

Abstract
GATA2 deficiency syndrome (G2DS) is a rare autosomal dominant genetic disease predisposing to a range of symptoms, of which myeloid malignancy and immunodeficiency including recurrent infections are most common. In the last decade since it was first reported, there have been over 480 individuals identified carrying a pathogenic or likely pathogenic germline GATA2 variant with symptoms of G2DS, with 240 of these confirmed to be familial and 24 de novo. For those that develop myeloid malignancy (75% of all carriers with G2DS disease symptoms), the median age of onset is 17 years (range 0–78 years) and myelodysplastic syndrome is the first diagnosis in 75% of these cases with acute myeloid leukemia in a further 9%. All variant types appear to predispose to myeloid malignancy and immunodeficiency. Apart from lymphedema in which haploinsufficiency seems necessary, the mutational requirements of the other less common G2DS phenotypes is still unclear. These predominantly loss‐of‐function variants impact GATA2 expression and function in numerous ways including perturbations to DNA binding, protein structure, protein:protein interactions, and gene transcription, splicing, and expression. In this review, we provide the first expert‐curated ACMG/AMP classification with codes of published variants compatible for use in clinical or diagnostic settings.
Keywords: GATA2 deficiency syndrome, germline variants, immunodeficiency, lymphedema, myeloid malignancy, predisposition
GATA2 mutation update including a comprehensive deconvoluted summary of all published germline GATA2 deficiency syndrome mutations to date, associated phenotypes, and expert ACMG/AMP classification (with criteria) of all 179 pathogenic and likely pathogenic unique variants.

1. BACKGROUND
GATA2 deficiency syndrome (G2DS) (MIM#s 601626, 614286, 614038, 614172; GATA2 deficiency with susceptibility to MDS/AML, MONDO:0042982) is a collective of hematological (malignant and nonmalignant) and nonhematological phenotypes due to germline predisposing variants in the GATA2 gene that act in a partially penetrant autosomal dominant manner. Symptoms may range from life‐threatening bone marrow (BM) failure, immunodeficiency, and/or myeloid malignancy, to no overt phenotype even at old age, although the latter is less common. Phenotypes that have been reported include myeloid malignancies (predominantly myelodysplastic syndrome [MDS] and acute myeloid leukemia [AML]), lymphedema (Emberger syndrome), and immune deficiency (DCML deficiency; combined deficit of DC, monocyte, B and NK lymphoid cells, MonoMAC; monocytopenia and mycobacterial infection) with associated recurrent infections (Dickinson et al., 2011; Donadieu et al., 2018; Emberger et al., 1979; Hahn et al., 2011; Hsu et al., 2011; Kazenwadel et al., 2012; Ostergaard et al., 2011; Spinner et al., 2014; Wlodarski et al., 2016). Other less common phenotypes include chronic neutropenia (Pasquet et al., 2013), cytopenia/BM failure (Ganapathi et al., 2015), pulmonary alveolar proteinosis (PAP), sensorineural deafness, neurological features, urogenital malformations (Donadieu et al., 2018; Spinner et al., 2014), thrombosis, autoimmune features, rheumatological features, premature labor, and miscarriage (Donadieu et al., 2018). Affected individuals may experience multiple phenotypes throughout their lifetime or even at a single time point, while others may present with only very mild symptoms.
To date, no single underlying common phenotype has been described in the majority of G2DS carriers, in contrast to platelet function disorder or thrombocytopenia in Familial platelet disorder with predisposition to acute myeloid leukemia (FPD/AML, MONDO:0011071) caused by germline pathogenic variants in the RUNX1 gene. Therefore, it is proposed that each G2DS phenotype arises due to biological or environmental stressors that act on particular cell types in which reduced functional GATA2 protein is at or near a threshold level, thereby creating a situation in which GATA2 activity becomes limiting for normal cellular function. These “stressor events” may occur during embryogenesis or after birth and their effects accumulate over time. A mechanism has been proposed for “hematopoietic stem cell exhaustion” due to recurrent or persistent infections resulting in BM failure (Hirabayashi et al., 2017; Hsu et al., 2015). One might propose similar mechanisms for each of the phenotypes such as physical, inflammatory, or infectious stresses on lymphatic vessels during development causing lymphedema (Kazenwadel et al., 2015) or more stochastic events leading to malformations of the auditory system or urinary tract. For myeloid malignancies, germline GATA2 variants may create a microenvironment that is conducive for the selection and clonal expansion of particular acquired mutations such as ASXL1, CEBPA, SETBP1, and −7/7q through a process coined “predestination” where a limited trajectory of disease evolution is imposed by pre‐existing variants (i.e., germline) or early (i.e., somatic) mutations (Papaemmanuil et al., 2013). Here we provide a comprehensive GATA2 variant update of published germline cases and include unpublished variants from our institution cohort.
1.1. Ascertainment criteria of GATA2 variants
Familial and de novo germline GATA2 variants were identified and extracted from peer‐reviewed literature and collated (Table 1 and Table S1). GATA2 variants are described according to Human Genome Variation Society nomenclature and annotated to GenBank accession number NM_032638.5. Variants were classified according to American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) guidelines (Richards et al., 2015) with ClinGen Sequence Variant Interpretation recommendations (Abou Tayoun et al., 2018). A total of 160 unique pathogenic or likely pathogenic GATA2 variants were identified, with an additional 19 describing partial or complete GATA2 gene deletions, resulting in haploinsufficiency. Phenotype expansion associated with G2DS has resulted in the duplication of individuals in the literature carrying germline GATA2 variants, as more clinical and genetic information has become available. In this review, we have attempted to remove duplication by combining genetic and clinical data for these individuals as extrapolated from the referenced publications. A total of 480 individuals carrying a germline GATA2 pathogenic or likely pathogenic variant were identified (Tables 1 and S1, Figures 1 and 2B). The available published data confirmed 240 individuals with inherited variants and 24 individuals carrying de novo variants. For the remaining 221 individuals with a G2DS phenotype, insufficient evidence was provided to conclude on the mode of GATA2 variant acquisition. Classified variants of uncertain significance (VUS) or likely benign were excluded from analysis, but are provided in Table S2. GATA2 pathogenic and likely pathogenic variants (ACMG/AMP classified) are very rarely seen in the general population (only 3 in total in gnomAD v2.1.1 and 3.0).
Table 1.
GATA2 germline variants reported in families or individuals displaying one or more GATA2 deficiency syndrome (G2DS) phenotypes
| Number individuals | Familial (F)/de novo (D) | GATA2 protein (NP_116027.2) | GATA2 cDNA (NM_032638.5) | Mutation effect | ACMG/AMP classification (curated) | ACMG/AMP criteria | Overall Phenotype | Chromosomal Abnormalities |
|---|---|---|---|---|---|---|---|---|
| 1 | N/A | p.(Glu6Alafs*178) | c.17_18del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | −7 |
| 1 | F | p.(Gln20*) | c.58C>T | NS | Pathogenic | PVS1, PS4_Supporting, PM2 | T‐ALL, HA, ID | 45,XX,dic(21;22)(p11.2;p11.2) |
| 7 | F |
p.(Gly28Alafs*52) (in cis p.(His26Pro)) |
c.77A>C/c.83delG | FS | Pathogenic | PVS1, PS4, PM2, PP1_Moderate | M/A, ID, L, HA | −7 |
| 3 | F | p.(Glu44*) | c.130G>T | NS | Pathogenic | PVS1, PS4_Moderate, PM2 | L, HA, ID | |
| 1 | N/A | p.(Ser54*) | c.161C>A | NS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | −7 |
| 1 | F |
p.(Tyr59*) (in cis p.?) |
c.177C>G (in cis c.[140T>G;142T>C;145T>C]) |
NS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, ID | +8 |
| 1 | D | p.(Arg69Leufs*115) | c.206_208delGCGinsT | FS | Pathogenic | PVS1, PS2, PS4_Supporting, PM2 | M/A | |
| 1 | N/A | p.(Val70Leufs*114) | c.207_208delCG | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | der(1;7) add |
| 1 | D | p.? | c.222_229+6del14ins21 | INDEL | Pathogenic | PVS1, PS2, PS4_Supporting, PM2 | M/A | +8 |
| 2 | F | p.? | c.229+13_229+14insGCCins203_229+13 | INDEL | Likely Pathogenic | PS4_Moderate, PM2, PP3 | M/A, ID | +8 |
| 3 | F | p.(Arg78Profs*107) | c.232dup [c.230‐1_230insC] | FS | Pathogenic | PVS1, PS4_ Moderate, PM2 | M/A, L, HA | −7 |
| 1 | N/A | p.(Gly82Argfs*103)[G81fs*] | c.243delinsGC | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, ID, HA | NK |
| 1 | N/A | p.(Arg86Profs*98) [C85fs*] | c.257_258del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, ID | −7 |
| 1 | N/A | p.(Arg86Alafs*33) | c.256del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | ID, M/A, HA | |
| 1 | N/A | p.(Gly101Alafs*18) [G101Afs*16] | c.302del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, CMML, ID, HA | del(11)(q13q23), −7, +8 |
| 3 | N/A | p.(Ala103Glnfs*16) | c.303del | FS | Pathogenic | PVS1, PS4_ Moderate, PM2 | M/A |
−7, der(1;7)(q10;p10), der(1;7) add |
| 1 | N/A | p.(Ala103Glnfs*16) | c.306del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | −7, add |
| 5 | F | p.(Leu105Profs*15) | c.312_313dup | FS | Pathogenic | PVS1, PS4, PM2 | M/A, ID, L, HA | ‐7 |
| 1 | N/A | p.(Ser106Cysfs*78) | c.317_318del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | ID | NK |
| 2 | F | p.(Ala107Cysfs*78) [S106fs] | c.318dup [c.318_319insT] | FS | Pathogenic | PVS1, PS4_ Moderate, PM2 | ID, L | |
| 15 | F | p.(Thr117=) | c.351C>G | SPL | Pathogenic | PS3_Very Strong, PS4, PM2, PP1_Strong, PP3 | M/A, ID, L, HA, NS |
−7, +8, Chr1 translocation |
| 1 | N/A | p.(Val118Glyfs*100) [V618fs] | c.353del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | −7 |
| 1 | N/A | p.(Gly136Argfs*49) | c.404dup | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, ID | |
| 1 | N/A | p.(Ser139Cysfs*78) | c.414_417del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | ID, M/A, HA, L | |
| 1 | N/A | p.(Ser139Cysfs*45) | c.416_417del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | −7 |
| 1 | N/A | p.(Val140Cysfs*45) [V140Cfs*44] | c.417dup | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, ID, HA | NK |
| 2 | F | p.(Tyr141*) | c.423C>A | NS | Pathogenic | PVS1, PS4_ Moderate, PM2 | M/A, ID | −7 |
| 1 | F | p.(Gly146Valfs*72) | c.437del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, ID | NK |
| 1 | N/A | p.(Glu180*) | c.538G>T | NS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, ID | NK |
| 1 | N/A | p.(Thr188Hisfs*14) | c.561dup | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | NK |
| 1 | D | p.(Ala194Serfs*8) | c.579dup | FS | Pathogenic | PVS1, PS2, PS4_Supporting, PM2 | L, M/A | −7 |
| 1 | N/A | p.(Ala198Glyfs*20) | c.593del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | HA, ID | |
| 1 | F | p.(Gly199Leufs*22) | c.586_593dup | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, ID, HA | −9q |
| 5 | F | p.(Gly200Valfs*18) [G199fs*] |
c.599del [c.594delG] |
FS | Pathogenic | PVS1, PS4, PM2 | M/A, ID, L | −7 |
| 11 | F/D | p.(Ser201*) [G200fs] | c.599dup [c.599_600insG] | NS | Pathogenic | PVS1, PS4, PM2 | M/A, ID, HA, L | +8, −7 |
| 1 | N/A | p.(Arg204*) | c.610C>T | NS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, ID, HA | 46,XX,der(9)t(1;9)(q12;q12), r(9)(q12q?34)[15]/46,XX [9] |
| 1 | N/A | p.(Val211Argfs*72) | c.627_630dup | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | −7 |
| 1 | N/A | p.(Glu224*) | c.670G>T | NS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, ID, HA | NK |
| 1 | N/A | p.(Leu229Cysfs*5) | c.685del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | −7 |
| 1 | N/A | p.(Gly237Alafs*89) [M236Ifs325*] | c.710del [c.708delC] | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | HA, L, ID | |
| 2 | N/A | p.(Ile246Hisfs*36) [P245fs*] | c.735dup [c.735_736insC] | FS | Pathogenic | PVS1, PS4_ Moderate, PM2 | ID, M/A, HA | |
| 1 | F | p.(Tyr260Cysfs*25) [Y260fs*24, D259fs*] | c.769_778dup | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, ID, HA | −7 |
| 1 | N/A | p.(Phe265Glufs*58) | c.793_802del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | |
| 3 | F | p.(Gly268*) | c.802G>T | NS | Pathogenic | PVS1, PS4_ Moderate, PM2 | M/A, ID, L |
−13q ‐7 +8 |
| 2 | N/A | p.(Gly273Thrfs*8) | c.817_818del [c.814_815del] | FS | Pathogenic | PVS1, PS4_ Moderate, PM2 | M/A |
‐7 +8 |
| 1 | N/A | p.(Pro274Thrfs*8) [G273fs*] | c.818dupG [c.819insG] | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | Other |
| 1 | F | p.(Ala286Val) | c.857C>T | MS | Likely pathogenic | PS3, PS4_Supporting, PM2, PP3 | CMML | |
| 1 | N/A | p.(Ser290*) | c.869C>A | NS | Pathogenic | PVS1, PS4_Supporting, PM2 | HA/ID | |
| 3 | F | p.(Cys298Leufs*86) | c.892dup | FS | Pathogenic | PVS1, PS4_ Moderate, PM2 | ID, M/A |
Chr1‐7 translocation +8 |
| 2 | F | p.(Trp306Alafs*77) | c.915_916del | FS | Pathogenic | PVS1, PS4_ Moderate, PM2 | M/A, ID, HA | −7 |
| 1 | N/A | p.(Trp306*) | c.917G>A | NS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, HA, L, ID | −7 |
| 1 | N/A | p.(Thr311Argfs*71) | c.932_937delinsG | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | −7 |
| 1 | F | p.(His313Tyr) | c.937C>T | MS | Likely Pathogenic | PS4_Supporting, PM1, PM2, PP3 | M/A, ID | −5, −7, +8, add10, −12, −18, −21 |
| 1 | F | p.(Tyr314Cysfs*66) | c.941_951del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, ID | −7 |
| 1 | N/A | p.(Leu315Pro) | c.944T>C | MS | Likely Pathogenic | PS4_Supporting, PM1, PM2, PP3 | ID | |
| 1 | N/A | p.(Ala318Thrfs*12) | c.941_951dup | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, ID, HA | −6 |
| 1 | F | p.(Ala318Thr) | c.952G>A | MS | Pathogenic | PS3, PS4_Supporting, PM1, PM2, PP3 | M/A, ID, HA | +1,der(1:7)(q10:p10), +8 |
| 1 | F | p.(Cys319Serfs*5) | c.956_962del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | ID, HA | |
| 1 | N/A | p.(His323Glnfs*61) | c.968dup | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | −7 |
| 1 | N/A | p.(Gln328*) | c.982C>T | MS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, ID | dic(1;15)(?;?) |
| 13 | F/D | p.(Arg330*) | c.988C>T | NS | Pathogenic | PVS1, PS4, PM2 | M/A, ID, L, HA, JMML |
−7, +8, +1 |
| 1 | N/A | p.(Leu332Thrfs*53) | c.989_992dup [c.992_993insGACC] | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, HA, L | −7, +8 |
| 1 | N/A | p.(Leu332Glnfs*60) | c.970_994dup | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | −7 |
| 7 | F | p.(Arg337*) | c.1009C>T | NS | Pathogenic | PVS1, PS4, PM2 | M/A, ID, L, HA | −7 |
| 1 | N/A | p.? | c.1017+1del | SPL | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | −7 |
| 2 | N/A | p.? [S340Gfs*99] | c.1017+2T>G | SPL | Pathogenic | PVS1, PS4_ Moderate, PM2 | M/A, ID, HA | −7 |
| 1 | D | p.? [S340Afs*49] | c.1017+2T>C | SPL | Pathogenic | PVS1, PS2, PS4_Supporting, PM2 | M/A, HA, L, ID | |
| 2 | F |
p.? INT enhancer (del ETS site) |
c.1017+513_1017+540del (c.1017+512del28) | REG | Likely Pathogenic | PS3_Supporting, PS4_ Moderate, PM1, PM2 | M/A, ID, HA | |
| 30 | F |
p.? INT enhancer (ETS site) |
c.1017+572C>T | REG | Pathogenic | PS3, PS4, PM1, PM2, PP1_Strong | M/A, ID, L, HA, CMML, ALL, B‐ALL |
−7 +8 +1 der(Y)t(Y;1)(q11.23;q21) der(1;7)(q10;p10) |
| 1 | N/A | p.? | c.1018‐11_1027del | SPL | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | −7 |
| 2 | N/A | p.? | c.1018‐10_1037del | SPL | Pathogenic | PVS1, PS4_ Moderate, PM2 | M/A | −7 |
| 2 | N/A | p.? [A341Rfs*38] | c.1018‐3_1031del | SPL | Pathogenic | PVS1, PS4_ Moderate, PM2 | ID, L, HA, HL | |
| 2 | F | p.? | c.1018‐2A>G | SPL | Pathogenic | PVS1, PS4_ Moderate, PM2 | M/A, L | −7 |
| 1 | N/A | p.? | c.1018‐2A>T | SPL | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | −7 |
| 2 | N/A | p.? [S340‐N381del] | c.1018‐1G>A | DEL | Likely Pathogenic | PVS1_Strong, PS4_ Moderate, PM2 | M/A,HA, ID | der(22)t(1;22)(q12;p13)/der(15)t(1;15)(q12;p13) |
| 1 | N/A | p.? [S340‐N381del] | c.1018‐1G>T | DEL | Likely Pathogenic | PVS1_Strong, PS4_Supporting, PM2 | HA,ID | |
| 2 | F | p.(Ser340Lysfs*40) | c.1018_1028del | FS | Pathogenic | PVS1, PS4_ Moderate, PM2 | M/A, ID | NK |
| 1 | N/A | p.(Ser340Trpfs*47) | c.1019del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | −7 |
| 1 | N/A | p.(Ala341Aspfs*53) | c.1019_1020insCGACTGGGAGGGCAAGGCAG | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, HA, L, ID | |
| 1 | N/A | p.(Ala341Profs*46) | c.1021del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | HA, ID, L | |
| 1 | N/A | p.(Ala341Profs*45) | c.1021_1024del [c.1019_1022del] | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | L, M/A, HL | |
| 3 | F | p.(Ala341Serfs*39) | c.1021_1031del | FS | Pathogenic | PVS1, PS4_ Moderate, PM2 | M/A, ID | −7, +8 |
| 1 | N/A | p.(Ala342Argfs*42) | c.1023dup | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, ID | NK |
| 2 | F | p.(Ala342Profs*45) | c.1023del | FS | Pathogenic | PVS1, PS4_ Moderate, PM2 | ID, L | NK |
| 2 | N/A | p.(Ala342del)l [341delA] | c.1024_1026del [c.1021_1023] | DEL | Pathogenic | PVS1, PS4_ Moderate, PM2 | M/A | −7 |
| 1 | N/A | p.(Arg343Profs*42) [A342Gfs*41] | c.1025_1026insGCCG | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | ID, HA | |
| 2 | F | p.(Arg344Glnfs*41) | c.1023_1026dup | FS | Pathogenic | PVS1, PS4_ Moderate, PM2 | M/A, ID, HA | −7 |
| 3 | N/A | p.(Arg344Glyfs*43) | c.1020_1029dup | FS | Pathogenic | PVS1, PS4_ Moderate, PM2 | M/A, ID, L, HA | NK |
| 1 | D | p.(Arg344Lysfs*37) | c.1031_1049del | FS | Pathogenic | PVS1, PS2, PS4_Supporting, PM2 | M/A | NK |
| 1 | N/A | p.(R344Kfs*40) | N/A | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | NK |
| 1 | N/A | p.(Gly346Serfs*40) |
c.1035_1036ins TCTGGCC |
FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, ID, HA | |
| 1 | D | p.(Thr347Argfs*38) | c.1035_1038dup | FS | Pathogenic | PVS1, PS2, PS4_Supporting, PM2 | M/A | −7 |
| 1 | N/A | p.(Thr347Argfs*42) | c.1023_1038dup (c.1038_1039insCGCCAGAAGAGCCGGC) | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | −7 |
| 2 | F | p.(T347fs) | 16bp tandem dup | FS | Pathogenic | PVS1, PS4_ Moderate, PM2 | ID, HA | |
| 1 | N/A | p.(Cys348Valfs*39) | c.1041del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | ID, M/A, HA | −7 |
| 1 | D | p.(Cys349Phe) [C348F] | c.1046G>T | MS | Likely Pathogenic | PS2, PS4_Supporting, PM1, PM2, PM5, PP3 | M/A | −7 |
| 1 | D | p.(Cys349Arg) | c.1045T>C | MS | Pathogenic | PS2, PS4_Supporting, PM1, PM2, PM5, PP3 | M/A, ID |
46,XY,der(3)t dic(1;3)(p11; p25) |
| 1 | N/A | p.(Cys349Gly) | c.1045T>G | MS | Likely Pathogenic | PS4_Supporting, PM1, PM2, PM5, PP3 | ID, M/A,L | |
| 1 | N/A | p.(Asn351Ser) | c.1052A>G | MS | Likely Pathogenic | PS4_Supporting, PM1, PM2, PP3 | M/A | −7 |
| 1 | F | p.(Cys352Arg) | c.1054T>C | MS | Likely Pathogenic | PS4_Supporting, PM1, PM2, PM5, PP3 | M/A | |
| 1 | N/A | p.(Cys352Gly) | c.1054T>G | MS | Likely Pathogenic | PS4_Supporting, PM1, PM2, PM5, PP33 | M/A | −7 |
| 1 | N/A | p.(Cys352Valfs*35) | c.1054del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | ID, M/A, HA, L | NK |
| 1 | N/A | p.(Cys352Phe) | c.1055G>T | MS | Likely Pathogenic | PS4_Supporting, PM1, PM2, PM5, PP3 | M/A | der(1;7)(q10;p10),+1 |
| 2 | F/D | p.(Thr354Pro) | c.1060A>C | MS | Pathogenic | PS2, PS4_Moderate, PM1, PM2, PM5, PP3 | M/A, ID, L, HA | NK |
| 53 | F | p.(Thr354Met) | c.1061C>T | MS | Pathogenic | PS3, PS4, PM1, PM2, PM5, PP1_Strong, PP3 | M/A, ID, L, HA |
−7, +8, +21, −5q, 1q abnormality, isochromosome 17, F100 t(1q:7p) +8 replaced by monodicentric 6 |
| 1 | F | p.(Thr354Arg) | c.1061C>G | MS | Likely Pathogenic | PS4_Supporting, PM1, PM2, PM5, PP3 | M/A, ID | 46,XX [20]/92,XXXX [2] |
| 2 | F | p.(Thr358del) [355delT] |
c.1065_1067del [c.1063_1065delACA] |
DEL | Pathogenic | PS3, PS4_Moderate, PM2, PM4 | M/A |
−7 +8 |
| 1 | N/A | p.(Thr358del) | c.1072_1074del | DEL | Pathogenic | PS3, PS4_Moderate, PM2, PM4 | ID | |
| 1 | N/A | p.(Thr356_Asn365del) | c.1066_1095del | DEL | Likely Pathogenic | PS4_Supporting, PM2, PM4_Strong | M/A | −7 |
| 2 | N/A | p.(Thr357Ala) | c.1069A>G | MS | Likely Pathogenic | PS4_ Moderate, PM1, PM2, PM5, PP3 | M/A |
der(1;7), add +8 |
| 1 | N/A | p.(Thr357Ile) | c.1070C>T | MS | Likely Pathogenic | PS4_Supporting, PM1, PM2, PM5, PP3 | M/A, ID | NK |
| 1 | F |
p.(Thr358Asn), p.(Leu359Val) (in‐cis) |
c.1073C>A c.1075T>G |
MS | Pathogenic/VUS | PS3, PS4_Supporting, PM1, PM2, PP3/PM1, PM2, PM5, PP3, BP2 | M/A, HA, ID | |
| 1 | F | p.(Leu359Val) | c.1076T>C | MS | Likely Pathogenic | PS4_Supporting, PM1, PM2, PP3 | M/A, ID, L |
46,XX,del(5)(q2?3q 3?3) [18]/46,XX |
| 3 | F | p.(Trp360Arg) | c.1078T>A | MS | Likely Pathogenic | PS4_Moderate, PM1, PM2, PM5, PP3 | ID, L, HA | |
| 1 | F | p.(Trp360Leu) | c.1079G>T | MS | Likely Pathogenic | PS4_Supporting, PM1, PM2, PM5, PP3 | M/A, HA, ID | −7 |
| 2 | F | p.(Arg361Gly) | c.1081C>G | MS | Likely Pathogenic | PS4_Moderate, PM1, PM2, PM5, PP3 | M/A, ID, L | |
| 7 | F | p.(Arg361Cys) | c.1081C>T | MS | Likely Pathogenic | PS4, PM1, PM5, PP3 | MA, ID, L, HA | −7 |
| 1 | D | p.(Arg361Leu) | c.1082G>T | MS | Pathogenic | PS2, PS3, PS4_Supporting, PM1, PM2, PM5, PP3 | L, HA, ID | |
| 8 | F | p.(Arg361His) | c.1082 G>A | MS | Pathogenic | PS4, PM1, PM2, PM5, PP3 | M/A, NS, ID | −7, +8 |
| 1 | N/A |
p.(Arg362_Asn365 del) [R361del4] |
c.1084_1095del [c.1083_1094del12] |
DEL | Likely Pathogenic | PS4_Supporting, PM2, PM4_Strong | M/A, ID, HA | −7, +21, +8 |
| 17 | F/D | p.(Arg362*) | c.1084C>T | NS | Pathogenic | PVS1, PS4, PM2 | M/A, ID, L, HA |
−7, add +8 −20q |
| 2 | F | p.(Arg362Pro) | c.1085G>C | MS | Likely Pathogenic | PS4_ Moderate, PM1, PM2, PP3 | M/A, ID, L |
−7 +8 |
| 1 | N/A | p.(Asp367Thrfs*20) | c.1099del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, HA, ID | |
| 3 | F | p.(Asp367Glyfs*17) | c.1099dup [c.1099insG] | FS | Pathogenic | PVS1, PS4_ Moderate, PM2 | M/A, ID, HA | NK |
| 1 | N/A | p.(Pro368Argfs*15) | c.1103_1104del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | NS | |
| 1 | N/A | p.(Cys370Trp) | c.1110C>G | MS | Likely Pathogenic | PS4_Supporting, PM1, PM2, PP3 | M/A | −7 |
| 4 | N/A | p.(Asn371Lys) | c.1113C>A | MS | Pathogenic | PS1, PS4, PM1, PM2, PP3 | MA, L, ID |
−7 +8 |
| 2 | N/A | p.(Asn371Lys) | c.1113C>G | MS | Pathogenic | PS1, PS4_Moderate, PM1, PM2, PP3 | M/A, HA,ID | −7, +mar |
| 1 | N/A | p.(Asn371Lysfs*16) | c.1113del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | −7, +8 |
| 6 | F | p.(Ala372Thr) | c.1114G>A | MS | Likely Pathogenic | PS4, PM1, PM2, PP3 | M/A, ID, L | −7, +15, +20 t(11;19), +8 |
| 3 | F |
p.(Cys373_Tyr377del) [C373del5] |
c.1117_1131del [c.1116_1130del15] | DEL | Likely Pathogenic | PS4_ Moderate, PM2, PM4_Strong* | M/A, ID | −7 |
| 1 | N/A | p.(Cys373Tyr) | c.1118G>A | MS | Likely Pathogenic | PS4_Supporting, PM1, PM2, PM5, PP3 | M/A, ID, L |
+1 −15 |
| 1 | N/A | p.(Cys373Arg) | c.1117T>C | MS | Likely Pathogenic | PS3_Supporting, PS4_Supporting, PM1, PM2, PM5, PP3 | L, M/A, ID | −7 |
| 1 | N/A |
p.(Leu375Val), p.? |
c.1123C>G in‐cis with 355 bp of GATA2 locus (part of last intron & exon) that has been duplicated and inserted into the last exon |
MS, INS |
Likely Pathogenic | PM1, PM2, PM5, PP3 | M/A | NK |
| 3 | F | p.(Leu375Phe) | c.1123C>T | MS | Likely Pathogenic | PS4_Moderate, PM1, PM2, PM5, PP3 | M/A, ID, L, HA | −7, +8, +20 |
| 2 | D | p.(Leu375Profs*12) | c.1124del | FS | Pathogenic | PVS1, PS2, PS4_ Moderate, PM2 | M/A | −7 |
| 1 | D | p.(Tyr376*) | c.1128C>G | NS | Pathogenic | PVS1, PS2, PS4_Supporting, PM2 | M/A | NK |
| 1 | N/A | p.(Tyr377Asp) | c.1129T>G | MS | Likely Pathogenic | PS4_Supporting, PM1, PM2, PP3 | ID, HA | |
| 1 | N/A | p.(Lys378*) | c.1132A>T | NS | Pathogenic | PVS1, PS4_Supporting, PM2 | HA, ID | hyperdiploidy (>80 chr), +8 |
| 2 | F | p.(Lys378Asnfs*12) | c.1126_1133dup | FS | Pathogenic | PVS1, PS4_ Moderate, PM2 | HA, ID, M/A | NK |
| 1 | N/A | p.(Asn381Metfs*6) | c.1142del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, ID | NK |
| 1 | D |
p.? [S340_N381del, V382Gfs*23 or N381_V382ins41] |
c.1143+2T>A | SPL | Pathogenic | PVS1, PS2, PS4_Supporting, PM2 | M/A, ID, HA | +8 |
| 3 | D | p.? [N381fs*20] | c.1143+5G>A | SPL | Likely Pathogenic | PS3, PS4_Moderate, PM2, PM6_Supporting | ID, M/A | |
| 2 | N/A | p.? | c.1143+5G>C | SPL | Likely Pathogenic | PS1_Supporting, PS4_Moderate, PM2, PP3 | M/A, ID | +8 |
| 1 | N/A | p.? [N381fs*] | c.1143+200_1198del | DEL | Pathogenic | PVS1, PS4_Supporting, PM2 | HA, ID, L | NK |
| 3 | F | p.(Pro385Gln) | c.1154C>A | MS | Likely Pathogenic | PS4_ Moderate, PM1, PM2, PP3 | M/A, ID, L, T‐ALL | −7 |
| 1 | N/A | p.(Thr387Asn) | c.1160C>A | MS | Likely Pathogenic | PS4_Supporting, PM1, PM2, PP3 | M/A | +8 |
| 2 | F | p.(Met388Val) | c.1162A>G | MS | Likely Pathogenic | PS4_ Moderate, PM1, PM2, PM5, PP3 | M/A, HA, ID |
−7 +8 |
| 5 | F | p.(Met388Thr) | c.1163T>C | MS | Likely Pathogenic | PS4, PM1, PM2, PM5, PP3 | ID, HA | |
| 1 | N/A | p.(Lys390Glu) | c.1168A>G | MS | Likely Pathogenic | PS4_Moderate, PM1, PM2, PP3 | M/A | der(1;7) add |
| 1 | N/A | p.(Glu391Glyfs*85) | c.1172_1175del | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | ID, HA | |
| 12 | F | p.(Arg396Trp) | c.1186C>T | MS | Pathogenic | PS4, PM1, PM2, PM5, PP3 | M/A, ID, L, HA | +8, +mar, −7, −21 |
| 24 | F/D | p.(Arg396Gln) | c.1187G>A | MS | Pathogenic | PS2, PS3, PS4, PM1, PM2, PM5, PP1_Moderate, PP3 | M/A, ID, L, HA | +8, −7, +11, der(1;16), add |
| 3 | F | p.(Arg396Leu) | c.1187G>T | MS | Likely Pathogenic | PS3_Supporting, PS4_ Moderate, PM1, PM2, PM5, PP1_Supporting, PP3 | M/A, HA, ID | |
| 22 | F | p.(Arg398Trp) | c.1192C>T | MS | Pathogenic | PS3, PS4, PM1, PM2, PM5, PP1_Moderate, PP3 | M/A, ID, HA, CMML, JMML, L | +1, −7, +8, ‐X |
| 4 | F | p.(Arg398Gln) | c.1193G>A | MS | Likely Pathogenic | PS4, PM1, PM5, PP3 | M/A, ID | −7, +8 |
| 1 | N/A | p.(Lys406Serfs*77) |
c.1200_1216dup [c.1216_1217ins17] |
FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | −7 |
| 1 | N/A | p.(Phe428Leufs*108) | c.1281dup [c.1281_1282insC] | FS | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | |
| 1 | N/A | p.(His442Glnfs*95) | c.1322_1325dup | FS | Likely pathogenic | PVS1_Moderate, PS4_Supporting, PM2 | M/A | |
| 8 | F | p.(Ser447Arg) | c.1339A>C | MS | Pathogenic | PS1, PS4, PM2, PP1_Moderate, PP3 | M/A, ID, HA | +8 |
| 2 | N/A | p.(Ser447Arg) | c.1341C>A | MS | Likely pathogenic | PS1, PS4_Moderate, PM2 | M/A | −7 |
| 1 | N/A |
Deletion (whole protein) |
DEL | Pathogenic | PVS1, PS4_Supporting, PM2 | L, M/A, ID, DF, NS | +21 | |
| 1 | N/A |
Deletion (whole protein) |
DEL | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, ID, DF, NS, HA | ||
| 1 | N/A |
Deletion (whole protein) |
DEL | Pathogenic | PVS1, PS4_Supporting, PM2 | HA, ID | ||
| 1 | N/A |
Deletion (whole protein) |
DEL | Pathogenic | PVS1, PS4_Supporting, PM2 | ID, HA | ||
| 2 | N/A |
Deletion (whole protein) |
c.1‐?_1443+?del | DEL | Pathogenic | PVS1, PS4_ Moderate, PM2 | M/A, ID | NK |
| 1 | N/A |
Deletion (whole protein) |
DEL | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, ID, DF, NS, HA | −7 | |
| 1 | N/A |
Deletion (whole protein) |
DEL | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, HA, ID, L | −7, +8 | |
| 1 | N/A |
Deletion (whole protein) |
DEL | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, HA, ID | ||
| 1 | N/A |
Deletion (whole protein) |
DEL | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | −7 | |
| 1 | N/A | del3q21 | DEL | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, ID, NS | del3q21, −7 | |
| 1 | N/A | Deletion | DEL | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, HA, ID | 45,XY,‐7[7]/46,XY,‐7,+mar[10]/46,XY[8] | |
| 1 | N/A | Deletion | DEL | Pathogenic | PVS1, PS4_Supporting, PM2 | HA, ID | ||
| 1 | F |
Delete ATG start codon |
DEL | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | t(2;12)(p21;p13) | |
| 3 | F | p.? (M1del290) | c.‐45‐155_871+527del | DEL | Pathogenic | PVS1, PS4_ Moderate, PM2 | M/A, ID, L, HA | NK |
| 1 | N/A | p.? | DEL | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, HA | ||
| 1 | F | p.? | DEL | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A | −7 | |
| 1 | F | p.? | DEL | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, ID | (7q22)x1,(7q31)x1 [260/400] | |
| 1 | F | del ZF2 & C‐terminus | c.1018‐? | DEL | Pathogenic | PVS1, PS4_Supporting, PM2 | M/A, ID | |
| 1 | N/A | del ZF2 & C‐terminus | c.1018‐?_1443+?del | DEL | Pathogenic | PVS1, PS4_Supporting, PM2 | ID, M/A |
Note: This table summarizes germline GATA2 likely pathogenic/pathogenic variants with full details available in Table S1. All mutations are numbered from the ATG start codon of GATA2 NM_032638.5 and NP_116027.2. Where the original published nomenclature differs, the original published mutation is indicated in square brackets []. For each mutation, the observed phenotypes for each individual were combined to generate the overall phenotype. Overall phenotypes: CMML, chronic myelomonocytic leukemia; DF, dysmorphic features; HA, hematological abnormality; HL, hearing loss; ID, immunodeficiency; JMML, juvenile myelomonocytic leukemia; L, lymphedema; M/A, MDS and/or AML; NS, neurologic symptoms; T‐ALL, T‐cell acute lymphoblastic leukemia. Mutation effect: DEL, large deletion; FS, frameshift; INDEL, small insertion/deletion; MS, missense; NS, nonsense; REG, regulatory; SPL, splice site.
Figure 1.
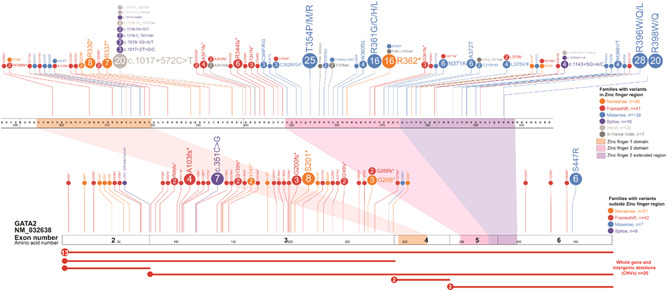
Germline GATA2 variants. All ascertained germline GATA2 variants are visualized using the ProteinPaint web application (https://pecan.stjude.cloud/home) (Zhou et al., 2016). Variants (displayed as protein changes where possible) are color‐coded according to mutation effect. The number of probands for each variant is indicated within the circle where the number is greater than one. All variants are annotated to NM_032638.5
Figure 2.
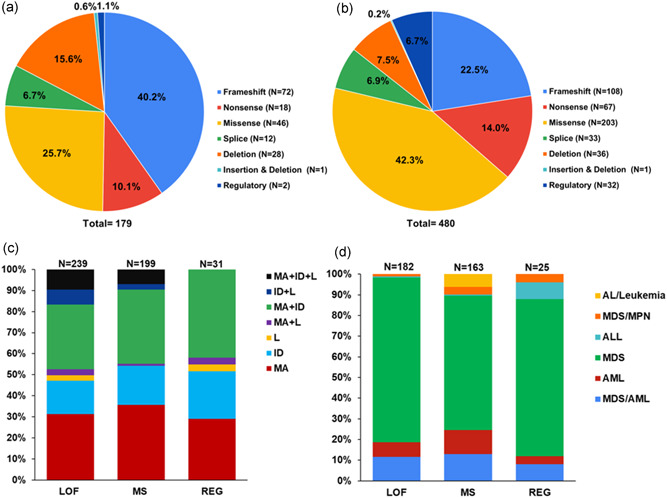
Prevalence of GATA2 variant type and associated G2DS phenotypes. (a) Number and type of unique GATA2 variants. (b) Total number and variant type of GATA2 individuals. (c) Percentage of individuals with each GATA2 variant type associated with each phenotype. (d) Percentage of individuals with each GATA2 variant type with associated hematological malignancies. Loss‐of‐function (LOF), frameshift (FS), nonsense (NS), missense (MS), splice site (SPL), large deletion (DEL), small insertion/deletion (INDEL), regulatory (REG), all variants combined (total). Overall phenotypes: L, lymphedema; M/A, MDS and/or AML; ID, immunodeficiency; malignancy types: myelodysplastic syndrome (MDS), acute myeloid leukemia (AML), myeloproliferative neoplasm (MPN). B‐ or T‐cell acute lymphoblastic leukemia (ALL), acute leukemia (AL).
1.2. GATA2 cohort characteristics
In the literature, 359 different families with germline GATA2 variants have been reported which encompasses 179 different variants (Tables 1 and Table S1, Figures 1 and 2A). There is an equal representation of both males and females presenting with G2DS phenotypes (1:1.08). In the MDS and MDS/AML population, the gender ratio was 1:1.04 suggesting no gender bias is associated with this mechanism of predisposition. This is interesting given a male bias of 1.8:1 for sporadic adult MDS (Greenberg et al., 2012; Stauder et al., 2018) and ~6:1 for germline DDX41‐driven MDS (Lewinsohn et al., 2016; Polprasert et al., 2015; Quesada et al., 2019; Sébert et al., 2019), and likely reflects the earlier onset of G2DS MDS associated with mechanistic specificities of disease pathobiology between different drivers of malignancy. The median age of GATA2‐driven myeloid malignancy (GDMM) onset is 17 years, with onset ranging from 0 to 78 years of age, with ~75% of identified GATA2 carriers developing a malignancy. The median age of onset is marginally earlier in males as compared to females (17 and 19, respectively). The majority of individuals presented with MDS (56.4%) followed by MDS/AML (9.12%) and AML (6.9%), with the median age of onset being 17 years for both MDS and MDS/AML and 20 years for AML. Four carriers presented with lymphoid malignancy (T‐ or B‐cell acute lymphoblastic leukemia), although this was rare in comparison to the myeloid malignancy (1.1% vs. 98.8%), and may represent sporadic cases. Notably, within this small lymphoid malignancy cohort, two individuals (50%) developed ALL with monosomy 7 (common in GDMM).
GATA2‐related immunodeficiency is also known as immunodeficiency 21 (MIM# 614172). These immunodeficient patients suffer from recurrent viral (Human papillomavirus leading to warts, Epstein‐Barr virus), fungal (commonly within the respiratory tract), and bacterial (most commonly Mycobacterial strains) infections, often as a result of various cytopenias, predominantly reduced or absent monocytes, B cells, NK cells, neutrophils, and/or dendritic cells (Spinner et al., 2014; Tangye et al., 2020). These patients are seen in primary immunodeficiency cohorts and are often treated with hematopoietic stem cell transplantation (HSCT) due to the severity of immunodeficiency. It is not clear if and to what extent such BM failure contributes to the development of GDMM (Arts et al., 2019; Spinner et al., 2014; Stray‐Pedersen et al., 2016).
The range in age of onset and penetrance of disease types, even within the same family, remains an intriguing mystery, one that if better understood may lead to beneficial prevention strategies. One possible answer is that of acquired monoallelic expression due to epigenetic silencing as a mechanism for further lowering of GATA2 activity levels (Al Seraihi et al., 2018).
1.3. Type of GATA2 variants
The majority of unique germline pathogenic and likely pathogenic variants in GATA2 seen in G2DS are loss‐of‐function (LOF) either through frameshift (40.2%), nonsense (10.1%), splicing (6.7%), or deletions (15.6%) throughout the gene. Missense variants (25.7%), mostly in the zinc finger 2 (ZF2) domain, also contribute a large proportion of the germline GATA2 variants, likely disrupting DNA binding (Chong et al., 2018; Hahn et al., 2011) or protein–protein interactions (Bresnick et al., 2020; Chong et al., 2018). The other important group, accounting for 1.1% G2DS variants, are regulatory, which affect the GATA2 intron 4 enhancer site and lead to reduced GATA2 expression in a range of hematopoietic stem and progenitor cells (Gao et al., 2013; Johnson et al., 2012, 2020; Mehta et al., 2017). Elegant studies in mice have shown the temporal and cell‐specific roles of the −77 kb and +9.5 kb (i.e., intron 4) Gata2 enhancers in development and cell differentiation fates (Bresnick et al., 2020). Although the repositioning of the equivalent −77 kb GATA2 enhancer in humans has been shown to be important in t(3;3)(q21;q26) and inv(3)(q21;q26) in AML in activating MECOM (Bresnick & Johnson, 2019; Gröschel et al., 2014; Yamaoka et al., 2020), no germline variants have been reported in this enhancer to date, possibly a result of this region not being included in most screening protocols.
Interestingly, there are only a few reports of confirmed missense germline variants in zinc finger 1 (ZF1). One of these is p.Ala318Thr (Kurata et al., 2017) which is seen 13 times as a somatic mutation in COSMIC (v 92) (also p.Ala318Asp/Gly/Val, 25 times) and occurs in a somatic mutational hotspot (p.Asn317‐Leu321, 95 times) with all in myeloid malignancies. In vitro transactivation studies demonstrated partial LOF (Greif et al., 2012; Katsumura et al., 2018). The p.Ala318Thr case was a 10‐year‐old girl with MDS and she had an unaffected mother and sister who were both carriers. Of note, a der(1;7)(q10;p10) was seen in this MDS case, which is a rare translocation that has been reported multiple times in pediatric/childhood (An et al., 2019; Wang et al., 2015; Wlodarski et al., 2016) and young adult MDS (Ganapathi et al., 2015) with pathogenic germline GATA2 variants; this results in chromosome 7q deletion analogous to −7/7q that is the most common acquired cytogenetic event in GDMM (Brown et al., 2020). Other ZF1 variants include p.His313Tyr and p.Leu315Pro, where p.His313Tyr has typical G2DS phenotypes (Donadieu et al., 2018). Intriguingly, a recent study confirmed a germline p.Asn317Ser variant (somatic in COSMIC –4 times; p.Asn317Ile/His, 5 times) in the first reported case of GATA2‐related primary myelofibrosis, which later progressed to pancytopenia (Rütsche et al., 2021). It is not clear whether this germline GATA2 variant contributed to this JAK2 positive myeloproliferative neoplasm and/or if it drove the pancytopenia. Notably, for each of these four ZF1 variants, there is only one affected individual. Hence, there is a small but growing number of germline GATA2 ZF1 variants associated with G2DS.
The only missense variant outside of ZF1 and the extended ZF2 domain is A286V that is predicted to impact splicing. p.Ala286Val has been shown to generate a cryptic splice donor site leading to a 16 nt deletion in RNA studies (Guidugli et al., 2017); hence this variant may better be described as a frameshift variant. In addition, a seemingly innocuous synonymous variant (p.Thr117=) has been shown by at least three groups to strongly alter splicing, generating a predominant transcript (Kozyra et al., 2020; Wehr et al., 2018) (and our unpublished data) that would result in the variant being more accurately called p.Val118Glnfs*55 (Fox et al., 2020). Hence, for some variants, there is an obvious disconnect between their annotation at the DNA level and their functional effect or impact. With the incorporation of appropriate bioinformatic tools (e.g., SpliceAI) into variant annotation pipelines, such cases can be highlighted, but the actual impact on splicing often requires functional studies for accurate interpretation.
Recurrent variants in the extended ZF2 domain have been reported including p.Thr354Met (53 individuals and 22 families), p.Arg396Trp (11 individuals and 11 families), p.Arg396Gln (24 individuals and 16 families), and p.Arg398Trp (22 individuals and 17 families). These variants seem predominantly to be passed on through familial inheritance although there are two cases of de novo p.Arg396Gln. Using haplotype mapping, p.Thr354Met was shown to be both a founder variant (with a common haplotype in an Australian and USA family) as well as independently derived in another family in the United States (Hahn et al., 2011). Notably, many of the most commonly seen germline variants represent potential mutational “hotspots.” For instance, recurrent GATA2 variants (p.Thr354Met, p.Arg396Gln/Trp, p.Arg398Gln/Trp, p.Arg362*, p.Arg361His/Cys, c.1017+572C>T) represent C>T (or G>A) changes at CpG dinucleotides that may be generated by spontaneous or enzymatic deamination of 5‐methylcytosines deposited during normal gene silencing and regulation. Such mutations (Signature SBS1) are considered part of the “normal” aging process (Alexandrov et al., 2020), and may contribute to generation of de novo and “founder” mutations/variants.
This abundance of Signature SBS1‐like variants in germline GATA2 differs from another myeloid malignancy predisposition gene, DDX41, where causal variants predominate within ethnicities or regions (e.g., p.Met1Ile/p.Met1? and p.Asp140Glyfs*2 in Europeans; p.Val152Gly, p.Ala500Cysfs*9 and p.Tyr259Cys in East Asians) (Choi et al., 2019; Kim et al., 2020; Qu et al., 2020; Yasuda et al., 2020) suggesting that these are derived from founder variants that are not actively selected against in the general population. This is consistent with the late age of onset of myeloid malignancy for DDX41 predisposition. The often early age of onset of malignancy, immunodeficiency, or other phenotypes for G2DS and apparent anticipation seen in some GATA2 families (Hahn et al., 2011) may explain a lower level of founder effect and lower predominance of germline GATA2 variants in sporadic adult MDS or AML cohorts compared to DDX41 (Kim et al., 2020; Qu et al., 2020; Quesada et al., 2019; Yasuda et al., 2020).
1.4. Genotype/phenotype correlations of germline GATA2 pathogenic and likely pathogenic variants
Among the reported phenotypes associated with germline GATA2 variants, myeloid malignancy is the most common phenotype (74.3%) with a median age of onset of 17 years (Figure 2c). This is in stark contrast with the median age of sporadic disease (MDS, 76 years; AML, 68 years (Appelbaum et al., 2006; Ma et al., 2007). Immunodeficiency was reported in 62% of affected carriers. All variant types are able to predispose to the three major phenotypes (Figure 2c).
To date, different types of GATA2 variants have not been linked strongly to particular subtypes of myeloid malignancy or age of onset or clinical outcomes. Interestingly, there is a predominance of myeloid malignancy with p.Thr354Met (83%; 44/53 cases) and immunodeficiency with p.Arg398Trp/Gln variants (85% cases). It remains to be established if variable clinical presentation is intrinsic to particular variants or is a consequence of ascertainment bias from recruitment criteria of various patient cohorts or whether there may be more prolific local environmental stressors in communities in which some founder variants are more prevalent.
There have been observations for the requirement of haploinsufficiency for lymphedema development (Kazenwadel et al., 2012). The majority of variants reported in lymphedema patients are premature termination variants (mainly frameshift or nonsense) resulting in haploinsufficiency (Figure 2c). Missense variants associated with lymphedema are predicted to be LOF and have high REVEL scores (range 0.879–0.989, median REVEL score 0.954). In vitro DNA binding and transactivation assays on three of the missense variants found in Emberger syndrome (p.Arg361Leu, p.Cys373Arg, p.Arg396Gln) demonstrated complete or almost complete LOF due to markedly diminished DNA binding and transactivation capacity (Chong et al., 2018; Kazenwadel et al., 2015). In keeping with haploinsufficiency, of 52 individuals carrying p.Thr354Met (retains residual GATA2 activity) (Chong et al., 2018; Hahn et al., 2011), there are no reported cases of lymphedema apart from an isolated case of vulvar lymphedema (Álvarez‐Chinchilla et al., 2017). The reason that haploinsufficient GATA2 mutations cause lymphedema is due to a crucial role for GATA2 in the development and maintenance of lymphatic vessel valves (Kazenwadel et al., 2015). In the setting of valve dysfunction, lymph is not efficiently returned to the bloodstream and lymphedema ensues. Mechanical stimuli including fluid flow and extracellular matrix‐induced tension, to which endothelial cells are exposed, have been demonstrated to regulate GATA2 levels in lymphatic endothelial cells (Frye et al., 2018, Kazenwadel et al., 2015). Moreover, these distinct mechanical signals induce specific transcriptional outputs; while oscillatory flow drives lymphatic vessel valve morphogenesis (Kazenwadel et al., 2018), exposure of cells to soft matrix drives lymphatic vessel sprouting and migration (Frye et al., 2018). Although hematopoietic transcription factors including SCL/TAL1 (Janardhan et al., 2017), ERG (Kazenwadel and Harvey, unpublished), and LMO2 (Coma et al., 2013) are present in lymphatic endothelial cells, the specific effects of the GATA2 mutations associated with Emberger syndrome on lymphatic vascular development suggest that unique transcriptional complexes comprising GATA2 are present in lymphatic endothelial cells compared to hematopoietic cells and that these are differentially impacted by distinct GATA2 mutations. The identification of GATA2 interacting transcriptional components, together with the signals that regulate their assembly in distinct cell types, will provide novel insight to the mechanisms by which GATA2 variants result in the varied phenotypes described in patients with G2DS.
Interestingly, certain GATA2 variants have been shown to display LOF and gain‐of‐function in different contexts. Though p.Thr354Met and p.Cys373Arg both disrupt the ZF2 structure in a way that reduces or abolishes DNA binding, respectively, they display an increased affinity for a known GATA2‐binding partner protein SPI1 (PU.1) (Chong et al., 2018). This has implications for downstream targets of both proteins in normal hematopoiesis and may contribute to leukemogenesis via mechanisms in addition to simple LOF mutations. A similar situation has been reported for certain somatic GATA2 mutations; p.Arg307Trp displays increased ability to skew towards granulocytic differentiation and induce cell cycle progression (Katsumura et al., 2018), and p.Leu359Val shows context‐dependent increased transactivation and DNA binding and the subsequent impact on downstream target genes (Chong et al., 2018; Hahn et al., 2011; Zhang et al., 2008).
In vitro differentiation studies of certain GATA2 variants showed increased granulocytic differentiation and a concurrent reduction in macrophage differentiation consistent with monocytopenia (Chong et al., 2018; Cortés‐Lavaud et al., 2015) and a case of noninfectious granulomatous dermatosis in patients (Polat et al., 2018). Also, patients with GATA2‐driven MDS display better maintenance of neutrophil counts than unselected MDS patients (Collin et al., 2015) and GATA2 deficient patients display a skewing of B‐cell to T‐cell differentiation (Nováková et al., 2016).
1.5. Somatic mutations associated with germline GDMM
There are a number of recurrent somatic cytogenetic aberrations and gene mutations that are seen with germline GDMM. Cytogenetically, the most common is monosomy 7, deletion of 7q and to a lesser extent der(1;7)(q10;p10), all of which effectively results in a loss of one copy of 7q (at least 49.5% (141/285) cases collectively; note, not all cases reported cytogenetics). Interestingly, in childhood MDS, these cytogenetic events occur more frequently than in sporadic cases with monosomy 7 seen in 37% pediatric and 72% adolescent MDS with germline GATA2 variants (Wlodarski et al., 2016) and der(1;7) is also more prevalent (Kurata et al., 2017; Wlodarski et al., 2016). Trisomy 8 is the next most common cytogenetic event occurring in at least 22.8% (65/285) of cases. Given the relative rarity of germline GATA2 cases and the lack of routine screening for somatic mutations, estimation of the prevalence and range of somatic gene mutations and those that are mutually exclusive is likely to be only indicative. At the gene level, ASXL1, NRAS/KRAS, STAG2, and SETBP1 are most commonly seen somatically mutated in MDS and AML. Interestingly, SF3B1, U2AF1, NPM1, and FLT3 mutations are uncommon in GATA2‐driven AML. FLT3 mutations may be rare as the FLT3 ligand is often elevated in symptomatic G2DS providing elevated stimulus for FLT3 signaling that may negate selection of spontaneous FLT3 mutations (Dickinson et al., 2011, 2014).
Both monoallelic and biallelic CEBPA somatic mutations are often associated with somatic GATA2 mutation in sporadic AML (Fasan et al., 2013; Greif et al., 2012). Although somatic CEBPA mutations have been reported in germline GDMM (5 cases, Table 1), because of the past difficulties in sequencing this highly GC‐rich gene, it is unclear as to the exact frequency of concurrent CEBPA mutations.
Somatic GATA2 mutations rarely occur in GDMM (i.e., biallelic) unlike for germline RUNX1, CEBPA, and DDX41‐driven myeloid malignancies where biallelic mutations are common (Brown et al., 2020; Cheah et al., 2017). Notably, while germline GATA2 premature termination mutations (frameshift, nonsense, and splice) and ZF2 variants are most common in GDMM, GATA2 mutations in sporadic myeloid malignancies are predominantly missense (mainly in ZF1, but also throughout the C‐terminus) or in‐frame indels in the C‐terminus, and premature termination mutations are seen to a lesser extent (COSMIC v92). This suggests that there may be a fundamental difference in the role of mutant GATA2 in the leukemogenic process between germline cases where mutant GATA2 is present in all cells throughout development including in the BM microenvironment, and somatic cases where the mutant protein is confined to hematopoietic cells and is rarely the first mutation acquired (Martignoles et al., 2018).
There is a need for a more systematic and comprehensive screening of somatic mutations in these rare cases of GDMM to better understand the range and frequency of concurrently mutated genes and why mutations in mutually exclusive genes are not required or selected for.
1.6. Clinical and diagnostic relevance
Due to the often early onset of G2DS symptoms and the potential severity of these, it is important to screen for and identify GATA2 germline pathogenic variants to help guide clinical decisions and facilitate family counseling. Although a family history of myeloid malignancies or immunodeficiency may help in the decision to screen for GATA2 variants, it is important to note that GATA2 de novo variants constitute a significant proportion of cases in some phenotypes such as in children or young adults with MDS (Wlodarski et al., 2016). Interestingly, there is even a report of a de novo c.1143+5G>A variant occurring in monozygotic twins both with typical G2DS phenotypes (Stray‐Pedersen et al., 2016). Depending on the severity of the disease at the time of molecular diagnosis, there may be an urgent need for therapeutic intervention. There are numerous reports of the requirement for early HSCT for G2DS patients with immunodeficiency or myeloid malignancies (Cuellar‐Rodriguez et al., 2011; McReynolds et al., 2018).
Considering the variable phenotypes in G2DS, family cascade testing of an inherited pathogenic variant may identify carriers that are asymptomatic or have a subclinical presentation within the spectrum of the disorder. Since the first report of GATA2 as a predisposition gene for myeloid malignancies in 2010 (Scott et al., 2010), screening for germline variants in relatives has been increasingly implemented to select for HSCT donors that do not carry the variant to avoid donor‐derived MDS/AML (Galera et al., 2018) or for individuals that may need surveillance for malignancy or immunodeficiency, and this is becoming routine in many clinics (DiNardo et al., 2016; University of Chicago Hematopoietic Malignancies Cancer Risk Team, 2016). In cases of myeloid malignancy, it is becoming widely recognized that different somatic mutations can have different prognostic values. In children and adolescents with MDS and monosomy 7, germline GATA2 variants do not confer poorer outcomes (Wlodarski et al., 2016).
Currently, HSCT is the most common treatment for the blood‐related phenotypes in G2DS (immunodeficiency and myeloid malignancy). Considering the high risk of developing these conditions, providing early genetic diagnosis to patients and/or their relatives before onset could allow more time to identify optimal HSCT donors, and lead to improved outcomes of transplantations.
Although GATA2 variants can predispose to G2DS symptoms, other influences may be important in expression of these variants such as epigenetics. One report provided evidence of monoallelic expression of the p.Thr354Met allele in symptomatic patients while asymptomatic individuals display equal expression from both p.Thr354Met and wildtype alleles (Al Seraihi et al., 2018). Such silencing of the wildtype allele may be selected to further reduce GATA2 activity to even more “favorably” low levels for aberrant cell expansion or survival under certain conditions. Whether the stimulus for such epigenetic changes is physiological or environmentally driven is unknown.
The ability to correct GATA2 pathogenic variants using gene therapeutic or editing approaches, particularly in the hematopoietic system, is an attractive idea. A recent paper reported evidence of a spontaneous GATA2 somatic genetic rescue event in the hematopoietic system of an elderly asymptomatic individual, opening up the exciting possibility of therapeutic strategies to facilitate and expedite correction of pathogenic variants to enable hematopoietic recovery or normalization (Catto et al., 2020).
1.7. In vivo models to dissect the functional role of GATA2 in hematopoietic and nonhematopoietic tissues
Before the discovery of GATA2 as a disease gene in humans, the first Gata2 knockout (KO‐Gata2 −/−) mice generated showed embryonic lethality at 10.5 days post coitum due to lack of definitive hematopoiesis and severe anemia (Tsai et al., 1994). Further characterization on mouse embryonic stem cells showed that GATA2 is required for proliferation of early hematopoietic stem cells (HSC) and that loss of GATA2 expression interrupts normal embryonic development (Tsai & Orkin, 1997; Tsai et al., 1994). Heterozygous Gata2 +/− mice survive until adulthood and are normal and fertile despite reduced expression (~50%) of Gata2 compared to wild‐type mice (Tsai & Orkin, 1997; Tsai et al., 1994). In the setting of stress hematopoiesis, BM from Gata2 +/ − mice exhibited a reduction in the abundance and functionality of immunophenotypically defined HSC (Guo et al., 2013; Rodrigues et al., 2005) displaying a phenotype resembling patients with G2DS immunodeficiency (Brown et al., 2020). Gata2 haploinsufficiency also reduced granulocyte‐macrophage progenitor (GMP) cell function while leaving other myeloid committed progenitors intact (Rodrigues et al., 2008). Together, these observations show that GATA2 plays a stage‐specific differentiation role not only in the HSC but also the GMP compartment and that germline GATA2 LOF variants in humans may act similarly to predispose to distinct diseases such as MDS/AML, immunodeficiency, and lymphedema (Brown et al., 2020; Pasquet et al., 2013).
Interestingly, a study on the effect of hypomorphic Gata2 variants in mice showed that reduction of GATA2 expression to ~20% induced development of chronic myelomonocytic leukemia‐like leukemia (Harada et al., 2019). Furthermore, animal models have shown the importance of the mouse Gata2 −77 kb upstream and +9.5 kb intronic enhancers in hematopoietic cell production and differentiation in embryonic, fetal, and adult hematopoiesis (reviewed in Bresnick et al., 2020; Johnson et al., 2020; Soukup & Bresnick, 2020). Germline variants in the +9.5 kb enhancer in humans predispose to myeloid malignancies, aplastic anemia, immunodeficiency, and lymphedema (Hsu et al., 2013; Johnson et al., 2012).
Tissue‐specific conditional KO models of Gata2 have been used to interrogate its roles in various tissues during later stages of development. For instance, conditional KO of Gata2 under control of VECre (i.e., VE‐Cadherin‐Cre: Gata2 deletion in vascular endothelial cadherin‐expressing endothelial cells before HSC formation) and Vav‐Cre (Gata2 deletion in hematopoietic cells after HSC generation) showed that complete deletion of Gata2 in these tissues was embryonic lethal due to defects in endothelial to hematopoietic transition during HSC formation and HSC survival (de Pater et al., 2013). The VECre mediated Gata2 loss resulted in death from anemia, hemorrhage, and edema due to lymphatic dysfunction (Lim et al., 2012). In addition, conditional loss of Gata2 in lymphatic endothelial cells during development results in dermal lymphatic vessel mispatterning and loss of lymphovenous valves (Frye et al., 2018; Kazenwadel et al., 2015). These elegant mouse studies have established the crucial role of GATA2 not only in hematopoiesis but lymphatic development. Another interesting finding from tissue‐specific knockdown of Gata2 was using an MSC‐specific Prx1 gene promoter in mice (Hasegawa et al., 2017). Their results suggested that GATA2 regulates cell adhesion and chemotaxis in BM to maintain the BM microenvironment, and that loss of GATA2 in mesenchymal stromal cells (MSC) may contribute to aberrant HSC colony formation (Hasegawa et al., 2017). This finding suggested that GATA2‐associated BM disorders may not solely result from HSC intrinsic processes, but also the BM niche. This is correlated with evidence that GATA2 expression is decreased in BM MSC from patients with aplastic anemia (Xu et al., 2009). In a different context, rescue of hematopoietic deficiency of GATA2 using a Gata2 yeast artificial chromosome transgene in mice led to the discovery of a contribution to urogenital development (Zhou et al., 1998). These mice suffered from megaureter and hydronephrosis caused by ureters that ended blindly or were aberrantly connected to the seminal vesicle or vas deferens and resulted in perinatal lethality. Interestingly, these findings are consistent with urogenital abnormalities being seen in 5%–12% of G2DS patients (Donadieu et al., 2018; Wlodarski et al., 2016).
Overall, mouse models have contributed greatly to our understanding of the role(s) of GATA2 in the genesis and function of hematopoietic and nonhematopoietic tissues. To date, there are no knockin mouse models that mimic any of the commonly seen human missense variants. Furthermore, few have faithfully modeled the initiation and progression of G2DS phenotypes as seen in human disease (e.g., MDS or AML), possibly due to the short murine lifespan and the different range of challenges and stressors experienced during a human lifetime.
2. CONCLUSION
In just one decade, the impact of germline GATA2 pathogenic variants has been noted around the world, and clinical practice has changed to help patients and their families. This collation of published GATA2 variants is a powerful resource for helping health professionals in ACMG/AMP classification of identified variants and subsequent clinical management. A better understanding of the types of GATA2 variants and their impacts on the cellular processes and environmental stressors leading to G2DS phenotypes will enable better surveillance measures, treatments, and ultimately strategies to prevent disease onset.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
Claire C. Homan, Parvathy Venugopal, Peer Arts, Nur H. Shahrin, Christopher N. Hahn, Natasha L. Harvey wrote the manuscript and prepared tables and figures. SF and CNH performed ACMG/AMP classifications. Parvathy Venugopal, David M. Lawrence, James Andrews, Sarah L. King‐Smith, and Christopher N. Hahn configured and transferred data to ClinVar. SF, ALB, NLH, and HSS reviewed and edited the manuscript.
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENTS
This study was supported by funding from the National Health and Medical Research Council (APP1086302, APP1086302, APP1164601) and the Cancer Council of South Australia Beat Cancer (APP1125849). PA was supported by a fellowship from The Hospital Research Foundation.
Homan, C. C. , Venugopal, P. , Arts, P. , Shahrin, N. H. , Feurstein, S. , Rawlings, L. , Lawrence, D. M. , Andrews, J. , King‐Smith, S. L. , Harvey, N. L. , Brown, A. L. , Scott, H. S. , & Hahn, C. N. (2021). GATA2 deficiency syndrome: A decade of discovery. Human Mutation, 42, 1399–1421. 10.1002/humu.24271
Claire C. Homan and Parvathy Venugopal contributed equally to this study.
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article.
REFERENCES
- Abou Tayoun, A. N. , Pesaran, T. , DiStefano, M. T. , Oza, A. , Rehm, H. L. , Biesecker, L. G. , & Harrison, S. M. ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI) . (2018). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Human Mutation, 39, 1517–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Seraihi, A. F. , Rio‐Machin, A. , Tawana, K. , Bödör, C. , Wang, J. , Nagano, A. , Heward, J. A. , Iqbal, S. , Best, S. , Lea, N. , McLornan, D. , Kozyra, E. J. , Wlodarski, M. W. , Niemeyer, C. M. , Scott, H. , Hahn, C. , Ellison, A. , Tummala, H. , Cardoso, S. R. , … Fitzgibbon, J. (2018). GATA2 monoallelic expression underlies reduced penetrance in inherited GATA2‐mutated MDS/AML. Leukemia, 32, 2502–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov, L. B. , Kim, J. , Haradhvala, N. J. , Huang, M. N. , Tian Ng, A. W. , Wu, Y. , Boot, A. , Covington, K. R. , Gordenin, D. A. , Bergstrom, E. N. , Islam, S. M. A. , Lopez‐Bigas, N. , Klimczak, L. J. , McPherson, J. R. , Morganella, S. , Sabarinathan, R. , Wheeler, D. A. , Mustonen, V. , PCAWG Mutational Signatures Working Group , … PCAWG Consortium . (2020). The repertoire of mutational signatures in human cancer. Nature, 578, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Álvarez‐Chinchilla, P. , Poveda, I. , Marco, F. M. , López‐Fernández, J. A. , Peiro, G. , Illán, F. , & Guijarro, J. (2017). Vulvar lymphedema and refractory VIN‐III heralding GATA2 deficiency syndrome. European Journal of Obstetrics, Gynecology and Reproductive Biology, 218, 138–140. [DOI] [PubMed] [Google Scholar]
- An, W. B. , Liu, C. , Wan, Y. , Chen, X. Y. , Guo, Y. , Chen, X. J. , Yang, W. Y. , Chen, Y. M. , Zhang, Y. C. , & Zhu, X. F. (2019). [Clinical and molecular characteristics of GATA2 related pediatric primary myelodysplastic syndrome]. Zhonghua Xue Ye Xue Za Zhi, 40, 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appelbaum, F. R. , Gundacker, H. , Head, D. R. , Slovak, M. L. , Willman, C. L. , Godwin, J. E. , Anderson, J. E. , & Petersdorf, S. H. (2006). Age and acute myeloid leukemia. Blood, 107, 3481–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arts, P. , Simons, A. , AlZahrani, M. S. , Yilmaz, E. , AlIdrissi, E. , van Aerde, K. J. , Alenezi, N. , AlGhamdi, H. A. , AlJubab, H. A. , Al‐Hussaini, A. A. , AlManjomi, F. , Alsaad, A. B. , Alsaleem, B. , Andijani, A. A. , Asery, A. , Ballourah, W. , Bleeker‐Rovers, C. P. , van Deuren, M. , van der Flier, M. , … Hoischen, A. (2019). Exome sequencing in routine diagnostics: A generic test for 254 patients with primary immunodeficiencies. Genome Medicine, 11, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresnick, E. H. , & Johnson, K. D. (2019). Blood disease–causing and –suppressing transcriptional enhancers: General principles and GATA2 mechanisms. Blood Advances, 3, 2045–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresnick, E. H. , Jung, M. M. , & Katsumura, K. R. (2020). Human GATA2 mutations and hematologic disease: How many paths to pathogenesis? Blood Advances, 4, 4584–4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, A. L. , Hahn, C. N. , & Scott, H. S. (2020). Secondary leukemia in patients with germline transcription factor mutations (RUNX1, GATA2, CEBPA). Blood, 136, 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catto, L. F. B. , Borges, G. , Pinto, A. L. , Clé, D. V. , Chahud, F. , Santana, B. A. , Donaires, F. S. , & Calado, R. T. (2020). Somatic genetic rescue in hematopoietic cells in GATA2 deficiency. Blood, 136, 1002–1005. [DOI] [PubMed] [Google Scholar]
- Cheah, J. J. C. , Hahn, C. N. , Hiwase, D. K. , Scott, H. S. , & Brown, A. L. (2017). Myeloid neoplasms with germline DDX41 mutation. International Journal of Hematology, 106, 163–174. [DOI] [PubMed] [Google Scholar]
- Choi, E.‐J. , Cho, Y.‐U. , Jang, S. , Park, C. , Park, H.‐S. , Lee, J.‐H. , Lee, K. H. , Hur, E.‐H. , Kim, N. , Ouk, H. J. , & Lee, J. H. (2019). DDX41 mutation in patients with idiopathic cytopenia of undetermined significance, myelodysplastic syndrome, and acute myeloid leukemia. Blood, 134, 3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong, C. E. , Venugopal, P. , Stokes, P. H. , Lee, Y. K. , Brautigan, P. J. , Yeung, D. T. O. , Babic, M. , Engler, G. A. , Lane, S. W. , Klingler‐Hoffmann, M. , Matthews, J. M. , D'andrea, R. J. , Brown, A. L. , Hahn, C. N. , & Scott, H. S. (2018). Differential effects on gene transcription and hematopoietic differentiation correlate with GATA2 mutant disease phenotypes. Leukemia, 32, 194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin, M. , Dickinson, R. , & Bigley, V. (2015). Haematopoietic and immune defects associated with GATA2 mutation. British Journal of Haematology, 169, 173–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coma, S. , Allard‐Ratick, M. , Akino, T. , van Meeteren, L. A. , Mammoto, A. , & Klagsbrun, M. (2013). GATA2 and Lmo2 control angiogenesis and lymphangiogenesis via direct transcriptional regulation of neuropilin‐2. Angiogenesis, 16, 939–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortés‐Lavaud, X. , Landecho, M. F. , Maicas, M. , Urquiza, L. , Merino, J. , Moreno‐Miralles, I. , & Odero, M. D. (2015). GATA2 germline mutations impair GATA2 transcription, causing haploinsufficiency: Functional analysis of the p.Arg396Gln mutation. Journal of Immunology, 194, 2190–2198. [DOI] [PubMed] [Google Scholar]
- Cuellar‐Rodriguez, J. , Gea‐Banacloche, J. , Freeman, A. F. , Hsu, A. P. , Zerbe, C. S. , Calvo, K. R. , Wilder, J. , Kurlander, R. , Olivier, K. N. , Holland, S. M. , & Hickstein, D. D. (2011). Successful allogeneic hematopoietic stem cell transplantation for GATA2 deficiency. Blood, 118, 3715–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pater, E. , Kaimakis, P. , Vink, C. S. , Yokomizo, T. , Yamada‐Inagawa, T. , Linden, R. , , van der Kartalaei, P. S. , Camper, S. A. , Speck, N. , & Dzierzak, E. (2013). Gata2 is required for HSC generation and survival. Journal of Experimental Medicine, 210, 2843–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson, R. E. , Griffin, H. , Bigley, V. , Reynard, L. N. , Hussain, R. , Haniffa, M. , Lakey, J. H. , Rahman, T. , Wang, X.‐N. , McGovern, N. , Pagan, S. , Cookson, S. , McDonald, D. , Chua, I. , Wallis, J. , Cant, A. , Wright, M. , Keavney, B. , Chinnery, P. F. , … Collin, M. (2011). Exome sequencing identifies GATA‐2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood, 118, 2656–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson, R. E. , Milne, P. , Jardine, L. , Zandi, S. , Swierczek, S. I. , McGovern, N. , Cookson, S. , Ferozepurwalla, Z. , Langridge, A. , Pagan, S. , Gennery, A. , Heiskanen‐Kosma, T. , Hämäläinen, S. , Seppänen, M. , Helbert, M. , Tholouli, E. , Gambineri, E. , Reykdal, S. , Gottfreðsson, M. , … Collin, M. (2014). The evolution of cellular deficiency in GATA2 mutation. Blood, 123, 863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNardo, C. D. , Bannon, S. A. , Routbort, M. , Franklin, A. , Mork, M. , Armanios, M. , Mace, E. M. , Orange, J. S. , Jeff‐Eke, M. , Churpek, J. E. , Takahashi, K. , Jorgensen, J. L. , Garcia‐Manero, G. , Kornblau, S. , Bertuch, A. , Cheung, H. , Bhalla, K. , Futreal, A. , Godley, L. A. , & Patel, K. P. (2016). Evaluation of patients and families with concern for predispositions to hematologic malignancies within the Hereditary Hematologic Malignancy Clinic (HHMC). Clinical Lymphoma, Myeloma & Leukemia, 16, 417–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donadieu, J. , Lamant, M. , Fieschi, C. , De Fontbrune, F. S. , Caye, A. , Ouachee, M. , Beaupain, B. , Bustamante, J. , Poirel, H. A. , Isidor, B. , Van Den Neste, E. , Neel, A. , Nimubona, S. , Toutain, F. , Barlogis, V. , Schleinitz, N. , Leblanc, T. , Rohrlich, P. , Suarez, F. , … French GATA2 study group (2018). Natural history of GATA2 deficiency in a survey of 79 French and Belgian patients. Haematologica, 103, 1278–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emberger, J. M. , Navarro, M. , Dejean, M. , & Izarn, P. (1979). Surdi‐mutite, lymphoedeme des membres inferieurs et anomalies hematologiques (leucose aigue, cytopenies) a transmission autosomique dominante [Deaf‐mutism, lymphedema of the lower limbs and hematological abnormalities (acute leukemia, cytopenia) with autosomal dominant transmission]. Journal de genetique humaine, 27, 237–245. [PubMed] [Google Scholar]
- Fasan, A. , Eder, C. , Haferlach, C. , Grossmann, V. , Kohlmann, A. , Dicker, F. , Kern, W. , Haferlach, T. , & Schnittger, S. (2013). GATA2 mutations are frequent in intermediate‐risk karyotype AML with biallelic CEBPA mutations and are associated with favorable prognosis. Leukemia, 27, 482–485. [DOI] [PubMed] [Google Scholar]
- Fox, L. C. , Tan, M. , Brown, A. L. , Arts, P. , Thompson, E. , Ryland, G. L. , Lickiss, J. , Scott, H. S. , Poplawski, N. K. , Phillips, K. , Came, N. A. , James, P. , Ting, S. B. , Ritchie, D. S. , Szer, J. , Hahn, C. N. , Schwarer, A. , & Blombery, P. (2020). A synonymous GATA2 variant underlying familial myeloid malignancy with striking intrafamilial phenotypic variability. British Journal of Haematology, 190, e297–e301. [DOI] [PubMed] [Google Scholar]
- Frye, M. , Taddei, A. , Dierkes, C. , Martinez‐Corral, I. , Fielden, M. , Ortsäter, H. , Kazenwadel, J. , Calado, D. P. , Ostergaard, P. , Salminen, M. , He, L. , Harvey, N. L. , Kiefer, F. , & Mäkinen, T. (2018). Matrix stiffness controls lymphatic vessel formation through regulation of a GATA2‐dependent transcriptional program. Nature Communications, 9, 1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galera, P. , Hsu, A. P. , Wang, W. , Droll, S. , Chen, R. , Schwartz, J. R. , Klco, J. M. , Arai, S. , Maese, L. , Zerbe, C. , Parta, M. J. , Young, N. S. , Holland, S. M. , Hickstein, D. D. , & Calvo, K. R. (2018). Donor‐derived MDS/AML in families with germline GATA2 mutation. Blood, 132, 1994–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathi, K. A. , Townsley, D. M. , Hsu, A. P. , Arthur, D. C. , Zerbe, C. S. , Cuellar‐Rodriguez, J. , Hickstein, D. D. , Rosenzweig, S. D. , Braylan, R. C. , Young, N. S. , Holland, S. M. , & Calvo, K. R. (2015). GATA2 deficiency‐associated bone marrow disorder differs from idiopathic aplastic anemia. Blood, 125, 56–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, X. , Johnson, K. D. , Chang, Y.‐I. , Boyer, M. E. , Dewey, C. N. , Zhang, J. , & Bresnick, E. H. (2013). Gata2 cis‐element is required for hematopoietic stem cell generation in the mammalian embryo. Journal of Experimental Medicine, 210, 2833–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg, P. L. , Tuechler, H. , Schanz, J. , Sanz, G. , Garcia‐Manero, G. , Solé, F. , Bennett, J. M. , Bowen, D. , Fenaux, P. , Dreyfus, F. , Kantarjian, H. , Kuendgen, A. , Levis, A. , Malcovati, L. , Cazzola, M. , Cermak, J. , Fonatsch, C. , Le Beau, M. M. , Slovak, M. L. , … Haase, D. (2012). Revised international prognostic scoring system for myelodysplastic syndromes. Blood, 120, 2454–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greif, P. A. , Dufour, A. , Konstandin, N. P. , Ksienzyk, B. , Zellmeier, E. , Tizazu, B. , Sturm, J. , Benthaus, T. , Herold, T. , Yaghmaie, M. , Dörge, P. , Hopfner, K.‐P. , Hauser, A. , Graf, A. , Krebs, S. , Blum, H. , Kakadia, P. M. , Schneider, S. , Hoster, E. , … Bohlander, S. K. (2012). GATA2 zinc finger 1 mutations associated with biallelic CEBPA mutations define a unique genetic entity of acute myeloid leukemia. Blood, 120, 395–403. [DOI] [PubMed] [Google Scholar]
- Gröschel, S. , Sanders, M. A. , Hoogenboezem, R. , de Wit, E. , Bouwman, B. A. M. , Erpelinck, C. , van der Velden, V. H. J. , Havermans, M. , Avellino, R. , van Lom, K. , Rombouts, E. J. , van Duin, M. , Döhner, K. , Beverloo, H. B. , Bradner, J. E. , Döhner, H. , Löwenberg, B. , Valk, P. , Bindels, E. , … Delwel, R. (2014). A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell, 157, 369–381. [DOI] [PubMed] [Google Scholar]
- Guidugli, L. , Johnson, A. K. , Alkorta‐Aranburu, G. , Nelakuditi, V. , Arndt, K. , Churpek, J. E. , Godley, L. A. , Townsley, D. , Young, N. S. , Fitzpatrick, C. , Del Gaudio, D. , Das, S. , & Li, Z. (2017). Clinical utility of gene panel‐based testing for hereditary myelodysplastic syndrome/acute leukemia predisposition syndromes. Leukemia, 31, 1226–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, G. , Luc, S. , Marco, E. , Lin, T.‐W. , Peng, C. , Kerenyi, M. A. , Beyaz, S. , Kim, W. , Xu, J. , Das, P. P. , Neff, T. , Zou, K. , Yuan, G. C. , & Orkin, S. H. (2013). Mapping cellular hierarchy by single‐cell analysis of the cell surface repertoire. Cell Stem Cell, 13, 492–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn, C. N. , Chong, C.‐E. , Carmichael, C. L. , Wilkins, E. J. , Brautigan, P. J. , Li, X.‐C. , Babic, M. , Lin, M. , Carmagnac, A. , Lee, Y. K. , Kok, C. H. , Gagliardi, L. , Friend, K. L. , Ekert, P. G. , Butcher, C. M. , Brown, A. L. , Lewis, I. D. , To, L. B. , Timms, A. E. , … Scott, H. S. (2011). Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nature Genetics, 43, 1012–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada, N. , Hasegawa, A. , Hirano, I. , Yamamoto, M. , & Shimizu, R. (2019). GATA2 hypomorphism induces chronic myelomonocytic leukemia in mice. Cancer Prevention Research, 110, 1183–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa, S. , Fujiwara, T. , Okitsu, Y. , Kato, H. , Sato, Y. , Fukuhara, N. , Onishi, Y. , Shimizu, R. , Yamamoto, M. , & Harigae, H. (2017). Effects of in vivo deletion of GATA2 in bone marrow stromal cells. Experimental Hematology, 56(31–45), e2–e45. [DOI] [PubMed] [Google Scholar]
- Hirabayashi, S. , Wlodarski, M. W. , Kozyra, E. , & Niemeyer, C. M. (2017). Heterogeneity of GATA2‐related myeloid neoplasms. International Journal of Hematology, 106, 175–182. [DOI] [PubMed] [Google Scholar]
- Hsu, A. P. , Johnson, K. D. , Falcone, E. L. , Sanalkumar, R. , Sanchez, L. , Hickstein, D. D. , Cuellar‐Rodriguez, J. , Lemieux, J. E. , Zerbe, C. S. , Bresnick, E. H. , & Holland, S. M. (2013). GATA2 haploinsufficiency caused by mutations in a conserved intronic element leads to MonoMAC syndrome. Blood, 121(3830–7), S1–S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, A. P. , McReynolds, L. J. , & Holland, S. M. (2015). GATA2 deficiency. Current Opinion in Allergy and Clinical Immunology, 15, 104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, A. P. , Sampaio, E. P. , Khan, J. , Calvo, K. R. , Lemieux, J. E. , Patel, S. Y. , Frucht, D. M. , Vinh, D. C. , Auth, R. D. , Freeman, A. F. , Olivier, K. N. , Uzel, G. , Zerbe, C. S. , Spalding, C. , Pittaluga, S. , Raffeld, M. , Kuhns, D. B. , Ding, L. , Paulson, M. L. , … Holland, S. M. (2011). Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood, 118, 2653–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janardhan, H. P. , Milstone, Z. J. , Shin, M. , Lawson, N. D. , Keaney, J. F. Jr. , & Trivedi, C. M. (2017). Hdac3 regulates lymphovenous and lymphatic valve formation. Journal of Clinical Investigation, 127, 4193–4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, K. D. , Conn, D. J. , Shishkova, E. , Katsumura, K. R. , Liu, P. , Shen, S. , Ranheim, E. A. , Kraus, S. G. , Wang, W. , Calvo, K. R. , Hsu, A. P. , Holland, S. M. , Coon, J. J. , Keles, S. , & Bresnick, E. H. (2020). Constructing and deconstructing GATA2‐regulated cell fate programs to establish developmental trajectories. Journal of Experimental Medicine, 217, e20191526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, K. D. , Hsu, A. P. , Ryu, M.‐J. , Wang, J. , Gao, X. , Boyer, M. E. , Liu, Y. , Lee, Y. , Calvo, K. R. , Keles, S. , Zhang, J. , Holland, S. M. , & Bresnick, E. H. (2012). Cis‐element mutated in GATA2‐dependent immunodeficiency governs hematopoiesis and vascular integrity. Journal of Clinical Investigation, 122, 3692–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsumura, K. R. , Mehta, C. , Hewitt, K. J. , Soukup, A. A. , Fraga de Andrade, I. , Ranheim, E. A. , Johnson, K. D. , & Bresnick, E. H. (2018). Human leukemia mutations corrupt but do not abrogate GATA‐2 function. Proceedings of the National Academy of Sciences of the United States of America, 115, E10109–E10118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazenwadel, J. , Betterman, K. L. , Chong, C.‐E. , Stokes, P. H. , Lee, Y. K. , Secker, G. A. , Agalarov, Y. , Demir, C. S. , Lawrence, D. M. , Sutton, D. L. , Tabruyn, S. P. , Miura, N. , Salminen, M. , Petrova, T. V. , Matthews, J. M. , Hahn, C. N. , Scott, H. S. , & Harvey, N. L. (2015). GATA2 is required for lymphatic vessel valve development and maintenance. Journal of Clinical Investigation, 125, 2979–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazenwadel, J. , & Harvey, N. L. (2018). Lymphatic endothelial progenitor cells: Origins and roles in lymphangiogenesis. Current Opinions in Immunology, 53, 81–87. [DOI] [PubMed] [Google Scholar]
- Kazenwadel, J. , Secker, G. A. , Liu, Y. J. , Rosenfeld, J. A. , Wildin, R. S. , Cuellar‐Rodriguez, J. , Hsu, A. P. , Dyack, S. , Fernandez, C. V. , Chong, C. E. , Babic, M. , Bardy, P. G. , Shimamura, A. , Zhang, M. Y. , Walsh, T. , Holland, S. M. , Hickstein, D. D. , Horwitz, M. S. , Hahn, C. N. , … Harvey, N. L. (2012). Loss‐of‐function germline GATA2 mutations in patients with MDS/AML or MonoMAC syndrome and primary lymphedema reveal a key role for GATA2 in the lymphatic vasculature. Blood, 119, 1283–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, B. , Yun, W. , Lee, S.‐T. , Choi, J. R. , Yoo, K. H. , Koo, H. H. , Jung, C. W. , & Kim, S. H. (2020). Prevalence and clinical implications of germline predisposition gene mutations in patients with acute myeloid leukemia. Scientific Reports, 10, 14297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozyra, E. J. , Pastor, V. B. , Lefkopoulos, S. , Sahoo, S. S. , Busch, H. , Voss, R. K. , Erlacher, M. , Lebrecht, D. , Szvetnik, E. A. , Hirabayashi, S. , Pasaulienė, R. , Pedace, L. , Tartaglia, M. , Klemann, C. , Metzger, P. , Boerries, M. , Catala, A. , Hasle, H. , de Haas, V. , … European Working Group of MDS in Childhood (2020). Synonymous GATA2 mutations result in selective loss of mutated RNA and are common in patients with GATA2 deficiency. Leukemia, 34, 2673–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurata, T. , Shigemura, T. , Muramatsu, H. , Okuno, Y. , & Nakazawa, Y. (2017). A case of GATA2‐related myelodysplastic syndrome with unbalanced translocation der(1;7)(q10;p10). Pediatric Blood & Cancer, 64(8), e26419. [DOI] [PubMed] [Google Scholar]
- Lewinsohn, M. , Brown, A. L. , Weinel, L. M. , Phung, C. , Rafidi, G. , Lee, M. K. , Schreiber, A. W. , Feng, J. , Babic, M. , Chong, C.‐E. , Lee, Y. , Yong, A. , Suthers, G. K. , Poplawski, N. , Altree, M. , Phillips, K. , Jaensch, L. , Fine, M. , D'Andrea, R. J. , … Scott, H. S. (2016). Novel germ line DDX41 mutations define families with a lower age of MDS/AML onset and lymphoid malignancies. Blood, 127, 1017–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, K.‐C. , Hosoya, T. , Brandt, W. , Ku, C.‐J. , Hosoya‐Ohmura, S. , Camper, S. A. , Yamamoto, M. , & Engel, J. D. (2012). Conditional Gata2 inactivation results in HSC loss and lymphatic mispatterning. Journal of Clinical Investigation, 122, 3705–3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, X. , Does, M. , Raza, A. , & Mayne, S. T. (2007). Myelodysplastic syndromes: Incidence and survival in the United States. Cancer, 109, 1536–1542. [DOI] [PubMed] [Google Scholar]
- Martignoles, J.‐A. , Delhommeau, F. , & Hirsch, P. (2018). Genetic hierarchy of acute myeloid leukemia: From clonal hematopoiesis to molecular residual disease. International Journal of Molecular Sciences, 19, 3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McReynolds, L. J. , Calvo, K. R. , & Holland, S. M. (2018). Germline GATA2 mutation and bone marrow failure. Hematology/Oncology Clinics of North America, 32, 713–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta, C. , Johnson, K. D. , Gao, X. , Ong, I. M. , Katsumura, K. R. , McIver, S. C. , Ranheim, E. A. , & Bresnick, E. H. (2017). Integrating enhancer mechanisms to establish a hierarchical blood development program. Cell Reports, 20, 2966–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nováková, M. , Žaliová, M. , Suková, M. , Wlodarski, M. , Janda, A. , Froňková, E. , Campr, V. , Lejhancová, K. , Zapletal, O. , Pospíšilová, D. , Černá, Z. , Kuhn, T. , Švec, P. , Pelková, V. , Zemanová, Z. , Kerndrup, G. , van den Heuvel‐Eibrink, M. , van der Velden, V. , Niemeyer, C. , … Mejstříková, E. (2016). Loss of B cells and their precursors is the most constant feature of GATA‐2 deficiency in childhood myelodysplastic syndrome. Haematologica, 101, 707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostergaard, P. , Simpson, M. A. , Connell, F. C. , Steward, C. G. , Brice, G. , Woollard, W. J. , Dafou, D. , Kilo, T. , Smithson, S. , Lunt, P. , Murday, V. A. , Hodgson, S. , Keenan, R. , Pilz, D. T. , Martinez‐Corral, I. , Makinen, T. , Mortimer, P. S. , Jeffery, S. , Trembath, R. C. , & Mansour, S. (2011). Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nature Genetics, 43, 929–931. [DOI] [PubMed] [Google Scholar]
- Papaemmanuil, E. , Gerstung, M. , Malcovati, L. , Tauro, S. , Gundem, G. , Van Loo, P. , Yoon, C. J. , Ellis, P. , Wedge, D. C. , Pellagatti, A. , Shlien, A. , Groves, M. J. , Forbes, S. A. , Raine, K. , Hinton, J. , Mudie, L. J. , McLaren, S. , Hardy, C. , Latimer, C. , … Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium (2013). Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood, 122, 3616–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquet, M. , Bellanné‐Chantelot, C. , Tavitian, S. , Prade, N. , Beaupain, B. , Larochelle, O. , Petit, A. , Rohrlich, P. , Ferrand, C. , Van Den Neste, E. , Poirel, H. A. , Lamy, T. , Ouachée‐Chardin, M. , Mansat‐De Mas, V. , Corre, J. , Récher, C. , Plat, G. , Bachelerie, F. , Donadieu, J. , & Delabesse, E. (2013). High frequency of GATA2 mutations in patients with mild chronic neutropenia evolving to MonoMac syndrome, myelodysplasia, and acute myeloid leukemia. Blood, 121, 822–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polprasert, C. , Schulze, I. , Sekeres, M. A. , Makishima, H. , Przychodzen, B. , Hosono, N. , Singh, J. , Padgett, R. A. , Gu, X. , Phillips, J. G. , Clemente, M. , Parker, Y. , Lindner, D. , Dienes, B. , Jankowsky, E. , Saunthararajah, Y. , Du, Y. , Oakley, K. , Nguyen, N. , … Maciejewski, J. P. (2015). Inherited and somatic defects in DDX41 in myeloid neoplasms. Cancer Cell, 27, 658–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu, S. , Li, B. , Qin, T. , Xu, Z. , Pan, L. , Hu, N. , Huang, G. , Peter Gale, R. , & Xiao, Z. (2020). Molecular and clinical features of myeloid neoplasms with somatic DDX41 mutations. British Journal of Haematology, 192, 1006–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesada, A. E. , Routbort, M. J. , DiNardo, C. D. , Bueso‐Ramos, C. E. , Kanagal‐Shamanna, R. , Khoury, J. D. , Thakral, B. , Zuo, Z. , Yin, C. C. , Loghavi, S. , Ok, C. Y. , Wang, S. A. , Tang, Z. , Bannon, S. A. , Benton, C. B. , Garcia‐Manero, G. , Kantarjian, H. , Luthra, R. , Medeiros, L. J. , & Patel, K. P. (2019). DDX41 mutations in myeloid neoplasms are associated with male gender, TP53 mutations and high‐risk disease. American Journal of Hematology, 94, 757–766. [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , & Gastier‐Forster, J. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genetics in Medicine, 17, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues, N. P. , Boyd, A. S. , Fugazza, C. , May, G. E. , Guo, Y. , Tipping, A. J. , Scadden, D. T. , Vyas, P. , & Enver, T. (2008). GATA‐2 regulates granulocyte‐macrophage progenitor cell function. Blood, 112, 4862–4873. [DOI] [PubMed] [Google Scholar]
- Rodrigues, N. P. , Janzen, V. , Forkert, R. , Dombkowski, D. M. , Boyd, A. S. , Orkin, S. H. , Enver, T. , Vyas, P. , & Scadden, D. T. (2005). Haploinsufficiency of GATA‐2 perturbs adult hematopoietic stem‐cell homeostasis. Blood, 106, 477–484. [DOI] [PubMed] [Google Scholar]
- Rütsche, C. V. , Haralambieva, E. , Lysenko, V. , Balabanov, S. , & Theocharides, A. P. A. (2021). A patient with a germline GATA2 mutation and primary myelofibrosis. Blood Advances, 5, 791–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, H. S. , Hahn, C. N. , Carmichael, C. L. , Wilkins, E. J. , Chong, C.‐E. , Brautigan, P. J. , Li, X.‐C. , Stankovic, M. , Lin, M. , Carmagnac, A. , Butcher, C. M. , Friend, K. L. , Ekert, P. G. , Kok, C. H. , Brown, A. L. , Lewis, I. D. , To, L. B. , Timms, A. E. , Storek, J. , … Horwitz, M. S. (2010). GATA2 is a new predisposition gene for familial myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). Blood, 116, LBA‐3. [Google Scholar]
- Sébert, M. , Passet, M. , Raimbault, A. , Rahmé, R. , Raffoux, E. , Sicre de Fontbrune, F. , Cerrano, M. , Quentin, S. , Vasquez, N. , Da Costa, M. , Boissel, N. , Dombret, H. , Peffault de Latour, R. , Socié, G. , Itzykson, R. , Fenaux, P. , Soulier, J. , Adès, L. , & Clappier, E. (2019). Germline DDX41 mutations define a significant entity within adult MDS/AML patients. Blood, 134, 1441–1444. [DOI] [PubMed] [Google Scholar]
- Soukup, A. A. , & Bresnick, E. H. (2020). GATA2 +9.5 enhancer: From principles of hematopoiesis to genetic diagnosis in precision medicine. Current Opinions in Hematology, 27, 163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinner, M. A. , Sanchez, L. A. , Hsu, A. P. , Shaw, P. A. , Zerbe, C. S. , Calvo, K. R. , Arthur, D. C. , Gu, W. , Gould, C. M. , Brewer, C. C. , Cowen, E. W. , Freeman, A. F. , Olivier, K. N. , Uzel, G. , Zelazny, A. M. , Daub, J. R. , Spalding, C. D. , Claypool, R. J. , Giri, N. K. , … Holland, S. M. (2014). GATA2 deficiency: A protean disorder of hematopoiesis, lymphatics, and immunity. Blood, 123, 809–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauder, R. , Yu, G. , Koinig, K. A. , Bagguley, T. , Fenaux, P. , Symeonidis, A. , Sanz, G. , Cermak, J. , Mittelman, M. , Hellström‐Lindberg, E. , Langemeijer, S. , Holm, M. S. , Mądry, K. , Malcovati, L. , Tatic, A. , Germing, U. , Savic, A. , van Marrewijk, C. , Guerci‐Bresler, A. , … de Witte, T. (2018). Health‐related quality of life in lower‐risk MDS patients compared with age‐ and sex‐matched reference populations: A European LeukemiaNet study. Leukemia, 32, 1380–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stray‐Pedersen, A. , Sorte, H. S. , Samarakoon, P. , Gambin, T. , Chinn, I. K. , Coban Akdemir, Z. H. , Erichsen, H. C. , Forbes, L. R. , Gu, S. , Yuan, B. , Jhangiani, S. N. , Muzny, D. M. , Rødningen, O. K. , Sheng, Y. , Nicholas, S. K. , Noroski, L. M. , Seeborg, F. O. , Davis, C. M. , Canter, D. L. , … Lupski, J. R. (2016). Primary immunodeficiency diseases—Genomic approaches delineate heterogeneous Mendelian disorders. Journal of Allergy and Clinical Immunology, 139, 232–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tangye, S. G. , Al‐Herz, W. , Bousfiha, A. , Chatila, T. , Cunningham‐Rundles, C. , Etzioni, A. , Franco, J. L. , Holland, S. M. , Klein, C. , Morio, T. , Ochs, H. D. , Oksenhendler, E. , Picard, C. , Puck, J. , Torgerson, T. R. , Casanova, J. L. , & Sullivan, K. E. (2020). Human inborn errors of immunity: 2019 update on the classification from the International Union of Immunological Societies Expert Committee. Journal of Clinical Immunology, 40, 24–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, F. Y. , Keller, G. , Kuo, F. C. , Weiss, M. , Chen, J. , Rosenblatt, M. , Alt, F. W. , & Orkin, S. H. (1994). An early haematopoietic defect in mice lacking the transcription factor GATA‐2. Nature, 371, 221–226. [DOI] [PubMed] [Google Scholar]
- Tsai, F.‐Y. , & Orkin, S. H. (1997). Transcription factor GATA‐2 is required for proliferation/survival of early hematopoietic cells and mast cell formation, but not for erythroid and myeloid terminal differentiation. Blood, 89, 3636–3643. [PubMed] [Google Scholar]
- University of Chicago Hematopoietic Malignancies Cancer Risk Team . (2016). How I diagnose and manage individuals at risk for inherited myeloid malignancies. Blood, 128, 1800–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Muramatsu, H. , Okuno, Y. , Sakaguchi, H. , Yoshida, K. , Kawashima, N. , Xu, Y. , Shiraishi, Y. , Chiba, K. , Tanaka, H. , Saito, S. , Nakazawa, Y. , Masunari, T. , Hirose, T. , Elmahdi, S. , Narita, A. , Doisaki, S. , Ismael, O. , Makishima, H. , … Kojima, S. (2015). GATA2 and secondary mutations in familial myelodysplastic syndromes and pediatric myeloid malignancies. Haematologica, 100, e398–e401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehr, C. , Grotius, K. , Casadei, S. , Bleckmann, D. , Bode, S. F. N. , Frye, B. C. , Seidl, M. , Gulsuner, S. , King, M.‐C. , Percival, M.‐B. , Pritchard, C. C. , Walsh, T. , Wu, D. , Keel, S. , & Salzer, U. (2018). A novel disease‐causing synonymous exonic mutation in GATA2 affecting RNA splicing. Blood, 132, 1211–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarski, M. W. , Hirabayashi, S. , Pastor, V. , Starý, J. , Hasle, H. , Masetti, R. , Dworzak, M. , Schmugge, M. , Van Den Heuvel‐Eibrink, M. , Ussowicz, M. , De Moerloose, B. , Catala, A. , Smith, O. P. , Sedlacek, P. , Lankester, A. C. , Zecca, M. , Bordon, V. , Matthes‐Martin, S. , Abrahamsson, J. , … EWOG‐MDS . (2016). Prevalence, clinical characteristics, and prognosis of GATA2‐related myelodysplastic syndromes in children and adolescents. Blood, 127, 1387–1397. [DOI] [PubMed] [Google Scholar]
- Wu, D. , Luo, X. , Feurstein, S. , Kesserwan, C. , Mohan, S. , Pineda‐Alvarez, D. E. , & Godley, L. A. , the collaborative group of the American Society of Hematology—Clinical Genome Resource Myeloid Malignancy Variant Curation Expert Panel . (2020). How I curate: Applying American Society of Hematology‐Clinical Genome Resource Myeloid Malignancy Variant Curation Expert Panel rules for RUNX1 variant curation for germline predisposition to myeloid malignancies. Haematologica, 105, 870–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Y. , Takahashi, Y. , Wang, Y. , Hama, A. , Nishio, N. , Muramatsu, H. , Tanaka, M. , Yoshida, N. , Villalobos, I. B. , Yagasaki, H. , & Kojima, S. (2009). Downregulation of GATA‐2 and overexpression of adipogenic gene‐PPARγ in mesenchymal stem cells from patients with aplastic anemia. Experimental Hematology, 37, 1393–1399. [DOI] [PubMed] [Google Scholar]
- Yamaoka, A. , Suzuki, M. , Katayama, S. , Orihara, D. , Engel, J. D. , & Yamamoto, M. (2020). EVI1 and GATA2 misexpression induced by inv(3)(q21q26) contribute to megakaryocyte‐lineage skewing and leukemogenesis. Blood Advances, 4, 1722–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda, T. , Sanada, M. , Nishijima, D. , Kanamori, T. , Iijima, Y. , Hattori, H. , Saito, A. , Miyoshi, H. , Ishikawa, Y. , Asou, N. , Usuki, K. , Hirabayashi, S. , Kato, M. , Ri, M. , Handa, H. , Ishida, T. , Shibayama, H. , Abe, M. , Iriyama, C. , … Horibe, K. (2020). Clinical utility of target capture‐based panel sequencing in hematological malignancies: A multicenter feasibility study. Cancer Prevention Research, 111, 3367–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S.‐J. , Ma, L.‐Y. , Huang, Q.‐H. , Li, G. , Gu, B.‐W. , Gao, X.‐D. , Shi, J.‐Y. , Wang, Y.‐Y. , Gao, L. , Cai, X. , Ren, R.‐B. , Zhu, J. , Chen, Z. , & Chen, S. J. (2008). Gain‐of‐function mutation of GATA‐2 in acute myeloid transformation of chronic myeloid leukemia. Proceedings of the National Academy of Sciences of the United States of America, 105, 2076–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, X. , Edmonson, M. N. , Wilkinson, M. R. , Patel, A. , Wu, G. , Liu, Y. , Li, Y. , Zhang, Z. , Rusch, M. C. , Parker, M. , Becksfort, J. , Downing, J. R. , & Zhang, J. (2016). Exploring genomic alteration in pediatric cancer using ProteinPaint. Nature Genetics, 48, 4–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y. , Lim, K.‐C. , Onodera, K. , Takahashi, S. , Ohta, J. , Minegishi, N. , Tsai, F.‐Y. , Orkin, S. H. , Yamamoto, M. , & Engel, J. D. (1998). Rescue of the embryonic lethal hematopoietic defect reveals a critical role for GATA‐2 in urogenital development. EMBO Journal, 17, 6689–6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article.