2 | 2 | 0 |
| ULK4 | 3 | 41 756 779 | 2 | 0 | 2 |
| ZNF880 | 19 | 52 888 070 | 2 | 2 | 0 |
Detailed information of each of the PTVs is presented in Table S3 with one row for each sample carrying the mutation. A “familial” PTV is carried by samples from the same family, while “private” PTVs can only be found in one sample (row). As can be seen in Table S3, many of the genes are enriched by PTVs with a high PHRED score and a high voting of functional prediction (dbNSFP Functional Prediction Voting), indicating high significance in functional implications. The trend of positive correlation (Spearman correlation coefficient 0.26, p < 1.87e‐06) between PHRED score and dbNSFP functional prediction is clearly shown in Figure S2.
3.3. Rare variants association analysis
A total of 12 297 variants with MAF < 0.01 are available for association analysis. These variants are from 7229 genes, among them 531 genes have at least one functional nonsynonymous mutation and one synonymous mutation which were tested using ProxECAT. The Q‐Q plot in Figure 3 displays significant genes with p‐values deviating from the random distribution. The Manhattan plot in Figure 4 shows the genes along chromosomal locations. There are 20 significant genes with FDR < 0.05 (p < 1.5e‐03) (Table 2), 84 genes with p < 0.05 (Table S4). The top 6 genes (p < 1e‐05, FDR < 1e‐03) include PALD1, LRP1B, COL4A2, CYLC2, ZFYVE9, BRD3. PALD1 (paladin) on chromosome 10 is highly significant (p < 1.53e‐33) as it stands out from the other genes in Figures 3, 4.
FIGURE 3.
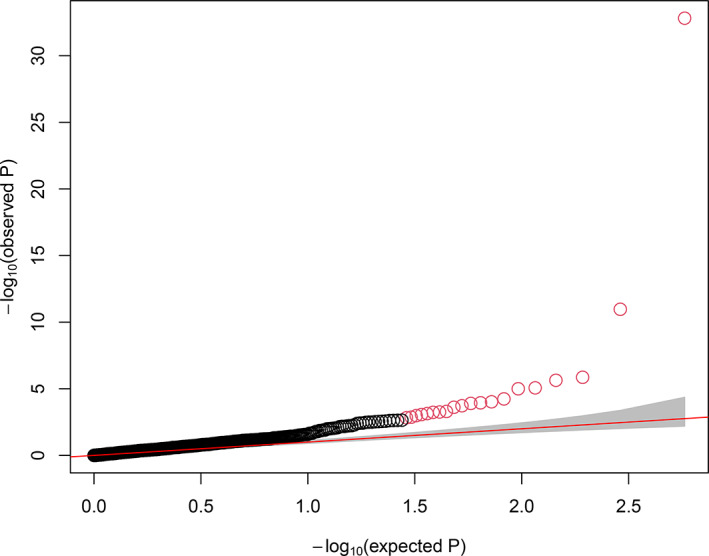
QQ plot for p‐values (negative logarithm) of genes from association test using ProxECAT. The significant genes deviate from the diagonal line of randomly distributed p‐values. The red dots are the 20 genes with FDR < 0.05 [Colour figure can be viewed at wileyonlinelibrary.com]
FIGURE 4.
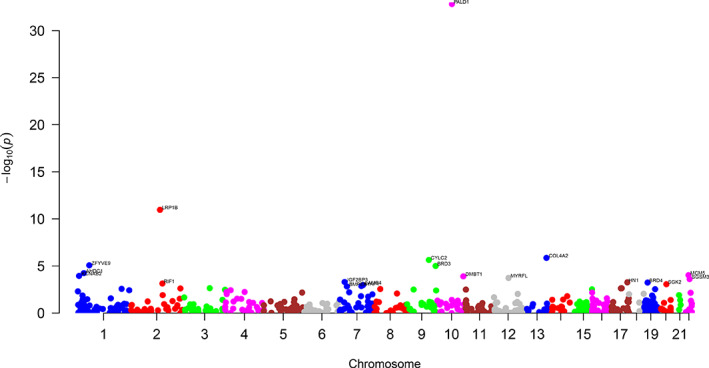
Manhattan plot plotting p‐values (negative logarithm) of genes from association test against their genomic locations [Colour figure can be viewed at wileyonlinelibrary.com]
TABLE 2.
The 20 identified genes with FDR < 0.05 using ProxECAT
| Gene name | Chrom | Start position (basepair) | Number of functional variants | Number of synonimous variants | ch2 | p‐value | FDR |
|---|---|---|---|---|---|---|---|
| PALD1 | 10 | 72 288 965 | 1 | 1 | 145.67 | 1.53e‐33 | 8.85e‐31 |
| LRP1B | 2 | 141 115 595 | 5 | 1 | 46.18 | 1.08e‐11 | 3.11e‐09 |
| COL4A2 | 13 | 111 082 772 | 1 | 1 | 23.32 | 1.37e‐06 | 2.64e‐04 |
| CYLC2 | 9 | 105 767 091 | 1 | 2 | 22.33 | 2.30e‐06 | 3.32e‐04 |
| ZFYVE9 | 1 | 52 729 426 | 1 | 1 | 19.81 | 8.55e‐06 | 9.60e‐04 |
| BRD3 | 9 | 136 899 924 | 1 | 1 | 19.52 | 9.96e‐06 | 9.60e‐04 |
| AHDC1 | 1 | 27 875 385 | 2 | 1 | 16.18 | 5.76e‐05 | 4.75e‐03 |
| MCM5 | 22 | 35 796 605 | 1 | 1 | 15.23 | 9.50e‐05 | 6.87e‐03 |
| KCNAB2 | 1 | 6 086 512 | 1 | 1 | 14.91 | 1.13e‐04 | 7.24e‐03 |
| DMBT1 | 10 | 124 339 274 | 6 | 1 | 14.71 | 1.26e‐04 | 7.26e‐03 |
| MYRFL | 12 | 70 329 887 | 1 | 1 | 13.95 | 1.88e‐04 | 9.88e‐03 |
| SGSM3 | 22 | 40 797 647 | 2 | 1 | 13.45 | 2.45e‐04 | 1.18e‐02 |
| IGF2BP3 | 7 | 23 358 880 | 1 | 1 | 12.11 | 5.03e‐04 | 2.24e‐02 |
| HN1 | 17 | 73 132 258 | 1 | 1 | 11.93 | 5.53e‐04 | 2.24e‐02 |
| BRD4 | 19 | 15 350 200 | 2 | 1 | 11.83 | 5.82e‐04 | .24e‐02 |
| RIF1 | 2 | 152 300 231 | 1 | 1 | 11.41 | 7.30e‐04 | 2.64e‐02 |
| SGK2 | 20 | 42 196 593 | 1 | 1 | 11.12 | 8.56e‐04 | 2.91e‐02 |
| LAMB4 | 7 | 107 720 041 | 1 | 1 | 10.75 | 1.05e‐03 | 3.36e‐02 |
| NYAP1 | 7 | 100 082 846 | 3 | 1 | 10.26 | 1.36e‐03 | 4.14e‐02 |
| BMPER | 7 | 34 010 036 | 1 | 1 | 10.08 | 1.50e‐03 | 4.33e‐02 |
There were 7 SNVs observed in PALD1, 6 missense and 1 splice region variants. Only 1 missense mutation (10:72294183, Ref/Alt: C/T) and the splice region variant met MAF < 0.01. Sixteen SNVs were observed for LRP1B (low‐density lipoprotein receptor‐related protein 1B, chromosome 2) after filtering, among them 6 had MAF < 0.01 with 5 missense mutations and 1 splice region variant. We observed 12 SNVs in COL4A2 (collagen type IV alpha2 chain), 4 of them had MAF < 0.01. After filtering, 1 missense variant (13:111155578, Ref/Alt: G/A) and 1 splice region variant remained. For CYLC2 (cylicin 2), there were 6 SNVs and 3 of them had MAF < 0.01, with 1 missense variant (9:105767091, Ref/Alt: C/A) and 2 splice region variants. The ZFYVE9 (zinc finger FYVE‐type containing 9) gene has 3 SNVs among which 2 SNVs had MAF < 0.01, with 1 splice acceptor variant (1:52729440, Ref/Alt: ‐/CA) and 1 splice region variant. There were 5 SNVs for BRD3 (bromodomain containing 3) gene, only 2 of them had MAF < 0.01 with 1 missense mutation (9:136899924, Ref/Alt: T/C) and 1 synonymous splice region variant. Table 2 also has another BRD gene, BRD4 with 2 missense mutations (19:15350625, Ref/Alt: C/T; 19:15350625, Ref/Alt: C/T) and 1 splice region variant.
Detailed information of each rare nonsynonymous variant in the significant genes in Table 2 can be found in Table S5 showing all genes with p < 0.05. In general, these variants have high PHRED scores (>20), although low scores are also observed in some of the variants. Notably, the vast majority of the nonsynonymous variants are missense mutations.
Similar to Figure S2, the PHRED scores show an obvious correlation with dbNSFP Functional Prediction Voting (Figure S3), suggesting that high PHRED scores are associated with high functionality of the variants. However, the degree of correlation is much higher than that for the PTVs (Spearman correlation coefficient 0.78 vs 0.26) with even higher statistical significance (p‐value 3.07e‐75 vs 1.87e‐06). From Figure S3, more variants with high PHRED scores are predicted as damaging as compared with Figure S2 suggesting that variants detected by the association test are functionally more relevant.
3.4. Over‐represented gene clusters
The 448 genes harbouring at least one PTV were submitted to GSEA for over‐representation analysis of canonical pathways using the hypergeometric test. Five canonical pathways are significantly over‐represented (FDR < 0.05) among the 2868 pathways from the GSEA databases encompassing 40 071 genes in universe (Table 3). The gene set “transport of small molecules” consisting of 728 genes has 26 genes overlapping with the list of 448 PTV genes resulting in a FDR < 2.44e‐03. The small gene set “butyrophilin (BTN) family interactions” has only 12 genes. Four of them can be found in the PTV genes with FDR < 1.29e‐02. Gene set “ion channel transport” is formed by 183 genes. Eleven of them can be found in the list of PTV genes leading to a significant over‐representation with FDR < 1.29e‐02. “Innate immune system” is a large gene set containing 1117 genes. Among them, 30 overlapped with the PTV genes (FDR < 2.35e‐02). There are 8 PTV genes in overlap with the 106 genes in “Stimuli‐sensing channels” resulting in a FDR < 2.35e‐02.
TABLE 3.
Functional pathways over‐represented by all PTV genes and ProxECAT significant genes
| Gene set name | # Genes in Gene Set (K) | Description | # Genes in overlap (k) | k/K | p‐value | FDR |
|---|---|---|---|---|---|---|
| PTV genes: | ||||||
|
REACTOME Transport of small molecules |
728 | Transport of small molecules | 26 | 0.04 | 8.36e‐07 | 2.44e‐03 |
|
REACTOME Butyrophilin (BTN) family interactions |
12 | Butyrophilin (BTN) family interactions | 4 | 0.33 | 8.96e‐06 | 1.29e‐02 |
|
REACTOME Ion channel transport |
183 | Ion channel transport | 11 | 0.06 | 1.33e‐05 | 1.29e‐02 |
|
REACTOME Innate immune system |
1117 | Innate immune system | 30 | 0.03 | 3.43e‐05 | 2.35e‐02 |
|
REACTOME Stimuli‐sensing channels |
106 | Stimuli‐sensing channels | 8 | 0.08 | 4.03e‐05 | 2.35e‐02 |
| ProxECAT genes: | ||||||
|
NABA Core matrisome |
275 | Ensemble of genes encoding core extracellular matrix including ECM glycoproteins, collagens and proteoglycans | 4 | 0.01 | 9.42e‐06 | 2.75e‐02 |
Next, we submitted the 20 significant genes tested by ProxECAT in Table 2 to GSEA. One pathway concerning the extracellular matrix (ECM) was significantly over‐represented (Table 3). Among the 275 genes in the pathway, 4 genes (LAMB4, DMBT1, BMPER, COL4A2) are represented in the list of significant genes in Table 2, displaying a significant overlap with a hypergeometric p value of 9.42e‐06 (FDR < 2.75e‐02). The pathway is an ensemble of genes encoding core extracellular matrix including ECM glycoproteins, collagens, and proteoglycans.
3.5. Enrichment analysis of cancer driver genes
We finally tested overlap of cancer driver genes in the list of genes hosting heterogenous PTVs and in the list of significant genes tested using ProxECAT, respectively. To do that, the number of overlapping genes between the detected gene lists and a collection of 460 cancer driver genes identified by Dietlein et al (2020) 23 was counted and tested using hypergeometric test. Among the 84 genes in Table S4, one gene, the KIT Proto‐Oncogene (KIT), overlapped with the cancer driver gene list. Although only one overlap, the hypergeometric test reported a p < 4.2e‐03, suggesting significant enrichment of cancer driver genes. The missense variant in KIT gene (4:55595566, Ref/Alt: C/T) has a PHRED score of 32 and 6 out of 6 votes predicted as damaging.
Likewise, among the 448 PTV genes in Table S2, 12 overlaps were found, TCHH, TMPRSS15, CHIT1, ZNF233, AIM2, SPATA31E1, PRDM2, DIS3, ATP11A, CCDC66, NFKBIE, TNFRSF10C, resulting in a hypergeometric p < 2.2e‐16, an extremely highly significant enrichment of cancer driver genes.
4. DISCUSSION
FDRs of FPC patients are at increased risk of developing PDAC and other cancers due to high degree of genetic relatedness and aggregation of risk genes within family members. Through performing the first WGS on FDRs of FPC patients in a nationwide cohort, we have found enrichment of rare genetic mutations in genes with significant implication in cancer pathogenesis.
4.1. The relevance of rare premature‐truncating variants
By shortening the protein‐coding sequence of genes, PTVs can lead to altered function of the hosting genes including gain or loss of gene function through for example nonsense mediated mRNA decay. 24 Identifying PTV associations to human diseases is a useful way to detect drug targets and to gain disease insights. Using the same approach as Roberts et al (2016), 16 we were able to examine PTV genes in FDRs and prioritize them for further assessment.
On top of Table 1 is CCDC40 (coiled‐coil domain containing protein) harbouring 6 heterogenous PTVs, all of which are “private” mutations. One study showed that the gene is associated with primary immunodeficiency diseases, 25 which may be related to PDAC development. 26 The MUC (mucin) genes encode a family of high molecular weight, heavily glycosylated proteins that form a protective coat around cancer cells. They are critical in the pathogenesis of pancreatic cancer and are associated with resistance to cytotoxic drugs, cancer invasiveness, metastases, and cell proliferation. 27 Expression of MUC genes has been shown to be associated with precursor lesions (pancreatic intraepithelial neoplasia, PanIN) in pancreatic cancer and overall survival. 28 Among the other genes in Table 1, somatic mutations in DNAH (dynein axonemal heavy chain) were reportedly associated with gastric cancer survival and treatment response. 29 A SNP in ANO7 was found to be associated with risk of aggressive prostate cancer with elevated expression of the gene correlating with disease severity and outcome. 30
Besides the observed PTVs, we also found pathogenic variants in known FPC susceptibility genes 16 that met our PTV definition but with allele frequency missing from the Ensembl Variation database. Among the 12 known FPC predisposition genes, we observed two pathogenic variants in BRCA2 gene in two families (one in each family), one frameshift variant in ATM gene in one family, one frameshift variant in CDKN2A gene in one family, and one stop gained variant in MSH6 in one family. These pathogenic variants together with the above well‐defined PTVs can serve as candidates for segregation analysis in the FPC families, when similar sequencing data are available on the probands (i.e. FPC patients in the respective families). Such analysis will help with identifying novel causal variants and further characterize the functional profiles of known variants in FPC.
4.2. The relevance of significant genes by association test
The gene showing the highest statistical significance in association analysis using ProxECAT is PALD1 (Table 2). By using machine learning, Deeb et al (2015) 31 found PALD1 as one of their four most predictive proteins for classifying diffuse large B‐cell lymphoma patients. In another study, knockdown of the expression of PALD1 was found to enhance the angiogenesis of immortalized human endothelial cells to promote cancer development. 32 By screening for base‐specific mutations, Tuupanen et al (2014) 33 found a hotspot mutation in PALD1 for colorectal cancer.
Mutational and transcriptional changes in some of the significant genes in Table 2 have previously been shown to be associated with PDAC. Brar et al (2019) 34 found that LRP1B (low‐density lipoprotein receptor‐related protein 1B) mutations are more frequent in metastatic lesions than in primary pancreatic tissue suggesting that mutation in this tumor suppressor gene may promote PDAC metastasis. Two genes in Table 2, BRD3 and BRD4, belong to the bromodomain and extraterminal (BET) family of proteins, which is one of the most prominent transcriptional vulnerabilities in human cancer – serving as potential therapeutic targets in cancer treatment. 35 It has been shown that BET bromodomain inhibitors can block growth of pancreatic cancer cells. 36 A recent study showed that AHDC1 gene is upregulated by competing endogenous RNA (ceRNA) interactions between lncRNAs and miRNAs to promote the progression of cervical cancer. 37 Over‐expression of MCM (mini‐chromosome maintenance) genes was significantly associated with PDAC progression and prognosis, 38 and expression of MCMs could serve as prognostic and therapeutic biomarkers for PDAC. 39 The missense mutation found in MCM5 in this study could function as cis‐regulatory mutation affecting expression of the gene (as an expression quantitative trait locus, eQTL) and contribute to the potential risk of PDAC. DMBT1 (deleted in malignant brain tumors 1) is a tumor suppressor gene. Secretion of COOH‐terminal fragment of DMBT1 has been revealed from PDAC cell lines, 40 while the gene was also found to be differentially expressed in PDAC. 41 The presently detected missense mutations in the gene may cause dysregulation of its tumor suppressor activity, thereby promoting the development of PDAC. Likewise, increased expression of IGF2BP3 (insulin‐like growth factor 2 mRNA‐binding protein 3) was found to promote invasiveness and metastasis of PDAC, 42 , 43 while dysregulation of SGK2 (serum/glucocorticoid regulated kinase 2) affected treatment response in PDAC. 44 The RIF1 gene has emerged as a conserved regulator of chromosome maintenance, acting to control DNA replication and repair. The gene is found highly upregulated in pancreatic cancer cell lines and is considered as a potential biomarker for diagnosis and therapeutic treatment of pancreatic cancer. 45
In summary, we identified multiple genes that have previously been shown to be associated with cancer development, progression and metastasis – with some of the genes being directly linked with PDAC pathogenesis. The expression of our detected significant genes could have been affected by the germline mutations through, for example, DNA‐transcription factors resulting in altered expression of the corresponding protein or by increased affinity for binding of the micro‐RNAs. 46 , 47 Given the high importance of gene expression in PDAC, it will be tempting to elucidate the underlying regulatory mechanisms involved.
Notably, the rare variants in the detected significant genes are predominantly missense variants (Table S5). The effects of missense mutations in cancer predisposition genes have been long discussed 48 and it is suggested that efforts to identify new susceptibility genes should not ignore missense variations considering their important roles in cancer susceptibility. 49 Large regions of BRCA1 and BRCA2 carry missense variations, although those occurring in the cold‐spot regions have been recently shown to be unlikely pathogenic. 50 It has been found that the majority of pathogenic variants in the breast cancer gene TP53 are missense variants, while the missense variants in two other breast cancer genes, ATM and CHEK2 are probably equally or even more important than PTVs in terms of their frequencies. 49
As indicated in Figure S3, the PHRED score is significantly correlated with predicted functionality, thus suggesting strong functional relevance of the rare missense variants from our association test. For all rare nonsynonymous variants (mostly missense) in genes tested with p < 0.05 (Table S5), the median PHRED score is 15.1, while variants in genes tested with FDR < 0.05 (p < 1.5e‐03) have a median PHRED score of 23. This again indicates that the missense variants in highly significant genes are more functionally relevant than the variants in less significant ones.
4.3. The relevance of enriched pathways
In Table 3, the only pathway enriched by the 20 significant genes from the ProxECAT association analysis is the core matrisome pathway comprising ECM glycoproteins, collagens and proteoglycans. In a recent network‐based analysis of gene expression data on FPC and sporadic pancreatic cancer, increased activity in extracellular structure and ECM organization was found. 51 It is interesting that significant association of ECM pathways was found by two different omics approaches (WGS and transcriptomics) suggesting that the detected rare variants could be involved in the regulation of genes of the ECM pathways. Among the 4 overlapping genes in the pathway, LAMB4 (subunit of the laminin gene family) is one of the most widely expressed ECM proteins and exerts many important functions in multiple organs. 52 COL4A2, DMBT1, BMPER are also highly involved in ECM pathways as COL4A2 constitutes one of the most abundant components of nearly all basement membranes – which is a thin, pliable sheet‐like type of ECM, that provides cell and tissue support. 53 DMBT1 encodes for an ECM protein responsible for epithelial to mesenchymal transition and differentiation. 54 BMPER (BMP binding endothelial regulator) has been shown to be highly expressed in multiple malignant tumors (lung, colon, and cervix). 55
One of the five pathways enriched by PTV genes, the transport of small molecules, is known to impact cancer development, metastasis and response to treatment. 56 The second enriched pathway is butyrophilin (BTN) family interactions. The butyrophilins are viewed as an emerging family of immune regulators. 57 The BTN genes are functionally implicated in T cell inhibition and in the modulation of epithelial cell–T cell interactions, thus being genetically associated with inflammatory diseases. One of the BTN member genes, BTN3A2, was identified as an independent prognostic marker of triple‐negative breast cancer. 58
Another significantly enriched pathway is related to the innate immune system. Cells of the innate immune system including: granulocytes, monocytes, macrophages, and dendritic cells, play important roles in cancer cell recognition, as well as the initiation of inflammation and antitumor immune responses. 59 However, persistent inflammation has been shown to be a driver of tumor progression in many malignancies by promoting immune suppression and cancer metastasis, as in the case of PDAC. 60
Other two significantly enriched pathways in the PTV genes are the ion channel transport and stimuli‐sensing channels. Both involve signaling transduction mechanisms and the capacity of cells to detect a specific stimulus depending on a characteristic combination of different ion channels. It is well known that ion channels regulate multiple cellular functions and are involved in the communication between extracellular events and intracellular signaling pathways. Altered activity of ion channels can have an impact on uncontrolled proliferation, promotion of invasion and migration of cancer. Research has indicated that certain ion channels are involved in the aberrant tumor growth and metastatic processes of PDAC. 61 The significant enrichment of multiple pathways involved in cancer development suggests that the rare PTVs in the FDRs could impact a broad range of functional processes that jointly contribute to the increased risk of PDAC in the FPC families.
4.4. The relevance of overlapping cancer‐driver genes
It is interesting that the genes from Table S2 (mainly representing PTVs of private mutations) are enriched for cancer driver genes, whose mutations give a growth advantage to cancer cells. The highly significant overlap of the 12 PTV genes with known cancer driver genes 23 indicates that rare germline mutations could constitute a potential risk for PDAC development. Among the 12 genes, DIS3 has allele‐specific expressions with decreased expression observed in carriers of pancreatic cancer risk‐increasing alleles, which could therefore affect nuclear RNA processing. 62 In another gene, TNFRSF10C (TNF Receptor Superfamily Member 10c), aberrant methylation at the promotor region has been frequently observed in pancreatic cancer cell lines suggesting that genetic variations of the gene could regulate gene activity through epigenetic mechanisms. 63 It is interesting that the expression levels of two members of the same gene family, TNFRSF11A and TNFRSF17, have been recently shown to correlate with PDAC sub‐grouping in regards to progression and therapeutic response. 64 TMPRSS15 (transmembrane serine protease 15) encodes an enzyme that converts the pancreatic proenzyme trypsinogen to trypsin, which activates other proenzymes including chymotrypsinogen and procarboxypeptidases. Strong genetic heterogeneity was found in the gene in patients with chronic pancreatitis. 65 ZNF233 (zinc finger protein 233) has been found to be associated with pancreatic cancer in a global genomic analysis of core signaling pathways. 66
Overall, the observation of rare germline PTVs in cancer driver genes provides novel data in support of potentially increased risk of cancer development in FDRs in FPC predisposed families and reveal a high degree of genetic heterogeneity in FPC susceptibility.
4.5. Strengths and limitations
The identification of multiple mutations associated with pancreatic cancer and other cancer types validates the ProxECAT test as a useful tool for rare variants association studies using existing large external sequencing databases as controls. Most importantly, the identified rare variants in FDRs of FPC patients could jointly contribute to the co‐aggregation of the disease within families. A limitation of the study design is that a direct association of the detected rare variants with FPC cannot be established by such analysis, as not all FDRs may develop PDAC. Prospective follow‐up data on development of PDAC among the FDRs will provide useful information to verify potential association and predictive value of the identified variants on risk of PDAC.
ProxECAT is a burden test that collapses rare variant data to estimate their enrichment within a gene region to achieve statistical power as compared to a single‐variant test. 67 By comparing difference in the ratio of functional to synonymous variants between cases and controls, ProxECAT enables the use of external controls in statistical testing. However, this also comes with a price as such a comparison can be underpowered in contrast to direct comparison of the functional variants between case and control groups. Nevertheless, the high quality of DNA samples obtained from peripheral blood and the large sample size of the external controls using the gnomAD database compensate the power issue, thus ensuring significant statistical testing by the ProxECAT analysis.
5. CONCLUSIONS
Relatives of FPC patients are at high risk of developing PDAC. Analyzing the genetic variants underlying cancer susceptibility is critical for risk assessment and early intervention. Our WGS analysis of rare variants in FDRs of FPC patients identified germline mutations and PTVs recurrent in cancer related genes and driver genes. The identified rare germline variants could contribute towards understanding the genetic basis of cancer susceptibility in relatives of FPC patients.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/cge.14038.
Supporting information
Appendix S1: Supporting information
ACKNOWLEDGEMENT
This project was jointly supported by: The Danish Cancer Society (Kræftens Bekæmpelse) (ref.: R218‐A13150), NEYE Fonden, Odense Pancreas Center (OPAC), Fabrikant Einar Willumsens Mindelegat, The Danish Cancer Research Fund (Dansk Kræftforskningsfond), Fonden til Lægevidenskabens Fremme (A.P. Møller Fonden), the Rigshospitalet (RH)/Odense University Hospital (OUH) Research Fund (ref.: A3183), Aase og Ejnar Danielsens Fond (ref.: 18‐10‐0686).
Tan M, Brusgaard K, Gerdes A‐M, et al. Whole genome sequencing identifies rare germline variants enriched in cancer related genes in first degree relatives of familial pancreatic cancer patients. Clinical Genetics. 2021;100(5):551-562. 10.1111/cge.14038
Funding information Rigshospitalet (RH)/Odense University Hospital (OUH) Research Fund, Grant/Award Number: A3183; Aase og Ejnar Danielsens Fond, Grant/Award Number: 18‐10‐0686; Dansk Kræftforsknings Fond (The Danish Cancer Research Fund); Fabrikant Einar Willumsens Mindelegat; Fonden til Lægevidenskabens Fremme (A.P. Møller Fonden); Kræftens Bekæmpelse (The Danish Cancer Society), Grant/Award Number: R218‐A13150; NEYE Fonden; Odense Pancreas Center (OPAC)
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Klein AP. Genetic susceptibility to pancreatic cancer. Mol Carcinog. 2012;51(1):14‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Petersen GM. Familial Pancreatic Adenocarcinoma. Hematol Oncol Clin North Am. 2015;29(4):641‐653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Permuth‐Wey J, Egan KM. Family history is a significant risk factor for pancreatic cancer: results from a systematic review and meta‐analysis. Fam Cancer. 2009;8(2):109‐117. [DOI] [PubMed] [Google Scholar]
- 5. Brand RE, Lerch MM, Rubinstein WS, et al. Advances in counselling and surveillance of patients at risk for pancreatic cancer. Gut. 2007;56(10):1460‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goggins M, Overbeek KA, Brand R, et al. Management of patients with increased risk for familial pancreatic cancer: updated recommendations from the International Cancer of the Pancreas Screening (CAPS) Consortium. Gut. 2020;69(1):7‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chaffee KG, Oberg AL, McWilliams RR, et al. Prevalence of germ‐line mutations in cancer genes among pancreatic cancer patients with a positive family history. Genet Med. 2018;20(1):119‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tan M, Brusgaard K, Gerdes A‐M, Mortensen MB, Detlefsen S. Schaffalitzky de Muckadell OB, et al. Cohort profile and heritability assessment of familial pancreatic cancer: a nation‐wide study. Scand J Gastroenterol. 2021;56(8):965‐971. [DOI] [PubMed] [Google Scholar]
- 9. Amundadottir L, Kraft P, Stolzenberg‐Solomon RZ, et al. Genome‐wide association study identifies variants in the ABO locus associated with susceptibility to pancreatic cancer. Nat Genet. 2009;41(9):986‐990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Petersen GM, Amundadottir L, Fuchs CS, et al. A genome‐wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nat Genet. 2010;42(3):224‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wolpin BM, Rizzato C, Kraft P, et al. Genome‐wide association study identifies multiple susceptibility loci for pancreatic cancer. Nat Genet. 2014;46(9):994‐1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Childs EJ, Chaffee KG, Gallinger S, et al. Association of Common Susceptibility Variants of Pancreatic Cancer in Higher‐Risk Patients: A PACGENE Study. Cancer Epidemiol Biomarkers Prev. 2016;25(7):1185‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen F, Childs EJ, Mocci E, et al. Analysis of Heritability and Genetic Architecture of Pancreatic Cancer: A PanC4 Study. Cancer Epidemiol Biomarkers Prev. 2019;28(7):1238‐1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747‐753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhen DB, Rabe KG, Gallinger S, et al. BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: a PACGENE study. Genet Med. 2015;17(7):569‐577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roberts NJ, Norris AL, Petersen GM, et al. Whole Genome Sequencing Defines the Genetic Heterogeneity of Familial Pancreatic Cancer. Cancer Discov. 2016;6(2):166‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roberts NJ, Jiao Y, Yu J, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012;2(1):41‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jorgensen MT, Mortensen MB, Gerdes AM, De Muckadell OB. Familial pancreatic cancer. Scand J Gastroenterol. 2008;43(4):387‐397. [DOI] [PubMed] [Google Scholar]
- 19. Joergensen MT, Gerdes AM, Sorensen J. Schaffalitzky de Muckadell O, Mortensen MB. Is screening for pancreatic cancer in high‐risk groups cost‐effective? ‐ Experience from a Danish national screening program. Pancreatology. 2016;16(4):584‐592. [DOI] [PubMed] [Google Scholar]
- 20. Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: A One‐Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice‐Site SNVs. Hum Mutat. 2016;37(3):235‐241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886‐d94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hendricks AE, Billups SC, HNC P, et al. ProxECAT: Proxy External Controls Association Test. A new case‐control gene region association test using allele frequencies from public controls. PLoS Genet. 2018;14(10):e1007591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dietlein F, Weghorn D, Taylor‐Weiner A, et al. Identification of cancer driver genes based on nucleotide context. Nat Genet. 2020;52(2):208‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. DeBoever C, Tanigawa Y, Lindholm ME, et al. Medical relevance of protein‐truncating variants across 337,205 individuals in the UK Biobank study. Nat Commun. 2018;9(1):1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stray‐Pedersen A, Sorte HS, Samarakoon P, et al. Primary immunodeficiency diseases: Genomic approaches delineate heterogeneous Mendelian disorders. J Allergy Clin Immunol. 2017;139(1):232‐245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yan Y, Gao R, Trinh TLP, Grant MB. Immunodeficiency in Pancreatic Adenocarcinoma with Diabetes Revealed by Comparative Genomics. Clin Cancer Res. 2017;23(20):6363‐6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Suh H, Pillai K, Morris DL. Mucins in pancreatic cancer: biological role, implications in carcinogenesis and applications in diagnosis and therapy. Am J Cancer Res. 2017;7(6):1372‐1383. [PMC free article] [PubMed] [Google Scholar]
- 28. Jonckheere N, Auwercx J, Hadj Bachir E, et al. Unsupervised Hierarchical Clustering of Pancreatic Adenocarcinoma Dataset from TCGA Defines a Mucin Expression Profile that Impacts Overall Survival. Cancers. 2020;12(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhu C, Yang Q, Xu J, et al. Somatic mutation of DNAH genes implicated higher chemotherapy response rate in gastric adenocarcinoma patients. J Transl Med. 2019;17(1):109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kaikkonen E, Rantapero T, Zhang Q, et al. ANO7 is associated with aggressive prostate cancer. Int J Cancer. 2018;143(10):2479‐2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deeb SJ, Tyanova S, Hummel M, Schmidt‐Supprian M, Cox J, Mann M. Machine Learning‐based Classification of Diffuse Large B‐cell Lymphoma Patients by Their Protein Expression Profiles. Mol Cell Proteomics. 2015;14(11):2947‐2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ren Y, Ji N, Kang X, et al. Aberrant ceRNA‐mediated regulation of KNG1 contributes to glioblastoma‐induced angiogenesis. Oncotarget. 2016;0. [Google Scholar]
- 33. Tuupanen S, Hänninen UA, Kondelin J, et al. Identification of 33 candidate oncogenes by screening for base‐specific mutations. Br J Cancer. 2014;111(8):1657‐1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brar G, Blais EM, Joseph Bender R, et al. Multi‐omic molecular comparison of primary versus metastatic pancreatic tumours. Br J Cancer. 2019;121(3):264‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xu Y, Vakoc CR. Targeting Cancer Cells with BET Bromodomain Inhibitors. Cold Spring Harb Perspect Med. 2017;7(7):a026674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sahai V, Kumar K, Knab LM, et al. BET bromodomain inhibitors block growth of pancreatic cancer cells in three‐dimensional collagen. Mol Cancer Ther. 2014;13(7):1907‐1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Feng Y, Qu L, Wang X, Liu C. LINC01133 promotes the progression of cervical cancer by sponging miR‐4784 to up‐regulate AHDC1. Cancer Biol Ther. 2019;20(12):1453‐1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Peng YP, Zhu Y, Yin LD, et al. The Expression and Prognostic Roles of MCMs in Pancreatic Cancer. PLoS One. 2016;11(10):e0164150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liao X, Han C, Wang X, et al. Prognostic value of minichromosome maintenance mRNA expression in early‐stage pancreatic ductal adenocarcinoma patients after pancreaticoduodenectomy. Cancer Manage Res. 2018;10:3255‐3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sasaki K, Sato K, Akiyama Y, Yanagihara K, Oka M, Yamaguchi K. Peptidomics‐based approach reveals the secretion of the 29‐residue COOH‐terminal fragment of the putative tumor suppressor protein DMBT1 from pancreatic adenocarcinoma cell lines. Cancer Res. 2002;62(17):4894‐4898. [PubMed] [Google Scholar]
- 41. Hustinx SR, Cao D, Maitra A, et al. Differentially expressed genes in pancreatic ductal adenocarcinomas identified through serial analysis of gene expression. Cancer Biol Ther. 2004;3(12):1254‐1261. [DOI] [PubMed] [Google Scholar]
- 42. Taniuchi K, Furihata M, Hanazaki K, Saito M, Saibara T. IGF2BP3‐mediated translation in cell protrusions promotes cell invasiveness and metastasis of pancreatic cancer. Oncotarget. 2014;5(16):6832‐6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cui XH, Hu SY, Zhu CF, Qin XH. Expression and prognostic analyses of the insulin‐like growth factor 2 mRNA binding protein family in human pancreatic cancer. BMC Cancer. 2020;20(1):1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yue W, Wang T, Zachariah E, et al. Transcriptomic analysis of pancreatic cancer cells in response to metformin and aspirin: an implication of synergy. Sci Rep. 2015;5:13390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. GursesCila HE, Acar M, Barut FB, Gunduz M, Grenman R, Gunduz E. Investigation of the expression of RIF1 gene on head and neck, pancreatic and brain cancer and cancer stem cells. Clin Invest Med. 2016;39(6):27500. [PubMed] [Google Scholar]
- 46. Zhang Z, Miteva MA, Wang L, Alexov E. Analyzing effects of naturally occurring missense mutations. Comput Math Methods Med. 2012;2012:805827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McGeary SE, Lin KS, Shi CY, et al. The biochemical basis of microRNA targeting efficacy. Science. 2019;366(6472):eaav1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Scott RJ, Meldrum CJ. Missense mutations in cancer predisposing genes: can we make sense of them? Hered Cancer Clin Pract. 2005;3(3):123‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tavtigian SV, Chenevix‐Trench G. Growing recognition of the role for rare missense substitutions in breast cancer susceptibility. Biomark Med. 2014;8(4):589‐603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dines JN, Shirts BH, Slavin TP, et al. Systematic misclassification of missense variants in BRCA1 and BRCA2 “coldspots”. Genet Med. 2020;22(5):825‐830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tan M. Schaffalitzky de Muckadell OB, Jøergensen MT. Gene Expression Network Analysis of Precursor Lesions in Familial Pancreatic Cancer. J Pancreat Cancer. 2020;6(1):73‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yang C, Liu Z, Zeng X, et al. Evaluation of the diagnostic ability of laminin gene family for pancreatic ductal adenocarcinoma. Aging. 2019;11(11):3679‐3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kuo DS, Labelle‐Dumais C, Gould DB. COL4A1 and COL4A2 mutations and disease: insights into pathogenic mechanisms and potential therapeutic targets. Hum Mol Genet. 2012;21(R1):R97‐R110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mollenhauer J, Herbertz S, Holmskov U, et al. DMBT1 encodes a protein involved in the immune defense and in epithelial differentiation and is highly unstable in cancer. Cancer Res. 2000;60(6):1704‐1710. [PubMed] [Google Scholar]
- 55. Heinke J, Kerber M, Rahner S, et al. Bone morphogenetic protein modulator BMPER is highly expressed in malignant tumors and controls invasive cell behavior. Oncogene. 2012;31(24):2919‐2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nizzero S, Ziemys A, Ferrari M. Transport Barriers and Oncophysics in Cancer Treatment. Trends Cancer. 2018;4(4):277‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Abeler‐Dörner L, Swamy M, Williams G, Hayday AC, Bas A. Butyrophilins: an emerging family of immune regulators. Trends Immunol. 2012;33(1):34‐41. [DOI] [PubMed] [Google Scholar]
- 58. Cai P, Lu Z, Wu J, et al. BTN3A2 serves as a prognostic marker and favors immune infiltration in triple‐negative breast cancer. J Cell Biochem. 2020;121(3):2643‐2654. [DOI] [PubMed] [Google Scholar]
- 59. Woo SR, Corrales L, Gajewski TF. Innate immune recognition of cancer. Annu Rev Immunol. 2015;33:445‐474. [DOI] [PubMed] [Google Scholar]
- 60. Stone ML, Beatty GL. Cellular determinants and therapeutic implications of inflammation in pancreatic cancer. Pharmacol Ther. 2019;201:202‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schnipper J, Dhennin‐Duthille I, Ahidouch A, Ouadid‐Ahidouch H. Ion Channel Signature in Healthy Pancreas and Pancreatic Ductal Adenocarcinoma. Front Pharmacol. 2020;11:568993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hoskins JW, Ibrahim A, Emmanuel MA, et al. Functional characterization of a chr13q22.1 pancreatic cancer risk locus reveals long‐range interaction and allele‐specific effects on DIS3 expression. Hum Mol Genet. 2016;25(21):4726‐4738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cai HH, Sun YM, Miao Y, et al. Aberrant methylation frequency of TNFRSF10C promoter in pancreatic cancer cell lines. Hepatobiliary Pancreat Dis Int. 2011;10(1):95‐100. [DOI] [PubMed] [Google Scholar]
- 64. Rasmussen LG, Verbeke CS, Sørensen MD, et al. Gene expression profiling of morphologic subtypes of pancreatic ductal adenocarcinoma using surgical and EUS‐FNB specimens. Pancreatology. 2021;21(3):530‐543. [DOI] [PubMed] [Google Scholar]
- 65. Sofia VM, Da Sacco L, Surace C, et al. Extensive molecular analysis suggested the strong genetic heterogeneity of idiopathic chronic pancreatitis. Mol Med. 2016;22:300‐309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801‐1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Povysil G, Petrovski S, Hostyk J, Aggarwal V, Allen AS, Goldstein DB. Rare‐variant collapsing analyses for complex traits: guidelines and applications. Nat Rev Genet. 2019;20(12):747‐759. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting information
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.