

Abstract
Introduction
The incidence of Frontotemporal Lobar Degeneration (FTLD)–related disorders and their characteristics are not well known. The “FRONTotemporal dementia Incidence European Research Study” (FRONTIERS) is designed to fill this gap.
Methods
FRONTIERS is a European prospective, observational population study based on multinational registries. FRONTIERS comprises 11 tertiary referral centers across Europe with long‐lasting experience in FTLD‐related disorders and comprehensive regional referral networks, enabling incidence estimation over well‐defined geographical areas.
Endpoints
The primary endpoints are (1) the incidence of FTLD‐related disorders across Europe; (2) geographic trends of FTLD‐related disorders; (3) the distribution of FTLD phenotypes in different populations and ethnicities in Europe; (4) inheritance of FTLD‐related disorders, including the frequencies of monogenic FTLD as compared to overall disease burden; and (5) implementation of data banking for clinical and biological material.
Expected impacts
FRONTIERS will improve the understanding of FTLD‐related disorders and their epidemiology, promoting appropriate public health service policies and treatment strategies.
Keywords: epidemiology, frontotemporal dementia, frontotemporal lobar degeneration, incidence, rare diseases, registry
1. INTRODUCTION
Frontotemporal lobar degeneration (FTLD) encompasses a spectrum of focal neurodegenerative diseases with progressive atrophy of the frontal and temporal lobes. 1 , 2 , 3 FTLD‐related disorders are heterogeneous clinical conditions, characterized by social dysfunctions and personality change along with impairment in language, executive, and motor functions. 4 Current clinical diagnostic criteria characterize specific presentations of FTLD, including the behavioral variant frontotemporal dementia (bvFTD) 5 and the primary progressive aphasias, with the agrammatic variant (avPPA) and semantic variant (svPPA) subtypes. 6 Progressive supranuclear palsy (PSP) 7 corticobasal syndrome (CBS), 8 and FTD with amyotrophic lateral sclerosis (FTD‐ALS) are also usually caused by subtypes of FTLD. There is limited correlation between these clinical syndromes and neuropathological features. 9 The neuropathological signature is associated mainly with either tau or transactive response (TAR)‐DNA‐binding protein‐43 (TDP‐43) protein inclusions. 10 Despite limited clinicopathological correlations, some types of FTLD have a strong genetic background. Up to ≈30% of FTD patients have positive family history, with mutations in the chromosome 9 open reading frame 72 (C9orf72), progranulin (GRN), or microtubule‐associated protein tau (MAPT) genes each accounting for 5% to 10% of patients. 11
In the last 10 years, in depth clinical characterization of FTLD‐associated features improved and operationalized clinical diagnostic criteria, and the use of biomarkers aimed at excluding Alzheimer's disease (AD), 12 have prompted a wider recognition of the diseases in clinical settings. This in turn increases the estimated incidence. However, epidemiological studies assessing the FTLD‐related disorders overall incidence and the relative frequency of the different phenotypes in the general population are scarce, with marked variation in results. 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 This variation can be attributed in part to narrow disease‐specific and phenotype based focus of individual studies; restriction to different age‐delimited sections of the population; and use of historical diagnostic criteria (before current consensus guidelines for each FTLD‐related disorder). Data on genetic correlates in different population‐representative cohorts remain sparse.
The FTLD‐related disorders are individually relatively rare, and so accurate epidemiological studies require surveillance of large populations with a registry‐based approach. This has been effective in the ALS field. 22 , 23 , 24 , 25 A recent Italian retrospective research registry (the Salento‐Brescia registry) aimed to assess the incidence of different FTLD phenotypes in the general population, demonstrating that FTLD‐related disorders are more common than previously recognized. There was a progressive increase in FTLD incidence across age groups, reaching its peak in individuals older than 70 years of age. 26 Comparable results have been provided in a survey study conducted with similar methodology in the UK. 15
Improved knowledge of FTLD‐related disorders and their epidemiology will help to promote appropriate public health service policies and treatment approaches. We have established the FRONTotemporal dementia Incidence European Research Study (FRONTIERS) as a collaborative European research initiative to assess FTLD epidemiology across Europe, aligning prospective multinational disease population‐based disease registries.
1.1. Rationale of FRONTIERS
The rationale of FRONTIERS is to assess the epidemiology of FTLD‐related disorders across Europe, resolving some of the current issues in the field.
First, FTLD has often been considered a presenile dementia. 11 However, it is clear that FTLD should not be confined exclusively within the early onset dementias, as peak incidence may well be above 65 years, and may continue to rise if case ascertainment were complete. Second, the distribution of clinical phenotypes in the general population is not yet known. Estimates are based on clinical series from tertiary referral centers, which are liable to be biased and nonrepresentative. Third, the distribution of monogenic FTLD‐related disorders in Europe has been deduced by ad hoc observational studies, with C9orf72 expansion being more prevalent in Northern Europe, whereas GRN is in Southern Europe, in areas like Northern Italy and Basque Country. 27 However, unbiased estimates of the incidence of monogenic FTLD causes disorders have not yet been established.
To address these issues, a population‐based approach through multinational registries has several advantages: (1) in each registry there is a prospective ascertainment of cases, seeking to minimize missing cases; (2) registries are generated from multiple sources of case ascertainment; (3) there is a continuous surveillance of the disease; and (4) matching diagnostic work‐up and diagnostic criteria are adopted. 25
1.2. End points of FRONTIERS
The primary aims of FRONTIERS are (1) to assess the incidence of FTLD‐related disorders across Europe, considering presenile and more advanced age groups; (2) to assess the geographic distribution of the disease; (3) to describe the distribution of FTLD phenotypes and their heterogeneity in different populations and different ancestral origins in Europe; (4) to define FTLD‐related disorders inheritance, and the frequencies of monogenic FTLD‐related disorders across Europe, including but not limited to the most frequent causes (ie., mutations within GRN, MAPT, and C9orf72 genes); (5) to implement data banking for clinical and biological material.
2. METHODS
2.1. Setting
FRONTIERS was established in February 6‐7, 2020 at a consensus meeting in Tricase, Italy. It was established with 11 centers across Europe, each with long‐lasting experience in the FTLD field and with the ability to cover a well‐defined geographical area. The pre‐requisite of a population‐based registry is the correct definition of the population at risk and the identification of proper sources of cases (see Figure 1 and Table 1 for centers and Principal Investigators).
FIGURE 1.
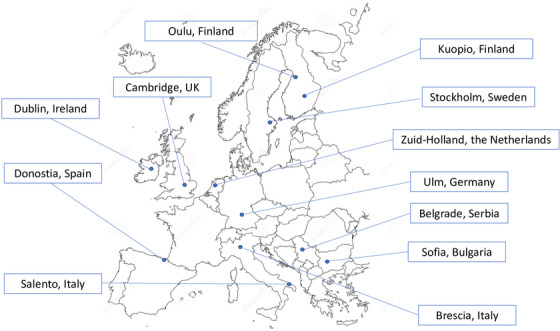
List of participating geographical areas
TABLE 1.
Centers and reference geographical area
| Country | Center | Principal investigator(s) | Reference geographical area | Reference population |
|---|---|---|---|---|
| Italy | Center for Neurodegenerative Disease and the Aging Brain, University of Bari | Giancarlo Logroscino | Salento area (Lecce province) | 795,134 a |
| Italy | Centre for Neurodegenerative Disorders, University of Brescia | Barbara Borroni | Brescia province | 1,265,954 b |
| Sweden | Karolinska University Hospital and Institutet, Stockholm | Caroline Graff | Stockholm area (Huddinge site) | 967,160 c |
| Bulgaria | Department of Neurology, University Hospital Alexandrovska, Sofia | Latchezar Traykov | Sofia region | 1,557,161 d |
| Spain | Neurology Department, Hospital Universitario Donostia | Fermin Moreno | Donostialdea area (Basque Country) | 388,142 e |
| Germany | Neurology, University Hospital of Uhlm | Markus Otto, Albert C. Ludolph | Ulm/Neu‐Ulm area | 184,711 f |
| UK | Centre for Frontotemporal Dementia and related disorders, Cambridge University | James Rowe | East England | 1,753,964 g |
| Finland | Oulu University Hospital | Anne Remes | Northern Osthrobothnia, Oulu and Northern Savonia, Kuopio | 657,763 h |
| Netherlands | Alzheimer Center, Erasmus, Rotterdam | Harro Seelaar | Zuid‐Holland | 3,673,893 i |
| Serbia | Neurology, University of Belgrade | Elka Stefanova | Belgrade area | 1,690,193 j |
| Ireland | Neurology, Trinity College, Dublin | Orla Hardiman | Dublin province | 1,904,806 k |
| Europe (11 areas) (Total Reference Population) | 14,838,881 | |||
RESEARCH IN CONTEXT
Systematic Review: Frontotemporal lobar degeneration (FTLD) is the second most common cause of early onset dementias. However, studies assessing the incidence of clinically manifest FTLD‐related disorders and the related clinical and genetic features are still limited. The “FRONTotemporal dementia Incidence European Research Study” (FRONTIERS) is designed to fill this gap.
Interpretation: FRONTIERS will allow us to determine the epidemiology of FTLD‐related disorders across Europe as well as the distribution of clinical phenotypes and genotypes. Furthermore, FRONTIERS will represent a source of cases for pragmatic trials and analytic population‐based studies, as well as a population‐based biobanking.
Future Directions: FRONTIERS will improve the understanding of FTLD‐related disorders and their epidemiology, promoting appropriate public health service policies and treatment strategies.
2.2. Participants and protocols
Patients with a new FTLD‐related diagnosis in the defined ascertainment time window and in the defined geographical boundaries will be considered in FRONTIERS. The diagnosis will be made according to current clinical criteria and supportive features for bvFTD, PPA (semantic and agrammatic variants), PSP, CBS, and FTD‐ALS diagnoses. 5 , 6 , 7 , 8 FRONTIERS will consider mixed or indeterminate PPA with negative AD markers, but will not consider the logopenic variant of PPA, as the majority of logopenic PPA cases have AD pathology. Moreover, FRONTIERS will not include patients in their prodromal stage and asymptomatic subjects carrying pathogenetic mutations, and FRONTIERS will not consider prevalent cases.
FRONTIERS participants will be assessed with a set of clinical, neuropsychological, imaging and biomarkers protocols, according to standard diagnostics in each participating center, following which diagnostic criteria of one of the FTLD phenotypes will be fulfilled as reported above. 5 , 6 , 7 , 8 A standardized set of data will be considered and recorded by each center, including demographic characteristics and clinical features, lifestyle and family history data, genetic screening results, and biomarkers testing.
Inclusion criteria will be the followings: (1) ≥18‐years‐old; (2) participants must fulfill current clinical criteria of FTD, PSP, or CBS 5 , 6 , 7 , 8 ; (3) diagnosis of the FTLD‐related disorder must be made in the referral period of time of the current prospective study; (4) participants must be living in the referral geographical area selected in each country for the purpose of the present study; and (5) participants must have an identified informant if necessary.
Exclusion criteria will be the followings: other medical or psychiatric illness that would interfere in completing assessments/diagnosis.
To minimize missing cases, the facilities of the referral geographical area, lay local associations, and charities involved in dementia care will be contacted by each center.
2.3. Demographic, clinical, and biomarkers data collection
Place of birth and residence as well as date of FTLD‐related disorders diagnosis will be recorded. We will consider age, age at disease onset, sex, clinical phenotype, the onset of symptoms, and disease duration (in the recall of the patient or family). Age at onset will be defined as the age at which first symptoms consistent with FTLD were observed by patients or caregivers. Years of full‐time education and occupation attainment will be also recorded. Family history for neurodegenerative dementias, ALS, or parkinsonism will be assessed by using a standardized family history questionnaire. 28 , ,
A standardized recording of extrapyramidal signs or motor neuron disease will be carried out. Disease severity at diagnosis will be assessed by the Clinical Dementia Rating plus NACC,FTLD, 29 and with the PSP‐Rating Scale (PSPRS) 30 or the Cortico Basal ganglia Functional Scale,(CBFS), 31 when appropriate.
Additional instrumental evaluations aimed at accomplishing a diagnosis will be recorded. In particular, we will consider cerebrospinal fluid (CSF) markers (tau, phospho‐tau and amyloid beta [Aβ]42 and Aβ40 dosage levels) or positron emission tomography (PET) amyloid scan, to exclude AD, and PET‐tau tracer or brain PET– fluorodeoxyglucose (FDG) scan. For each of these markers, the result will be recorded as “supporting FTLD diagnosis,” “uninformative,” or “not supporting FTLD diagnosis.” We will also record FTLD mimickers, namely, those subjects fulfilling FTLD criteria but with positive AD biomarkers and likely with mixed pathologies, especially in the older group, to determine their incidence in population‐based setting.
Finally, we will additionally measure neurofilament light in plasma and CSF in FTLD patients. 32
2.4. Genetic testing and sample biobanking
Each patient will undergo blood sampling for genetic screening and biobanking. The most frequent monogenic causes are C9orf72, MAPT, and GRN pathogenetic mutations. These will be assessed locally, according to standard procedures. 27 Where the genetic testing is locally unavailable, genetic analyses will be provided by other centers in the network. Biological samples, namely DNA, plasma, and serum, will be stored locally for additional future analyses, such as genome‐wide association studies or exome sequencing analyses. CSF storage for future analyses will be considered optional.
2.5. Data handling and record keeping
Data will be pseudo‐anonymized, and each participant will be assigned a unique investigation number (progressive number—country code), and the patient identity will be accessible only to the referral center where the data is acquired. Web‐based data acquisition will be carried out.
2.6. Ethical and regulatory considerations
The study will be performed on the basis of International Conference on Harmonisation/WHO (World Health Organization) Good Clinical Practice (ICH‐GCP) standards (www.ema.europa.eu/en/ich‐e6‐r2‐good‐clinical‐practice). Ethical approval for the study will be obtained from the coordinating center and from each local ethical review committees. Informed consent will cover participation in all the aspects of the study, except where a participant has lost the mental capacity to consent, in which case site‐specific national laws will be followed regarding the procedures to include legal incapacity participants in research. Data transfer agreements will be introduced for transfer of clinical data and biosamples.
2.7. FRONTIERS lifespan, data access and publication policy
FRONTIERS will have a duration of 3 years from the starting date, which will be the same across the sites; a first end point will occur at 12 months and a second end point will be set at 24 months for preliminary analyses. An extension of study duration may be considered.
Each PI will have access to anonymized data, and formal requests for either data analyses or biological samples will be submitted in writing to the other PIs, along with a brief description of the project that includes specific aims, study design, and characteristics of the data requested. The data request will be reviewed by the PIs and approved.
Publication policy agreement has already been defined and articles will list as authors all PIs and all other researchers who have made a scientific and/or clinical contribution. Co‐investigators will be quoted under a final corporate author reflecting the FRONTIERS collaboration.
2.8. Statistics
The following sets of variables will be recorded: (1) demographic data; (2) family history; (3) clinical phenotype; and (4) genetic mutations. Overall, FRONTIERS will cover defined geographical areas of interest across 10 European Countries (see Table 1), with a total estimated overall population of about 15 million people. In a first step, incidence rates will be computed separately for each region. The numerator of each rate will be the number of cases from the region, collected by each center during the study period. The denominator of each rate (person‐years spent at risk during the study period) will be estimated using the information on the number of residents living in the covered geographic area at a specific time point. 33 Age‐sex standardized incidence rates will be also computed using the European population as standard. Heterogeneity of incidence rate estimates across geographical regions will be assessed. In a second step, results from different geographical regions will be combined using methods to properly account for between‐region variability such as mixed‐effect models, 34 to obtain a European summary incidence estimate.
3. EXPECTED RESULTS AND IMPACT
Registries are powerful tools to develop clinical research, to improve patients care by appropriate planning of public policies for health care and to facilitate the correct design for clinical trials. 25 This is particularly true in case of relatively rare neurodegenerative disorders, characterized by clinical complexity and heterogeneity as well as low incidence and prevalence. In this setting, population‐based registries represent the only cost‐effective way to estimate the incidence of a disease. 25 , 26 Assessing disease incidence and studying how the frequency changes over time in a given geographical area and among subgroups are crucial in epidemiology to understand disease causation and to investigate possible risk factors. 35
FRONTIERS, a multinational prospective population‐based registry, will provide multi‐source case ascertainment and will allow the description of FTLD‐related phenotypes across Europe as well as the distribution of causative genetic traits. We will work with other cohort studies of related disorders, including the Genetic Frontotemporal Dementia Initiative (GENFI; seven FRONTIERS sites are members of GENFI consortium), and UK, German and Italian research cohorts for PSP and CBS (ie., PROgressive Supranuclear Palsy Cortico‐Basal Syndrome, PROSPECT study), FTLD consortium (www.ftld.de), Treatment Research Initiative to Cure ALS, TRICALS Consortium for ALS.
FRONTIERS will represent a source of cases for pragmatic trials and analytic population‐based studies, as well as a population‐based biobanking. A major impact of FRONTIERS will be an accurate understanding of epidemiology of FTLD‐related disorders, to answer a broad range of questions: (1) Are FTLD‐related disorders common? (2) Is incidence of FTLD‐related disorders comparable across European ancestral populations and countries? (3) Should FTLD‐related disorders be considered a disorder of midlife? (4) Is bvFTD more common than PPAs, or vice versa? (5) Are FTLD‐related disorders associated with specific risk factors? (6) Is distribution of FTLD‐related disorders comparable across Europe? (7) Which is the impact of monogenic FTLD to the overall burden of disease? (8) Is distribution of monogenic FTLD‐related disorders comparable across ethnicities/countries? (9) What are the characteristics of the diagnostic journey for FTLD‐related disorders? (10) Can we confirm suggestions for the staging of FTLD, both neuropathologically and clinically?. 36 , 37
FRONTIERS will be equipped to address these questions, as the study is population based, and all the new cases in a defined period of time and in a defined geographical area will be collected for comprehensive analysis of demographics, phenotypes, and outcomes across different geographic regions. 25
FRONTIERS will allow us to determine the distribution of clinical phenotypes and genotypes, as well as the relative weight of each in each country in Europe. Moreover, we seek new insights into the spreading of FTLD‐related mutations throughout Europe. 38
High quality data of registries and near‐completeness of case ascertainment is one of key elements to obtain reliable data. FRONTIERS will attempt to minimize misclassification biases with the inclusion of tertiary referral centers with long‐lasting experiences in FTLD diagnosis, with the rigorous definition of geographic boundaries of areas of interest, with the standardization of diagnostic and inclusion/exclusion criteria, and with the implementation of effective case identification strategies by referrals through networks of clinical professionals and charities. Furthermore, if suitable sources like administrative data are available, we are going to implement capture‐recapture analyses to estimate the rate of missing cases, 39 as previously has been succesfully done in ALS. 40 An important effort will be devoted to the harmonization of data among centers with periodic meetings with special focus on diagnosis of different phenotypes.
We recognize that this project does not involve systematic collection and storage of CSF and brain tissue, thus representing a limitation to the study. Moreover, assessment of biological markers that have recently aroused interest in the FTLD field have not been included in the FRONTIERS primary outcomes; however, additional studies may be carried out in the next future on stored biosamples.
Finally, we acknowledge that population‐based registers require a number of years of activity to ensure that early biases are resolved; this is generally a bias toward enrollment of prevalent cases, wrongly classified as incident within the first year of the study. 25
In conclusion, FRONTIERS will be a unique contribution to the field, primarily in the area of epidemiology, and will allow improving our understanding of clinical and biological FTLD heterogeneity within and across different populations, which may help to guide future regulatory policies and clinical decision‐making. FRONTIERS will be an exceptional platform to develop new pharmacological and non‐pharmacological trials.
ACKNOWLEDGMENTS
Frontiers has been partially granted for the first meeting held in Tricase, Italy by the following: Biogen, Cambridge, MA, USA; Alector, South San Francisco, California, USA; and Tecnopolo Puglia per la Medicina di Precisione D.G.R. Regione Puglia n. 2117 of 21.11.2018. Barbara Borroni received honoraria from Alector and Wave Pharmaceuticals; she reported a patent pending on Non Invasive Brain Stimulation method. Caroline Graff received funds from Schörling Foundation, Swedish FTD Initiative, Swedish Dementia Foundation, Swedish Research Council, Swedish Brain Foundation, and Stohnes Foundation Gamla Tjänarinnor; and she received travel funds for participation in a congress as speaker. Orla Hardiman received funds from Science Foundation Ireland Health Research Board, MND Association UK ALSA, and TRICALS Research Motor Neuron; and she received personal fees from Biogen Idec, Cytokinetics Orion, Wave Pharmaceuticals, Biogen, and Novartis. Albert C. Ludolph received funds from State of Baden‐Württemberg. Fermin Moreno received funds from Tau Consortium (Grant, Institution) and Instituto de Salud Carlos III (Grant, Institution). Markus Otto received funds from the German Ministry of Science and Technology, EU, Thierry Latran foundation, and FTLD CSF society; he received personal fees from Roche and Axon. Marco Piccininni received funds from Novartis Pharma for a self‐initiated research project, unrelated to this work, on migraine remission. He was awarded with a research grant from the Center for Stroke Research Berlin (private donations) for a self‐initiated project, unrelated to this work, on causal diagrams. He worked as a consultant for SANITANOVA S.r.l. in 2018; he received travel funds from Department of Clinical Research in Neurology, University of Bari “Aldo Moro” at Pia Fondazione Card. G. Panico and Institution Charité—Universitätsmedizin Berlin. Anne M Remes received funds from Academy of Finland, no. 315460, AMR. James B Rowe received funds from the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre (BRC‐1215‐20014), Medical Research Council (SUAG/051 G101400), Welcome Trust, Evelyn Trust, Alzheimer's Research UK, PSP Association, Cambridge Centre for Parkinson‐plus; he had a pro bono relationship with Guarantors of Brain, Trustee PSP Association, Trustee, Darwin College; he received personal fees from Asceneuron, UCB, Wave Pharmaceuticals, Biogen, Astex, SV Health, GE Healthcare, Copenhagen University. Harro Seelaar received funds from the ZonMW (project 733050513). Elka Stefanova received funds from the Alzheimer's Association; she received personal fees from Actavis Teva Pharma Swiss KRKA, Actavis Teva Krka. Latchezar Traykov has nothing to disclose. Giancarlo Logroscino received funds from Roche, Amplifon; he received travel funds for participation in a congress as speaker.
Open Access Funding provided by Universita degli Studi di Bari Aldo Moro within the CRUI‐CARE Agreement.
FRONTIERS members
Antonella Alberici, Centre for Neurodegenerative Disorders, Neurology Unit, ASST Spedali Civili Brescia, Italy;
Sarah Anderl‐Straub, Department of Neurology, University Hospital Ulm, Ulm, Germany;
Myriam Barandiaran, Cognitive Disorders Unit, Department of Neurology, Hospital Universitario Donostia, San Sebastian, Spain, and Neuroscience Area, Biodonostia Health Research Institute, San Sebastian, Spain;
Diyana Belezhanska, UH “Alexandrovska,” Department of Neurology, Medical University Sofia, Sofia, Bulgaria;
Alberto Benussi, Centre for Neurodegenerative Disorders, Neurology Unit, ASST Spedali Civili Brescia and University of Brescia, Italy;
Angelo Bianchetti, General Medicine and Rehabilitation, Istituto Clinico S.Anna, Brescia, Italy;
Rosa Capozzo, Center for Neurodegenerative Diseases and the Aging Brain, Pia Fondazione di Culto e Religione, Cardinale Giovanni Panico, University of Bari‐Aldo Moro, Bari, Italy;
Maria Sofia Cotelli, Neurology Unit, Ospedale Vallecamonica, Esine, Brescia, Italy;
Maria Teresa Dell'Abate, Center for Neurodegenerative Diseases and the Aging Brain, Pia Fondazione di Culto e Religione, Cardinale Giovanni Panico, University of Bari‐Aldo Moro, Bari, Italy;
Irena Dreharova, UH “Alexandrovska,” Department of Neurology, Medical University Sofia, Sofia, Bulgaria;
Emma Ehn, Unit for Hereditary Dementia, Theme Aging, Karolinska University Hospital‐Solna, Stockholm, Sweden;
Alazne Gabilondo, Neuroscience Area, Biodonostia Health Research Institute, San Sebastian, Spain, and Hospital Bidasoa, Irun, Spain;
Begoña Indakoetxea, Cognitive Disorders Unit, Department of Neurology, Hospital Universitario Donostia, San Sebastian, Spain, and Neuroscience Area, Biodonostia Health Research Institute, San Sebastian, Spain;
Vesna Jelic, Karolinska Institutet, Department NVS, Division of Neurogeriatrics, Stockholm, Sweden;
Johanna Krüger, Unit of Clinical Neuroscience, Neurology, University of Oulu and Medical Research Center, Oulu University Hospital, Oulu, Finland;
Jose Laffita Mesa, Unit for Hereditary Dementia, Theme Aging, Karolinska University Hospital‐Solna, Stockholm, Sweden;
Jolina Lombardi, Department of Neurology, University Hospital Ulm, Ulm, Germany;
Eugenio Magni, Neurology, Poliambulanza Hospital, Brescia, Italy;
Shima Mehrabian, UH “Alexandrovska”, Department of Neurology, Medical University Sofia, Sofia, Bulgaria;
Alex Murley, Department of Clinical Neurosciences, MRC Cognition and Brain Sciences Unit, and Cambridge University Hospitals NHS Trust, University of Cambridge, Cambridge, United Kingdom;
Genoveva Nacheva, UH “Alexandrovska”, Department of Biology Medical University Sofia, Sofia, Bulgaria;
Gabriele Nagel, Department of Neurology, University Hospital Ulm, Ulm, Germany;
Ivana Novakovic, Faculty of Medicine, University of Belgrade, Neurology Clinic, University Clinical Center Serbia;
Linn Öijerstedt, Unit for Hereditary Dementia, Theme Aging, Karolinska University Hospital‐Solna, Stockholm, Sweden;
Margarita Raycheva, UH “Alexandrovska,” Department of Neurology, Medical University Sofia, Sofia, Bulgaria;Timothy Rittman, Department of Clinical Neurosciences, MRC Cognition and Brain Sciences Unit, and Cambridge University Hospitals NHS Trust, University of Cambridge, Cambridge, United Kingdom;
Catharina Roman, Unit for Hereditary Dementia, Theme Aging, Karolinska University Hospital‐Solna, Stockholm, Sweden;
Angela Rosenbohm, Department of Neurology, University Hospital Ulm, Ulm, Germany;
Dietrich Rothenbacher, Department of Neurology, University Hospital Ulm, Ulm, Germany;
Niall Pender, Academic Unit of Neurology Trinity College Dublin and Deppartment of Neuropsychology, Beaumont Hospital, Dublin
Ivo Popivanov, UH “Alexandrovska,” Department of Neurology, Medical University Sofia, Sofia, Bulgaria;
Eino Solje, Neurology, University of Eastern Finland and Neuro center, Neurology, Kuopio University Hospital, Kuopio, Finland;
Katherine Stockton, Department of Clinical Neurosciences, MRC Cognition and Brain Sciences Unit, and Cambridge University Hospitals NHS Trust, University of Cambridge, Cambridge, United Kingdom;
Gorana Mandic Stojmenovic, Faculty of Medicine, University of Belgrade, Neurology Clinic, University Clinical Center Serbia;
Katya Stoyanova, UH “Alexandrovska,” Department of Neurology, Medical University Sofia, Sofia, Bulgaria;
Noora Suhonen, Medical Research Center, Oulu University Hospital, Oulu, Finland;
Mikel Tainta, Neuroscience Area, Biodonostia Health Research Institute, San Sebastian, Spain;
Emma L. van der Ende, Department of Neurology and Alzheimer center, Erasmus University Medical Center Rotterdam, The Netherlands;
John C. van Swieten, Department of Neurology and Alzheimer center, Erasmus University Medical Center Rotterdam, The Netherlands;
Chiara Zecca, Center for Neurodegenerative Diseases and the Aging Brain, Pia Fondazione di Culto e Religione, Cardinale Giovanni Panico, University of Bari‐Aldo Moro, Bari, Italy;
Dora Zlatareva, UH “Alexandrovska,” Department of Radiology, Medical University Sofia, Sofia, Bulgaria;
Miren Zulaica, Neuroscience Area, Biodonostia Health Research Institute, San Sebastian, Spain.
Borroni B, Graff C, Hardiman O, et al. FRONTotemporal dementia Incidence European Research Study—FRONTIERS: Rationale and design. Alzheimer's Dement. 2022;18:498–506. 10.1002/alz.12414
the list of FRONTIERS members is provided in the Appendix at the end of the manuscript.
[Correction added on May 12, 2022, after first online publication: CRUI‐CARE funding statement has been added.]
REFERENCES
- 1. Kertesz A, McMonagle P, Blair M, Davidson W, Munoz DG. The evolution and pathology of frontotemporal dementia. Brain. 2005;128:1996‐2005. [DOI] [PubMed] [Google Scholar]
- 2. MacKenzie IRA, Neumann M, Bigio EH, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119:1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rohrer JD, Lashley T, Schott JM, et al. Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration. Brain. 2011;134:2565‐2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Murley AG, Coyle‐Gilchrist I, Rouse MA, et al. Redefining the multidimensional clinical phenotypes of frontotemporal lobar degeneration syndromes. Brain. 2020;143:1555‐1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134:2456‐2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gorno‐Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76:1006‐1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Mov Disord. 2017;32:853‐864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013;80:496‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Irwin DJ, Cairns NJ, Grossman M, et al. Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathol. 2015;129:469‐491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Perry DC, Brown JA, Possin KL, et al. Clinicopathological correlations in behavioural variant frontotemporal dementia. Brain. 2017;140:3329‐3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Benussi A, Padovani A, Borroni B. Phenotypic heterogeneity of monogenic frontotemporal dementia. Front Aging Neurosci. 2015;7:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Simrén J, Ashton NJ, Blennow K, Zetterberg H. An update on fluid biomarkers for neurodegenerative diseases: recent success and challenges ahead. Curr Opin Neurobiol. 2020;61:29‐39. [DOI] [PubMed] [Google Scholar]
- 13. Borroni B, Alberici A, Grassi M, et al. Prevalence and demographic features of early‐onset neurodegenerative dementia in Brescia County, Italy. Alzheimer Dis Assoc Disord. 2011;25:341‐344. [DOI] [PubMed] [Google Scholar]
- 14. Chiari A, Vinceti G, Adani G, et al. Epidemiology of early onset dementia and its clinical presentations in the province of Modena, Italy. Alzheimers Dement. 2021;17(1):81‐88. [DOI] [PubMed] [Google Scholar]
- 15. Coyle‐Gilchrist ITS, Dick KM, Patterson K, et al. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology. 2016;86:1736‐1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harvey RJ, Skelton‐Robinson M, Rossor MN. The prevalence and causes of dementia in people under the age of 65 years. J Neurol Neurosurg Psychiatry. 2003;74:1206‐1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gislason TB, Sjögren M, Larsson L, Skoog I. The prevalence of frontal variant frontotemporal dementia and the frontal lobe syndrome in a population based sample of 85 year olds. J Neurol Neurosurg Psychiatry. 2003;74:867‐871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Knopman DS, Petersen RC, Edland SD, Cha RH, Rocca WA. The incidence of frontotemporal lobar degeneration in Rochester, Minnesota, 1990 through 1994. Neurology. 2004;62:506‐508. [DOI] [PubMed] [Google Scholar]
- 19. Onyike CU, Diehl‐Schmid J. The epidemiology of frontotemporal dementia. Int Rev Psychiatry. 2013;25:130‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Turcano P, Stang CD, Mielke MM, et al. Incidence of frontotemporal disorders in Olmsted County: a population‐based study. Alzheimers Dement. 2020;16:482‐490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rosso SM, Kaat LD, Baks T, et al. Frontotemporal dementia in The Netherlands: patient characteristics and prevalence estimates from a population‐based study. Brain. 2003;126:2016‐2022. [DOI] [PubMed] [Google Scholar]
- 22. Beghi E, Logroscino G, Chiò A, et al. The epidemiology of ALS and the role of population‐based registries. Biochim Biophys Acta. 2006;1762:1150‐1157. [DOI] [PubMed] [Google Scholar]
- 23. Logroscino G, Traynor BJ, Hardiman O, et al. Descriptive epidemiology of amyotrophic lateral sclerosis: new evidence and unsolved issues. J Neurol Neurosurg Psychiatry. 2008;79(1):6‐11. [DOI] [PubMed] [Google Scholar]
- 24. Hardiman O, Al‐Chalabi A, Brayne C, et al. The changing picture of amyotrophic lateral sclerosis: lessons from European registers. J Neurol Neurosurg Psychiatry. 2017;88:557‐563. [DOI] [PubMed] [Google Scholar]
- 25. Rooney JPK, Brayne C, Tobin K, Logroscino G, Glymour MM, Hardiman O. Benefits, pitfalls, and future design of population‐based registers in neurodegenerative disease. Neurology. 2017;88:2321‐2329. [DOI] [PubMed] [Google Scholar]
- 26. Logroscino G, Piccininni M, Binetti G, et al. Incidence of frontotemporal lobar degeneration in Italy: the Salento‐Brescia Registry study. Neurology. 2019;92:E2355‐63. [DOI] [PubMed] [Google Scholar]
- 27. Moore KM, Nicholas J, Grossman M, et al. Age at symptom onset and death and disease duration in genetic frontotemporal dementia: an international retrospective cohort study. Lancet Neurol. 2020;19:145‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Byrne S, Heverin M, Elamin M, et al. Aggregation of neurologic and neuropsychiatric disease in amyotrophic lateral sclerosis kindreds: a population‐based case‐control cohort study of familial and sporadic amyotrophic lateral sclerosis. Ann Neurol. 2013;74:699‐708. [DOI] [PubMed] [Google Scholar]
- 29. Miyagawa T, Brushaber D, Syrjanen J, et al. Utility of the global CDR® plus NACC FTLD rating and development of scoring rules: data from the ARTFL/LEFFTDS Consortium. Alzheimers Dement. 2020;16:106‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Golbe LI, Ohman‐Strickland PA. A clinical rating scale for progressive supranuclear palsy. Brain. 2007;130:1552‐1565. [DOI] [PubMed] [Google Scholar]
- 31. Lang AE, Stebbins GT, Wang P, Jabbari E, Lamb R, Morris H. The Cortical Basal ganglia Functional Scale (CBFS): development and preliminary validation. Parkinsonism Relat Disord. 2020;79:121‐126. [DOI] [PubMed] [Google Scholar]
- 32. Verde F, Steinacker P, Weishaupt JH, et al. Neurofilament light chain in serum for the diagnosis of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2019;90:157‐164. [DOI] [PubMed] [Google Scholar]
- 33. Logroscino G, Kurth T, Piccininni M. The reconstructed cohort design: a method to study rare neurodegenerative diseases in population‐based settings. Neuroepidemiology. 2020;54:114‐122. [DOI] [PubMed] [Google Scholar]
- 34. Basagaña X, Pedersen M, Barrera‐Gómez J, et al. Analysis of multicenter epidemiological studies: contrasting fixed or random effects modelling and meta‐analysis. Int J Epidemiol. 2018;47:1343‐1354. [DOI] [PubMed] [Google Scholar]
- 35. Logroscino G, Piccininni M. Amyotrophic lateral sclerosis descriptive epidemiology: the origin of geographic difference. Neuroepidemiology. 2019;52:93‐103. [DOI] [PubMed] [Google Scholar]
- 36. Brettschneider J, Arai K, Del Tredici K, et al. TDP‐43 pathology and neuronal loss in amyotrophic lateral sclerosis spinal cord. Acta Neuropathol. 2014;128:423‐437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lulé DE, Ludolph AC. In vivo tracking of TDP43 in ALS: cognition as a new biomarker for brain pathology. J Neurol Neurosurg Psychiatry. 2020;91:125. [DOI] [PubMed] [Google Scholar]
- 38. Pliner HA, Mann DM, Traynor BJ. Searching for Grendel: origin and global spread of the C9ORF72 repeat expansion. Acta Neuropathol. 2014;127:391‐396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hook & Regal . Capture‐Recapture methods in epidemiology: methods and limitations. Epidemiol Rev. 1995;17:243‐264. [DOI] [PubMed] [Google Scholar]
- 40. Preux PM, Druet‐Cabanac M, Couratier P, et al. Estimation of the amyotrophic lateral sclerosis incidence by capture‐recapture method in the Limousin region of France. J Clin Epidemiol. 2000;53:1025‐1029. [DOI] [PubMed] [Google Scholar]