

Abstract
The molecular basis for the propensity of a small number of environmental proteins to provoke allergic responses is largely unknown. Herein, we report that mite group 13 allergens of the fatty acid binding protein (FABP) family are sensed by an evolutionary conserved acute phase protein, serum amyloid A1 (SAA1), that promotes pulmonary type 2 immunity. Mechanistically, SAA1 interacted directly with allergenic mite FABPs (Der p 13/Blo t 13). The interaction between mite FABPs and SAA1 activated the SAA1-binding receptor, formyl peptide receptor 2 (FPR2), which drove the epithelial release of the type 2-promoting cytokine interleukin 33 (IL-33) in a SAA1-dependent manner. Importantly, the SAA1-FPR2-IL-33 axis was upregulated in nasal epithelial cells from chronic rhinosinusitis patients. These findings identify an unrecognized role for SAA1 as a soluble pattern recognition receptor for conserved FABPs found in common mite allergens that initiate type 2 immunity at mucosal surfaces.
Introduction
Allergic diseases of the respiratory system such as asthma are initiated by aberrant type 2 immunity to an exclusive group of otherwise innocuous environmental proteins, called allergens. Though extensive efforts have been focused on defining the common motifs underlying allergenicity1, the immunological mechanisms by which allergens induce type 2 immune responses are still not well understood. Recent evidence suggests that dysregulated allergen sensing through pattern recognition receptors (PRRs) and the aberrant release of innate type 2 mediators, such as interleukin 25 (IL-25), IL-33, and thymic stromal lymphopoietin (TSLP) at mucosal surfaces2, 3, 4, are central to the initiation or amplification of existing allergic respiratory diseases in genetically susceptible individuals.
While PRRs are classically thought of as membrane bound Toll-like receptors (TLRs) or cytoplasmic (NOD-like receptors/RIG-like receptors), several other evolutionarily ancient but secreted molecules constitute a distinct class of soluble PRRs (sPRR). sPRRs, including pentraxins, are constitutively expressed at low basal concentrations by a variety of cells and can rapidly increase to signal the presence of pathogens, regulate the migration of leukocytes, and modulate tissue responses in extracellular compartments such as the alveolar space5. As such, they are part of humoral innate defense mechanisms that protect barrier tissues from microorganisms and potential injuries. Although sPRRs have been linked to allergic inflammation in the lung, their role in type 2 immunity at this environmentally exposed site remains poorly understood.
In the respiratory tract, airway epithelial cells (AECs) are a major source of sPRRs that include the acute phase reactant serum amyloid A (SAA)6. The products of the SAA1/2 genes, the SAA1 and SAA2 proteins, have been classically viewed as highly inducible, liver-derived factors in response to infection or trauma7, but baseline expression has been documented in many histologically normal human extrahepatic tissues including the alveolar epithelial lining6. As sPRRs, SAA proteins may serve as opsonins that enhance bacterial clearance and block viral entry into cells8, 9. In contrast to high-density lipoprotein (HDL)-bound SAA1 in the serum10, locally produced, lipid-free SAA1 can promote proinflammatory cytokine production, the recruitment of immune cells and epithelial wound repair11 through a number of potential SAA receptors12. These SAA1 functions, however, are not operative under physiological conditions13, 14, probably requiring a ligand-induced activation of SAA1.
In vitro, lipid-free SAA1 is prone to self-assemble into hexamers, which may have implications for SAA1 functions and may be modulated by ligand binding and/or the extent of SAA expression15, 16. While SAA recognition of bacteria in the gut is essential for TH17 immunity17, much less is known about SAA innate immune sensing mechanisms in the lung18. Although a positive association between elevated sputum and airway SAA1 concentrations and asthma prevalence has been established in several independent studies19, 20, 21, 22, a causative role for SAA1 in allergic airway inflammation has remained elusive. Herein, we demonstrate a previously unrecognized role for SAA1 as an innate environmental sensor in type 2 immunity through its interaction with allergenic fatty acid binding proteins (FABPs) resulting in formyl peptide receptor 2 (FPR2)-dependent initiation of type 2 inflammation.
Results
SAA1 drives type 2 immune responses in vivo.
To examine whether SAA1 (the predominant SAA isoform) is critical for allergen-driven aspects of type 2 immunity in the lungs, wild-type and Saa–/– (lacking expression of SAA1.1 and its less abundant isoform, SAA2.1) C57BL/6 mice were exposed to an extract of the house dust mite (HDM) species Dermatophagoides pteronyssinus, a complex source of allergenic patterns associated with type 2 responses in the lung3 (Extended Data Fig. 1a). While HDM-exposed wild-type mice developed increased airway hyperresponsiveness (AHR) in response to methacholine challenge compared to PBS-exposed mice, Saa–/– mice were largely protected from HDM-induced AHR (Fig. 1a). In addition, Saa–/– mice had reduced total serum IgE concentrations, bronchoalveolar lavage (BAL) eosinophilia, and goblet cell metaplasia of the airway lumen as compared to control animals (Fig. 1b-e). The suppression of the allergic phenotype was associated with significantly lower numbers of CD11c+CD11bhiMHCIIhi dendritic cells (DCs), IL-13-secreting CD3+CD4+ (TH2) and type 2 innate lymphoid cells (ILC2) and reduced TH2-cytokine production by HDM–restimulated lung cells (Fig. 1f-h, Extended Data Fig. 2-3). The contribution of SAA1 to allergen-driven type 2 immunity was confirmed in an independent approach locally administering SAA1 neutralizing antibodies in the lungs of BALB/c mice (Extended Data 1b). Consistent with genetic deficiency, SAA1 neutralization throughout the allergen exposure regiment reduced cardinal markers of allergic asthma including AHR and numbers of type 2 effector cells (Extended Data Fig. 4a-g). To determine whether SAA signaling was also necessary for the innate manifestation of type 2 immunity we analyzed the secretion of epithelial type 2 cytokines that are central drivers of the allergic response23. BAL concentrations of IL-33 were significantly increased after a single inhalational HDM exposure in wild-type mice, while baseline BAL IL-33 amounts in Saa–/– mice were unchanged. HDM exposure failed to augment IL-25 concentrations in wild-type or Saa–/– mice, and TSLP was not detected in the BAL (Extended Data Fig. 1c, Fig. 2a-c). Allergen-triggered epithelial IL-33 release24 leads to the expansion of IL-13-secreting lung-resident ILC225, 26 that initiate and maintain local allergic responses. Wild-type C57BL/6 mice had significantly elevated numbers of IL-13+ ILC2 cells in their lungs compared with Saa–/– mice (Fig. 2d). Similarly, inhibiting the local effects of SAA1 through HDL- or antibody-mediated neutralization in BALB/c mice caused significantly reduced numbers of HDM-induced lung IL-13+ ILC2s (Extended Data Fig. 1d, Fig. 2e). Local overexpression of SAA1, which raised baseline SAA1 concentrations, was not sufficient to induce spontaneous expansion of IL-13-producing ILC2 cells in BALB/c mice. Only in the presence of HDM, overexpression of SAA1 resulted in enhanced numbers of IL-13+ ILC2 cells as compared to control mice (Extended Data Fig. 1e, Fig. 2f). This indicates that, even at increased local concentrations, SAA1 requires the interaction with HDM allergens to boost its type 2-mediating properties and the expansion of IL-13+ ILC2.
Fig. 1: SAA drives HDM-induced allergic airway inflammation.
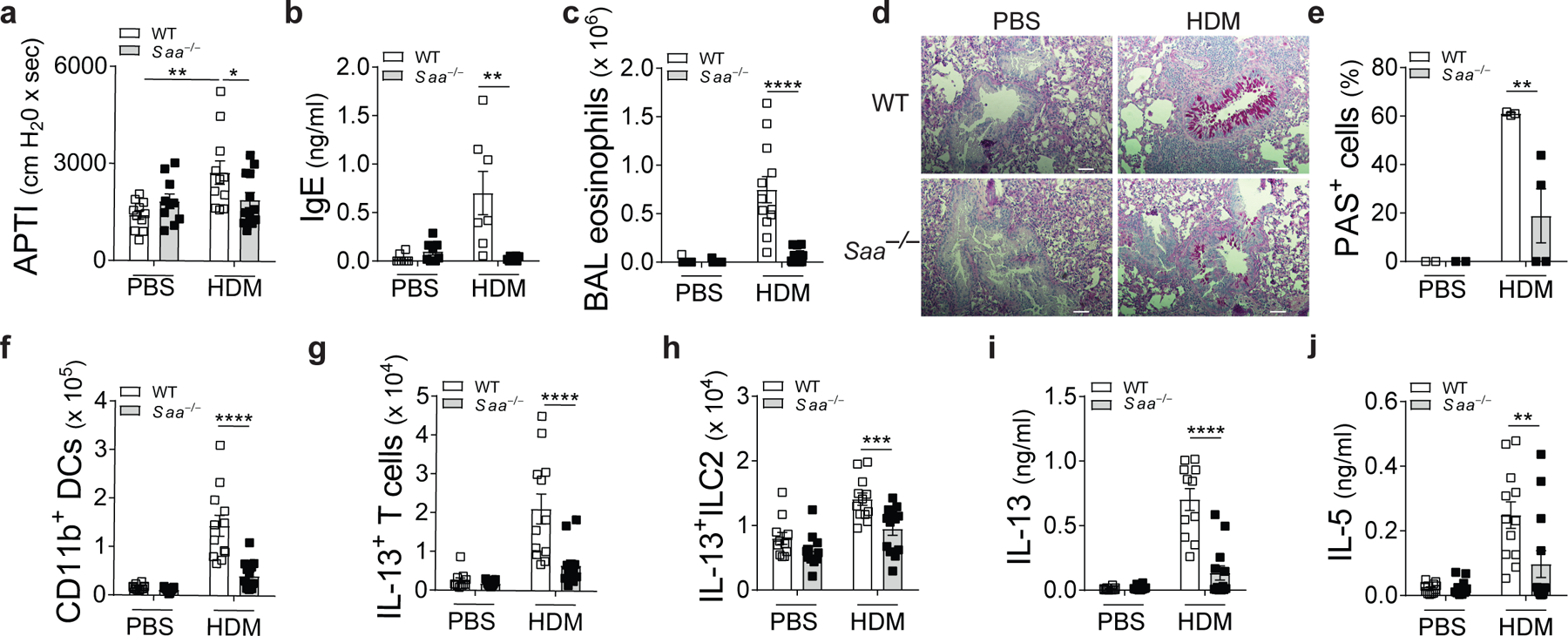
Allergic phenotype in WT and Saa–/– C57BL/6 mice sensitized and challenged with HDM was analyzed seventy-two hours after the last challenge. Control mice received PBS at both sensitization and challenge. (a) Airway responses to cholinergic stimulation shown as airway pressure over time (APTI, *P = 0.0378, **P = 0.0019), (b) total serum IgE concentrations (**P = 0.0019), (c) bronchoalveolar lavage (BAL) eosinophilia and (d and e) histological examination of airway inflammation. Sections were stained for mucus production with periodic acid Schiff (PAS; Scale bars 100 μm) and bar graphs represent percentage of mucus positive cells in the airways (*P = 0.0241). Numbers of (f) CD11b+ DCs, (g) IL-13+ CD3+CD4+ and (h) IL-13+ILC2 cells (***P = 0.0003) in the lungs and (i and j) TH2 cytokine production from HDM-restimulated lung cells (**P = 0.0018). Data represent means ± SEM of pooled data from 2 independent experiments containing n=11 WT PBS, n=12 WT HDM, n=11 Saa–/– PBS and n=13 Saa–/– HDM animals per group (a, c, and f-j), n=2 WT PBS, n=3 WT HDM, n=2 Saa–/– PBS and n=4 Saa–/– HDM animals per group (d, e), or are representative of 2 experiments (b) with n=8 WT PBS, n=7 WT HDM, n=8 Saa–/– PBS and n=8 Saa–/– HDM animals per group. P values were calculated with a two-tailed test using one-way analysis of variance (ANOVA) with Dunett’s post hoc (b, c, e-j) or Holm-Sidak’s (a) analysis that compares WT HDM to counterparts. ***P ≤ 0.001; ****P ≤ 0.0001.
Fig. 2: SAA signaling was also necessary for the innate manifestation of type 2 immunity.
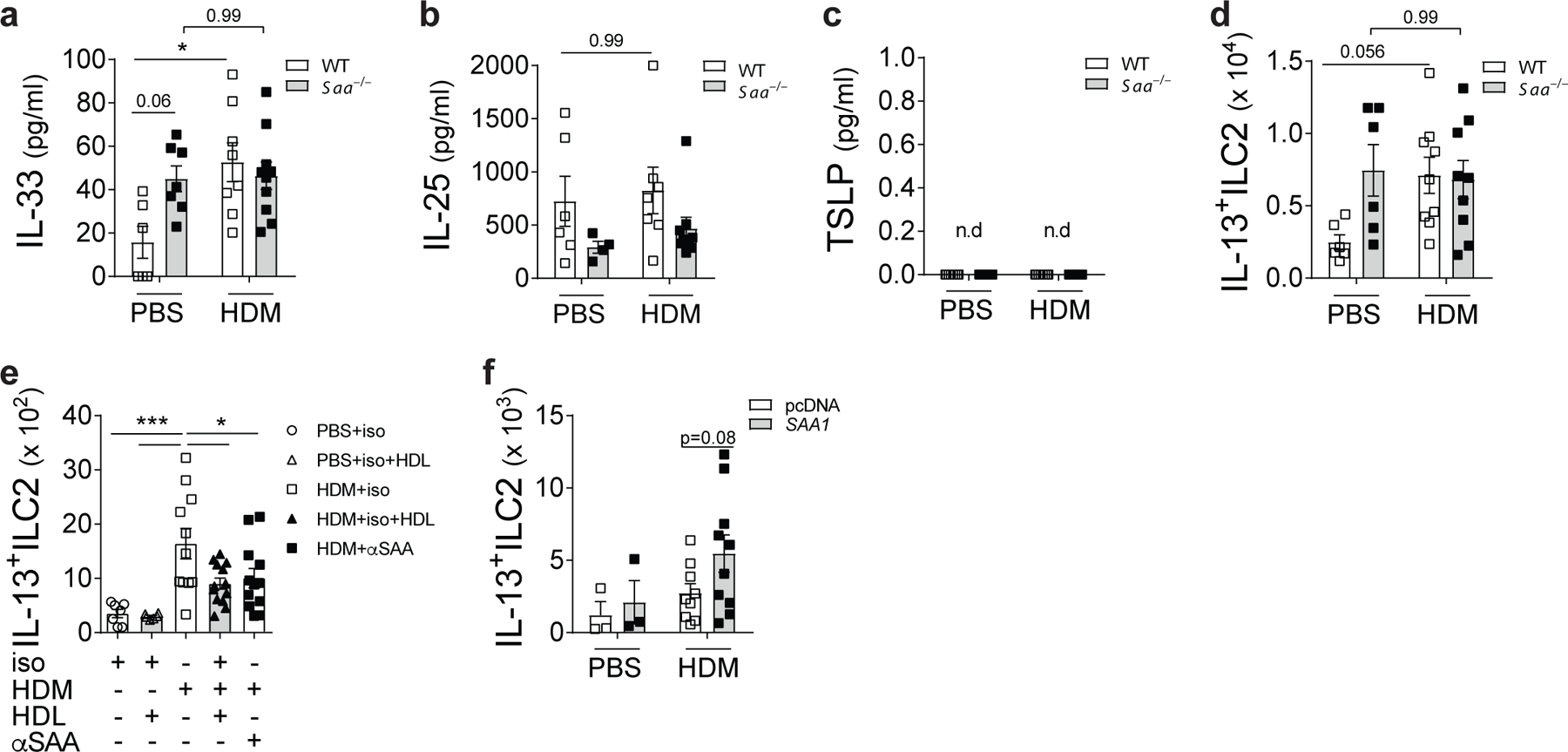
Increase of the type 2 cytokines (a) IL-33 (*P = 0.0138), (b) IL-25, and (c) TSLP in the BAL of WT and Saa–/– mice 24h after a single dose of PBS or HDM. (d) Numbers of IL-13+ ILC2 cells in the lungs of these mice. (e) Effects of local SAA1 neutralization in WT BALB/c mice through HDL (*P = 0.0117) or local SAA antibody blockade (αSAA, *P = 0.0392) of WT BALB/c receiving isotype (iso), HDM+isotype (HDM+iso), or HDM+αSAAab-treated (HDM+αSAA). (f) Effects of SAA1 overexpression on numbers of Lin-CD45+CD25+ST2+IL-13+ ILC2s in the lungs. For overexpression, WT mice were injected retro-orbitally with a SAA1 overexpression plasmid (SAA1, grey bars) or a non-coding control vector (pcDNA, open bars). Data represent means ± SEM of pooled data from 2 independent experiments containing (a-c) n= 6 WT PBS, n=8 WT HDM, n=7 Saa–/– PBS and n=10 Saa–/– HDM animals per group; (d) n= 5 WT PBS, n=9 WT HDM, n=6 Saa–/– PBS and n=9 Saa–/– HDM animals per group; (e) n=7 PBS+iso, n=6 PBS+iso/HDL, n= 11 HDM+iso, n=13 HDM+iso/HDL, n=12 HDM+αSAA animals per group; (f) n=3 pcDNA PBS, n=9 pcDNA HDM, n=3 SAA1 PBS and n=10 SAA1 HDM animals per group. P values were calculated with a two-tailed test using one-way analysis of variance (ANOVA) with Dunett’s post hoc analysis (a-e) or two-sided Student’s t-test (f). ***P ≤ 0.001
SAA1 is a sPRR for fatty acid-binding proteins.
In the lung, SAA is predominantly synthesized by the airway epithelium6 and secreted into the alveolar space5, where it has important immune-protective functions13, 27. A functional interaction of SAA1 has been described for the bacterial ligand outer membrane protein A (OmpA), facilitating clearance of Gram-negative bacteria9. Notably, the β-barrel structure of OmpA is related to those of lipocalins28, a protein family that includes the highly conserved arthropod group 13 allergens of FABPs29. Thus, we hypothesized that, given the structural homology to OmpA, epithelial-derived SAA1 may function as a secreted sensor for mite FABPs that activates epithelial effector functions.
To evaluate a potential interaction between SAA1 and HDM allergens, HDM extract was blotted following SDS-PAGE and probed with recombinant human SAA1 (rSAA1) and a SAA-specific monoclonal antibody . This analysis revealed an immunoreactive band of approximately 15 kDa, the reported molecular weight of Der p 13, the HDM FABP (Fig. 3a, HDM). Owing to their high structural similarities and sequence identity (> 80%), we used recombinant Blo t 13 (rBlot 13) from the tropical mite Blomia tropicalis as a representative of the mite FABP family in subsequent experiments. SAA was able to interact with rBlo t 13, (14.8 kDa), but not with the other major HDM allergens (rDer p 2 and rDer p 23) that have a similar migratory behavior in SDS-PAGE (15.9 and 14 kDa, respectively; Fig. 3a). To confirm a direct SAA1-mite FABP interaction, we used native PAGE. Binding of other proteins alters the electrophoretic mobility of the protein that depends on both the protein’s charge and hydrodynamic size. In the presence of Blo t 13, SAA1 shifted to higher masses and a SAA1 species that overlapped with Blo t 13 was detected by anti-SAA1 in a concentration-dependent manner, suggesting co-migration of SAA1 with Blo t 13 and the formation of SAA1-Blo t 13 complexes (Fig. 3b). Similarly, using sequence-specific antisera against the C-terminal tail of human SAA1 (aa 89–104)30, altered electrophoretic mobility and stronger binding of the antisera was detected when Blo t 13 was present, despite similar loading (Extended Data Fig. 5a). This result suggests that SAA1 ligand binding might lead to an altered conformation of the protein, making the C-terminal tail more available for antibody binding. Also, when chemically cross-linked by glutaraldehyde, which offers a direct method of identifying transient and stable protein interactions, the formation of SAA1 dimers and lower molecular weight oligomers was reduced by increasing amounts of Blot 13 (Fig. 3c and Supplementary table 1). No higher molecular weight complexes of SAA1/Blo t 13 could be detected in this approach.
Fig. 3: SAA1 is a soluble pattern recognition molecule for mite fatty acid binding proteins.
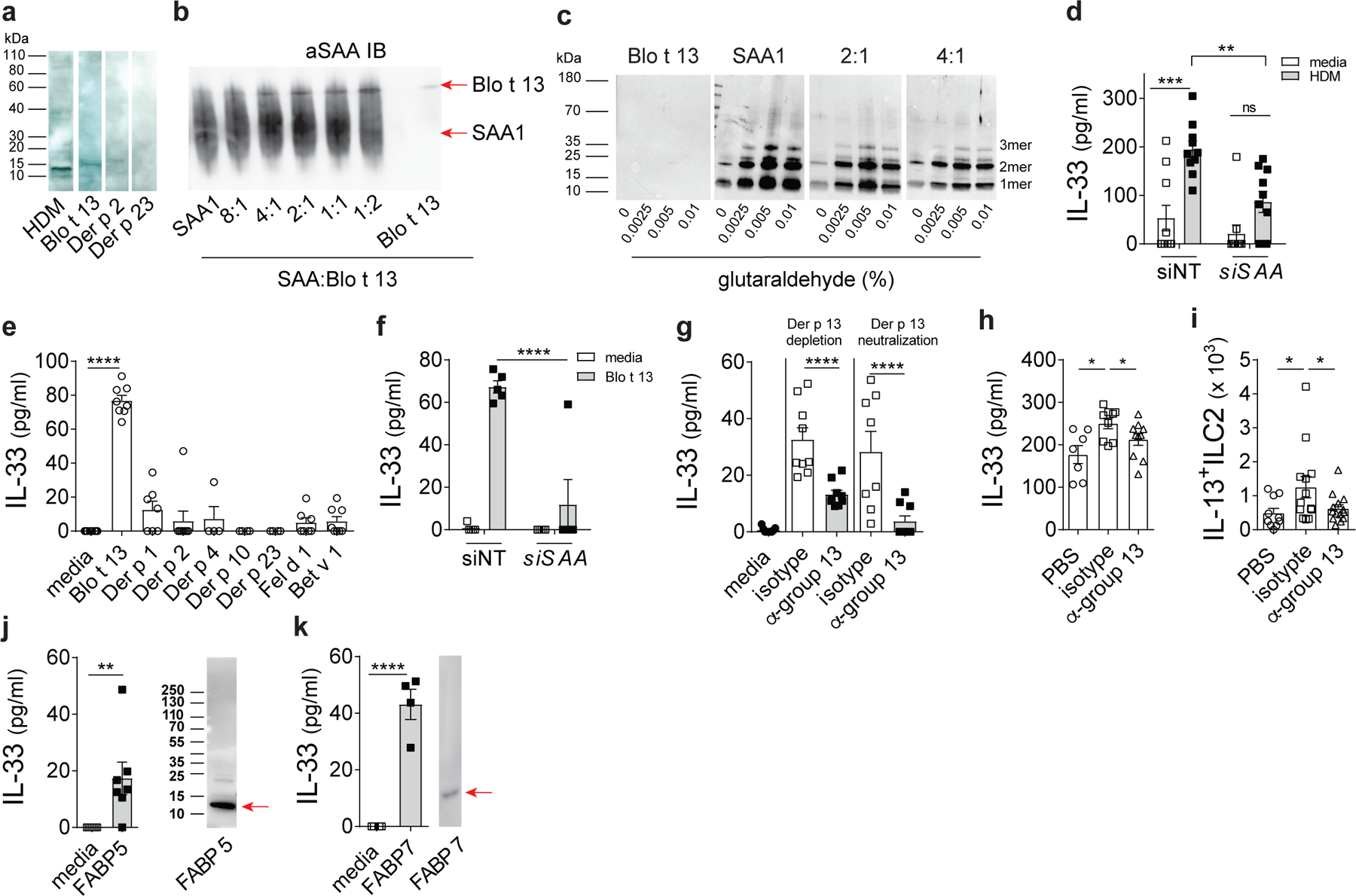
(a) HDM, rBlo t 13, rDer p2 or rDer p 23 were separated by SDS-PAGE followed by detection with rSAA1 and a mouse monoclonal antibody specific for human SAA1. (b) Migration of SAA1 (1 mg/ml) in PBS was analyzed in the presence of increasing amounts of the mite FABP Blo t 13 (4:1 (0.25 mg/ml), 2:1 (0.5 mg/ml), 1:1 (1 mg/ml) and 1:2 (2 mg/ml) of Blo t 13) by native PAGE followed by immunoblot analysis using mouse monoclonal antibody specific for human SAA1 (R&D systems; MAB30196). (c) SAA1 (1 mg/ml) was chemically cross-linked in the presence of Blo t 13 added at a ratio of 1:2 or 1:4 (as indicated) using 0.0025%, 0.005%, and 0.01% glutaraldehyde and analysed by immunoblot using sequence-specific (amino acid 14–30) rabbit antiserum for human SAA1 (densitometric analysis shown in supplementary table 1). (d) HDM-induced IL-33 concentrations in BEAS-2B cells after siRNA-mediated silencing of SAA1 (siSAA1) or non-targeting scrambled siRNA (siNT) (**P = 0.0002, ns = 0.1911). (e) IL-33 amounts induced by individual mite or unrelated major cat (Fel d 1) and birch pollen (Bet v 1) allergens (10 μg/ml) (. (f) Blo t 13-induced IL-33 in BEAS-2B cells with siRNA-mediated silencing of SAA1 (siSAA1) or transfected with non-targeting scrambled siRNA (siNT). (g) IL-33 amounts induced by Der p 13-depleted HDM extract and HDM extract where Der p 13 was neutralized (100 μg/ml). (h) BAL IL-33 levels (*P = 0.0209, ***P = 0.006) as well as (i) Lin-CD45+ST2+IL-13+ ILC2s in the lungs of WT BALB/c mice receiving a single i.t. challenge with 100 μg Der p 13-depleted HDM extract as compared to isotype-treated control extract (*P = 0.0423). IL-33 amounts induced in the BEAS-2B cell line and SAA1 binding by the human fatty acid binding proteins (j) FABP5 (**P = 0.0207) and (k) FABP7. Cropped images are shown. Data are shown as means ± SEM and are pooled data from 2 (d, e, g, j) or 3 (h, i) independent experiments or representative of 2 (k) or 3 independent experiments (f) each containing at least n=4 biologically independent samples or n=8 PBS, n=12 HDM-isotype and n= 14 HDM-α-group 13 (h) and or n=10 PBS, n=13 HDM-isotype and n= 14 HDM-α-group 13 (i) animals per group. Immunoblots are representative of an experimental n=2 (a-c, j, k). P values were calculated with a two-tailed test using one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test (d, e, f), Dunett’s (h) or Holm-Sidaks (i) post hoc analysis, and two-tailed Student’s t-test with Welch’s correction (g, j, k) ****P ≤ 0.0001.
To determine whether SAA1 and FABP-SAA1 interactions were necessary for HDM-driven epithelial IL-33 release, we analyzed IL-33 secretion in the human airway epithelial cell line BEAS-2B. SiRNA-targeted SAA1 knockdown in BEAS-2B cells significantly impaired HDM-triggered IL-33 release (Fig. 3d), which was not associated with de novo IL-33 protein synthesis or a loss of cell viability (Extended Data Fig. 5b,c). Other HDM-induced mediators such as IL-6 and IL-8 remained unchanged by loss of SAA1 expression (Extended Data Fig. 5d-f). ndand consistent with the ability to interact with SAA1, only Blot 13 out of several major and minor mite allergens tested, induced IL-33 release in BEAS-2B cells which was dependent on SAA1 protein (Fig. 3e,f). In addition, the major allergens from cat dander (Fel d 1) and birch pollen (Bet v 1) failed to induce IL-33 release (Fig. 3e). HDM is a complex mixture of many allergenic proteins, but not all might have IL-33-releasing ability. To more directly test the HDM FABP homologue, Der p 13, and its individual contribution to HDM-induced IL-33 release we utilized two complementary approaches. Monoclonal antibody-mediated neutralization or depletion of Der p 13 in HDM extract decreased IL-33 to near baseline amounts in BEAS 2B cells (Fig. 3g) but did not affect other HDM-induced cytokines such as IL-6 (Extended Data Fig. 5g). Similarly, BALB/c mice challenged with Der p 13-depleted HDM extract showed significantly reduced BAL IL-33 concentrations and lower numbers of IL-13+ ILC2 in the lungs as compared to mice receiving isotype-treated extract (Fig. 3h,i). Der p 13 not only contributed to acute airway inflammation, but also critically modulated the magnitude of the allergic phenotype in the full asthma protocol (Extended Data Fig. 6). These results show that SAA1 was necessary for HDM-driven epithelial IL-33. Blo t 13/Der p 13 were sufficient to recapitulate the effects of HDM on IL-33 release which supports our hypothesis that arthropod group 13 allergens are sensed by the SAA1 innate immune recognition pathways to induce a strong type 2 response at mucosal surfaces.
Conserved surface residues are not only shared among mite homologues but also other FABPs, e.g. from humans, which have received attention lately as potential regulators of allergic airway inflammation31. Like group 13 allergens, human epidermal and brain FABPs, FABP5 and FABP7, respectively, triggered IL-33 release in BEAS-2B cells and were recognized by SAA1 in immunoblots (Fig. 3j,k). Cytoplasmic FABPs have been also identified in excretory/secretory products of a variety of parasites including Schistosoma mansoni (S. mansoni)32. The adult stages of S. mansoni release FABP-containing exosome-like particles into host tissues to acquire essential fatty acids for their survival33. We found that Saa–/– mice showed impaired lung inflammatory responses following exposure to an adult worm-derived extract containing the S. mansoni FABP, Sm1432 (Extended Data Fig. 1f), associated with significantly lower numbers of CD11b+ DCs in the lungs, reduced effector T cell expansion and impaired TH cytokine secretion (Fig. 4). Our data indicate specificity of SAA1 for FABPs and provide evidence of a broader context of the SAA1-FABP interaction in regulating IL-33 release which might have implications for other chronic inflammatory diseases associated with increased SAA1 concentrations or helminth infections.
Fig. 4: Schistsoma mansoni worm extract-triggered TH2 skewing is impaired in Saa–/– mice.
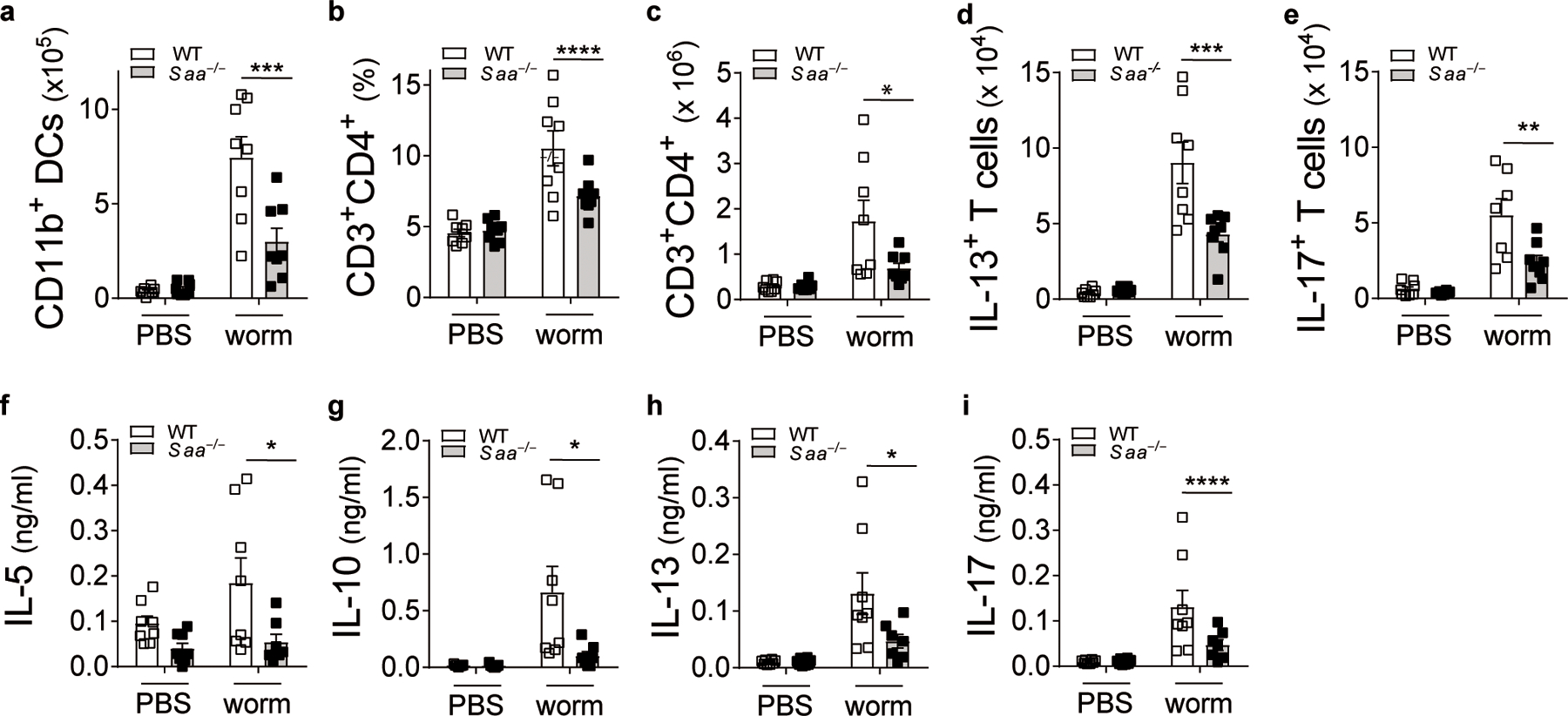
The parasitic worm Schistosoma mansoni (a Puerto Rican isolate) routinely maintained in the laboratory by cycling between the intermediate snail host, Biomphalaria glabrata, and outbred ICR mice as definitive hosts. (a) Numbers of CD11b+ DCs in the lungs of PBS or Schistosoma mansoni worm extract-treated WT and Saa–/– C57BL/6 mice (***P = 0.0002). (b) Frequency and (c) total numbers of CD3+CD4+ T cells (*P = 0.014). (d) TH2 (***P = 0.0003) and (e) TH17 (**P = 0.0014) cells in the lungs of these mice. (f-i) Cytokine production from worm extract-restimulated lung cells (IL-5: *P = 0.019, IL-10: *P = 0.011, IL-13: *P = 0.017, IL-17: *P = 0.017). Data represents means ± SEM of pooled data from 2 independent experiments containing a total of n= 8 animals in each group. P values were calculated with a two-tailed test using one-way analysis of variance (ANOVA) with Dunett’s post hoc that compares WT worm to counterparts. ****P ≤ 0.0001.
SAA1 dissociation initiates the release of IL-33 from airway epithelial cells.
SAA1 can adopt multiple conformations (monomer, dimer, hexamer) through a number of hydrogen bonding interactions between the C-terminal tail (aa 89–104) and the α-helix bundles15 (Fig. 5a). These interacting residues contribute to the overall protein stability and generate numerous surface structures to bind antigenic ligands and cell surface receptors. Hence, we hypothesized that the structure of SAA1 might be related to its ability to interact with allergens and drive type 2 responses.
Fig. 5: HDM-induced IL-33 is dependent on SAA1 dissociation.

(a) Sequence of SAA1. Indicated are: secondary structure α-helices (α 1–4) and loops (connecting lines between α-helices), C-terminal tail (CTL). Amino acids located within the hydrophobic core of the SAA1 hexamer are shaded in green. The epitopes recognized by affinity-purified IgGs raised against residues 27–44, 40–63, 68–84, and 89–104 are indicated by yellow lines (used in panel e). The mutation site at the hydrophobic core is indicated by an orange dot (used in panel f). C-terminal deletion of amino acids (Δ1–11) is shaded in blue. (b) BEAS-2B supernatants were cleared of lipid-bound SAA1 pulling down HDL-bound SAA1 using a polyclonal goat anti-ApoA1 antibody. Cleared supernatants were immunoblotted with a monoclonal mouse anti-human SAA1 antibody (Acris). (c) IL-33 secretion induced by rSAA1 alone (open bars) or in complex with a mouse monoclonal antibody specific for human SAA1 (αSAA ab, closed bars) in the BEAS-2B cell line. (d) Supernatants of cells left either untreated (media) or stimulated with recombinant SAA1 (rSAA) and immunoblotted with a monoclonal antibody specific for human SAA (R&D Systems; MAB30196). (e) IL-33 release from BEAS-2B cells after incubation with sequence-specific rabbit antisera specific for human SAA1 (**P = 0.0029). (f) HDM-triggered IL-33 release in BEAS-2B cells transfected with empty plasmid control (EV), wildtype SAA1 (WT) overexpression plasmid or a SAA1 plasmid with a Trp to Ala mutation at position 53 of the amino acid sequence (W53A) to mutate the hydrophobic core of SAA1 (WT to EV **P = 0.0032; WT to W53A **P = 0.0085). (g) HDM-induced IL-33 amounts in epithelial cells grown in media supplemented with charcoal-stripped FBS (**P = 0.0027). Cropped images are shown. Data shown as means ± SEM represent are representative of 2 independent experiments (c, d, f, g) or 3 (e) each containing at least n=4 biologically independent samples. Blots are representative of 2 (d) and 4 (b) independent experiments. P values were calculated with a two-tailed test using one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test (e, g) or Dunett’s post hoc analysis (f). ****P ≤ 0.0001
Indeed, we found that HDM-triggered SAA1 functions were not regulated by de novo protein translation and/or secretion (Extended Data Fig. 7a,b). Rather, untreated BEAS-2B cells secreted SAA1 as a lipid-free oligomer of a calculated molecular weight between 60 and 80 kDa into the supernatant (Fig. 5b, media) that likely corresponds to a SAA1 hexamer as described previously15, 16. Within minutes after HDM treatment, hexameric SAA1 dissociated into dimers and monomers (Fig. 5b, HDM), suggesting that IL-33 release is associated with SAA1 dissociation. Similarly, the mite FABP Blo t 13 triggered a concentration-dependent decrease in SAA1 hexamer that was associated with increasing IL-33 concentrations (Extended Data Fig. 7c-e). Also, when using lipid-free rSAA1, increased IL-33 concentration could only be detected in the presence of dissociated forms of SAA1. While SAA1 at 0.1–1 µg/ml exclusively separated as a hexamer and induced only very low amounts of IL-33 (Fig. 5c,d), higher SAA1 concentrations allowed the recovery of dimers and monomers along with increased IL-33 concentrations. In this setting, SAA1 hexamer might be stabilized by binding of lipids (e.g. retinoic acid) contained in the cell culture media34. Increasing the SAA1 concentration, however, could limit the availability of stabilizing lipids, resulting in hexamer dissociation and the exposure of IL-33-triggering epitopes. Complexing rSAA1 to anti-SAA synergistically enhanced the release of IL-33, suggesting that binding might favor SAA1 monomers by preventing formation of lipid-stabilized hexamers. This indicates a structure-function relationship of SAA1 that depends on ligand interaction to activate its type 2-promoting effects. Differences in the quaternary structure might thus also help explain the pleiotropic physiological functions of SAA1 and the presence of multiple SAA receptors12.
Notably, antisera raised against amino acids 40–63 and 68–84 of SAA130 induced a robust IL-33 release from BEAS-2B cells (Fig. 5e). These surface patches are involved in SAA hexamerization and the binding of lipids via the hydrophobic pocket15, 16, 34, that contribute to the overall stability of hexameric SAA. Perturbing this stability through binding of FABPs might facilitate SAA1 dissociation and the amplification of IL-33 release. Consistent with this assumption, introducing a Trp53Ala (W53A) mutation in the hydrophobic core of hSAA1 resulted in significantly higher IL-33 amounts after HDM stimulation in cells overexpressing SAA1-W53A as compared to wild-type SAA1 (Fig. 5f). Similarly, epithelial cells that had been grown in media depleted of lipophilic molecules34, showed higher IL-33 release in response to HDM (Fig. 5g) compared to cells maintained in non-depleted media. These findings provide a potential structural basis for the type 2 immunity-promoting effects of SAA1.
SAA1 signals via FPR2 to induce type 2 immune responses.
We next asked which of the commonly known SAA1 receptors (FPR2, TLR4, CD36, P2YR) were involved in mediating IL-33 release. Both FPR2 and TLR4 significantly contributed to the HDM-induced IL-33 release (Fig. 6a,b). However, when BEAS-2B cells overexpressed SAA1, blocking of TLR4 with lipopolysaccharide (LPS) from Rhodobacter sphaeroides (LPS-RS) could no longer suppress IL-33 release, while HDM-induced IL-33 was still inhibited by WRW4 (71.5 ± 34.4% inhibition P < 0.0001), a selective and potent antagonist of FPR2 (Fig. 6c). Moreover, targeting CD36 or P2Y receptors had no inhibitory effect on HDM-triggered IL-33 secretion from BEAS-2B cells, while inhibition of TLR2 induced only a marginal reduction in IL-33 (Fig. 6d-f). Consistent with these findings, when we overexpressed an N-terminal truncated form of SAA1 (Δ1–11) that shows reduced avidity for FPR235, the IL-33-enhancing capability of wild-type SAA1 was lost (Fig. 6g). These results suggested that FPR2 is the dominant receptor mediating SAA1-induced IL-33 release in the airway epithelium.
Fig. 6: SAA1 signals via the FPR2 receptor to induce IL-33.

IL-33 concentrations in BEAS-2B cells blocking the SAA-binding receptors (a) FPR2 (WRW4, 12 μM; ***P = 0.0006) and (b) TLR4 (LPS from Rhodobacter sphaeroides; LPS-RS, 10 μg/ml; ). (c) Effects of FPR2 or TLR4 blockade in cells transfected with empty plasmid control (EV) or SAA1 overexpression plasmid. **P = 0.0047 § indicates p = 0.056; # indicates p = 0.22 between EV and SAA plasmid. Blockade of (d) CD36 (anti-human CD36 blocking antibody, aCD36; 10 μg/ml; ***P = 0.0005), (e) the P2 receptor antagonist suramin (100 μM; *P = 0.028), and (f) TLR2 (anti-human TLR2 IgA, aTLR2, 10 μg/ml; **P = 0.0055). (g) HDM-triggered IL-33 release in BEAS-2B cells transfected with EV, WT SAA1 overexpression plasmid or a SAA1 plasmid with a deletion of the C-terminal amino acids 1–11 (Δ1–11; *P = 0.01, **P = 0.0078 ). Data are representative of 2–3 independent experiments (b, d-f) or pooled data from 2 independent experiments (a, c, d, g) each containing at least n=4 biologically independent samples and depicted as means ± SEM. P values were calculated with a two-tailed test using one-way analysis of variance (ANOVA) with Dunett’s (a, b, d-g) or Tukey’s (c) post hoc analysis.****P ≤ 0.0001.
Three closely related FPR isoforms have been identified in humans: FPR1–3. While FPR1 and FPR2 showed comparable mRNA expression in BEAS-2B cells after HDM stimulation, FPR3 mRNA abundance was very low (Extended Data Fig. 8a). Its predominant intracellular distribution36 also makes FPR3 an unlikely target for HDM-induced IL-33 release. While FPR1 overexpression significantly decreased IL-33 release after HDM exposure, FPR2 overexpression enhanced IL-33 release as expected (Extended Data Fig. 8c,b). This suggests that overexpression of FPR1 could override type 2 immunity-promoting signals delivered through FPR237. The effects of FPR2 gain or loss of function were highly selective for IL-33, as the concentrations of other HDM-triggered cytokines such as IL-6 or IL-8 were either not or only minimally modulated (Extended Data Fig. 8d-g).
To further demonstrate that the SAA1-FPR2 axis is critical for HDM-induced responses, we blocked FPR2 signaling in vivo in BALB/c mice using WRW4. Similar to SAA1 deficiency, local delivery of WRW4 (Extended Data Fig. 1g,h) improved the pulmonary function in WRW4-treated mice, which was accompanied by reduced HDM-induced IgE synthesis, diminished eosinophilic lung inflammation and mucus production concomitant with decreased HDM-induced Il5 and Il13 mRNA expression in WRW4-treated mice (Fig. 7a-f). WRW4-treated mice also showed significantly reduced IL-33 BAL concentrations, reduced lung IL-13+ ILC2 numbers and IL-13 released into the BAL (Fig. 7g-i) in an innate model of HDM exposure as compared to HDM only treated mice. Importantly, the SAA1-FPR2 axis seemed to be specific for the recognition of allergenic FABPs, as an extract of Alternaria alternata (Alt a), a mold causing respiratory allergy, had no effect on SAA1 hexamer dissociation in epithelial cell culture supernatants and the Alt a-triggered IL-33 release occurred independently of the SAA1-FPR2 signaling axis (Extended Data Fig. 9a-e). Supporting this observation, Saa deficiency had no effect on Alt a-induced type 2 responses in vivo (Extended Data Fig. 1i and Extended Data Fig. 9f-j). These data are consistent with recent findings that the IL-33 releasing capacity of Alt a is due to its serine proteolytic cleavage of the protease-activated receptor 2 leading to ATP signaling38.
Fig. 7: FPR2 axis controls sensitization to HDM by regulating IL-33-mediated ILC2 activation and TH2 cytokine production.
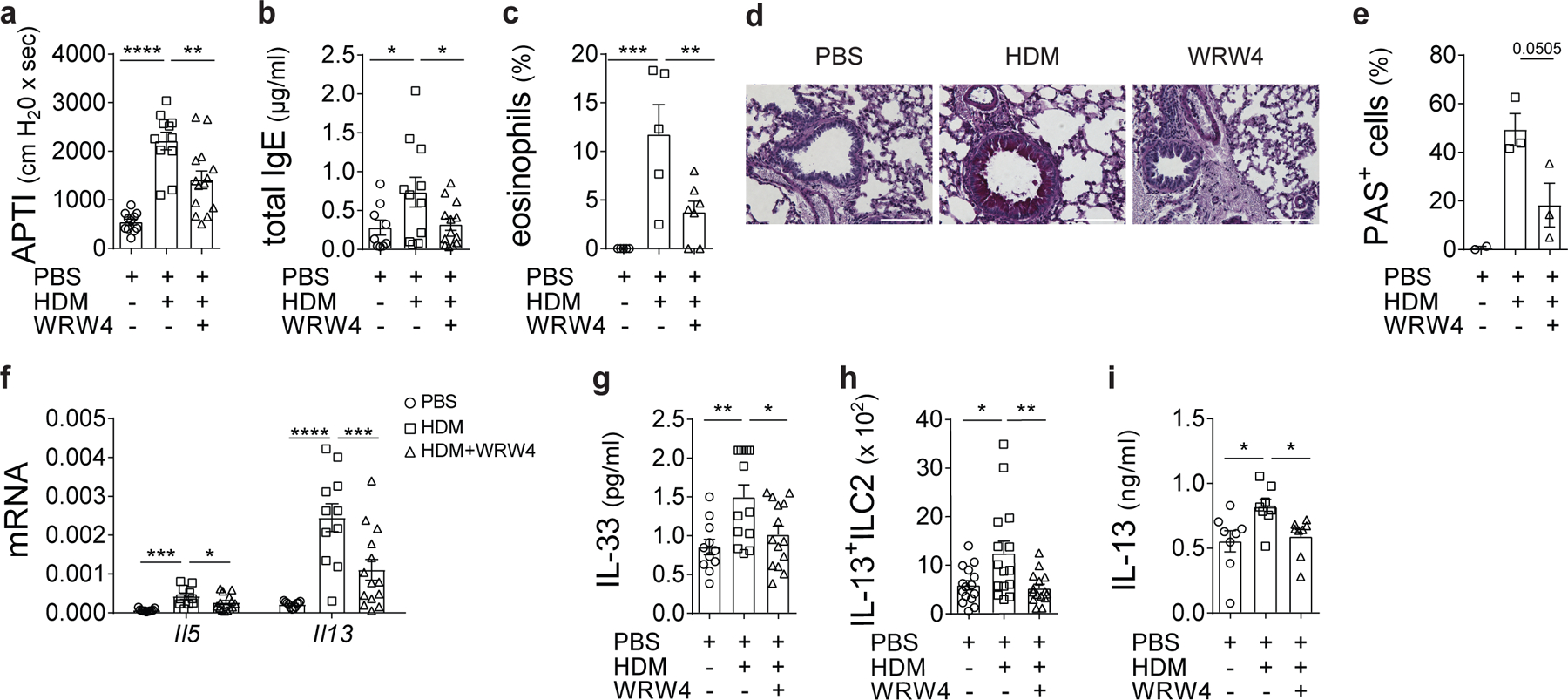
(a) Airway responses to cholinergic stimulation shown as airway pressure over time (APTI) measured on day 17 in PBS, HDM, or HDM+WRW4-treated (4 mg/kg) WT BALB/c mice (**P = 0.0022). (b) Total IgE serum concentrations of treated mice (HDM to PBS **P = 0.0483; HDM to WRW4 **P = 0.0486). (c) Eosinophilic infiltration into the lungs (**P = 0.0094 and ***P = 0.0009). (d and e) Histological examination of airway inflammation. Sections were stained for mucus production with periodic acid Schiff (PAS; Scale bars 100 μm). (f) Lung expression of Il5 (*P = 0.0421 and ***P = 0.0029) and Il13 (***P = 0.0009) mRNA. (g) BAL IL-33 (*P = 0.0175 and **P = 0.0009) and (h) Frequency of Lin-CD45+ST2+IL-13+ ILC2s (*P = 0.0125 and **P = 0.0058) and (i) BAL IL-13 (HDM to PBS *P = 0.0169; HDM to WRW4 *P = 0.0398) in the lung 24 h after a single i.t. challenge with PBS, HDM (100 μg), or HDM in combination with the FPR2 inhibitory peptide WRW4 (4 mg/kg). Data represents means ± SEM of pooled data from 2 experiments containing (a, b, f) n=11 PBS, n=11 HDM, n=14 HDM+WRW4 animals per group; (d, e) n=2 PBS, n=3 HDM, n=3 HDM+WRW4 animals per group; (g) n=11 PBS, n=13 HDM, n=14 HDM+WRW4 animals per group; (h) n=15 PBS, n=15 HDM, n=15 HDM+WRW4 animals per group; or are representative of 2 (c) or 3 (i) independent experiments with (c) n=5 PBS, n=5 HDM, n=7 HDM+WRW4 animals per group and (i) n=8 animals in each group, respectively. P values were calculated with a two-tailed test using one-way analysis of variance (ANOVA) with Dunett’s post hoc (a-c, e, g-i) or Holm-Sidak’s (f) post hoc analysis that compares HDM to PBS and WRW4 counterparts. ****P ≤ 0.0001.
Dysregulated SAA-FPR2 axis in type 2 allergic diseases in humans.
To test whether the SAA1-FPR2 axis was dysregulated in primary human tissue, we examined expression of SAA1 and FPR2 in primary nasal epithelial cells from chronic rhinosinusitis (CRS) patients, who are commonly sensitized to house dust mites39. We found highly elevated SAA1 and FPR2 expression in epithelial cells from individuals with CRS (Fig. 8a, b and Supplementary Table 2) as compared to cells from controls. Upon HDM stimulation, CRS donor nasal epithelial cells (Supplementary Table 3) showed significantly increased IL-33 secretion along with a more robust SAA1 hexamer dissociation as compared to control donor ECs (Fig. 8c-e). Together these data suggested that dysregulated IL-33 release in patients with CRS might result from aberrant SAA1 dissociation. A similar trend of upregulated SAA1/FPR2 expression was observed in primary bronchial epithelial cells from asthmatics (Extended Data Fig. 10a,b and Supplementary Table 4). Also, concentrations of circulating SAA1 were higher in the serum of HDM allergic patients (Fig. 8f and Supplementary Table 5). These results support a role for a dysregulated SAA1-FPR2 axis in allergic diseases.
Fig. 8: Epithelial SAA1 dysregulation in human type 2 immune responses.
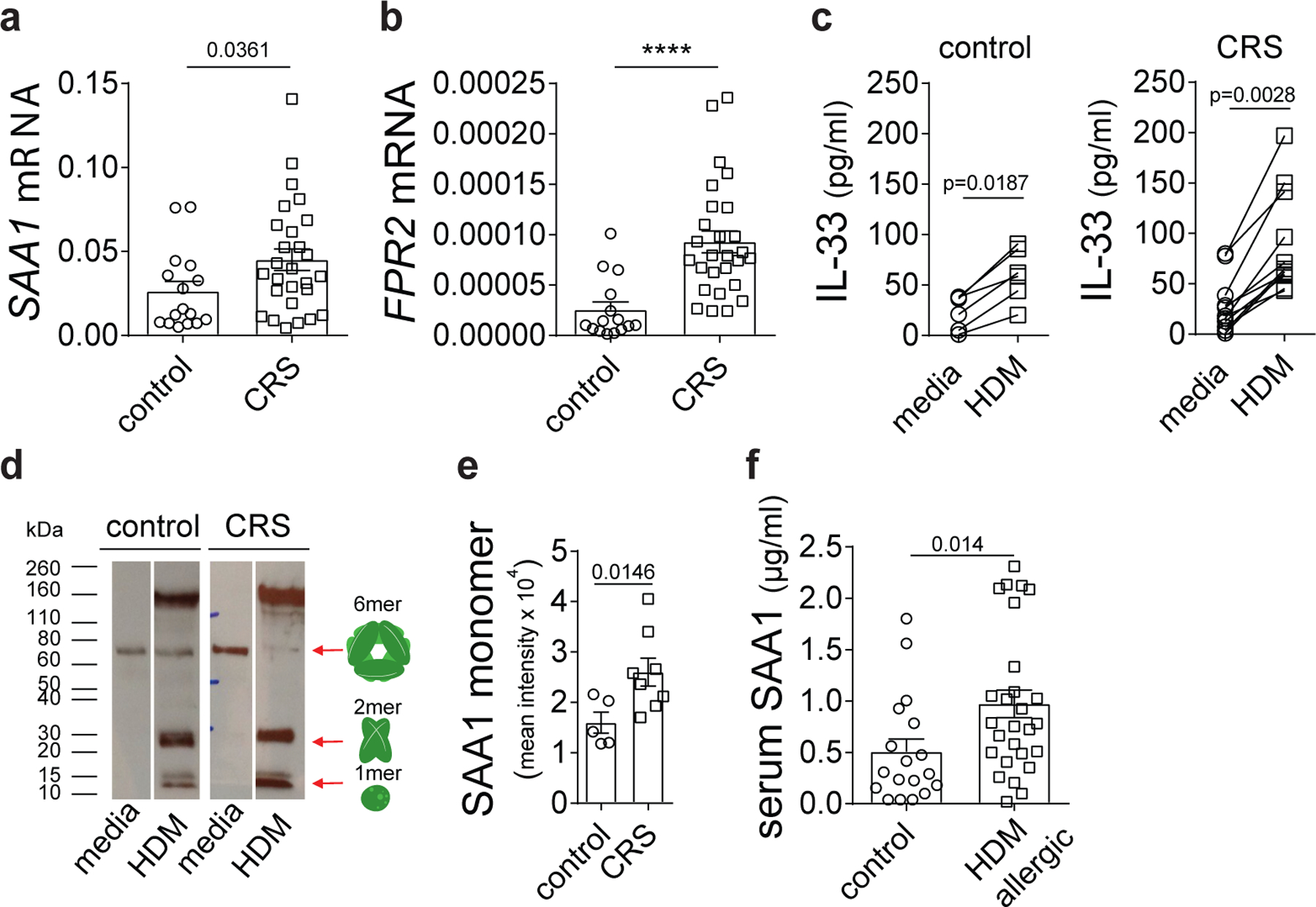
Basal (a) SAA1 and (b) FPR2 mRNA expression in nasal epithelial cells of CRS patients and matched control donors. (c) HDM (100 μg/ml)-triggered IL-33 concentration in primary nasal epithelial cells of CRS patients as compared to controls. (d) Dissociation of hexameric SAA was analyzed separating lipid-free and lipid-bound SAA by HDL pull down using a polyclonal goat antibody specific for human ApoA1 and immunoblotting with a monoclonal mouse antibody specific for human SAA1 (Acris). (e) Bar graph represents quantitative analysis of SAA monomer band intensities (LI-COR Image Studio Software). (f) SAA1 protein amounts in sera of control donors and HDM allergic individuals. Data represents means ± SEM of (a and b) n=16 control and n=27 CRS, (c) n=6 control and n=12 CRS patients per group, (e) n=5 control and n=8 CRS patients per group and (f) n=18 control and n=27 HDM allergic patients per group. Representative immunoblot of one control and one CRS patient (d). Cropped images are shown. P values were calculated with a two-tailed test using Student’s t-test with Welch’s correction. ****P ≤ 0.0001.
Discussion
Pattern recognition at mucosal surfaces is essential to maintain tissue homeostasis. However, when dysregulated, innate sensing of otherwise harmless environmental proteins causes an acute innate immune response favoring allergic sensitization and type 2 inflammation in the lungs3, 4. Given the different classes of allergens that can elicit allergic responses, multiple unique innate allergen recognition mechanisms have likely evolved for their detection.
Our studies identify a previously unrecognized mechanism of allergen sensing by SAA1 that initiates type 2 immunity through the release of IL-33 which is among the earliest cytokines secreted and has been reproducibly associated with asthma susceptibility in several GWAS studies. Our results using Saa–/– mice or blocking SAA1-triggered signaling pathways indicated that, in the absence of SAA, the early release of IL-33 and the first wave of ILC2-derived IL-13 in response to initial allergen sensitization was decreased compared with wild-type or control mice. These mice were also resistant to the development of AHR concomitant with significant reductions in all the hallmarks of allergic asthma. Although SAA1 is known to bind several receptors, we demonstrate that its interaction with FPR2 specifically, leads to airway epithelial IL-33 release, and the development of allergic inflammation in vivo. IL-33 also recruits and activates DCs that skew naïve CD4+ T cell differentiation towards a TH2 phenotype40, 41. The decreased frequency of CD11b+CD11c+MHCIIhi DCs in the lungs and impaired TH2 responses in the Saa–/– mice likely results from both impaired IL-33 activation of DCs and lack of ILC2-derived IL-13 to support proper induction and maintenance of TH2 responses41, 42. These are likely to be additive effects. In summary, our findings broaden our understanding of how certain aeroallergens such as mite FABPs are sensed by the airway epithelium and reveal a role for local SAA1 in the initiation and maintenance of type 2 immune responses acting upstream of the IL-33–ILC2–IL-13 axis.
SAA1 has pleiotropic effects on the immune system, illustrating the need for tight molecular regulation of this protein. In addition to liver-derived SAA, basal expression of SAA proteins has been detected in several mucosal tissues including the lung6. Although the exact mechanisms of SAA1 regulation at these tissue sites are not well understood, the ability of SAA1 to adopt different oligomeric states may have important functional implications for promoting local inflammation15, 16, 34, 43, 44. Our results show that SAA1 produced by the airway epithelium is not activated via upregulation of its expression or release, but through dust mite allergen-driven dissociation of ‘low activity’ SAA1 oligomers. This was associated with active, non-cytolytic IL-33 release from AECs. In support of this hypothesis and similar to other studies45, 46, elevated amounts of SAA1 per se were not sufficient to trigger type 2 immune responses (e.g., IL-33 release and higher numbers of ILC2) but the presence of HDM causing its dissociation was necessary to fully activate its type 2 immunity-promoting effects. Complexing rSAA1 to anti-SAA1, which likely maintains SAA1 in its dissociated form, greatly enhanced its IL-33 releasing activity. These data support our contention that only SAA1 present in its dissociated form is able to drive type 2 immunity.
Our finding that changes in the oligomeric organization of SAA1 are important to unveil its full bioactivity is consistent with our understanding of the need for tight control of other acute phase proteins found in high quantities in serum, such as complement components and pentraxin family members, like C-reactive protein (CRP). The mechanisms whereby SAA1 oligomers, which are relatively unstable in vitro16, 44, gain stability in vivo are likely heterogeneous. A common feature seems to be the association with small, nonpolar molecules via its hydrophobic pocket34. Previously, monomeric SAA1 was identified as a sPRR that can directly target Gram-negative bacteria through the bacterial ligand OmpA9. One could speculate that this ‘fatty acid binding sink’ evolved to inhibit pathogen growth at mucosal surfaces through sequestration of essential nutrients. Notably, the β-barrel structure of OmpA is related to the structurally conserved family of cytosolic FABPs that are present in a spectrum of species including humans and have also been found as allergens in arthropods (mite FABPs)29, 47. Indeed, when group 13 allergens were depleted from HDM extracts, the IL-33/ILC2-inducing activity of HDM was abolished. Consistent with our findings, other HDM allergens do not induce IL-33 protein release from epithelial cells, but might enhance this process by proteolytic processing of IL-33 into highly active, shorter mature forms24, 48. Therefore, mite FABPs appear to be the main regulators of IL-33 release and initiation of innate type 2 immunity at mucosal surfaces. The requirement for allergenic components such as mite FABPs likely explains why no enhancement of type 2 immunity was observed in the study by Ather et al.18 in which mice sensitized to ovalbumin (a non FABP protein) in the presence of rSAA showed elevations in IL-17A49. Similarly, in the context of colonization with single filamentous bacteria, SAA1 drives Th17 immune responses. SAA-induced Th17 responses have been linked to TLR2 activation and it is currently unknown whether SAA is dissociated in this scenario.
Our discovery that the lung type 2 immune response can be activated by sensing of allergenic FABPs might have implications beyond the context of asthma in the setting of tissue injury or helminth infection. While a FABP containing S. mansoni extract induced type 2 polarized immune responses in wild-type mice, Saa–/– mice were significantly protected. Helminth FABPs show the typical β-barrel fold previously resolved for other FABP homologues, with the Schistosoma FABP being partially similar to that of the mite FABP Blo t 1350, 51. While SAA1 sensing of parasitic or endogenous FABPs is likely advantageous to ward off attack by fellow parasites and initiate an immediate tissue repair process, we found that the SAA1-FPR2 pathway was chronically upregulated in respiratory epithelial cells from patients with asthma or CRS. HDM stimulation further enhanced this signaling pathway through increased SAA dissociation. This provides evidence that the elevated SAA amounts may be linked to HDM sensitization and the hyperactivated IL-33/ILC2 axis previously reported in these patients39, 52, 53. Interestingly, proteins encoded by different alleles of SAA1 differ in two amino acids within the ligand binding hydrophobic pocket34, which may contribute to variations in stability/biological activity and receptor specificity. In fact, SAA1 allelic isoforms have altered PRR selectivity54 suggesting that differential allele usage may explain the enhanced SAA1-IL-33 responses in the presence of HDM in allergic individuals.
Collectively, we report that, at mucosal surfaces, SAA1 exists in physiologically and functionally different states. Encounter with allergenic FAPBs such as the group 13 mite allergens triggers the dissociation of a ‘low activity’ SAA1 hexamer with pro-type 2 signaling capacity that initiate type 2 immune responses and asthma pathogenesis. This pathway that evolved to recognize either pathogens or endogenous danger signals released upon cellular stress or damage, may have additional clinical implications. Our results suggest that, in spite of the low specific IgE-binding frequency in allergic individuals, FABPs might have sustained and widely distributed lung inflammatory effects in exposed subjects. Initiation of type 2 immune responses is a critical first step in the development of an allergic response upstream of IgE production and allergen/IgE-binding. Recognition by PRRs and initiation of innate immune responses are similarly important properties of allergens that should be included as novel molecular biomarkers. This classification should help inform a more precise endotype-driven asthma treatment55 and provide the basis for the design of preventive and/or therapeutic anti-inflammatory agents to effectively block allergen recognition and the ensuing inflammatory cascade. The link between SAA1 activation and the IL-33–IL-13 axis may also provide insights into the underlying pathogenesis of a plethora of chronic inflammatory diseases in which SAA1 plays an important pathogenic role including atherosclerosis, rheumatoid arthritis, Alzheimer’s or Crohn’s disease.
Methods
Mice
BALB/cJ mice were originally obtained from Taconic and bred in-house at the Bloomberg School of Public Health animal facility. Saa–/– mice (lacking expression of SAA1.1 and its less abundant isoform, SAA2.110 on the C57BL/6 background were a kind gift from Maria C. deBeer (University of Kentucky Medical Center, KY, US). Mice were housed in a specific pathogen-free animal facility in micro-isolator cages. For all experiments sex and age-matched mice were used. All in vivo experiments were approved by the Institutional Animal Use and Care Committee at Johns Hopkins Bloomberg School of Public Health and the Medical University of Vienna (BMWFW-66.009/0380-WF/V/3b/2017). All mice used for Schistosoma mansoni egg isolation were maintained by certified personal (certificate number CZ 02627) in accredited laboratories of the Institute of Immunology and Microbiology of the First Faculty of Medicine, Charles University and the General University Hospital in Prague (accreditation number 8615/2019-MZE-17214). S. mansoni (a Puerto Rican isolate) is routinely maintained in the laboratory under approval number MSMT - 7063/2017–2 for S. mansoni laboratory life cycle maintenance by cycling between the intermediate snail host, Biomphalaria glabrata, and outbred ICR mice as definitive hosts according to protocols published previously56. Briefly, mice were euthanized six weeks post infection by an intra-peritoneal injection of ketamine (Narkamon 5% - 1.2 ml/kg body weight) and xylazine (Rometar 2% - 0.6 ml/kg body weight) containing heparin (200 U/ml). Mice were dissected by opening of the abdominal and thoracic cavity. Adult female and male worms were recovered from the mice by perfusion from via the hepatic portal vein. Collected worms were repeatedly washed in RPMI medium before homogenization. Extracts were sterile filtered before use.
Treatment protocols
Allergic airway inflammation was induced in 6–8-week-old wild-type C57BL/6 or Saa–/– C57BL/6 mice following allergen sensitization on day 0 with HDM, Alt a or worm extract (1 μg in 30 μl of PBS) intratracheally (i.t.). Mice, anesthetized using sevoflurane (AbbVie), were subsequently challenged intranasally (i.n.) with 10 μg of HDM, Alt a (or worm extract using in 30 μl of PBS on days 7–11 and mice were sacrificed on day 11. Control mice received PBS in all experiments at time of sensitization and challenge phase. In receptor blockade experiments, wild-type BALB/cJ mice were treated with HDM (100 μg) or HDM in combination with WRW4 (4 mg/kg mouse) or vehicle control (DMSO) in a total of 40 μl i.t. In early time point experiments, mice received a single HDM dose or HDM plus WRW4 and were sacrificed as indicated. For in vivo SAA1 neutralization, wild-type BALB/cJ mice were exposed i.t. to PBS, HDM (100 μg), or HDM in combination with a goat anti-mouse SAA1 antibody (R&D Systems, 5 μg/mouse) were sacrificed as indicated. Normal goat IgG was used as isotype control (BioXcell). For the in vivo overexpression of SAA1, a SAA1 expression plasmid was complexed to polyethylenimine (PEI; In vivo-JetPEI) and administered i.v. to mice. Successful transfection was monitored by qRT-PCR and secreted SAA1 protein in the bronchoalveolar lavage (BAL). Control mice were transfected with an empty vector control. Mice were exposed i.t. to PBS or HDM 48 h after initial installation of plasmid DNA.
Assessment of allergen-induced AHR
To evaluate airway responses, mice were anesthetized by intraperitoneal (i.p.) administration of ketamine (100 mg/kg)/xylazine (5 mg/kg), and paralyzed with decamethonium bromide (25 mg/kg; Sigma-Aldrich), intubated and respirated at a rate of 120 breaths/min with a constant tidal volume (0.2 ml) 72 h after final allergen challenge. Methacholine (30 mg/ml for BALB/cJ and 200 mg/ml for C57BL/6J; Sigma-Aldrich) was injected into the inferior vena cava or delivered by nebulization and dynamic airway pressure (cm H20 × sec) was followed for 5 min. Serum and BAL fluid were collected, processed, and analyzed as described previously57. In brief, blood was collected to measure total serum IgE concentrations. To collect BAL fluid, lungs were lavaged with Hank’s Balanced Salt Solution (HBSS, Invitrogen). Cells were recovered by centrifugation and total cells were counted (trypan blue exclusion). Slides were prepared and stained with Siemens Diff-Quik stain (Siemens Healthcare) for differential cell counts.
Lung cell isolation and in vitro culture
Murine lungs were excised, minced, and digested in serum-free RPMI containing Liberase CI (0.5 mg/ml, Roche) and DNase I (0.5 mg/ml, Sigma) at 37 °C for 45 min. Cells were washed with RPMI containing 10% (v/v) FBS and viable cells were counted by trypan blue exclusion. For HDM-specific restimulation, whole lung cells were cultured at 250,000 cells per well of a 96-well plate (250 μl final volume) with HDM (30 μg/ml) and cell culture supernatants were harvested for cytokine measurements 72 h later.
FACS analysis and cell sorting
All staining reactions were performed at 4°C following incubation with FcBlock (TruStain fcX antibody, BioLegend) for 30 min. Dead cells were excluded using the fixable viability dye Zombie Aqua (BioLegend). Monocytes (CD11bhiCD11c–Ly6G–Ly6Chi), neutrophils (CD11bhiLy6G+), CD11b+ DCs (CD11c+CD11b+Ly6G–MHChi), alveolar macrophages (CD11c+CD11bloCD317–Ly6C–SSChi), plasmacytoid dendritic cells (DC, CD11c+CD11b−CD317+Ly6C+), and CD103+ DCs (CD11c+CD11b–CD103+) were quantified using anti-CD11c-Alexa Fluor 700 (HL3; BD Pharmingen) or anti-CD11c-PE-Cy7 (HL3; BD Pharmingen), anti-CD11b-PE-Cy7 (M1/70; BD Pharmingen) or anti-CD11b-APC-Cy7 (M1/70; BioLegend), and anti-Ly6C-Pacific Blue (HK1.4, BioLegend), anti-Ly6G-APC-Cy7 (1A8, BioLegend), anti-MHCII-PE (M5/114.15.2, BioLegend), anti-CD103-PerCP-Cy5.5 (2E7), anti-CD317-APC (eBio129c). CD3e (145–2C11; BD Pharmingen) and CD4 (RM4–5; eBioscience) staining was used to identify T cells. For measurement of intracellular cytokines, cells were stimulated with phorbol 12-myristate 13-acetate (PMA; 100 ng/ml) and ionomycin (1 μg/ml) for 16 h at 37°C. Monensin and brefeldin A (eBiosciences) were added for the last 4 h of incubation. Cells were fixed, permeabilized and stained with antibodies against IL-13 (eBio13A).
For ILC2 flow, cells were plated at 2 × 106 cells/ml, restimulated with PMA/ionomycin overnight as described above before staining with PerCP-Cy5.5 or APC-Cy7-conjugated lineage marker monoclonal antibodies (FcεRI, MAR-1, BioLegend; CD3ε, 145–2C11, BD Pharmingen; TCRβ, H57–597, BD Pharmingen; B220, RA3–682, BD Pharmingen; CD49b, DX5, eBioscience; CD11b, M1/70, BD Pharmingen; CD11c, N418; BioLegend; Gr1, RB6–85C, BD Pharmingen; NK1.1, PK136, BioLegend), Alexa Fluor700-conjugated CD45 (30-F11; BD Pharmingen), BV605-conjugated CD90.2 (53–2.1, BD), APC-e780-conjugated CD25 (PC615, eBioscience), PE-Cy7-conjugated ICOS (C398.4A; BioLegend), BV421-conjugated IL-33Rα (DIH9, BioLegend) and FITC-conjugated T1/ST2 (DJ8, MD Bioproducts). After cell surface staining, cells were fixed, permeabilized and stained for intracellular IL-13-PE. Cells were analyzed using a LSRII flow cytometer (BD Biosciences) equipped with three laser lines (405 nm, 488 nm, and 633 nm). Compensation of spectral overlap and data analysis was performed using the FACSDiva software (BD Biosciences).
Primary human nasal ECs
Primary human nasal ECs were prepared as described previously58. The research protocol was approved through the Johns Hopkins Institutional Review process, and all subjects gave signed informed consent. Inclusion criteria included continuous symptoms of rhinosinusitis as defined by the AAO-HNS Chronic Rhinosinusitis Task Force for greater than 12 weeks, computed tomography of the sinuses revealing isolated or diffuse sinus mucosal thickening or air-fluid levels, and nasal polyps visible on diagnostic endoscopy58 Mucosal tissue removed during endoscopic sinus surgery was digested at 4 °C overnight in Ham’s F12 media containing 0.01% protease Sigma type XIV (Sigma) and supplemented with antibiotics (100 U/ml penicillin, 100 μg/ml streptomycin, 2.5 μg/ml amphotericin B, and 50 μg/ml gentamicin, Gibco). Digestion in protease was performed, with separation of cells by agitation the following day. Cells were then separated by straining, washed, and seeded at a density of ≥1.5 × 104 cells/cm2, onto Vitrogen 100-coated (1:75 in sterile water; Cohesion) P-100 dishes in bronchial epithelium growth medium (BEGM). ECs were then grown at 37°C for 24 h and washed with HBSS to remove debris. The media was then changed every 48 h until the cells reached confluence. Primary sinonasal ECs were grown in BEGM media. At 80% confluency, cells were trypsinized and seeded into a 96-well culture plate pre-coated with fibronectin-collagen. Cells were starved overnight in media without bovine pituitary extract at 90% confluency and treated with HDM (200 μg/ml) for 2 h the following day. The supernatants were collected for IL-33 ELISAs and cells for RNA isolation. The patients’ characteristics are listed in Supplementary Tables 2 and 3.
Bronchial EC culture
ECs were isolated from bronchial biopsies of healthy subjects, mild steroid naive asthmatics and severe asthmatic subjects (Supplementary Table 4). Subjects were recruited from the Asthma Clinic at Institut Universitaire de Cardiologie et de Pneumologie de Québec (Québec, QC, Canada). The ethics committee board approved the study and all subjects gave written informed consent. The asthmatic subjects were diagnosed according to the American Thoracic Society criteria. Asthmatic subjects were atopics, and had a positive skin prick test to common aeroallergens, methacholine PC20 of <8 mg/ml, and FEV1 >70% predicted, with only intermittent use of β2- agonists. Control subjects were non-atopics, non-smokers, with no history of asthma or airway and systemic diseases. None of the asthmatic subjects had an asthma exacerbation or respiratory tract infection in the preceding 4 weeks before entry into the study. Bronchial epithelial cells were characterized and cultured as previously described59, 60. RNA was extracted from near-confluent cultures.
HDM allergic patient sera
Approval from the ethics committee of the Medical University of Vienna was obtained for studies with human plasma and informed consent was obtained from the volunteer donors (EK1538/2014). Allergic individuals diagnosed with HDM allergy were included in this study. The diagnosis of allergy was based on a characteristic history of clinical symptoms (allergic conjunctivitis, rhinitis, urticaria, and asthma) and on at least one of the following diagnostic criteria: a positive skin prick test or a positive ImmunoCAP. The characteristics of the patients are summarized in Supplementary Table 5.
Proteins, expression constructs, and cell lines.
House dust mite (HDM, extracted from Dermatophagoides pteronyssinus) and Alternaria alternata (Alt a) extracts were from Greer (Greer Laboratories). The FPR2 selective agonist WRW4 was from Calbiochem (Calbiochem, Merck). LPS from Rhodobacter sphaeroides (LPS-RS) was from InVivoGen. Recombinant (r)FABP5 and rFABP7 were from Sino Biological, lipoxin A 4 (LXA4) from Cayman Chemical, and rBlo t 13 as well as a monoclonal antibody against Blo t 13 were produced as previously described47, 61. High-density lipoprotein (HDL, isolated from human plasma was from Sigma (Sigma-Aldrich). Human FPR2 (#SC322591, Origene) and human SAA1 (#RC202738, Origene); murine rFPR1 (#MR221675, Origene), pcDNA3.1 (Invitrogen, Thermo Scientific). hFPR2, hSAA1, and On-TARGET plus Non-targeting siRNA (Smart pool: ON-TARGETplus siRNA, GE Dharmacon,); BEAS-2B and Caco-2 cell line was from American Type Culture Collection (ATCC). BEAS-2B were maintained in complete media composed of BEBM base medium (Lonza) containing bovine pituitary extract (52 μg/ml), hydrocortisone (0.5 μg/ml), human endothelial growth factor (0.5 ng/ml), epinephrine (0.5 μg/ml), transferrin (10 μg/ml), insulin (5 μg/ml), retinoic acid (0.1 ng/ml), triiodothyronine (6.5 ng/ml), gentamycin (50 μg/ml) (BEGM SingleQuots; Lonza) on collagen/fibronectin (BD Biosciences) coated flasks in an atmosphere of 5% CO2 at 37 °C. Cells were sub-seeded at the second–third passage and used for subsequent experiments. Cell viability was determined by morphology and trypan blue exclusion. Fetal bovine serum albumin (FBS) and charcoal-stripped FBS were from Gibco (Gibco). Recombinant human SAA1 (rSAA1) was from Peprotech or produced in house. Codon optimized human SAA1 featuring a TEV cleavable N-terminal hexahistidine tag was expressed in BL21 DE3 star E. coli (Invitrogen). The culture was gown to an OD600 of 0.6 at 37°C in terrific broth (Carl Roth GmbH + Co. KG) shaking at 180 rpm before setting the incubator to 16°C and allowing the culture to cool and grow for another 45 min. Expression was induced by adding IPTG at a final concentration of 1 mM and left overnight. The protein was purified by nickel affinity chromatography from the soluble fraction before incubation with 1% (m/m) TEV protease at 4°C overnight to remove the purification tag. A second pass through on an IMAC column was performed to remove uncleaved SAA1, tags and the His-tagged TEV protease, before a final polishing step of size exclusion chromatography on an S200 Increase 10/300 (GE Healthcare Life Science) into PBS was performed.
Epithelial cell (EC) stimulation and transfections
For stimulation with allergen extracts, 3 × 104 BEAS-2B ECs were seeded per well of a 96-well plate in complete medium and grown until they reached confluence. For plasmid-directed overexpression studies, 1 × 104 BEAS-2B cells were transfected with 0.1 μg plasmid DNA per well coding for human FPR2 (hFPR2) and hSAA1, or empty pcDNA3.1 vector using FuGENE HD (Roche) for 48 h. The SAA1-W71A mutant was generated using the QuickChange II site-directed mutagenesis kit (Agilent Technologies). Oligonucleotides (F 5′- GGG TGT CAG CAG CGA TGG GGC TCG GG −3′) and (R 5′ CCC GAG CCC GAT CGC TGC TGA CAC CC −3′) were used to delete the wild-type sequence +1RSFFSFLGEAF+11, resulting in the deletion of these 11 residues to produce hSAA1mΔ1–11 leading to a SAA mutant with diminished FPR2 avidity. Sanger sequencing (Genetics resource core facility, Johns Hopkins School of Medicine, Baltimore, MD, USA) was used to confirm correct DNA sequences. siRNA-mediated knockdown was performed according to the manufacturer’s recommendation. Briefly, BEAS-2B ECs, plated at 1 × 104 per well of a 96-well plate in antibiotic-free medium, were transfected with 25 nM siRNA or non-targeting siRNA in a final volume of 100 µl using DharmaFECT transfection reagent (Thermo Scientific). In lipid-depleted media experiments, Caco-2 cells were used and maintained in IMDM media supplemented with charcoal-stripped FBS.
For all experiments, cells were serum starved overnight before allergen-stimulation (HDM used at 100 µg/ml; Alt a used at 25 μg/ml) for 30 min and 24 h. Cell culture supernatants were taken after indicated time points and analyzed for secreted cytokines by ELISA and cells lysed in TRIzol (Invitrogen) for RNA isolation. In selected experiments, pharmacological agents and proteins were used to investigate receptor dependency, including the FPR2 antagonist WRW4 (12 μM), the P2-purinergic receptor (P2YR) antagonist suramin (10 μM, Sigma) the Toll-like receptor 4 (TLR4) antagonist lipopolysaccharide (LPS, from Rhodobacter sphaeroides (LPS-RS, 10 μg/ml, InVivoGen), a TLR2 neutralizing antibody (anti-human TLR2 IgA, 10 µg/ml, InVivoGen), a CD36 blocking antibody (Clone JC63.1, 10 μg/ml, Cayman), LXA4 (100 nM), SAA1 (0.5 μg/ml), mouse anti-human SAA1 antibody (0.25–0.5 μg/ml, DM1004B, Acris). For Der p 13 neutralization and depletion experiments, a monoclonal antibody directed against its closely related homologue Blo t 13 was used61. Controls include vehicle and matched isotype control.
HDL pull-down experiments and SAA1 ligand binding
To determine whether HDM treatment influences the association/dissociation rate of SAA-HDL or its quaternary structure a straightforward procedure of HDL-immunoprecipitation via its major protein component, apolipoprotein A-I (ApoA1) was used. HDM and media control-treated cell culture supernatants were incubated overnight at 4 °C with a polyclonal goat anti-human ApoA1 antibody (10 μg/ml; R&D Systems). HDL-antibody complexes were separated from cell culture supernatants by magnetic separation using protein G-coated magnetic beads (SureBeads Protein G, Bio-Rad). HDL-depleted supernatants as well as HDL-precipitates were analyzed for their SAA1 content by immunoblotting; immunoreactivity was tested with mouse anti-human SAA1 antibody (DM1004B, Acris; MAB30196, R&D Systems) or sequence-specific antibodies as described previously 30. For SAA1 ligand binding experiments, the recombinant fatty acid binding proteins (rFABP) and extracts were first resolved by SDS-PAGE and blotted onto nitrocellulose membrane: the minor allergen of Blomia tropicalis, rBlo t 13, and the major HDM allergens rDer p 2 and rDer p 23, as well as whole HDM extract. Membranes were incubated with rSAA1 at 1 μg/ml pre-adsorbed to 0.5 μg/ml mouse monoclonal anti-SAA1 antibody (Invitrogen) overnight. Detection was by streptavidin-horseradish peroxidase and chemiluminescence using ECL (Bio-Rad) and Biomax film (Kodak) or detected with a chemiluminescence detection system (ChemiDoc Bio-Rad). For glutaraldehyde cross-linking experiments, rSAA1 at 1 mg/ml in PBS was incubated at 20–22 °C for 1 h with increasing amounts of Blo t 13 (1 mg/ml (1:1); 2 mg/ml (1:2); and 4 mg/ml (1:4)). Samples were chemically cross-linked with glutaraldehyde (Sigma) concentrations ranging from 0.0025% to 0.01% (wt/vol) for 5 min at 20–22 °C. The cross-linking reaction was stopped by adding Laemmli sample buffer and samples were analyzed by SDS/PAGE followed by immunoblotting with an anti-human SAA1 antibody. In case of native SDS-PAGE experiments, a fixed concentration of SAA1 of 1 mg/ml was incubated with increasing amounts of Blo t 13 (4:1 (0.25 mg/ml Blo t 13),2:1 (0.5 mg/ml Blo t 13), 1:1 (1 mg/ml Blo t 13) and 1:2 (2 mg/ml Blo t 13) for 2 h at 20–22 °C, separated by native PAGE and immunoblotted with a polyclonal sequence-specific anti-SAA1 raised against amino acids 14–30 of human SAA130.
Determination of SAA and cytokine concentrations
Anti-human SAA1 antibody pairs were purchased from Novex (Novex, Thermo Fisher Scientific) or human SAA1 DuoSet ELISA development F (DY3019–05, R&D Systems). Mouse SAA1 capture antibody from Thermo Fisher (D9H4L41) and mouse SAA1 detection antibody from R&D Systems (BAF2948). Human IL-33 ELISA and mouse IL-33 cytokine ELISA kits were from R&D Systems. Mouse IL-25 and TSLP ELISA were from Invitrogen. ELISA kits specific for mouse IL-5, IL-10, IL-11, IL-13, IL-22, IL-17A, and IFN-γ, and GM-CSF from R&D Systems or were determined using the Luminex system (Luminex 100IS, Biomedica) described previously62.
Isolation of RNA and quantitative real-time PCR
Total RNA was isolated from whole mouse lung tissue samples or FACS sorted cell populations frozen individually in TRIzol reagent (Invitrogen) and total RNA was extracted according to the manufacturer’s instructions. Individual sample RNA (1 μg) was reverse-transcribed using Superscript II (Invitrogen) and a mixture of oligo (dT) and random primers. Real-time PCR was performed on a StepOnePlus PCR system (Applied Biosystems) using iTaq DNA polymerase (Bio-Rad). Data were analyzed by the ΔCT method using the SDS 2.1 software (Applied Biosystems). Expression of the target genes was normalized to mRNA abundance of the housekeeping gene Rps14. For human samples target gene expression was normalized to eukaryotic elongation factor (EEF) and ubiquitin c (UBC). Primer sequences used are listed in Supplementary table 6.
Statistical analysis
All data were analyzed using GraphPad Prism 6 software. Unless otherwise indicated, all values are represented as mean values ± SEM. Figure legends denote the specific statistical tests used for each experiment. P values of less than 0.05 were considered significant.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data Availability statement
The datasets supporting the conclusions of this article are available from the corresponding authors upon request.
Extended Data
Extended Data Fig. 1: Experimental design.

(a) Model of allergen-induced airway hyperresponsiveness (AHR) for wild-type (WT) and Saa–/– mice (both C57BL/6 background). Mice were sensitized i.t. on day 0 (1 μg) and i.n. on days 7–11 with 10 μg of HDM extract. Airway measurements were performed 72 h after the last allergen challenge (used in Fig. 1a-j and Extended Data Fig. 2 and 3). (b) For SAA1 antibody blockade, we used an established mouse model of allergen-induced AHR sensitizing WT BALB/cJ mice i.t. on day 0 and 14 with 100 µg of HDM extract + isotype control, or HDM + αSAAab. Airway measurements and tissue harvests were performed 72 h after the last allergen challenge (used in Extended Data Fig. 4). (c) In short-term exposure protocols WT and Saa–/– mice (both C57BL/6 background) received a single HDM challenge (100 μg) were sacrificed 16 h later (used in Fig. 2a-d). (d) In short-term exposure protocols BALB/cJ mice received a single HDM challenge (100 μg)+ isotype control, HDM challenge + HDL (200μg) and isotype control, or HDM + αSAAab and were sacrificed 24 h later (used in Fig. 2e). Contol mice received either PBS + isotype or PBS + HDL and isotype control. (e) For overexpression of SAA1 in vivo, mice were injected 20 µg of DNA complexed to polyethylenimine at day 0, exposed to PBS or HDM 48 h later and ILC2s as well as BAL cytokines measurements were performed on day 3 (used Fig. 2f). (f) WT and Saa–/– mice were sensitized i.t. on day 0 (1 μg) and i.n. on days 7–11 with 10 μg with extracts from the parasitic worm Schistosoma mansoni (a Puerto Rican isolate). Tissues were harvested 72 h after the last allergen challenge (used in Fig. 4). (g) For FPR2 blockade (WRW4, 2 mg/kg), WT BALB/cJ mice were sensitized and challenged i.t. on day 0 and 14 with 100 μg of HDM extract. Airway measurements were performed 72 h after the last allergen challenge (used in Fig. 7a-f). (h) In short term exposure experiments, BALB/cJ mice received a single HDM challenge (100 μg) or HDM + WRW4 and were sacrificed 24 h later (used in Fig. 7g and i). (i) Model of Alternaria alternata (Alt a )-induced airway inflammation for WT and Saa–/– mice. Mice were sensitized i.t. on day 0 (1 μg) and i.n. on days 7–11 with 10 µg of Alt a extract. Tissues were harvested 72 h after the last allergen challenge (used in Extended Data Fig. 9f-j).
Extended Data Fig. 2: Reduced recruitment of CD11b+ DCs in Saa–/– mice after allergen exposure.

Allergic phenotype in WT and Saa–/– mice sensitized and challenged with PBS or HDM was analyzed seventy-two hours after the last challenge. (a) Lung cell gating strategy used in Fig. 1, Fig. 4, Extended Data Fig. 6 and Extended Data Fig 9. Frequency of dendritic cell (DC) populations (b-d), monocytes (e-f) and granulocytes (g-h) in the lungs of WT and Saa–/– mice used in figure 1. Data represent means ± SEM of pooled data from 2 independent experiments containing n=11 WT PBS, n=12 WT HDM, n=11 Saa–/– PBS and n=13 Saa–/– HDM animals per group. P values were calculated with a two-tailed test using one-way analysis of variance (ANOVA) with Dunett’s post hoc analysis that compares WT HDM to counterparts. **P = 0.0039, ****P ≤ 0.0001.
Extended Data Fig. 3: Reduced migration of CD3+CD4+ T cells to the lungs of Saa–/– mice after allergen exposure.

Allergic phenotype in WT and Saa–/– mice sensitized and challenged with PBS or HDM was analyzed seventy-two hours after the last challenge. (a) ILC and T cell gating strategy (used in Fig. 1g and h, Fig. 4b-e, Extended Data Figure 6d and e and Extended Data Fig. 9h-j). (b) Total lung cell counts (**P = 0.0094), (c) frequency and (d) numbers of CD3+CD4+ T cells (**P = 0.0014) and (e) frequency and numbers of (f) ICOS+ST2+ ILCs in the lungs of WT and Saa–/– mice. Data represent pooled data presented as means ± SEM from 2 independent experiments containing n=11 WT PBS, n= 12 WT HDM, n=11 Saa–/– PBS and n=13 Saa–/– HDM animals per group. P values were calculated with a two-tailed test using one-way analysis of variance (ANOVA) with Dunett’s post hoc analysis that compares WT HDM to counterparts.
Extended Data Fig. 4: Airway SAA1-neutralization ameliorates the allergic phenotype.

(a) AHR (*P < 0.0105), (b) total serum IgE concentrations (*P = 0.0265), (c) eosinophil infiltration into the lungs, and (d) PAS stained lung sections of isotype (iso), HDM+isotype (HDM), or HDM+αSAAab-treated (αSAA) WT BALB/c mice. Antibodies were administered at 5 μg/i.t. Frequency of (e) Lin-CD45+ST2+IL-13+ ILC2s (**P = 0.0027, ***P = 0.0002), (f) TH2 and (*P = 0.0260, ***P = 0.0001) (g) TH17 cells (*P = 0.0149, ***P = 0.0005) in the lungs of these mice. Data represents means ± SEM of pooled data from 2 independent experiments containing (a) n=6 PBS+iso, n=7 HDM+iso, n=9 HDM+αSAA animals per group; (b, c, f, g) n=9 PBS+iso, n=11 HDM+iso, n=13 HDM+αSAA animals per group or are representative of 2 independent experiments with (e) n=4 PBS+iso, n=5 HDM+iso, n=7 HDM+αSAA animals per group. P values were calculated with a two-tailed test using one-way analysis of variance (ANOVA) with Dunett’s post hoc analysis that compares HDM to iso and αSAA counterparts. ****P ≤ 0.0001
Extended Data Fig. 5: SAA1 is a pattern recognition molecule for mite-derived and human cytosolic FABPs.

(a) Migration of SAA1 (1 mg/ml) in IMDM media was analyzed in the presence of the mite FABP rBlo t 13 (1 mg/ml) by native PAGE followed by immunoblot analysis using a sequence-specific antiserum (amino acid 89–104) raised against human SAA1. (b) Effects of the protein synthesis inhibitor cycloheximide on IL-33 concentrations in BEAS-2B cells treated for 30 min with HDM (100 μg/ml). (c) Cell viability of BEAS-2B cells after HDM exposure over time as measured by continuous reduction of a cell viability substrate by viable cells (**P = 0.0041). SAA1 concentrations in cells after (d) siRNA-mediated silencing of SAA1 (siSAA1) or non-targeting scrambled siRNA (siNT). Effects of SAA1 on HDM-induced IL-6 (**P = 0.0086, ***P = 0.0007) and IL-8 release in cells with siRNA mediated knockdown of SAA1 (siSAA1) (e and f). (g) HDM-induced IL-6 amounts after Der p 13-depletion and/or neutralization. Data are shown as means ± SEM and are representative of 2 (b, e) or 3 (c) independent experiments or pooled data from 2 independent experiments (d, f) each containing at least n=4 biologically independent samples. Representative analysis of SAA1 migration patterns in the presence of Blo t 13 using sequence-specific rabbit antiserum raised against human SAA1 (aa 89–104) (a). IL-6 amounts were analysed for one representative experiments with n=5 biologically independent samples (g). Cropped images are shown. P values were calculated with a two-tailed test using one-way analysis of variance (ANOVA) followed by Dunett’s post (b) or Tukey’s hoc analysis (e,f), two-way ANOVA followed by Dunnet correction (c) or Student’s t-test (d, g). ***P ≤ 0.001; ****P ≤ 0.0001.
Extended Data Fig. 6: Der p 13-depleted HDM has decreased TH2 skewing capacity.

(a) Total IgE serum concentrations and numbers of BAL (b) eosinophils and (c) T cells in mice undergoing a full allergen-exposure protocol using HDM that was depleted from the mite FABP Der p 13 using either an isotype control antibody or a group-13 specific antibody. Numbers of (d) mediastinal lymph node IL-13+ CD4+ T cells and (e) lung ICOS+ST2+ ILCs. Cytokine production from HDM-restimulated lung cells (*P = 0.024, ). Data represent mean ± SEM of n=2 PBS, n=4 HDM-isotype depleted, n=4 HDM- α-group 13-depleted comparing HDM-isotype to α-group 13-depleted extract group using two-tailed unpaired t-test.
Extended Data Fig. 7: HDM-induced IL-33 is dependent on SAA1 dissociation.
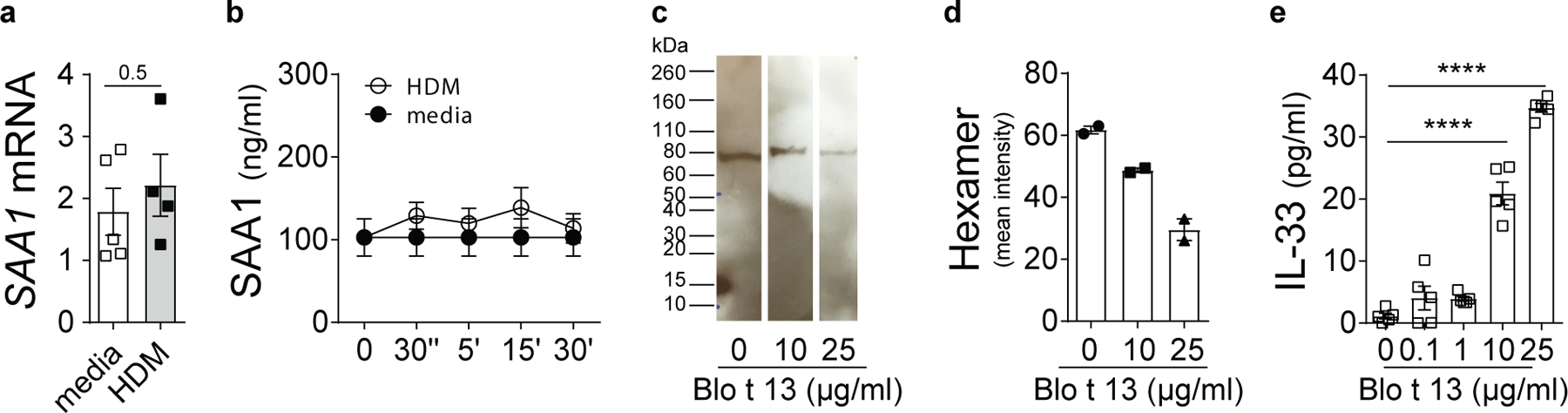
(a) mRNA and (b) protein amounts of SAA in response to HDM (100 μg/ml). (c) SAA1 hexamer after rBlo t 13 stimulation was analyzed as described in Fig. 3b. (d) Bar graph represents quantitative analysis of SAA hexamer using LI-COR Image Studio Software. (e) Concentration-dependent IL-33 release from BEAS-2B cells induced by rBlo t 13. Data are shown as means ± SEM and are pooled data from 2 independent experiments (b, e) each containing n=4–5 replicate wells or representative of 2–3 independent experiments (d). SAA1 mRNA expression, normalized to the average of housekeeping genes, is presented as mean value ± SEM (n = 5). Immunoblot is representative of an experimental n=2. Cropped images are shown. P values were calculated with a two-tailed test using Student’s t-test (a), two-way analysis of variance (ANOVA) followed by Dunnet’s correction (b) or one-way ANOVA with Dunett’s post hoc analysis (d, e). ****P ≤ 0.0001.
Extended Data Fig. 8: The SAA1-FPR2 axis regulates IL-33.

(a) mRNA expression of the FPR family members FPR1, FPR2 and FPR3 at baseline (open bars) or after 2 h of HDM stimulation (filled bars). (b) HDM-triggered IL-33 amounts in BEAS-2B cells overexpressing human FPR1 (**P = 0.0068, ***P = 0.0003). (c) IL-33 secretion in BEAS-2B cells overexpressing human FPR2 or cells transfected with an empty vector (EV; pcDNA3.1) (**P = 0.0068, ***P = 0.0003). HDM-induced IL-6 and IL-8 amounts in BEAS-2B cells overexpressing FPR2 (d and e) or blocking the FPR2 receptor (f (**P = 0.0043) and g (**P = 0.0021)) using WRW4. Data presented as means ± SEM and is representative of 2 independent experiments each containing at least n= 4 biologically independent samples (b, d, f) or pooled data from 2 independent experiments (c, e, g). mRNA expression, normalized to the average of housekeeping genes, is presented as mean values ± SEM (n = 5 biologically independent samples) performed in duplicates (a). P values were calculated with a two-tailed test using Student’s t-test (a) or one-way analysis of variance (ANOVA) with Tukeys multiple comparison test (b-e) or Dunett’s post hoc analysis (f, g). ****P ≤ 0.0001
Extended Data Fig. 9: SAA1-IL-33 axis is specific to HDM and not induced by Alternaria alternata:

(a) Immunoblot of SAA1 after Alternaria alternata (Alt a) stimulation of BEAS-2B cells performed as described in Fig. 3b. Alt a-induced IL-33 and IL-8 (**P = 0.0044) secretion in BEAS-2B cells after siRNA-mediated silencing of SAA1 (siSAA1) (b and c) or WRW4-mediated FPR2 blockade (d and e). (f) Total serum IgE concentrations, (g) eosinophil counts and frequency of (h) CD3+CD4+, (i) TH2 and (j) TH17 cells in the lungs of PBS or Alt a-treated WT and Saa–/– mice. Immunoblots are representative of an experimental n=2 (a). Data are presented as means ± SEM and represent pooled data from 2 independent experiments (b, d, e, f, g, i, j) or show one representative experiment (c) each containing at least n= 4 biologically independent samples or n=8 WT PBS, n=13 WT Alt a, n=8 Saa–/– PBS and n=11 Saa–/– Alt a animals per group (f, g, i, j) or n=5 WT PBS, n=9 WT Alt a, n=5 Saa–/– PBS and n=7 Saa–/– Alt a animals per group (h). Cropped images are shown. P values were calculated with a two-tailed test using one-way analysis of variance (ANOVA) with Tukeys multiple comparison test (a, b) or Dunett’s post hoc analysis (d-j). ****P ≤ 0.0001 siNT = non-targeting siRNA; siSAA1 = SAA1-targeting siRNA. ns=not significant.
Extended Data Fig. 10: Dysregulated SAA and FPR2 expression in patients with asthma.

Basal (a) SAA1 and (b) FPR2 expression in bronchial epithelial from asthmatic patients and matched controls. Data represents means ± SEM of (a and b) n= 6 control and n= 6 asthmatic patients per group. x.
Supplementary Material
Acknowledgments:
The authors thank M.C. deBeer (University of Kentucky Medical Center, KY, US) for kindly providing the Saa–/– mice. E. Schmidt and R. Zeiner for animal care, Z. Lijie, D. Trapin and S. Kickmaier for expert technical assistance. The authors also thank M .Marlovics (dsgn&cde; hq@dsgncde.com) for the graphic designs in Figure 3 and 4 and C. Zwicker for critically reading the manuscript and for helpful discussion and suggestions. This work was funded by the National Institute of Allergy and Infectious Diseases (grants U19AI070235 and R01 AI083315 to M.W.-K.) and the NIH (grants R56AI118791 and R01AI127644 to S.L. and R01AI132590 to A.P.L.) as well as the Austrian Science Fund (DK W1248 and SFB F4609 to W.F.P.). U.S. was supported by an Erwin Schrödinger Fellowship (J3332-B21) of the Austrian Science Fund, a research grant of the American Thoracic Society and DK W1248. N.G. was supported by an NIEHS T32ES007141 grant. E.M. was supported by the Austrian National Bank (17600). J.D. was supported by the COFUND-MSCA program (CZ.02.2.69/0.0/0.0/17_050/0008014). J.C. was supported by the Canadian Institutes of Health Research (CIHR, DC0190GP).
Footnotes
Competing Interests Statement: The authors have no competing interests to declare.
References
- 1.Wills-Karp M, Nathan A, Page K & Karp CL New insights into innate immune mechanisms underlying allergenicity. Mucosal Immunol 3, 104–110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Corrigan CJ et al. Allergen-induced expression of IL-25 and IL-25 receptor in atopic asthmatic airways and late-phase cutaneous responses. J. Allergy Clin. Immunol 128, 116–124 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Gour N et al. Dysregulated invertebrate tropomyosin-dectin-1 interaction confers susceptibility to allergic diseases. Sci Immunol 3 (20): eaam9841 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schuijs MJ et al. Farm dust and endotoxin protect against allergy through A20 induction in lung epithelial cells. Science 349, 1106–1110 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Smole U, Kratzer B & Pickl WF Soluble pattern recognition molecules: Guardians and regulators of homeostasis at airway mucosal surfaces. Eur. J. Immunol 50, 624–642 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Urieli-Shoval S, Cohen P, Eisenberg S & Matzner Y Widespread expression of serum amyloid A in histologically normal human tissues. Predominant localization to the epithelium. J. Histochem. Cytochem 46, 1377–1384 (1998). [DOI] [PubMed] [Google Scholar]
- 7.Malle E, Sodin-Semrl S & Kovacevic A Serum amyloid A: an acute-phase protein involved in tumour pathogenesis. Cell. Mol. Life Sci 66, 9–26 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai Z et al. Human serum amyloid A protein inhibits hepatitis C virus entry into cells. J. Virol 81, 6128–6133 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shah C, Hari-Dass R & Raynes JG Serum amyloid A is an innate immune opsonin for Gram-negative bacteria. Blood 108, 1751–1757 (2006). [DOI] [PubMed] [Google Scholar]
- 10.de Beer MC et al. Impact of serum amyloid A on high density lipoprotein composition and levels. J. Lipid Res 51, 3117–3125 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hinrichs BH et al. Serum Amyloid A1 Is an Epithelial Prorestitutive Factor. Am. J. Pathol 188, 937–949 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun L & Ye RD Serum amyloid A1: Structure, function and gene polymorphism. Gene 583, 48–57 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng N, Liang Y, Du X & Ye RD Serum amyloid A promotes LPS clearance and suppresses LPS-induced inflammation and tissue injury. EMBO Rep 19 (10): e45517, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim MH, de Beer MC, Wroblewski JM, Webb NR & de Beer FC SAA does not induce cytokine production in physiological conditions. Cytokine 61, 506–512 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu J, Yu Y, Zhu I, Cheng Y & Sun PD Structural mechanism of serum amyloid A-mediated inflammatory amyloidosis. Proc. Natl. Acad. Sci. U. S. A 111, 5189–5194 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y et al. Serum amyloid A 2.2 refolds into a octameric oligomer that slowly converts to a more stable hexamer. Biochem. Biophys. Res. Commun 407, 725–729 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sano T et al. An IL-23R/IL-22 Circuit Regulates Epithelial Serum Amyloid A to Promote Local Effector Th17 Responses. Cell 163, 381–393 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ather JL et al. Serum amyloid A activates the NLRP3 inflammasome and promotes Th17 allergic asthma in mice. J. Immunol 187, 64–73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buyukozturk S et al. Acute phase reactants in allergic airway disease. Tohoku J. Exp. Med 204, 209–213 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Jousilahti P et al. The association of sensitive systemic inflammation markers with bronchial asthma. Ann. Allergy. Asthma. Immunol 89, 381–385 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Ozseker F et al. Serum amyloid A (SAA) in induced sputum of asthmatics: a new look to an old marker. Int Immunopharmacol 6, 1569–1576 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Ricklefs I et al. ALX receptor ligands define a biochemical endotype for severe asthma. JCI Insight 2 (14): e93534. (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lambrecht BN, Hammad H & Fahy JV The Cytokines of Asthma. Immunity 50, 975–991 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Cayrol C et al. Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nat Immunol 19, 375–385 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Gold MJ et al. Group 2 innate lymphoid cells facilitate sensitization to local, but not systemic, TH2-inducing allergen exposures. J. Allergy Clin. Immunol 133, 1142–1148 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Halim TY et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 40, 425–435 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ather JL et al. Serum Amyloid A3 is required for normal lung development and survival following influenza infection. Sci Rep 8, 16571 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pautsch A & Schulz GE High-resolution structure of the OmpA membrane domain. J. Mol. Biol 298, 273–282 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Niemi MH et al. Dimerization of lipocalin allergens. Sci Rep 5, 13841 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malle E et al. Mapping of antigenic determinants of purified, lipid-free human serum amyloid A proteins. Scand. J. Immunol 48, 557–561 (1998). [DOI] [PubMed] [Google Scholar]
- 31.Suojalehto H et al. Level of Fatty Acid Binding Protein 5 (FABP5) Is Increased in Sputum of Allergic Asthmatics and Links to Airway Remodeling and Inflammation. PLoS One 10, e0127003 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thaumaturgo N, Vilar MM, Diogo CM, Edelenyi R & Tendler M Preliminary analysis of Sm14 in distinct fractions of Schistosoma mansoni adult worm extract. Mem. Inst. Oswaldo Cruz 96 Suppl, 79–83 (2001). [DOI] [PubMed] [Google Scholar]
- 33.Franchini GR et al. The unusual lipid binding proteins of parasitic helminths and their potential roles in parasitism and as therapeutic targets. Prostaglandins Leukot. Essent. Fatty Acids 93, 31–36 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Derebe MG et al. Serum amyloid A is a retinol binding protein that transports retinol during bacterial infection. Elife 3, e03206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou H, Chen M, Zhang G & Ye RD Suppression of Lipopolysaccharide-Induced Inflammatory Response by Fragments from Serum Amyloid A. J. Immunol 199, 1105–1112 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Rabiet MJ, Macari L, Dahlgren C & Boulay F N-formyl peptide receptor 3 (FPR3) departs from the homologous FPR2/ALX receptor with regard to the major processes governing chemoattractant receptor regulation, expression at the cell surface, and phosphorylation. J. Biol. Chem 286, 26718–26731 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cooray SN et al. Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses. Proc. Natl. Acad. Sci. U. S. A 110, 18232–18237 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Snelgrove RJ et al. Alternaria-derived serine protease activity drives IL-33-mediated asthma exacerbations. J. Allergy Clin. Immunol 134, 583–592 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Green BJ et al. Allergic sensitization in Canadian chronic rhinosinusitis patients. Allergy Asthma Clin Immunol 10, 15 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chu DK et al. IL-33, but not thymic stromal lymphopoietin or IL-25, is central to mite and peanut allergic sensitization. J. Allergy Clin. Immunol 131, 187–200 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Rank MA et al. IL-33-activated dendritic cells induce an atypical TH2-type response. J. Allergy Clin. Immunol 123, 1047–1054 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Besnard AG et al. IL-33-activated dendritic cells are critical for allergic airway inflammation. Eur. J. Immunol 41, 1675–1686 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Frame NM, Jayaraman S, Gantz DL & Gursky O Serum amyloid A self-assembles with phospholipids to form stable protein-rich nanoparticles with a distinct structure: A hypothetical function of SAA as a “molecular mop” in immune response. J. Struct. Biol 200, 293–302 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jayaraman S, Gantz DL, Haupt C & Gursky O Serum amyloid A forms stable oligomers that disrupt vesicles at lysosomal pH and contribute to the pathogenesis of reactive amyloidosis. Proc. Natl. Acad. Sci. U. S. A 114, E6507–E6515 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bjorkman L et al. The proinflammatory activity of recombinant serum amyloid A is not shared by the endogenous protein in the circulation. Arthritis Rheum 62, 1660–1665 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Christenson K et al. Endogenous Acute Phase Serum Amyloid A Lacks Pro-Inflammatory Activity, Contrasting the Two Recombinant Variants That Activate Human Neutrophils through Different Receptors. Front Immunol 4, 92 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Puerta L, Kennedy MW, Jim nez S & Caraballo L Structural and ligand binding analysis of recombinant blo t 13 allergen from Blomia tropicalis mite, a fatty acid binding protein. Int. Arch. Allergy Immunol 119, 181–184 (1999). [DOI] [PubMed] [Google Scholar]
- 48.Scott IC et al. Interleukin-33 is activated by allergen- and necrosis-associated proteolytic activities to regulate its alarmin activity during epithelial damage. Sci Rep 8, 3363 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ivanov II et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caraballo L et al. Cloning and IgE binding of a recombinant allergen from the mite Blomia tropicalis, homologous with fatty acid-binding proteins. Int. Arch. Allergy Immunol 112, 341–347 (1997). [DOI] [PubMed] [Google Scholar]
- 51.Zimmerman AW & Veerkamp JH New insights into the structure and function of fatty acid-binding proteins. Cell. Mol. Life Sci 59, 1096–1116 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miljkovic D et al. Association between group 2 innate lymphoid cells enrichment, nasal polyps and allergy in chronic rhinosinusitis. Allergy 69, 1154–1161 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Shaw JL et al. IL-33-responsive innate lymphoid cells are an important source of IL-13 in chronic rhinosinusitis with nasal polyps. Am. J. Respir. Crit. Care Med 188, 432–439 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen M, Zhou H, Cheng N, Qian F & Ye RD Serum amyloid A1 isoforms display different efficacy at Toll-like receptor 2 and formyl peptide receptor 2. Immunobiology 219, 916–923 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lotvall J et al. Asthma endotypes: a new approach to classification of disease entities within the asthma syndrome. J. Allergy Clin. Immunol 127, 355–360 (2011). [DOI] [PubMed] [Google Scholar]
Methods only references
- 56.Leontovyc A et al. SmSP2: A serine protease secreted by the blood fluke pathogen Schistosoma mansoni with anti-hemostatic properties. PLoS Negl Trop Dis 12, e0006446 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lajoie S et al. IL-21 receptor signalling partially mediates Th2-mediated allergic airway responses. Clin. Exp. Allergy 44, 976–985 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paris G, Pozharskaya T, Asempa T & Lane AP Damage-associated molecular patterns stimulate interleukin-33 expression in nasal polyp epithelial cells. Int Forum Allergy Rhinol 4, 15–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Darveau ME, Jacques E, Rouabhia M, Hamid Q & Chakir J Increased T-cell survival by structural bronchial cells derived from asthmatic subjects cultured in an engineered human mucosa. J. Allergy Clin. Immunol 121, 692–699 (2008). [DOI] [PubMed] [Google Scholar]
- 60.Goulet F et al. Morphologic and functional properties of bronchial cells isolated from normal and asthmatic subjects. Am. J. Respir. Cell Mol. Biol 15, 312–318 (1996). [DOI] [PubMed] [Google Scholar]
- 61.Labrada M et al. Monoclonal antibodies against Blo t 13, a recombinant allergen from Blomia tropicalis. Int. Arch. Allergy Immunol 129, 212–218 (2002). [DOI] [PubMed] [Google Scholar]
- 62.Kratzer B et al. Prevention of allergy by virus-like nanoparticles (VNP) delivering shielded versions of major allergens in a humanized murine allergy model. Allergy 74, 246–260 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting the conclusions of this article are available from the corresponding authors upon request.