

Abstract
Objective
To noninvasively assess myelin status in chronic white matter lesions of multiple sclerosis (MS), we developed and evaluated a simple classification scheme based on T1 relaxation time maps derived from 7‐tesla postmortem and in vivo MRI.
Methods
Using the MP2RAGE MRI sequence, we classified 36 lesions from 4 postmortem MS brains as “long‐T1,” “short‐T1,” and “mixed‐T1” by visual comparison to neocortex. Within these groups, we compared T1 times to histologically derived measures of myelin and axons. We performed similar analysis of 235 chronic lesions with known date of onset in 25 MS cases in vivo and in a validation cohort of 222 lesions from 66 MS cases, investigating associations with clinical and radiological outcomes.
Results
Postmortem, lesions classified qualitatively as long‐T1, short‐T1, and mixed‐T1 corresponded to fully demyelinated, fully remyelinated, and mixed demyelinated/remyelinated lesions, respectively (p ≤ 0.001). Demyelination (rather than axon loss) dominantly contributed to initial T1 prolongation. We observed lesions with similar characteristics in vivo, allowing manual classification with substantial interrater and excellent intrarater reliability. Short‐T1 lesions were most common in the deep white matter, whereas long‐T1 and mixed‐T1 lesions were prevalent in the juxtacortical and periventricular white matter (p = 0.02) and were much more likely to have paramagnetic rims suggesting chronic inflammation (p < 0.001). Older age at the time of lesion formation portended less remyelination (p = 0.007).
Interpretation
7‐tesla T1 mapping with MP2RAGE, a clinically available MRI method, allows qualitative and quantitative classification of chronic MS lesions according to myelin content, rendering straightforward the tracking of lesional myelination changes over time. ANN NEUROL 2021;90:612–626
Magnetic resonance imaging (MRI) is an invaluable tool in the diagnosis and monitoring of multiple sclerosis (MS), 1 accurately measuring disease activity and facilitating assessment of disease‐modifying therapies. However, MRI has not yet achieved its potential as a clinically applicable approach to estimate myelin content in chronic MS lesions, a prerequisite for assessing endogenous and exogenous lesion repair.
Histopathologically, chronic MS lesions can be broadly classified according to the extent of inflammation, myelin, and viable axons. 2 Lesions are typically described as chronic active (or “smoldering”), chronic inactive, or repaired/remyelinated, 3 though mixed demyelinated/remyelinated lesions are common. A corresponding, in vivo classification is currently not feasible.
For chronic lesions, arguably the most successful approach to subclassification has been visualization of iron‐laden phagocytes at the edge of chronic active lesions, best seen on gradient‐echo phase MRI at both 7 T and 3 T. 4 Atrophy measures capture neuroaxonal loss 5 and have gradually been incorporated into clinical research and practice, 6 but they tend to be either global or restricted to structures other than lesions. 7 With respect to remyelination, however, no approach has been accepted as the gold standard or adopted in widespread clinical practice. 8 Thus, imaging remyelination in MS lesions with high fidelity and at high resolution remains an unmet need.
The longitudinal MRI relaxation time constant (T1) is heavily determined by lipid‐water interactions, strongly dominated by myelin in white matter. 9 , 10 While T1 prolongation (“black hole”) has been considered to mark neuroaxonal loss within lesions, MRI physics suggests that changes in T1, which scales with field strength, might rather be a particularly good noninvasive marker of demyelination and remyelination. The MP2RAGE sequence is an efficient method of calculating approximate T1 times that can easily be implemented at high spatial resolution on clinical scanners. 11 Previous studies have found that MP2RAGE improved detection of MS‐induced tissue damage in vivo, at both 3 T and 7 T. 12 , 13 , 14 , 15 The automatic inclusion of MP2RAGE T1 maps that are naturally coregistered to T1‐weighted images in the set of images produced by the scanner renders feasible a qualitative MRI biomarker based on a simple visual rating scale. This is an important advantage over other myelin‐sensitive techniques, particularly in the clinical setting, where offline image postprocessing and prolonged acquisition times present a challenge.
Here, we aim to define the contribution of myelin loss to lesional T1 on 7 T MP2RAGE scans and to explore how this technique might be used as a myelin‐sensitive biomarker for application to individual patients in clinical practice. We first carried out a postmortem study with ex vivo, high‐resolution scans to characterize chronic MS lesions both qualitatively (using a heuristic classification) and quantitatively. Guided by the ex vivo MRI data, we performed a variety of stains to correlate key pathological processes, which are present to varying degrees in both repaired and unrepaired chronic MS lesions, with the MRI findings. Finally, we conducted an exploratory study to investigate the potential applicability of our visual classification to in vivo scans at 7 T and to assess the characteristics of lesions grouped by T1 time.
Materials and Methods
Our in vivo study was approved by the NIH Institutional Review Board. Informed consent was obtained from living participants and from next of kin for autopsies.
Postmortem Study
Whole brains from 4 MS cases were collected (Table 1). Brains were preserved in 10% formalin for 2 weeks and then underwent postmortem MRI prior to tissue processing using customized, 3D‐printed cutting boxes. 16 Lesions selected based on postmortem MRI were identified and blocked, embedded in paraffin, and cut into 7–10‐μm thick sections.
TABLE 1.
Autopsy Cases
| Case | Sex | Age (years) | Clinical phenotype | Disease duration (years) | EDSS prior to death | Cause of death | Post‐mortem delay | Blocks (n) |
|---|---|---|---|---|---|---|---|---|
| 1 | F | 78 | SPMS | 54 | 7 | Pneumonia | 12 h | 7 |
| 2 | M | 76 | SPMS | 46 | 9.5 | Pneumonia | 12 h | 7 |
| 3 | F | 60 | SPMS | 16 | 7 | Sepsis | 7 h | 3 |
| 4 | F | 78 | SPMS | 24 | 9.5 | Pneumonia | 26 h | 3 |
EDSS = Expanded Disability Status Scale; SPMS = secondary progressive multiple sclerosis.
Postmortem MRI used a Siemens 7 T system equipped with a birdcage transmit coil and a 32‐channel receive coil. T1‐weighted (T1w) images and quantitative T1 maps were obtained with a 3D magnetization‐prepared 2 rapid acquisition gradient echoes (MP2RAGE) sequence 11 with the following parameters: 0.35 mm isometric voxels; repetition time (TR) = 4000 ms; echo time (TE) = 4.6 ms, inversion time 1 (TI1) = 350 ms, TI2 = 1350 ms, flip angle (FA) = 4°. Well‐demarcated lesions were manually identified and selected on postmortem T1 maps. A representative sample of lesions was included, including lesions that were hyperintense, hypointense, or of mixed signal intensity relative to cortex.
Intensity‐based, qualitative classification of lesions was performed, independently, by two experienced raters, a neurologist (HK) and a neuroradiologist (DSR), who were masked to histological findings. Discrepancies were discussed and resolved. On postmortem T1 maps, lesions were classified visually into 3 groups, denoted “long‐T1,” “short‐T1,” and “mixed‐T1.” Long‐T1 lesions were substantially hyperintense to cortex and appeared as “black holes” on T1w images, and short‐T1 lesions were isointense or hypointense to cortex. Lesions with well‐defined long‐T1 and short‐T1 areas were categorized as mixed‐T1.
Adjacent tissue sections were deparaffinized and prepared with Luxol fast blue/periodic acid Schiff (LFB‐PAS) and Sudan black to visualize myelin, Bielschowsky's stain for axons, and diaminobenzidine (DAB)‐enhanced Turnbull stain for iron. For immunostaining, sections underwent heat‐induced antigen retrieval and protein‐blocking steps for 20 minutes and were then incubated with primary antibodies for 1 hour at room temperature. Primary antibodies anti‐myelin proteolipid protein (PLP; AbD Serotec MCA839G; 1:500) and anti‐CD68 (PGM1; Dako GA61361‐2; 1:100) were used to identify myelin and microglia/macrophages, respectively. Immunoreactions were visualized with DAB or alkaline phosphatase. 17 Sections were then counterstained with 10% hematoxylin or LFB. All stained sections were scanned (magnification 10X; Zeiss Axio Observer Z.1).
Lesions were histopathologically classified into three groups: remyelinated/shadow plaques, mixed remyelinated/demyelinated lesions, and homogenously demyelinated lesions. 2 , 18 Demyelinated lesions showed marked absence of normal myelin on all relevant myelin stains. Remyelinated lesions were identified as well‐defined, homogenously pale areas of reduced LFB‐PAS staining in comparison to normal‐appearing white matter (NAWM). Mixed demyelinated/remyelinated lesions required discrete areas of both types of myelin staining within the lesion. 18 Two raters with experience in MS histopathology (HK and MA) classified lesions independently and subsequently achieved consensus. Lesions were further classified as active, chronic active, or chronic inactive, 2 , 19 and axon loss was graded as mild, moderate, or severe based on intensity and coverage of the Bielschowsky stain.
Figure 1A provides an overview of the imaging and histology processing and analysis workflow. All image processing and quantification steps were performed using ImageJ/Fiji 20 and Medical Image Analysis and Visualization (MIPAV). 21 For each slide, regions of interest (ROI) were placed over lesions and the surrounding areas of NAWM and normal‐appearing cortex. Before quantification, all double staining went through color deconvolution using a custom vector to isolate each individual stain. 22 Quantification of histochemical staining involved computing optical density (OD) from all ROI. For MRI quantification, median T1 times were obtained for all ROI.
FIGURE 1.

Schematic outline of key steps in the magnetic resonance imaging (MRI) and histology processing and analysis. Top panel (A): matching ex vivo MRI with histology. Lesions of interest were identified on ex vivo MRI (A). Based on anatomical landmarks, each brain slab was then visually matched with the corresponding MRI slice (B), and tissue blocks were excised and embedded in paraffin. MRI slices were cropped and oriented to best match the corresponding histological section (C). Tissue samples were processed, stained for myelin (LFB‐PAS, Luxol fast blue–periodic acid Schiff) and axons (Bielschowsky's silver stain), digitized at 10× magnification, and then cropped in a similar fashion (D). Stained sections and MRI were coregistered using a 2D nonrigid thin‐plate spline approach (E). Scale bar: 1 cm. Bottom panel (B): in vivo workflow. Gadolinium‐enhancing lesions were identified on T1‐weighted images and confirmed on 3 T FLAIR images. Three tesla images were coregistered with 7 T MP2RAGE images. Hypointense lesions were manually segmented on 3 T T1‐weighted images. Finally, lesions were assessed visually for qualitative classification, and regions of interest were manually delineated for T1 time computation. [Color figure can be viewed at www.annalsofneurology.org]
In Vivo Study
A retrospective cohort of 25 patient participants in an MS natural history study (NCT00001248) was selected. Inclusion required confirmed MS, 23 ≥ 1 enhancing lesion on an archival scan (1992–2018), and a follow‐up 7 T MP2RAGE. Table 2 provides demographic and clinical details, collected by experienced research clinicians. Expanded Disability Status Scale (EDSS) 24 and MS Severity Scale (MSSS) 25 scores were available for all cases, Paced Auditory Symbol Addition Test (PASAT, 3‐second version) for 19, and Symbol Digit Modality Test (SDMT, paper‐based) for 20. MSSS normalizes EDSS for disease duration, which has been linked to remyelination efficiency. 26
TABLE 2.
In Vivo Cohort: Main Clinical and Demographic Characteristics
| Original cohort | Validation cohort | |
|---|---|---|
| Participants, n | 25 | 66 |
| Sex, n (%) | ||
| Female | 19 (76%) | 50 (76%) |
| Male | 6 (24%) | 16 (24%) |
| Age (years), mean (SD) | 39 (9.5) | 48 (12.5) |
| Clinical phenotype, n (%) | ||
| Clinically isolated syndrome | 2 (8%) | 0 (0%) |
| Relapsing–remitting | 22 (88%) | 47 (71%) |
| Secondary progressive | 1 (4%) | 15 (23%) |
| Primary progressive | 0 (0%) | 4 (6%) |
| Disease duration (years), median (IQR) | 4.5 (8) | 12 (14.7) |
| Disease‐modifying treatment | ||
| Untreated | 14 (56%) | 21 (32%) |
| Glatiramer acetate | 5 (20%) | 5 (8%) |
| Interferon beta | 4 (16%) | 8 (12%) |
| Dimethyl fumarate | 1 (4%) | 8 (12%) |
| Daclizumab | 1 (4%) | 1(1%) |
| Anti‐CD20 | 0 (0%) | 12 (18%) |
| Fingolimod | 0 (0%) | 6 (9%) |
| Teriflunomide | 0 (0%) | 3 (5%) |
| Natalizumab | 0 (0%) | 2 (3%) |
| EDSS score, median (IQR) | 1.5 (1) | 2 (4) |
| MSSS score, median (IQR) | 2 (2.8) | 1.8 (3.8) |
| PASAT score, median (IQR) | 53 (16) | 50 (17) |
| SDMT score, median (IQR) | 54 (16) | 49 (19) |
| # of CEL lesions per scan, median (IQR) | 2 (3) | NA |
| # of scans with CEL per case, median (IQR) | 2 (3) | NA |
| Total lesion volume (ml), median (IQR) | 11.4 (12.5) | 17 (32.9) |
| Follow‐up time (years), median (IQR) | 4.2 (9.8) | NA |
All values except for contrast‐enhancing lesions are computed at the time of 7‐tesla scanning.
CEL = contrast enhancing lesion; EDSS = Expanded Disability Status Scale; IQR = Interquartile range; MSSS = Multiple Sclerosis Severity Score; PASAT = Paced Auditory Serial Addition Test; SDMT = Symbol Digit Modalities Test.
In vivo MRI was performed on the same 7 T MRI scanner used for the postmortem scans and incorporated the following sequences: (1) 3D MP2RAGE, providing uniformized, denoised, T1w images (“UNIDEN”) and estimated T1 maps: TR = 6000 ms; TE = 3.02 ms; TI1 = 800 ms, TI2 = 2700 ms, FA1 = 4°; FA2 = 5°; acquisition time (AT) = 10 minutes; 224 sagittal slices; 0.7 mm isometric voxels. (2) 2D dual gradient‐echo providing T2*w magnitude and phase images: TR = 1300 ms; TE1 = 15 ms; TE2 = 32 ms; 25 axial slices; FA = 50°; AT = 8:36; in‐plane resolution = 0.2 × 0.2 mm; slice thickness = 1 mm. Three minimally overlapping slabs covered most of the supratentorial brain. Phase unwrapping and initial postprocessing were previously described. 27 Figure 1B summarizes the in vivo workflow. Image analysis was conducted in MIPAV. To allow accurate determination of the age of each lesion, only lesions demonstrating contrast enhancement on ≥1 prior scan in the archive, more than 3 months prior to the 7 T scan, were analyzed. Included lesions needed to be discrete and visible on ≥2 contiguous axial slices on postcontrast T1w images, where they were classified as juxtacortical, periventricular, deep white matter, or infratentorial, and their pattern of enhancement was classified as homogenous, nodular, or ring‐like. All available 7 T MP2RAGE scans were analyzed. For quantitative analysis of T1 times, only lesions hypointense to NAWM on unenhanced 7 T T1w images and visible on at ≥2 contiguous slices were included. MR images, including T1w with and without contrast, as well as 7 T MP2RAGE T1 maps and T1w images, were linearly registered to the latest 7 T MP2RAGE scan using customized scripts (MATLAB, The Mathworks). Identified lesions were manually segmented from all scans. Paramagnetic rim lesion (PRL) status (present/absent) was based on the T2*w phase images. 27
1.5 or 3 T FLAIR and T1w sequences were used to compute total white matter lesion and brain volumes. To estimate total lesion and brain volumes for the 7 T MP2RAGE scans, we used the closest available 3 T scan, as long as it was acquired within 6 months of the 7 T. Since the scans used for volumetric analysis occurred over 16 years, acquisition parameters varied. T2‐FLAIR and T1w images were coregistered, and lesion and brain volumes were obtained using Lesion‐TOADS. 28 Segmentation results were manually reviewed and corrected.
Because T1 times change with tissue fixation, and classification of postmortem lesions identified on 2D images differs from classification of in vivo lesions identified on 3D images, the 3‐group, T1 time‐based lesion classification was adjusted for in vivo application: long‐T1 lesions were isointense to CSF, short‐T1 lesions were isointense to cortex, and a heterogenous group of “mixed‐T1” lesions included lesions with well‐defined long‐T1 and short‐T1 areas as well as lesions with intermediate signal intensity (higher than cortex but substantially lower than CSF) throughout. All lesions were classified by a single investigator (HK). To determine interrater agreement, a subset of 110 lesions was randomly selected, and two raters (MA and ESB), both neurologists, performed independent lesion classifications that were then compared with those of HK. To determine intrarater agreement, HK reanalyzed the same 110 lesions 6 months after the initial rating.
For validation, in a second set of 66 MS cases scanned with the same protocol, 222 lesions (3–5 per case) were randomly selected on 3D FLAIR images and delineated on 7 T T1‐images by HK. The same classification procedures were applied to categorize the lesions both visually and quantitatively, using the T1 relaxation time cutoffs from the original cohort.
Statistical Analysis
For clinical variables, descriptive statistics included mean/standard deviation (SD) for normally distributed data, otherwise median/interquartile range for data. Normality was investigated by Shapiro–Wilk testing and visual inspection. A linear mixed‐effects model compared myelin OD, axon OD, and T1 times across the three lesion groups while accounting for multiple lesions per case and per slide. Pairwise comparisons were Bonferroni‐corrected. Linear mixed‐effects models described the relationships between log‐transformed myelin and axon OD and T1 time (the dependent variable). An F‐test was used for pairwise comparisons of variance. Receiver operating characteristic (ROC) curves assessed the predictive efficiency of T1 time for myelin content and axon loss, and the optimal cutoff was determined with Youden's index (J). To investigate misclassification, outlier observations were identified as those more than 1.5*interquartile range (IQR) below the 1st, or 1.5*IQR above the 3rd, T1 time quartile. Outliers falling within the corresponding 1.5*IQR range of a different lesion group were considered as potentially misclassified and carefully reviewed.
For in vivo lesion classification and characterization, linear mixed‐effects models with lesion‐specific random intercepts were used to account for correlations between lesion measures within the same participant. Initial models were fit to test potential associations of various covariates, including participant age, sex, enhancing lesion volume, enhancement pattern, lesion location, and PRL status. To describe predictors of lesion T1 time, models were also fit with predictors of participant age, disease duration, normalized gray matter and CSF volume, baseline PASAT, baseline MSSS, and follow up MSSS (separately). Paired‐sample t‐tests were used to assess differences in lesion T1 time for a subset of participants with more than one 7 T MP2RAGE. Differences in volume, PRL status, and location among lesions in the three lesion groups were assessed with analysis of variance (ANOVA) and chi‐squared tests. ROC curves were used to estimate the predictive efficiency of T1 time‐based visual classification. Intrarater and interrater reliability for the in vivo lesion classification was computed using Cohen's kappa (2 raters) and Kendall's coefficient of concordance (3 raters). P‐values are reported directly. 29 Statistical analysis used R (version 1.1.463, https://cran.r-project.org/) and Prism (version 8.4.3, GraphPad).
Results
Forty‐six lesions from four MS cases were identified on postmortem MP2RAGE MRI and included in the pathological study. Seven blocks (10 lesions) were excluded from further analysis due to partial lesions, failure to locate lesions identified on postmortem MRI, and/or technical problems. Based on the absence or presence of a CD68+ inflammatory infiltrate at the lesion border, 35 lesions were classified as chronic inactive and 1 as chronic active. The chronic active lesion was fully demyelinated. Of the 35 chronic inactive lesions, 13 were fully remyelinated/shadow plaques, 15 were fully demyelinated, and 7 were mixed (containing both demyelinated and remyelinated areas) (Fig 2A). Histological assessment revealed variable axon loss, most prominent in severely demyelinated lesions.
FIGURE 2.

Postmortem 7‐tesla T1 mapping is sensitive to both myelination level and axon loss in multiple sclerosis (MS) lesions. (A) Myelination status of lesions studied in postmortem samples, n (%). (B) Exemplary postmortem MRI and histopathology images of representative chronic MS lesions from a 78‐year‐old woman with secondary progressive MS. Ex‐vivo 7 T MP2RAGE recapitulates myelin status in longstanding MS lesions that are fully demyelinated (asterisk), with a “black hole” appearance on T1w images; partially demyelinated (dashed arrow); and fully remyelinated (arrowheads). Top row: MRI T1 map; second row: corresponding T1‐weighted (T1w) MRI; third row (myelin): Luxol fast blue–periodic acid Schiff (LFB‐PAS) stain; fourth row (myelin): Sudan black (SB) stain; fifth row (myelin): myelin proteolipid protein (PLP) immunohistochemistry; sixth row (axons): Bielschowsky's silver stain (Biel). Scale bar: 1 cm. (C) Receiver operating characteristic (ROC) curve demonstrating the sensitivity and specificity of T1 time cutoffs for differentiating fully demyelinated and fully remyelinated (circles) or severe vs. mild axon loss (stars). [Color figure can be viewed at www.annalsofneurology.org]
Figure 2B shows examples of the MRI and histopathological appearance of representative lesions. In all lesions, myelin and axon OD were tightly related (=0.67, p < 0.001). T1 time was longer in demyelinated (mean 951 ms, SD 181 ms) than remyelinated (mean 634, SD 47, p < 0.001) or mixed (mean 743, SD 187, p = 0.001) lesions. There was no difference in T1 time between mixed and fully remyelinated lesions (p = 0.34). T1 time was longer in lesions with severe (mean 982, SD 188) compared to moderate (mean 798, SD 206, p < 0.001) or mild (mean 642, SD 57, p < 0.001) axon loss. There was no difference in T1 time between lesions with moderate and mild axon loss (p = 0.14).
Based on ROC curves, the optimal T1 time threshold for differentiating lesions with complete or partial remyelination from fully demyelinated lesions was 779 ms, with 94% sensitivity and 95% specificity (AUC 0.94, p < 0.001). For axons, the same cut point of 779 ms optimally differentiated lesions with mild‐to‐moderate axon loss from lesions with severe axon loss, with 100% sensitivity and 86% specificity (AUC 0.90, p < 0.001) (Fig 2C).
Visual classification of lesions using 7 T MP2RAGE T1 maps successfully differentiated remyelinated (OD: mean 0.23, SD 0.07), partially remyelinated (mean 0.14, SD 0.05), and severely demyelinated (mean 0.07, SD 0.01) lesions (p < 0.001) (Fig 3A). Whereas OD of the axon stain was lower in long‐T1 (mean 0.21, SD 0.05) than short‐T1 (mean 0.38, SD 0.14; p < 0.001) or mixed‐T1 (mean 0.31, SD 0.11, p = 0.004) lesions, there was no difference between short‐T1 and mixed‐T1 lesions.
FIGURE 3.

Postmortem 7‐tesla T1 mapping more effectively discriminates myelination status than axon loss in remyelinated and mixed multiple sclerosis lesions but quantifies axon loss in fully demyelinated lesions. (A) Visual classification of ex vivo MRI clustered lesions into 3 groups with significantly different myelin content. Mean lesion T1 relaxation time, mean myelin stain optical density (OD), and mean axon stain OD for the three qualitatively assigned lesion groups (horizontal axis). (B) Correlation of mean T1 relaxation time with myelin and axon stains for the 3 lesion groups. Although well separated from other lesions, severely demyelinated lesions (low OD) showed a relatively large range of T1 relaxation times, suggesting a floor effect in the contribution of myelin to T1 time in this lesion group, and a regime in which T1 time is a more effective marker of axon loss.
Long‐T1 lesions clustered in a relatively narrow range of myelin OD (mean 0.07, SD 0.01, range 0.05–0.09) (Fig 3B). In contrast, the ranges of myelin OD for short and mixed‐T1 lesions were substantially larger (“short‐T1”: mean 0.23, SD 0.07, range 0.13–0.41; “mixed‐T1”: mean 0.14, SD 0.46, range 0.08–0.21). The variance for axon OD (0.0026) was >20‐fold larger than for myelin OD (0.00012) (p < 0.001) in long‐T1 lesions, and the same was true, to a lesser degree, in mixed‐T1 lesions (0.012 vs. 0.0021, p = 0.03). There was no apparent difference in the variance between axon and myelin OD within short‐T1 lesions (0.019 vs. 0.0056, p = 0.051). Three lesions met our prespecified outlier criteria, of which 2 were reclassified from short‐T1 to mixed‐T1 and 1 from mixed‐T1 to long‐T1. Following outlier reclassification, there were differences between the variance of axon and myelin OD for all 3 lesion groups (short‐T1: 0.02 vs. 0.002, p = 0.004; mixed‐T1: 0.014 v.s 0.01, p = 0.006; long‐T1: 0.003 vs. 0.0001, p < 0.001).
Twenty‐five adults with MS were studied in vivo (Table 2). After reviewing the natural history database, 78 postcontrast T1w scans, containing 268 gadolinium‐enhancing lesions, were analyzed in detail. On follow‐up 7 T MP2RAGE, 36 lesions were not visible, 7 were difficult to differentiate from surrounding abnormal white matter, and 10 were confluent at follow‐up. Lesions were confirmed on T2w images (2 scans) or pre‐ and postcontrast T1w images (6 scans) when FLAIR images were not available. Median time interval from lesion formation to assessment on 7 T MP2RAGE was 4.2 years (IQR: 7.9, range: 0.3–25).
Figure 4A shows examples of visual classification of chronic MS lesions, the characteristics of which were similar to lesions studied postmortem. Assessed in a randomly selected subset of 110 lesions, interrater reliability was substantial (Kendall W t = 0.88, p < 0.001) and intrarater reliability was excellent (Cohen κ = 0.96, p < 0.001). The three lesion groups differed in mean T1 time (p < 0.001) (Fig 4B). Separation between short‐T1 (mean 1617 ms, SD 156 ms) and long‐T1 (mean 2774, SD 274) lesions was excellent (p < 0.001), with no overlap of T1 time between groups.
FIGURE 4.
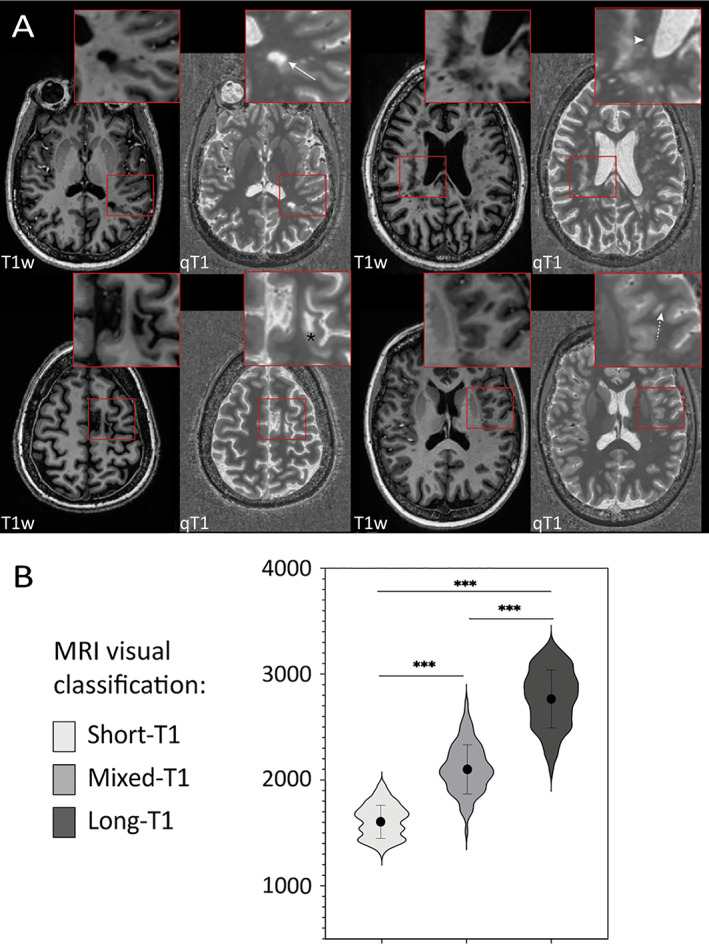
(A) Representative examples of the in vivo T1 relaxation time spectrum within MS lesions on high resolution MP2RAGE MRI at 7 T. Each panel includes T1‐weighted images (T1w, left) and quantitative T1 maps (qT1, right), scaled consistently across panels, and the insets show magnified views. Top row: Long‐T1 lesion in the left parietal white matter (arrow) and short‐T1 lesion in the right periventricular white matter (arrowhead). Bottom row: Spatially heterogenous, mixed‐T1 juxtacortical lesion in the left frontal lobe (asterisk) and left opercular subcortical lesion expressing intermediate signal intensity throughout (dashed arrow). (B) Violin plots showing the distribution of T1 times in the 3 lesion groups in vivo. The solid dot indicates the mean, and the vertical line the associated standard deviation. [Color figure can be viewed at www.annalsofneurology.org]
Based on ROC curves, the optimal T1 time threshold for differentiating short‐T1 from mixed‐T1 lesions was 1800 ms (sensitivity 91%, specificity 93%, AUC 0.96, p < 0.001), very similar to the average cortical T1 time of 1844 ms (SD 219 ms). For the mixed‐T1 and short‐T1 lesions, the optimal T1 time threshold was 2433 ms (sensitivity 91%, specificity 91%, AUC 0.96, p < 0.001), approximately 2.5 SD above the average cortical T1 time. Interrater agreement between the two methods (visual and quantitative lesion clustering) was substantial (W t = 0.90, p < 0.001). In a validation cohort of 222 lesions randomly selected from 66 MS cases (Table 2), interrater agreement between the visual and quantitative lesion classification was very good (W t = 0.87, p < 0.0001).
On 7 T phase images, 23 (8.6%) of the lesions were PRL; 11 of these (48%) were long‐T1, 11 (48%) mixed‐T1, and 1 (4%) short‐T1 (chi‐squared test, p < 0.001; Fig 5A). In the model controlling for covariates, only PRL status was associated with T1 time (p < 0.001), though it did not modify the relationship between T1 lesion group and T1 time. Thus, the final model included only PRL status as a covariate and explained 78% (marginal R 2) of the variance in T1 time (p < 0.001).
FIGURE 5.
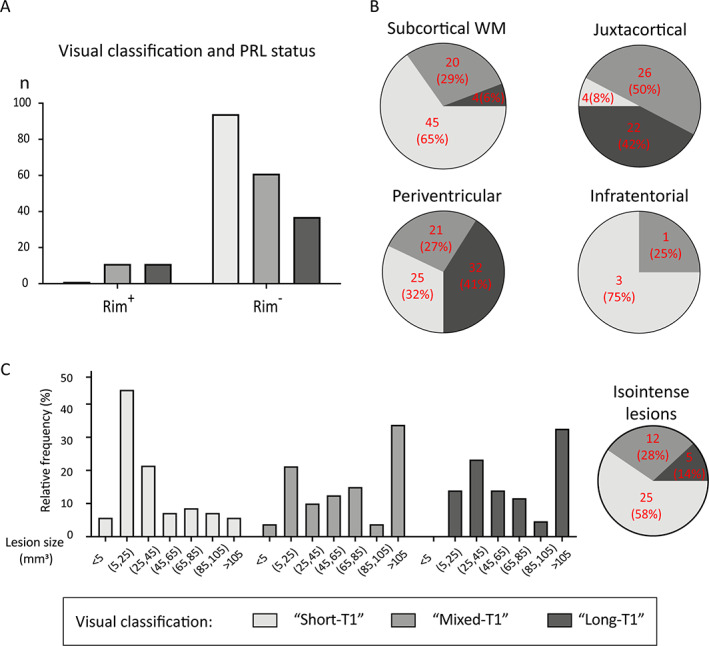
Small subcortical and periventricular lesions, and lesions that do not develop paramagnetic rims, are more likely to have T1 times associated with remyelination when evaluated a median of 4.2 years (range: 0.3–25) after lesion formation. (A) Paramagnetic rim lesions (PRL; n = 23) were nearly always categorized as “long‐T1” or “mixed‐T1” (chi‐squared test, p < 0.001). (B) Lesion location affects the relative proportions of lesion types (chi‐squared test, p = 0.02): long‐T1 lesions are most common in periventricular and juxtacortical regions, whereas short‐T1 lesions predominate in subcortical white matter but are also common in the periventricular region. (C) Histogram of sizes of lesions that were T1‐hypointense at the time of gadolinium enhancement showing that smaller lesions were more likely to evolve into “short‐T1” lesions, suggesting remyelination. Pie chart shows the fate of lesions that were T1‐isointense at the time of gadolinium enhancement; only 5 (14%) of these 42 lesions were in the long‐T1 group at follow‐up 7 T MRI. [Color figure can be viewed at www.annalsofneurology.org]
For a subset of 80 lesions from 14 patients, two MP2RAGE 7 T scans were available for analysis (mean interval 1.6, SD 0.4 years). For 67 lesions, classification was unchanged. Seven short‐T1 lesions were no longer identified on the follow‐up 7 T MP2RAGE: 5 lesions were difficult to differentiate from surrounding abnormal white matter, and 2 small lesions were not clearly visible. The remaining 6 lesions were all mixed‐T1 lesions on the earlier scan, of which 5 were reclassified as long‐T1 and 1 as short‐T1 at follow‐up.
A higher proportion of subcortical lesions had short T1. Long‐T1 lesions were almost all found in the periventricular or juxtacortical white matter, with very few in the deep white matter, and mixed‐T1 lesions were found in all supratentorial regions (chi‐squared test, p = 0.02, Fig 5B). Isointense and small hypointense lesions on baseline T1w images, corresponding to shorter T1 time, showed disproportionally more repair, than large hypointense lesions (chi‐squared test, p < 0.001, Fig 5C), and ring‐enhancing lesions were associated with both mixed‐T1 and long‐T1 lesion outcomes (p < 0.001). The total number of enhancing lesions per case did not correlate with T1 time and did not differ among the three lesion groups.
At the participant level, a strong predictor of lesion T1 was age at the time of lesion formation. For every 10‐year increase in age at the time of lesion formation, there was a 185 ms increase in mean lesion T1 time at 7 T follow‐up (p = 0.007), an association that remained strong after accounting for disease duration and disease‐modifying treatment status (treated vs. untreated). There was no association between mean T1 time and length of follow‐up or disease duration at time of enhancement or follow‐up. Mean lesion T1 time at follow‐up was associated with proportional gray matter (β= − 179 ms, p = 0.046) and cerebrospinal fluid‐space (β=318 ms, p = 0.02) volumes at the time of lesion formation. For every 10‐point decrease in PASAT score at baseline, there was a 151 ms increase in mean lesion T1 time at follow‐up (p = 0.008). There was no association between mean lesion T1 time and any other measure of physical or cognitive disability (MSSS, EDSS, SDMT, 9HPT) at baseline. MSSS was the only clinical measure at the time of 7 T scanning that was associated with T1 time: each 72 ms increase in mean lesion T1 time was associated with a 1‐point increase in MSSS (p = 0.02). There was no association between lesion T1 time and any structure volume or disability score at the time of 7 T scanning.
Discussion
Key findings of this research can be summarized as follows: (1) myelin strongly determines T1 time in MS lesions; (2) visual assignment of lesions into three groups based on postmortem 7 T T1 time recapitulates histopathologically assigned lesion status (remyelinated, demyelinated, mixed); (3) a similar in vivo classification is robust, with high interrater and intrarater reliability, and may be combined with susceptibility‐based MRI to differentiate chronic active, chronic inactive, and remyelinated lesions; (4) lesion remyelination may be predicted on the basis of clinical and radiological factors, especially patient age at the time of lesion development and lesion location.
White matter lesion T1w signal intensity is an important clinical trial outcome measure in MS, and hypointensity (“black hole,” defined on 1.5 T spin‐echo MRI) is generally taken to indicate irreversible axon loss. 30 However, since the introduction of high‐resolution, gradient‐echo T1w sequences, it has become clear that T1w hypointensity is a graded phenomenon; simple binary classifications are not appropriate. 30 , 31 , 32
Prior studies have shown the correlation of myelin and T1 signal, and proposed T1 relaxation time as a marker of lesion repair. 33 , 34 , 35 Our results suggest that myelin status is especially relevant at high magnetic field strength. Considering all lesions, the variance of OD measurements, which quantify staining intensity, was smaller for myelin than for axons, suggesting a more heterogenous relationship between axons and T1 time, especially for remyelinated and mixed lesions. Interestingly, in demyelinated lesions, myelin OD clustered in a narrow range, whereas T1 time varied widely, suggesting a floor effect in myelin's contribution to T1 time. Thus, the myelin/T1 time relationship may dominate in short‐T1/remyelinated and mixed lesions, and as such may be particularly useful for monitoring remyelination. On the other hand, in long‐T1 lesions, which are fully demyelinated, axons (and potentially other factors, such as gliosis, which was not studied here) may be determinative. These observations suggest that it may be possible to use T1 time to identify lesions with better repair potential. Longitudinal studies, including in the context of clinical trials of putatively remyelinating drugs, should test this possibility.
Adapting our postmortem visual lesion classification to in vivo MRI scans was straightforward, using the cortex as a suitable reference. Classification performance was robust in two datasets, recapitulating the postmortem results and highly consistent with results based on quantitative T1 time cutoffs. However, T1 quantification in individual lesions requires regions of interest or image segmentation, which is time‐consuming, subjective, and potentially inaccurate. Visual classification, on the other hand, is quick and amenable to the clinical neuroradiological workflow.
Several imaging approaches have been proposed as remyelination biomarkers, chiefly the semiquantitative magnetization transfer ratio (MTR). 36 MTR shows good sensitivity but questionable specificity for myelin and is limited by suboptimal signal‐to‐noise ratio and sensitivity to acquisition parameters and field inhomogeneities. An advantage of MP2RAGE is that it generates high‐quality images that allow lesion identification and delineation without additional dedicated acquisition. Direct comparison of the two methods with respect to quantification at clinical field strengths was beyond the scope of this study.
To understand determinants of lesion remyelination in vivo, we queried our long‐term natural history database to identify cases where age of individual lesions later studied at 7 T could be reliably defined on the basis of gadolinium enhancement. We then extracted clinical and radiological variables at the time of lesion development and assessed their contribution to lesion outcomes (Fig 6). Large lesions with ring enhancement had poor long‐term repair and fell into the long‐T1 group. 37 This is consistent with our prior work, which showed that larger, centripetally enhancing lesions were more likely to evolve into chronic active lesions, in which remyelination is impaired. 38 On the other hand, smaller lesions and those that were isointense to white matter at the time of lesion enhancement appear to remyelinate more effectively, also consistent with prior work. 39 Additionally, some anatomical locations may favor remyelination more than others. Subcortical lesions were more likely to evolve into short‐T1/remyelinated lesions, whereas juxtacortical and periventricular lesions were more likely to be long‐T1/demyelinated lesions. This comports with published human pathological studies 18 , 40 and the notion that periventricular lesions are less amenable to remyelination. 41
FIGURE 6.

Schematic illustrating the lesion and patient characteristics that associate with remyelination in vivo. Younger age, higher cognitive function, and higher brain volume at the time of lesion formation predict shorter follow‐up lesion T1 time a median of 4.2 years (range: 0.3–25) later. [Color figure can be viewed at www.annalsofneurology.org]
At the patient level, lesions forming at an older age were more likely to evolve into long‐T1/demyelinated lesions, consistent with our prior work 38 and with preclinical studies demonstrating that remyelination efficiency declines with age, similar to other regenerative processes. 42 Because remyelination mainly occurs within the first few months after lesion formation, age at the time of the acute insult seems critical and highlights the importance of early treatment. We also found that baseline cognitive impairment and higher brain atrophy predicted unfavorable lesion outcomes. Since cognitive impairment is consistently found to correlate with brain atrophy, 43 , 44 our observation may be interpreted as suggesting that newly formed lesions in patients with lower cognitive performance are less likely to remyelinate, possibly because of preexisting substantial axonal loss.
Predictably, PRL, which correspond to chronic active lesions, 3 , 45 were almost exclusively classified as long‐T1 or mixed‐T1. This is consistent with previous reports, which showed the destructive nature of these lesions and noted their association with long T1 times. 38 , 46 , 47 Our results imply that the combination of 7 T T1 mapping with MP2RAGE and PRL detection with susceptibility‐based MRI allows in vivo classification of most white matter lesions into the broad categories of chronic active, chronic inactive, and remyelinated lesions (Fig 7).
FIGURE 7.
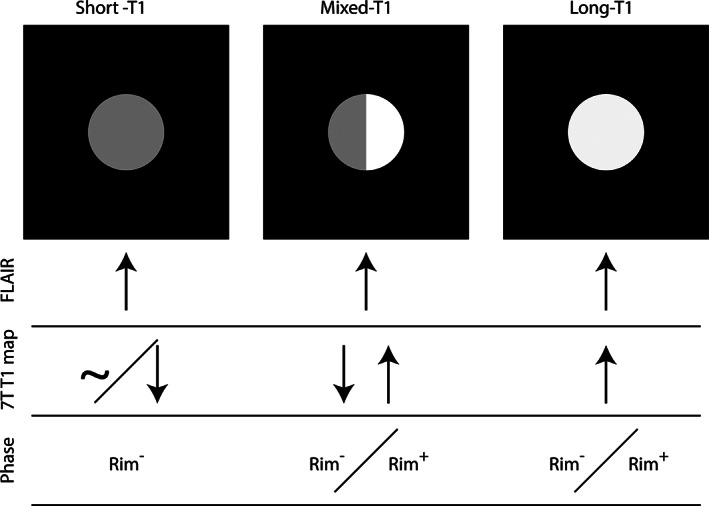
7 T T1 mapping and susceptibility‐based (phase) MRI allow noninvasive classification of most chronic white matter MS lesions: lesions with uniformly long T1 (relative to cortex) may be chronic active (paramagnetic rim) or chronic inactive (no paramagnetic rim), whereas lesions with uniformly short T1 (similar to or shorter than cortex) suggest remyelination. Mixed‐T1 lesions are more difficult to classify and may contain elements of all three pathological lesion types.
Limitations. Our postmortem study included four brains with a limited number of lesions from each group available for analysis, but those lesions included examples of MS lesion pathology described in the literature. Our in vivo study was performed in a research institution as part of natural history protocol, and as such may be affected by referral and enrollment biases that are common in this setting; thus, generalization awaits population‐based studies. Similarly, the long follow‐up of individual lesions means that measurement of some of our predictors used heterogeneous MRI methodology, and we did not attempt to correct for such variation. Additionally, one investigator (HK) analyzed both in vivo and histopathological data, but risk of unblinding was mitigated by a gap of at least 6 months between these analyses.
Prospective studies are needed to validate the predictors of remyelination we have identified and to determine the association with mediators of disability not explored here, including cortical, infratentorial, and spinal cord lesions. It will also be important to characterize individuals according to the relative proportion of different lesion types and to assess their medium‐ and long‐term clinical and radiological progression.
To conclude, we propose a straightforward approach for estimating tissue repair and remyelination in chronic MS lesions. Our results suggest that a qualitative classification of lesions on MP2RAGE T1 maps may be combined with susceptibility‐based MRI into a useful way to characterize MS lesions, identify individuals at risk for worse outcomes, and potentially stratify (and even track) participants in clinical trials for remyelination. Extension of the results to 3 T MRI systems that are far more prevalent in clinical practice will be an important next step.
Author Contributions
H.K., M.A., P.S., G.N.a., and D.S.R. contributed to the conception and design of the study. H.K., M.A., E.B., S.K.H., Y.S., G.N.o., I.C., P.S., G.N.a. and D.S.R. contributed to the acquisition and analysis of data. HK, GNo, and DSR contributed to drafting the text and preparing the figures.
Potential Conflicts of Interest
The authors report no conflict of interest.
Acknowledgments
We thank the staff and fellows of the NINDS Neuroimmunology Clinic for recruiting and evaluating study participants, Rose Cuento for protocol support, and the NIMH Functional Magnetic Resonance Facility for helping with MRI scanning. We thank Tobias Kober and Sunil Patil of Siemens Healthineers for assistance with implementation of the MP2RAGE sequence. The study was supported by Intramural Research Program of the National Institute of Neurological Disorders and Stroke (NINDS), National Institutes of Health (NIH); the Myelin Repair Foundation; and the Adelson Medical Research Foundation. Martina Absinta is also supported by the Conrad N. Hilton Foundation (Marylin Hilton Bridging Award for Physician‐Scientists, grant #17313), the Roche Foundation for Independent Research, the Cariplo Foundation (grant #1677) and the FRRB Early Career Award (grant#1750327). Erin Beck is also supported by a Career Transition Fellowship from the National Multiple Sclerosis Society.
MRI differentiates re/demyelinated MS lesions.
References
- 1. Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med 2018;378:169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kuhlmann T, Ludwin S, Prat A, et al. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol 2017;133:13–24. [DOI] [PubMed] [Google Scholar]
- 3. Frischer JM, Weigand SD, Guo Y, et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol 2015;78:710–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Absinta M, Sati P, Fechner A, et al. Identification of chronic active multiple sclerosis lesions on 3T MRI. AJNR Am J Neuroradiol 2018;39:1233–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Anderson VM, Fox NC, Miller DH. Magnetic resonance imaging measures of brain atrophy in multiple sclerosis. J Magn Reson Imaging 2006;23:605–618. [DOI] [PubMed] [Google Scholar]
- 6. Rocca MA, Battaglini M, Benedict RH, et al. Brain MRI atrophy quantification in MS: from methods to clinical application. Neurology 2017;88:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dwyer MG, Bergsland N, Ramasamy DP, et al. Atrophied brain lesion volume: a new imaging biomarker in multiple sclerosis. J Neuroimaging 2018;28:490–495. [DOI] [PubMed] [Google Scholar]
- 8. Wang CT, Barnett M, Barnett Y. Imaging the multiple sclerosis lesion: insights into pathogenesis, progression and repair. Curr Opin Neurol 2019;32:338–345. [DOI] [PubMed] [Google Scholar]
- 9. Stüber C, Morawski M, Schäfer A, et al. Myelin and iron concentration in the human brain: a quantitative study of MRI contrast. Neuroimage 2014;93:95–106. [DOI] [PubMed] [Google Scholar]
- 10. Koenig SH, Brown RD 3rd, Spiller M, Lundbom N. Relaxometry of brain: why white matter appears bright in MRI. Magn Reson Med 1990;14:482–495. [DOI] [PubMed] [Google Scholar]
- 11. Marques JP, Kober T, Krueger G, et al. MP2RAGE, a self bias‐field corrected sequence for improved segmentation and T1‐mapping at high field. Neuroimage 2010;49:1271–1281. [DOI] [PubMed] [Google Scholar]
- 12. Kober T, Granziera C, Ribes D, et al. MP2RAGE multiple sclerosis magnetic resonance imaging at 3 T. Invest Radiol 2012;47:346–352. [DOI] [PubMed] [Google Scholar]
- 13. Beck ES, Sati P, Sethi V, et al. Improved visualization of cortical lesions in multiple sclerosis using 7T MP2RAGE. AJNR Am J Neuroradiol 2018;8:459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fartaria MJ, Sati P, Todea A, et al. Automated detection and segmentation of multiple sclerosis lesions using ultra‐high‐field MP2RAGE. Invest Radiol 2019;54:356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Spini M, Choi S, Harrison DM. 7T MPFLAIR versus MP2RAGE for quantifying lesion volume in multiple sclerosis. J Neuroimaging 2020;30:531–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Absinta M, Nair G, Filippi M, et al. Postmortem magnetic resonance imaging to guide the pathologic cut: individualized, 3‐dimensionally printed cutting boxes for fixed brains. J Neuropathol Exp Neurol 2014;73:780–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Buchwalow IB, Boecker W. Immunohistochemistry basics and methods. Berlin: Springer, 2010. [Google Scholar]
- 18. Patrikios P, Stadelmann C, Kutzelnigg A, et al. Remyelination is extensive in a subset of multiple sclerosis patients. Brain 2006;129:3165–3172. [DOI] [PubMed] [Google Scholar]
- 19. Lassmann H, Raine CS, Antel J, Prineas JW. Immunopathology of multiple sclerosis: report on an international meeting held at the Institute of Neurology of the University of Vienna. J Neuroimmunol 1998;86:213–217. [DOI] [PubMed] [Google Scholar]
- 20. Schindelin J, Arganda‐Carreras I, Frise E, et al. Fiji: an open‐source platform for biological‐image analysis. Nat Methods 2012;9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McAuliffe MJ, Lalonde FM, McGarry D, Gandler W, Csaky K, Trus BL, Medical image processing, analysis and visualization in clinical research. Proceedings 14th IEEE symposium on computer‐based medical systems CBMS 2001; July 26–27, 2001.
- 22. Ruifrok AC, Johnston DA. Quantification of histochemical staining by color deconvolution. Anal Quant Cytol Histol 2001;23:291–299. [PubMed] [Google Scholar]
- 23. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol 2018;17:162–173. [DOI] [PubMed] [Google Scholar]
- 24. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 1983;33:1444–1452. [DOI] [PubMed] [Google Scholar]
- 25. Roxburgh RH, Seaman SR, Masterman T, et al. Multiple sclerosis severity score: using disability and disease duration to rate disease severity. Neurology 2005;64:1144–1151. [DOI] [PubMed] [Google Scholar]
- 26. Bramow S, Frischer JM, Lassmann H, et al. Demyelination versus remyelination in progressive multiple sclerosis. Brain 2010;133:2983–2998. [DOI] [PubMed] [Google Scholar]
- 27. Absinta M, Sati P, Gaitán MI, et al. Seven‐tesla phase imaging of acute multiple sclerosis lesions: a new window into the inflammatory process. Ann Neurol 2013;74:669–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shiee N, Bazin PL, Ozturk A, et al. A topology‐preserving approach to the segmentation of brain images with multiple sclerosis lesions. Neuroimage 2010;49:1524–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Perneger TV. What's wrong with Bonferroni adjustments. BMJ 1998;316:1236–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dupuy SL, Tauhid S, Kim G, et al. MRI detection of hypointense brain lesions in patients with multiple sclerosis: T1 spin‐echo vs. gradient‐echo. Eur J Radiol 2015;84:1564–1568. [DOI] [PubMed] [Google Scholar]
- 31. Lapucci C, Romano N, Schiavi S, et al. Degree of microstructural changes within T1‐SE versus T1‐GE hypointense lesions in multiple sclerosis: relevance for the definition of "black holes". Eur Radiol 2020;30:3843–3851. [DOI] [PubMed] [Google Scholar]
- 32. Thaler C, Faizy T, Sedlacik J, et al. T1‐ thresholds in black holes increase clinical‐radiological correlation in multiple sclerosis patients. PLoS One 2015;10:e0144693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mottershead JP, Schmierer K, Clemence M, et al. High field MRI correlates of myelin content and axonal density in multiple sclerosis–a post‐mortem study of the spinal cord. J Neurol 2003;250:1293–1301. [DOI] [PubMed] [Google Scholar]
- 34. Bot JC, Blezer EL, Kamphorst W, et al. The spinal cord in multiple sclerosis: relationship of high‐spatial‐resolution quantitative MR imaging findings to histopathologic results. Radiology 2004;233:531–540. [DOI] [PubMed] [Google Scholar]
- 35. Schmierer K, Scaravilli F, Altmann DR, et al. Magnetization transfer ratio and myelin in postmortem multiple sclerosis brain. Ann Neurol 2004;56:407–415. [DOI] [PubMed] [Google Scholar]
- 36. Oh J, Ontaneda D, Azevedo C, et al. Imaging outcome measures of neuroprotection and repair in MS. A consensus statement from NAIMS. Neurology 2019;92:519–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kermode AG, Tofts PS, Thompson AJ, et al. Heterogeneity of blood‐brain barrier changes in multiple sclerosis: an MRI study with gadolinium‐DTPA enhancement. Neurology 1990;40:229–235. [DOI] [PubMed] [Google Scholar]
- 38. Absinta M, Sati P, Schindler M, et al. Persistent 7‐tesla phase rim predicts poor outcome in new multiple sclerosis patient lesions. J Clin Invest 2016;126:2597–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Franklin RJ, Blakemore WF. To what extent is oligodendrocyte progenitor migration a limiting factor in the remyelination of multiple sclerosis lesions? Mult Scler (Houndmills, Basingstoke, England) 1997. Apr;3:84–87. [DOI] [PubMed] [Google Scholar]
- 40. Goldschmidt T, Antel J, König FB, et al. Remyelination capacity of the MS brain decreases with disease chronicity. Neurology 2009;72(22):1914–1921. [DOI] [PubMed] [Google Scholar]
- 41. Cunniffe N, Coles A. Promoting remyelination in multiple sclerosis. J Neurol 2021;268:30–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Neumann B, Segel M, Chalut KJ, Franklin RJ. Remyelination and ageing: reversing the ravages of time. Mult Scler (Houndmills, Basingstoke, England) 2019. Dec;25:1835–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sanfilipo MP, Benedict RH, Weinstock‐Guttman B, Bakshi R. Gray and white matter brain atrophy and neuropsychological impairment in multiple sclerosis. Neurology 2006;66:685–692. [DOI] [PubMed] [Google Scholar]
- 44. Zivadinov R, Sepcic J, Nasuelli D, et al. A longitudinal study of brain atrophy and cognitive disturbances in the early phase of relapsing‐remitting multiple sclerosis. J Neurol Neurosurg Psychiatry 2001;70:773–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Filippi M, Brück W, Chard D, et al. Association between pathological and MRI findings in multiple sclerosis. Lancet Neurol 2019;18:198–210. [DOI] [PubMed] [Google Scholar]
- 46. Dal‐Bianco A, Grabner G, Kronnerwetter C, et al. Slow expansion of multiple sclerosis iron rim lesions: pathology and 7 T magnetic resonance imaging. Acta Neuropathol 2017;133:25–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Absinta M, Sati P, Masuzzo F, et al. Association of chronic active multiple sclerosis lesions with disability in vivo. JAMA Neurol 2019;76:1474–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]