

Abstract
Metabolic stress contributes to the regulation of cell death in normal and diseased tissues. While different forms of cell death are known to be regulated by metabolic stress, how the cell engulfment and killing mechanism entosis is regulated is not well understood. Here we find that the death of entotic cells is regulated by the presence of amino acids and activity of the mechanistic target of rapamycin (mTOR). Amino acid withdrawal or mTOR inhibition induces apoptosis of engulfed cells and blocks entotic cell death that is associated with the lipidation of the autophagy protein microtubule‐associated protein light chain 3 (LC3) to entotic vacuoles. Two other live cell engulfment programs, homotypic cell cannibalism (HoCC) and anti‐CD47 antibody‐mediated phagocytosis, known as phagoptosis, also undergo a similar vacuole maturation sequence involving LC3 lipidation and lysosome fusion, but only HoCC involves mTOR‐dependent regulation of vacuole maturation and engulfed cell death similar to entosis. We further find that the regulation of cell death by mTOR is independent of autophagy activation and instead involves the 4E‐BP1/2 proteins that are known regulators of mRNA translation. Depletion of 4E‐BP1/2 proteins can restore the mTOR‐regulated changes of entotic death and apoptosis rates of engulfed cells. These results identify amino acid signaling and the mTOR‐4E‐BP1/2 pathway as an upstream regulation mechanism for the fate of live engulfed cells formed by entosis and HoCC.
Keywords: amino acids, cell death, entosis, metabolism, mTOR
Abbreviations
- 4E‐BP1/2
eukaryotic translation initiation factor 4E‐binding proteins 1 and 2
- Bcl‐2
B‐cell lympoma 2
- HoCC
homotypic cell cannibalism
- LC3
microtubule‐associated protein light chain 3
- mTOR
mechanistic/mammalian target of rapamycin
- mTORC1/2
mTOR complex 1 and 2
1. INTRODUCTION
Programmed cell death through apoptosis is well known to control cell turnover during metazoan development and in adult tissues. In addition to apoptosis, other non‐apoptotic forms of programmed cell death have been identified that can also contribute to cell turnover. 1 , 2 , 3 For example, forms of programmed necrosis have been shown to eliminate cells infected with viruses or that become damaged as a result of ischemia‐reperfusion. 4 In addition to these programs, it is also becoming clear that cells can be targeted for death by being engulfed by professional phagocytes or by neighboring cells. 5 , 6 Examples of cell death‐inducing engulfment mechanisms include entosis, 7 emperitosis, 8 suicidal emperipolesis, 9 phagoptosis, 5 and homotypic cell cannibalism (HoCC). 10 While cell engulfment appears to be the cause of cell death for these programs, little is known about the molecular mechanisms that regulate the execution of ingested cells. In some circumstances, engulfment per se appears insufficient to cause cell death, as entotic cells and those engulfed by HoCC have been shown to escape, post‐engulfment, and thereby avoid execution. 7 , 10
We have previously found that the death of epithelial cells ingested by entosis is executed, in part, through lipidation of microtubule‐associated protein light chain 3 (LC3), once thought to function exclusively in autophagy, to the single‐membrane entotic vacuole. 11 LC3 lipidation is followed by the fusion of lysosomes, whose enzymes appear to kill engulfed cells by a non‐apoptotic cell death program termed “entotic cell death” or “entotic death”. A similar activity of autophagy proteins occurs during phagocytosis and macropinocytosis and is thought to control vacuole maturation, most likely by facilitating lysosome fusion. 12 But while phagosome maturation involving LC3 lipidation occurs rapidly after engulfment, entotic vacuoles exhibit a maturation delay of hours or even days, suggesting that engulfment is insufficient to trigger LC3 lipidation and death of the internalized cell, and that there may be additional regulatory signals. 11 Why entotic vacuoles exhibit delayed maturation, and the nature of the signal that ultimately triggers maturation, are unknown. Like cells ingested by entosis, the predominant fate of cells ingested by suicidal emperipolesis or phagoptosis is considered to be cell death, 5 , 9 but detailed imaging‐based quantification of the fates of engulfed cells, including the kinetics of death and whether targeted cells can be rescued post‐engulfment, have not been performed. Here we investigate the regulation of cell death induced by these engulfment processes and identify the amino acid‐responsive mechanistic target of rapamycin (mTOR) kinase pathway as a regulator of entotic cell death.
2. MATERIALS AND METHODS
2.1. Cell culture and reagents
MCF10A cells were cultured in DMEM/F‐12 (11320‐033; Sigma‐Aldrich, St. Louis, MO, USA) supplemented with 5% horse serum (HS) (S12150, Atlanta Biologicals, Lawrenceville, GA, USA), 20 ng/ml epidermal growth factor (EGF) (AF‐100‐15, Peprotech, Princeton, NJ, USA), 10 µg/ml insulin (I‐1882, Sigma‐Aldrich, St. Louis, MO, USA), 0.5 µg/ml hydrocortisone (H‐0888, Sigma‐Aldrich, St. Louis, MO, USA), 100 ng/ml cholera toxin (C‐8052, Sigma‐Aldrich, St. Louis, MO, USA), and penicillin/streptomycin (30‐002‐Cl, Mediatech, Manassas, VA, USA). BxPC3 and Jurkat cells were cultured in RPMI‐1640 (11875‐101, Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) (F2442, Sigma‐Aldrich, St. Louis, MO, USA) and penicillin/streptomycin. RAW 264.7 cells were cultured in DMEM (11965‐092, Life Technologies, Carlsbad, CA, USA) supplemented with 10% FBS and penicillin/streptomycin. Amino acid‐free medium was prepared by dialyzing FBS for 4 h at 4℃ in PBS (P‐3813, Sigma‐Aldrich, St. Louis, MO, USA) in MWCO 3500 dialysis tubing (21‐152‐9, Thermo Fisher Scientific, Waltham, MA, USA), followed by overnight at 4℃ in fresh PBS and subsequent addition to base media prepared without amino acids to a 10% final concentration. When indicated, cells were treated with 0.5 µM Torin1 (4247, Tocris Bioscience, Bristol, UK), 2.5 µM PP242 (CD0258, Chemdea Pharmaceuticals, Ridgewood, NJ, USA), 10 µg/ml cycloheximide (C7698, Sigma‐Aldrich, St. Louis, MO, USA), or 1 µM Sytox Orange (S11368; Molecular Probes, Eugene, OR, USA) was added to cultures.
2.2. Virus production and infection
Cells expressing GFP‐LC3, H2B‐GFP, H2B‐mCherry, or Bcl2 were prepared by infecting cells with retroviruses made with pBabe‐GFP‐LC3, pBabe‐H2B‐GFP, pBabe‐H2B‐mCherry, or pBabepuro‐Bcl2 constructs, as described previously. 11 , 13 For virus infection, cells were seeded in a 6‐well plate at 1 × 105 per well. The next day, 1 ml viral supernatant was added with 10 µg/ml polybrene (NC9451129, Thermo Fisher Scientific, Waltham, MA, USA) for 24 h followed by a media change. Cells were then selected with the appropriate antibiotic, 2 µg/ml puromycin (P9620, Sigma‐Aldrich, St. Louis, MO, USA) or 10 µg/ml blasticidin (R210‐01, Thermo Fisher Scientific, Waltham, MA, USA).
2.3. siRNA and shRNA
siGenome SMART pool siRNAs against human FIP200 and mTOR were obtained from Dharmacon. MCF10A cells were seeded in a 6‐well plate at 1 × 105 per well and transfected with 100 nM siRNA using Oligofectamine (12252‐011, Thermo Fisher Scientific, Waltham, MA, USA). Cells were routinely assayed 48 hours post‐transfection. shRNAs against human Atg13 and 4E‐BP2 were obtained from Sigma. Stable cell lines expressing shRNAs were generated by virus production and infection mentioned above.
2.4. CRISPR/Cas9‐mediated knockout
For depletion of 4E‐BP1 in MCF10A cells by CRISPR‐Cas9, guide RNAs (gRNAs) were designed using the online CRISPR design tool from Feng Zhang’s laboratory (http://crispr.mit.edu). The vector encoding for Cas9 (pCDNA3.3‐TOPO‐hCas9, plasmid #41815, Addgene, Watertown, MA, USA), as well as the vector encoding the gRNA (pCR‐Blunt II‐TOPO, plasmid #41824, Addgene, Watertown, MA, USA), were introduced into control MCF10A cells by nucleofection (Cell Line Nucleofector Kit V, VCA‐1005, Lonza, Alpharetta, GA, USA). Single cell clones were selected and examined for 4E‐BP1 expression by western blotting.
2.5. Western blotting
Cells were lysed in ice‐cold RIPA buffer and western blotting was performed as described previously. 11 The following antibodies were used: Bcl2 (sc‐7382, Santa Cruz Biotechnology, Dallas, TX, USA), LC3 (4108, Cell Signaling, Beverly, MA, USA), 4E‐BP1 (9452; Cell Signaling, Beverly, MA, USA), 4E‐BP2 (2845, Cell Signaling, Beverly, MA, USA), mTOR (2983, Cell Signaling, Beverly, MA, USA), p53 (sc‐126, Santa Cruz Biotechnology, Dallas, TX, USA), anti‐β‐actin (A1978, Sigma‐Aldrich, St. Louis, MO, USA), anti‐rabbit IgG HRP‐linked antibody (7074, Cell Signaling, Beverly, MA, USA), and anti‐mouse IgG HRP‐linked antibody (7076, Cell Signaling, Beverly, MA, USA).
2.6. Immunofluorescence
Immunofluorescence was performed on cells cultured on glass‐bottom dishes (P35G‐1.5‐20‐C, MatTek, Ashland, MA, USA), as described previously. 7 Briefly, cells were fixed in 1:1 methanol/acetone for 5 min at −20℃, followed by three 5‐min PBS washes and blocking in 5% BSA, 100 mM glycine in PBS for 1 h, followed by incubation with primary antibodies at 4℃ overnight. Samples were then incubated with secondary antibodies and counterstained with DAPI (D1306, Life Technologies, Carlsbad, CA, USA). Confocal microscopy was performed with the Ultraview Vox spinning‐disk confocal system (PerkinElmer) equipped with a Yokogawa CSU‐X1 spinning disk head and an electron‐multiplying charge‐coupled device camera (Hamamatsu C9100‐13) coupled to a Nikon Ti‐E microsope; image analysis was done using Volocity software (PerkinElmer). The following antibodies were used for immunofluorescence: anti‐phospho‐mTOR (Ser2448) (5536, Cell Signaling, Beverly, MA, USA), anti‐LAMP1 (555798, BD Biosciences, San Jose, CA, USA), anti‐BrdU (5292, Cell Signaling, Beverly, MA, USA), Alexa Fluor 568 goat anti‐mouse secondary (A‐11031, Life Technologies, Carlsbad, CA, USA), and Alexa Fluor 488 goat anti‐rabbit secondary (A‐11034, Life Technologies, Carlsbad, CA, USA).
2.7. Time‐lapse microscopy
Cells were cultured on glass‐bottom dishes (P06G‐1.5‐20‐F, MatTek, Ashland, MA, USA), and time‐lapse microscopy was performed in 37℃ and 5% CO2 live‐cell incubation chambers, as described previously. 11 Fluorescence and differential interference contrast (DIC) images were acquired every 20 min for 20 h using a Nikon Ti‐E inverted microscope attached to a CoolSNAP charge‐coupled device camera (Photometrics) and NIS Elements software (Nikon).
2.8. Phagoptosis
RAW 264.7 cells were seeded onto glass‐bottom coverslip dishes in the presence of 200 U/ml IFNγ (315‐05, Peprotech, Princeton, NJ, USA) for 2 days. Jurkat cells were added to RAW 264.7 cultures at 4 × 106 per dish and imaged by time‐lapse microscopy as mentioned above. When indicated, 10 µg/ml anti‐CD47 antibody (B6H12.2; ab3283, Abcam, Cambridge, UK; 14‐0479‐82, eBioscience, San Diego, CA, USA), 0.5 µM Torin1, 100 nM concanamycin A (C9705, Sigma‐Aldrich, St. Louis, MO, USA), or 50 nM LysoTracker Red (L7528, Thermo Fisher Scientific, Waltham, MA, USA) was added to cultures.
2.9. BrdU uptake assay
For BrdU uptake assay, cells were seeded in a 12‐well plate at 5 × 104 per well, incubated with 10 µM BrdU (B23151, Thermo Fisher Scientific, Waltham, MA, USA) at 37℃ for 24 h. Cells were then fixed with 3.7% formaldehyde for 15 min and permeabilized with Triton X‐100 for 20 min with three 5‐min PBS washes between each step. Cells were then treated with 1N HCl for 10 min on ice, 2N HCl for 10 min in room temperature, phosphate/citric acid buffer, pH 7.4 for 10 min in room temperature, washed with three 2‐min Triton X‐100 permeabilization buffer washes, and proceeded with the immunofluorescence steps mentioned above.
2.10. Statistics
The indicated p values were obtained using Student’s t‐test (****p < .0001; ***p < .001, **p < .01, *p < .05; ns, not significant).
3. RESULTS
3.1. Amino acids and mTOR regulate the fate of live engulfed cells in entosis
Entosis leads to the formation of “cell‐in‐cell” structures that harbor viable cells that have been engulfed by their neighbors. Despite their viability when internalized, entotic cells have been shown to have restricted access to culture media, for example, growth factors, 11 and we also found that they also display low levels of mTOR activity compared to host cells or neighboring cells (Figure 1A), consistent with restricted access to exogenous nutrients. When observed by time‐lapse microscopy for 20 h, approximately 50% of internalized cells underwent non‐apoptotic cell death associated with LC3 lipidation and subsequent lysosome fusion to entotic vacuoles, similar to previous reports, 7 , 11 through a process henceforth referred to as entotic death (Figure 1B). On the other hand, a lower percentage of entotic cells underwent apoptosis instead, which was not associated with LC3 lipidation or lysosome fusion prior to death (Figure 1C), an observation that is also previously reported. 7 , 11 While we have shown that some cells can remain internalized and viable for prolonged periods, and a small percentage can even be released from their hosts, 7 , 11 we focused here on studying the regulation of entotic death. As internalized cells are shielded from the extracellular environment (Figure 1A), it was unexpected that the removal of amino acids from the culture medium would change the fates of the engulfed cells. However, we found that amino acid deprivation had a significant effect on internalized cell fate, reducing the frequency of entotic cell death and increasing the percentage that underwent apoptosis (Figure 1D). Furthermore, direct inhibition of the amino acid‐responsive kinase mTOR, by treatment with an ATP‐competitive inhibitor Torin1, changed the fate of engulfed cells in a similar manner, as fewer died by entotic death and more died through apoptosis (Figure 1E). These data demonstrate that the fate of engulfed cells is regulated, surprisingly, by the availability of extracellular amino acids and mTOR activity.
FIGURE 1.
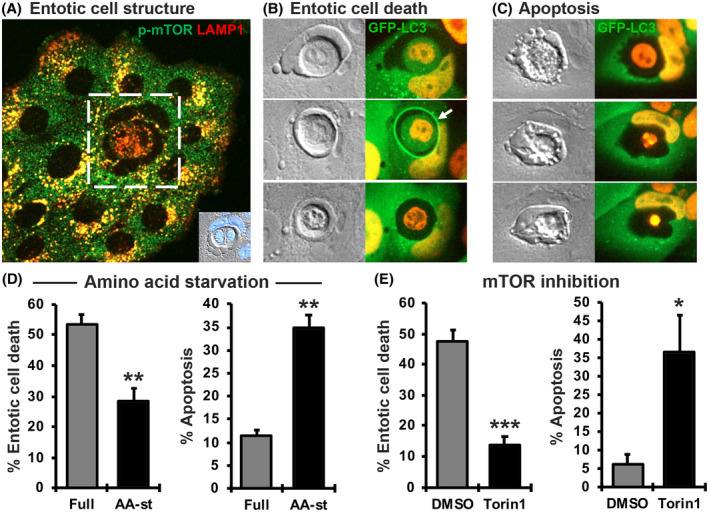
Amino acids and mTOR regulate the fate of live engulfed cells in entosis. (A) Engulfed MCF10A cells have lower mTOR activity indicated by levels of phospho‐mTOR (green) overlapping with LAMP1 (red), located on lysosome membranes, compared to host or neighboring cells in full media. Inset shows brightfield image and DAPI staining of the cell‐in‐cell structure in white box. (B) Representative images of entotic death in MCF10A cells expressing GFP‐LC3 and H2B‐mCherry through time (white arrow indicates LC3 lipidation to entotic vacuole preceding entotic death of engulfed cell). (C) Representative images of apoptosis in cells expressing GFP‐LC3 and H2B‐mCherry through time. Note distinct cellular and nuclear morphology of engulfed cell and absence of LC3 lipidation prior to cell death. (D) Quantification of entotic death and apoptosis of engulfed cells over 20 h in amino acid starvation. (E) Quantification of entotic death and apoptosis of engulfed cells over 20 h with 0.5 µM Torin1 treatment
Because amino acid starvation and mTOR inhibition had a dual effect to decrease entotic death and increase apoptosis, we sought to determine if these two results were linked, and the increased rate of apoptosis was inhibitory to entotic cell death. To examine this, the fates of entotic cells overexpressing an inhibitor of apoptosis, B‐cell lympoma 2 (Bcl‐2) (Figure 2A), which are resistant to apoptosis but undergo entotic death similar to control cells (Figure 2A,B), 11 were quantified by time‐lapse imaging, in the presence or absence of amino acids or treatment with inhibitors of mTOR. As shown in Figure 2C,D, starvation for exogenous amino acids or inhibition of mTOR activity (through treatment with Torin1 or PP242) led to decreased rates of entotic cell death even in the absence of apoptosis, demonstrating that nutrient availability and mTOR signaling regulate entotic death independently of apoptosis. The inhibition of mTOR also did not have an additive effect to inhibit entotic death when combined with amino acid starvation, suggesting that amino acids and mTOR regulate entotic death by acting through the same pathway (Figure 2C).
FIGURE 2.
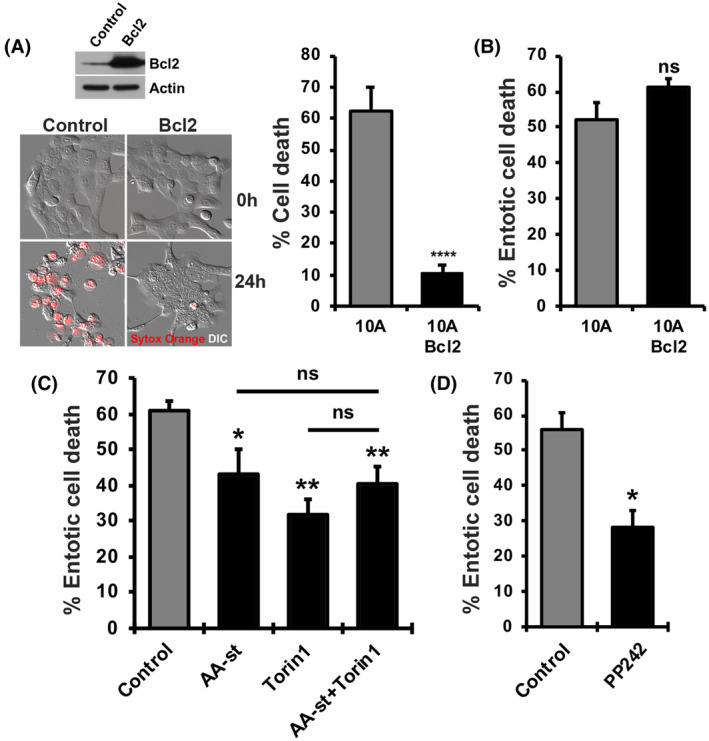
Amino acids and mTOR regulate entotic death independently of apoptosis. (A) Western blot showing Bcl2 expression and representative images and quantification of Sytox Orange‐positive cells in MCF10A control and Bcl2‐expressing cells treated with 10 µg/ml cycloheximide for 24 h. (B) Quantification of entotic death of engulfed cells in control and Bcl2‐expressing MCF10A cells. (C) Quantification of entotic death of engulfed cells of Bcl2‐expressing MCF10A cells over 20 h in amino acid starvation, 0.5 µM Torin1 treatment, or both. (D) Quantification of entotic death of engulfed cells in Bcl2‐expressing MCF10A cells over 20 h with 2.5 µM PP242 treatment
3.2. Amino acids and mTOR regulate engulfed cell fate in HoCC but not anti‐CD47 antibody‐mediated phagoptosis
To examine whether amino acid signaling and mTOR regulate other live cell engulfment processes, we investigated cell‐in‐cell structures formed by HoCC, 10 and live cell engulfments occurring as a result of anti‐CD47 antibody‐mediated phagoptosis mediated by macrophages. 5 Like entotic cell structures, cell‐in‐cell structures formed by HoCC exhibited two distinct types of cell death when examined by time‐lapse microscopy. Internalized cells either died by a non‐apoptotic mechanism associated with LC3 lipidation to vacuoles and lysosome fusion (Supplementary Figure S1A), or by apoptosis, which was not preceded by LC3 lipidation and was characterized by the appearance of condensed and/or fragmented nuclei. When cells were starved for amino acids or treated with Torin1, the rates of non‐apoptotic death decreased while rates of apoptosis increased (Supplementary Figure S1B). By contrast, during anti‐CD47 antibody‐mediated phagocytosis, or phagoptosis, 14 although phagosomes containing live lymphocytes also had a maturation process involving LC3 lipidation and lysosome‐dependent cell death (Figure 3A, Supplementary Figure S2, and [Link], [Link], [Link]), starvation for amino acids or treatment with Torin1 did not alter the fate of ingested cells (Figure 3B). These findings demonstrate that regulation of cell death through amino acid signaling and mTOR is specific to HoCC and entosis. We also found that while entotic cells underwent cell death at variable times, and often after a long delay after engulfment, live cells ingested by phagoptosis were killed quickly, suggesting the presence of additional regulatory steps during entotic cell death that may be controlled by amino acid signaling and mTOR (Figure 3C).
FIGURE 3.
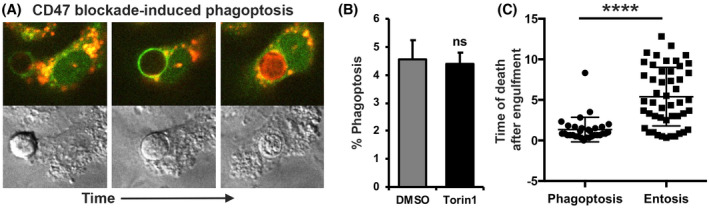
Engulfed cell fate in phagoptosis is not regulated by amino acids or mTOR. (A) Representative images of anti‐CD47 antibody‐mediated phagocytosis, or phagoptosis, of live Jurkat lymphocytes by RAW 264.7 macrophages associated with LC3 lipidation (white arrow) and lysosome fusion (lysotracker staining) to phagosomes. (B) Quantification of phagoptosis rates over 20 h with 0.5 µM Torin1 treatment. (C) Quantification of engulfed cell death timing of MCF10A after engulfment in phagoptosis by RAW 264.7 macrophages or entosis over 15 h
3.3. mTOR regulates entotic death by acting within host cells
To further elucidate the mechanism of cell fate regulation by mTOR, we first sought to determine in which cell, engulfed or host (inner or outer cell), mTOR regulated entotic cell death. mTOR expression was depleted in cells expressing a red fluorescent marker (H2B‐mCherry), and these cells were mixed at a 1:1 ratio with control cells expressing a green fluorescent marker (H2B‐GFP). The fates of ingested cells were then examined by time‐lapse imaging, with mTOR‐depleted cells either on the inside or the outside of entotic structures (Figure 4A,B). In all possible combinations of entotic structures, we found that the frequency of entotic death was reduced only when mTOR was depleted from host cells (Figure 4C), demonstrating that mTOR regulates entotic death by functioning within host cells and not within engulfed cells.
FIGURE 4.
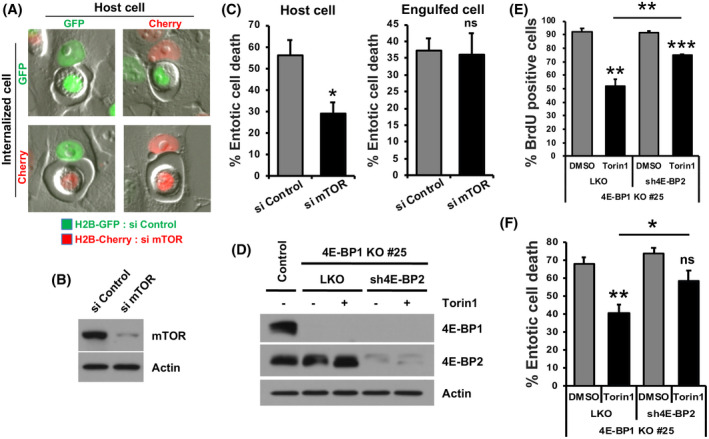
mTOR regulates entotic death within host cells through protein synthesis. (A) Representative images of MCF10A cell‐in‐cell structures in siRNA‐mediated control (H2B‐GFP) and mTOR knockdown (H2B‐mCherry) mixed cultures. (B) Western blot showing knockdown of mTOR. (C) Quantification of entotic death and apoptosis of engulfed cells over 20 h depending on host cell or engulfed cell status. (D) Western blot showing shRNA‐mediated 4E‐BP2 knockdown in the 4E‐BP1 knockout clone #25 with 0.5 µM Torin1 treatment. (E) Quantification of BrdU‐positive cells in 4E‐BP1/2‐depleted MCF10A cells with 0.5 µM Torin1 treatment. (F) Quantification of entotic death of 4E‐BP1/2‐depleted MCF10A cells over 20 h with 0.5 µM Torin1 treatment
3.4. mTOR regulates entotic cell death through protein synthesis
To identify how mTOR regulates entotic cell death, we first examined a possible role for the autophagy pathway that is induced in response to mTOR inhibition. As shown in Supplementary Figure S3A‒C, the knockdown of proteins from the autophagy pre‐initiation complex, including FIP200 and Atg13, which are required for autophagy induction but not entotic vacuole maturation, 11 had an inhibitory effect on autophagy induced by Torin1, but no effect on the reduction of entotic cell death. These data suggest that the regulation of autophagy by mTOR is not involved in entotic cell death. Another function of mTOR is to regulate translational machinery that controls the initiation of protein synthesis, and we next asked whether this pathway was involved in the regulation of entotic death. In potential support of this hypothesis, cycloheximide treatment, which blocks protein synthesis, potently inhibited the entotic death of ingested cells (Supplementary Figure S3D), demonstrating a general requirement for protein synthesis to support this process. To investigate whether mTOR regulates entotic death by controlling protein synthesis, we generated cells depleted for expression of eukaryotic translation initiation factor 4E‐binding proteins 1 and 2 (4E‐BP1/2). 4E‐BP1/2 proteins interfere with the interaction between eukaryotic translation initiation factor 4E (eIF4E) and eIF4G to block translation initiation and, when directly phosphorylated by mTOR, dissociate from eIF4E to allow protein synthesis to proceed. 15 Cells with CRISPR/Cas9‐mediated knockout of 4E‐BP1 were generated, and then expression of 4E‐BP2 was knocked‐down in these cells (Figure 4D). In cells depleted of 4E‐BP1/2, we observed that cell proliferation, as measured by BrdU uptake, was partially rescued in the context of mTOR inhibition, in a similar manner as reported previously for 4E‐BP1/2 knockout cells (Figure 4E). Using these cells, we examined whether depletion of 4E‐BP1/2 altered the entotic death frequencies regulated by mTOR. As shown in Figure 4F, we indeed found that the depletion of 4E‐BP1/2 partially rescued the frequency of entotic death in the context of mTOR inhibition, demonstrating that the decrease in entotic death caused by mTOR inhibition is due, at least in part, to the inhibition of mRNA translation.
4. DISCUSSION
Here we identify the amino acid‐mTOR‐4E‐BP1/2 pathway as a regulator of the non‐apoptotic cell death that is induced by the live cell engulfment process entosis. Amino acid withdrawal or mTOR inhibition reduces the frequency of host cell‐mediated killing of internalized cells, which occurs by a non‐apoptotic mechanism resulting from vacuole maturation and lysosome fusion. 11 The reduction of entotic death by mTOR inhibition is rescued by depletion of the 4E‐BP1/2 proteins, suggesting that amino acid‐mTOR signaling exerts control over this process by regulating protein synthesis. We have previously shown that autophagy proteins participate in entotic death by facilitating vacuole maturation, and that depletion of the machinery that controls LC3 lipidation also reduces entotic death frequencies. 11 However, we found that the regulation of entotic death by mTOR appears unrelated to its control over autophagy, consistent with previous findings that autophagy induction is unrelated to phagosome maturation which is also associated with LC3 lipidation. 16 It will be of interest to examine whether the amino acid‐mTOR‐4E‐BP1/2 pathway can regulate cell death in cell‐in‐cell structures formed by other cell lines, such as MCF7 17 or 16HBE. 18 While the methods of mTOR inhibition shown here (Torin 1 or PP242 treatment and siRNA‐mediated knockdown) both block mTOR complex 1 (mTORC1) and mTORC2 activity, the finding that amino acid withdrawal and mTOR inhibition act redundantly, and that 4E‐BP1/2 proteins, known mTORC1 effectors, play a role in cell death specifically implicate the mTORC1 complex as a regulator of entotic death.
We also find that the death of cells ingested by HoCC is regulated by the availability of extracellular amino acids and mTOR activity, similar to entotic death, while the death of tumor cells ingested by phagoptosis is not, although both cell death processes go through a similar process of LC3 lipidation and lysosome fusion. As entosis and HoCC are associated with an apparent vacuole maturation delay compared to phagoptosis, we speculate that the initiation of vacuole maturation in the context of epithelial cell‐in‐cell engulfment requires a secondary, post‐engulfment signal that requires mTORC1. Such a signal could participate in the initiation of endocytic maturation, or even in the final resolution of engulfment, as we have shown that entosis involves a scission event that removes a small piece of the internalizing cell and correlates with the initiation of entotic death. 19 The engagement of Fcγ receptors on macrophages by the anti‐CD47 antibody used to trigger phagoptosis could conceivably play a role to facilitate rapid phagosome maturation that bypasses similar regulation. 20 The identity of the protein, or proteins, required for vacuole maturation during entosis or HoCC that are synthesized in an mTOR‐dependent manner is an important topic for future studies.
DISCLOSURES
Memorial Sloan Kettering Cancer Center and one investigator involved in this study (M.O.) have financial interests in Elucida Oncology, Inc.
AUTHOR CONTRIBUTIONS
Sung Eun Kim and Michael Overholtzer designed and carried out experiments and wrote the paper. Justin Zhang carried out experiments associated with autophagy inhibition. Enoch Jiang analyzed the timing of phagoptosis and entosis. All authors participated in editing the manuscript.
Supporting information
Fig S1
Fig S2
Fig S3
Video S1
Video S2
Video S3
Legends
ACKNOWLEDGEMENTS
This work was supported by grants NCI RO1CA154649 (M.O.) and supported by a Korea University Grant K1926621, National Research Foundation of Korea (NRF) grant NRF‐2020R1C1C1013220 (S.E.K.). We thank members of the Overholtzer lab for discussions and critical reading of the manuscript.
Kim SE, Zhang J, Jiang E, Overholtzer M. Amino acids and mechanistic target of rapamycin regulate the fate of live engulfed cells. FASEB J. 2021;35:e21909. 10.1096/fj.202100870R
Contributor Information
Sung Eun Kim, Email: sek19@korea.ac.kr.
Michael Overholtzer, Email: overhom1@mskcc.org.
REFERENCES
- 1. Galluzzi L, Vitale I, Abrams JM, et al. Molecular definitions of cell death subroutines: recommendations of the nomenclature committee on cell death 2012. Cell Death Differ. 2012;19(1):107‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yuan J, Kroemer G. Alternative cell death mechanisms in development and beyond. Genes Dev. 2010;24(23):2592‐2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tait SWG, Ichim G, Green DR. Die another way – non‐apoptotic mechanisms of cell death. J Cell Sci. 2014;127(10):2135‐2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370(5):455‐465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brown GC, Neher JJ. Eaten alive! Cell death by primary phagocytosis: “phagoptosis”. Trends Biochem Sci. 2012;37(8):325‐332. [DOI] [PubMed] [Google Scholar]
- 6. Overholtzer M, Brugge JS. The cell biology of cell‐in‐cell structures. Nat Rev Mol Cell Biol. 2008;9(10):796‐809. [DOI] [PubMed] [Google Scholar]
- 7. Overholtzer M, Mailleux AA, Mouneimne G, et al. A nonapoptotic cell death process, entosis, that occurs by cell‐in‐cell invasion. Cell. 2007;131(5):966‐979. [DOI] [PubMed] [Google Scholar]
- 8. Wang S, He M‐F, Chen Y‐H, et al. Rapid reuptake of granzyme B leads to emperitosis: an apoptotic cell‐in‐cell death of immune killer cells inside tumor cells. Cell Death Dis. 2013;4(10):e856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Benseler V, Warren A, Vo M, et al. Hepatocyte entry leads to degradation of autoreactive CD8 T cells. Proc Natl Acad Sci. 2011;108(40):16735‐16740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cano CE, Sandí MJ, Hamidi T, et al. Homotypic cell cannibalism, a cell‐death process regulated by the nuclear protein 1, opposes to metastasis in pancreatic cancer. EMBO Mol Med. 2012;4(9):964‐979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Florey O, Kim SE, Sandoval CP, Haynes CM, Overholtzer M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat Cell Biol. 2011;13(11):1335‐1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Florey O, Overholtzer M. Autophagy proteins in macroendocytic engulfment. Trends Cell Biol. 2012;22(7):374‐380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Debnath J, Mills KR, Collins NL, Reginato MJ, Muthuswamy SK, Brugge JS. The role of apoptosis in creating and maintaining luminal space within normal and oncogene‐expressing mammary acini. Cell. 2002;111(1):29‐40. [DOI] [PubMed] [Google Scholar]
- 14. Chao MP, Weissman IL, Majeti R. The CD47–SIRPα pathway in cancer immune evasion and potential therapeutic implications. Curr Opin Immunol. 2012;24(2):225‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ma XM, Blenis J. Molecular mechanisms of mTOR‐mediated translational control. Nat Rev Mol Cell Biol. 2009;10(5):307‐318. [DOI] [PubMed] [Google Scholar]
- 16. Sanjuan MA, Dillon CP, Tait SWG, et al. Toll‐like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450(7173):1253‐1257. [DOI] [PubMed] [Google Scholar]
- 17. Hamann JC, Surcel A, Chen R, et al. Entosis is induced by glucose starvation. Cell Rep. 2017;20(1):201‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Durgan J, Tseng Y‐Y, Hamann JC, et al. Mitosis can drive cell cannibalism through entosis. eLife. 2017;11(6):e27134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee Y, Hamann JC, Pellegrino M, et al. Entosis controls a developmental cell clearance in C. elegans . Cell Rep. 2019;26(12):3212‐3220.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Métayer LE, Vilalta A, Burke GAA, Brown GC. Anti‐CD47 antibodies induce phagocytosis of live, malignant B cells by macrophages via the Fc domain, resulting in cell death by phagoptosis. Oncotarget. 2017;8(37):60892‐60903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Video S1
Video S2
Video S3
Legends