

Summary
Paroxysmal nocturnal haemoglobinuria (PNH) is characterized by complement‐mediated intravascular haemolysis, severe thrombophilia and bone marrow failure. While for patients with bone marrow failure the treatment follows that of immune‐mediated aplastic anaemia, that of classic, haemolytic PNH is based on anti‐complement medication. The anti‐C5 monoclonal antibody eculizumab has revolutionized treatment, resulting in control of intravascular haemolysis and thromboembolic risk, with improved long‐term survival. Novel strategies of complement inhibition are emerging. New anti‐C5 agents reproduce the safety and efficacy of eculizumab, with improved patient convenience. Proximal complement inhibitors have been developed to address C3‐mediated extra‐vascular haemolysis and seem to improve haematological response.
Keywords: paroxysmal nocturnal haemoglobinuria, intravascular haemolysis, extravascular haemolysis, proximal complement inhibitors, terminal complement inhibitors
Paroxysmal nocturnal haemoglobinuria (PNH) is a rare haematological disease characterized by the clinical triad: complement‐mediated intravascular haemolysis, severe thrombophilia and bone marrow failure. 1 PNH is due to somatic loss‐of‐function mutations in the phosphatidylinositol N‐acetylglucosaminyltransferase subunit A (PIGA) gene, 2 , 3 , 4 occurring in one or more haematopoietic stem cells (HSCs). The development of the disease is associated with the clonal dominance of PIGA‐mutated HSCs, which progressively replace normal haematopoiesis, probably because they are spared in the presence of immune‐mediated damage of haematopoietic progenitors. 5 , 6 , 7 The PIGA mutation precludes the biosynthesis of the glycosylphosphatidylinositol (GPI) anchor, resulting in the lack on the cell surface of all GPI‐anchored proteins (GPI‐AP), both on affected haematopoietic progenitors and on their mature progeny blood cells. 8 , 9 , 10 , 11
The hallmark of PNH — the typical complement‐mediated intravascular haemolysis — is due to the fact that the two key complement regulators, CD55 12 , 13 , 14 and CD59, 15 , 16 are expressed on the cell surface via the GPI anchor. Consequently, PNH erythrocytes are deficient for both CD55 and CD59, and are highly susceptible to complement‐mediated lysis, because they are unable to physiologically modulate complement activation on their surface. These complement regulators are lacking also on PNH granulocytes and platelets, and impaired complement regulation on these cells might contribute. Haemolysis of PNH is chronic, secondary to the physiologic spontaneous hydrolysis of complement component 3 (the so called “C3 tick‐over”), 17 with possible acute exacerbations triggered by transient complement activation associated with specific clinical events, such as infections. Thromboembolic events are the most feared complication in PNH; thrombosis is pathogenically linked to intravascular haemolysis of PNH erythrocytes, but impaired complement regulation on other affected blood cells might contribute to the thrombophilic status of PNH. 18
Historically, the diagnosis of PNH was based on the demonstration of an erythrocyte population susceptible to complement‐mediated haemolysis in vitro through the Ham test. 19 , 20 Today, flow cytometry is the established diagnostic assay, which enables high‐sensitivity detection of GPI‐AP‐deficient populations in all blood lineages. Guidelines describing panels of monoclonal antibodies and protocols have been published. 21 , 22 , 23 , 24 These techniques allow the detection of GPI‐AP‐deficient cell populations as low as 0·01%. Once a GPI‐AP‐deficient cell population (i.e. a PNH clone) has been detected, patients can be classified as having classic PNH, PNH in the context of another bone marrow disorder, or subclinical PNH. 25
More practically (and correctly, since PNH is by definition characterized by haemolysis), PNH patients may be purely haemolytic (i.e. florid PNH), or with broader pancytopenia fulfilling or not fulfilling the diagnostic criteria for aplastic anaemia (AA; AA/PNH syndrome and intermediate PNH respectively). 26 In contrast, patients defined as “subclinical PNH” simply harbour GPI‐AP‐deficient blood cell populations, which may have a pathophysiologic meaning in the context of other conditions, usually immune‐mediated bone marrow failures. 27
Standard treatment
Treatment indications are driven by the two clinical presentations: haemolytic, without overt marrow failure, referred to as classic, haemolytic PNH; and with marrow failure, often described as AA/PNH syndrome (Fig 1). Before specific therapy became available, PNH resulted in the death of approximately half of all patients, mainly through thrombotic complications, including a particularly grim prognosis for patients presenting with classic PNH. 26
Fig 1.
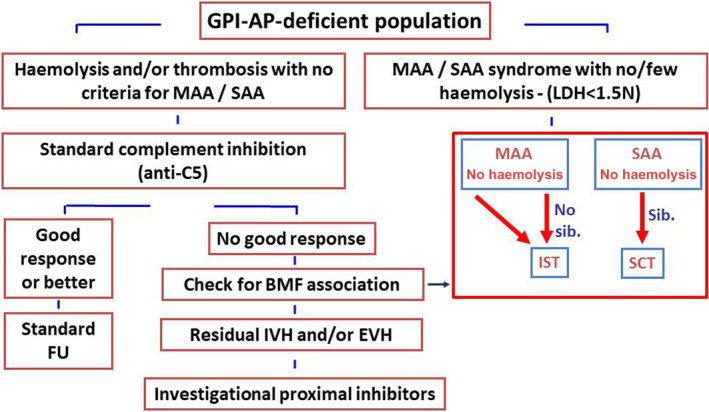
Treatment algorithm. A tentative treatment algorithm of PNH; based on disease presentation and response to available treatments. BMF, bone marrow failure; EVH, extra‐vascular haemolysis; FU, follow‐up; GPI‐AP, glycosylphosphatidylinositol‐anchored proteins; IST, immuno‐suppressive therapy; IVH, intravascular haemolysis; LDH, lactate dehydrogenase; MAA/SAA, moderate/severe aplastic anaemia; PNH, paroxysmal nocturnal haemoglobinuria; SCT, stem cell transplantation.
Treatment options for haemolytic PNH remained limited and often inadequate until the availability of eculizumab, a humanized monoclonal antibody (mAb) targeting component 5 (C5) of the complement cascade. 28 By disabling the complement cascade at the level of the terminal complement (i.e. membrane attack complex — MAC — formation), eculizumab prevents the lysis of PNH erythrocytes, which cannot properly curb complement activation on their surface.
The efficacy of eculizumab in PNH patients was first demonstrated in a pilot study from the UK, which showed robust inhibition of complement‐mediated intravascular haemolysis. 29 Two subsequent large international phase III randomized studies demonstrated that eculizumab prevents intravascular haemolysis in PNH, eventually leading to haemoglobin stabilization, reduction or eradication of red blood cell transfusions and resolution of most disease‐related symptoms. 30 , 31 These data were confirmed in longer follow‐up analysis, which showed further haematological improvement on continuous maintenance treatment with eculizumab, with no safety concerns. 32 Notably, eculizumab also reduced the thromboembolic risk, 33 the most feared complication in PNH, thereby impacting on the disease course, morbidity and long‐term survival. With the caveat of the relatively short follow‐up, two independent studies have shown that PNH patients receiving continuous treatment with eculizumab have a five‐year survival >90%. 34 , 35 These survival rates appear superior to well‐established data on the natural history of PNH, 26 , 36 , 37 elegantly shown in a retrospective comparison between eculizumab‐treated patients and historical controls. 35
Therapeutic complement inhibition by eculizumab has thus revolutionized the treatment of these patients and is hitherto considered the standard of care for PNH patients with haemolytic disease or thromboembolic complications. Eculizumab is symptomatic, with PNH‐related symptoms resurfacing upon withdrawal. Haemolysis‐related transfusions, clinical signs arising from intravascular haemolysis and thrombosis occurrence are all good reasons to begin therapy, whereas clone size per se is not. Conversely, patients with ongoing bone marrow failure from moderate or severe aplastic anaemia are less likely to derive any benefit from eculizumab. For these patients, therapy should address the underlying bone marrow failure (Fig 1).
All patients receiving eculizumab should be vaccinated, since the most severe risks of terminal complement blockade are life‐threatening neisserial infections. In some countries like France and the UK, penicillin prophylaxis is usually recommended, but this has not yet been formally evaluated. Despite the fact that eculizumab was a breakthrough therapy for PNH, recent efforts have been aimed at further improving the current standard therapy in PNH. In this manuscript, we review current issues in anti‐complement treatment for PNH, settling the goals for future complement inhibitors in development for PNH.
Response to anti‐complement treatment
The fact that eculizumab has drastically improved the survival of PNH patients, with the control of most severe signs and symptoms of disease, does not mean that eculizumab‐treated PNH patients are cured. Patients still require careful clinical and laboratory assessment to investigate possible changes in disease presentation, which appears to be the rule, rather than the exception, in PNH. This is especially true for bone marrow failure, which remains subtle in most patients, but may eventually become clinically meaningful any time during the disease course.
Periodical bone marrow analysis should be performed (every 18/24 months, or at even longer intervals if blood counts remain stable) to rule out the very low risk of evolution to myeloid malignancies. Similarly, the thrombophilic status characteristic of PNH may account for thromboembolic events, albeit rarely, even during eculizumab treatment. Thus, upon cases of signs or symptoms of such complications, PNH patients should be evaluated for possible thrombotic complications, with the most appropriate methods depending on the site and on clinical presentation.
However, eculizumab treatment has been designed to inhibit complement‐mediated haemolytic anaemia, the hallmark of PNH. The rest of the manuscript will thus aim to dissect the efficacy of eculizumab on haemolytic anaemia, trying to provide a guide for physicians in the forthcoming scenario of alternative anti‐complement agents available in the market or within clinical trials. Our guide is driven by the goal of maximizing therapeutic benefit for each individual PNH patient, based on their own clinical and biological data.
The efficacy of eculizumab has been largely demonstrated based on its effect on lactate dehydrogenase (LDH) and transfusion requirement, as shown in the pivotal registration trials; irrespective of haematological benefit, eculizumab has been proven biologically effective in reducing intravascular haemolysis in all PNH patients, with the exception of those carrying a rare C5 polymorphism. 38 Nevertheless, the actual haemoglobin levels in PNH patients on eculizumab have not been described in detail, 39 even if it is obvious that many patients remain variably anaemic. 40 Haematological response categories were initially proposed in 2009, 27 , 40 but they have not been formally established.
We and others have recently tried to define objective response categories during eculizumab treatment, based on haemoglobin and on biological evidence of disease activity (using LDH and reticulocyte count as biomarkers of haemolysis). 41 We have developed a six‐category classification (Table I), spanning from patients with normal blood counts with no laboratory sign of haemolysis to patients who remain transfusion‐dependent, despite eculizumab. In our experience, no more than 15% of PNH patients receiving eculizumab achieve the two top response categories (complete and major response, both characterized by normal‐like haemoglobin levels). 42 Thus, for the remaining 85% of patients, different reasons account for residual anaemia, irrespective of its clinical relevance.
Table I.
Haematological response to complement inhibitors in paroxysmal nocturnal haemoglobinuria.
| Response category | Red blood cell transfusions | Haemoglobin level | Residual haemolysis and breakthrough episodes* , ** |
|---|---|---|---|
| Complete response | None |
≥130 g/l (males) or ≥120 g/l (females) |
LDH ≤1.5 × ULN and ARC ≤150 000/µl,§ no breakthrough episodes |
| Major response | None |
≥130 g/l (males) or ≥120 g/l (females) |
LDH >1.5 × ULN and/or ARC >150 000/µl,§ only subclinical breakthrough episodes |
| Good response | None |
≥10 and <130 g/l (males) or ≥10 and <120 g/l (females) |
Any LDH and ARC value, only subclinical breakthrough episodes (rule out bone marrow failure)† |
| Partial response | None or occasional (≤2 every 6 months) | ≥8 and <100 g/l | |
| Minor response# | None or occasional (≤2 every 6 months) | <80 g/l | |
| Regular (3–6 every 6 months) | <100 g/l | ||
| Reduction by ≥50%^ | <100 g/l | ||
| No response# | Regular (>6 every 6 months) | <100 g/l |
ARC, absolute reticulocyte count; LDH, lactate dehydrogenase; ULN, upper limit of the normal.
The presence of clinically meaningful episodes of breakthrough haemolysis downgrades the response category by one degree.
To rule out increased erythropoietic response to compensate ongoing haemolysis; the value of 150 000/µl is a tentative index based on 1.5 × ULN (which in most laboratories is set at 100 000/µl).
To assess the relative contribution of the degree of bone marrow failure to any response less than complete: a value of ARC below 60 000/µl could be a tentative index to establish such a contribution.
For patients with previous transfusion history (with a pre‐treatment follow‐up of at least six months).
For patients who do not accept red blood cell transfusions, minor response can be defined based on haemoglobin level ≥60 and <80 g/l, and no response based on haemoglobin <60 g/l. All haemoglobin, LDH and ARC values should be assessed based on the median value over a period of six months.
These response categories apply to patients with adequate control of intravascular haemolysis; patients with clinically meaningful haemolysis (e.g., recurrent symptoms or LDH stably >2 × ULN) are considered as non‐responders according to this classification.
There are three major causes contributing to residual anaemia in PNH patients on eculizumab treatment (Table II), which are inadequate erythropoietic response, residual intravascular haemolysis and emerging C3‐mediated extra‐vascular haemolysis. 27 , 40 , 41 , 43 , 44 Inadequate erythropoietic response is possibly due to a concomitant bone marrow failure, but vitamin and iron deficiencies should also be ruled out. As discussed above, bone marrow failure is an expected presentation of PNH, which requires treatment different from complement inhibitors. In contrast, the other two causes imply a pathogenic role for the complement cascade and they represented an obvious trigger for the development of second‐generation complement inhibitors.
Table II.
Reasons for inadequate haematological response to anti‐C5 agents and possible actions.
| Cause | Cause | Prevalence | Mechanism | Clinical impact | Action |
|---|---|---|---|---|---|
| Intravascular haemolysis | Inherited C5 variants | Ultra‐rare (<1%, usually in Japanese patients) | Intrinsic resistance due to impaired binding of eculizumab (and of ALXN1210) | Minimal (but very significant for the few patients for whom there is no available treatment) | Switch to other investigational agents (mostly alternative C5 inhibitors) |
| Recurrent pharmacokinetic breakthrough | 10–15% of patients | Inadequate plasma level of eculizumab | Significant | Decrease interval of dosing (10–12 days) or increase dose of eculizumab (1 200 mg), or consider novel investigational agents | |
| Sporadic pharmacodynamic breakthrough | May occur in any patients | Massive complement activation due to concomitant clinical events | Irrelevant | None (treat the underlying cause) | |
| Extravascular haemolysis | C3‐mediated extra‐vascular haemolysis | 25–50% of patients (even more considering subclinical events) | Persistent uncontrolled activation of proximal complement, leading to C3‐fragment opsonization of PNH red blood cells and subsequent removal by professional hepato‐splenic phagocytes | Very significant | Consider investigational proximal inhibitors of the complement |
| Bone marrow disorders | Bone marrow failure | 10–35% (depending also on initial patient selection) | Inadequate production of red blood cells | Significant | Treat underlying aplastic anaemia with either immunosuppression or bone marrow transplantation |
| Clonal evolution to myeloid malignancies | 1–5% | Additional stochastic somatic mutations | Relevant | Treat the myeloid malignancy |
PNH, paroxysmal nocturnal haemoglobinuria.
Residual intravascular haemolysis is due to an incomplete C5 blockade, which can be chronic (leading to low‐grade continuous haemolysis), or more frequently transient (presenting as breakthrough haemolysis, Fig 2). The reasons accounting for this partial C5 blockade are not fully understood, with exception of the 15–20% of patients in whom recurrent breakthrough haemolysis is due to underdosage of eculizumab (pharmacokinetic breakthrough) 45 ; these patients usually benefit from either increased doses or decreased dosing intervals of eculizumab. 45 , 46
Fig 2.
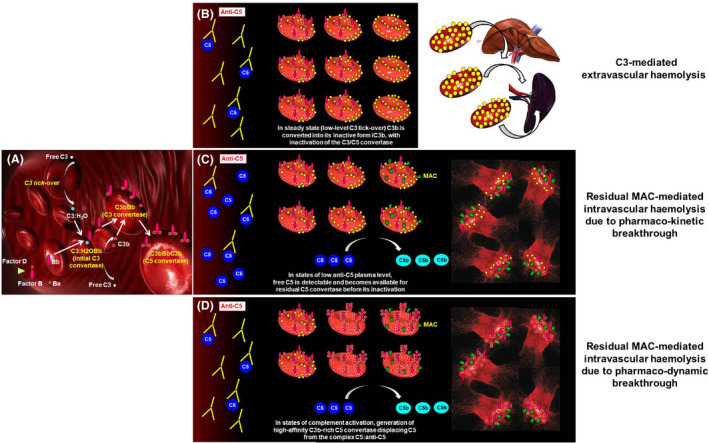
Biology of complement‐mediated haemolysis in PNH on terminal complement inhibitors. (A) Initial complement activation. C3:H2O generated by spontaneous hydrolysis of C3 (the so‐called “C3 tick‐over”) continuously initiates the complement cascade through its alternative pathway in the fluid phase. Due to the lack of CD55, PNH erythrocytes are unable to regulate complement activation on their surface, and C3bBb C3 convertase can be generated from C3 tick‐over and factor B cleavage operated by factor D. These C3 convertases generate further C3b, eventually self‐transforming into the C3bBbC3b C5 convertases. These steps are not affected by C5 inhibitors, which act downstream, making free C5 not available for the C5 convertases. (B) C3‐mediated extra‐vascular haemolysis. Terminal complement inhibitors (i.e. anti‐C5 agents) prevent the cleavage of C5 into C5a and C5b, thereby disabling the formation of the MAC and inhibiting intravascular lysis of PNH erythrocytes. Nevertheless, early steps of complement activation and upstream C5 cleavage remain uncontrolled, leading to opsonization of PNH erythrocytes with C3 fragments. C3‐opsonized erythrocytes can be recognised by C3‐specific receptors, expressed on professional macrophages in the liver and in the spleen, eventually resulting in extra‐vascular haemolysis. (C) Residual intravascular haemolysis due to pharmaco‐kinetic breakthrough. In the case of inadequate plasma levels of eculizumab (or any other anti‐C5 agent), free C5 becomes once again available for the C5 convertases. This eventually enables the terminal pathway of the complement, leading to MAC‐mediated residual intravascular haemolysis. (D) Residual intravascular haemolysis due to pharmaco‐dynamic breakthrough. During massive complement activation, C3 convertases may generate an excess of active forms of C3 (C3b), leading to the generation of “C3b‐rich” C5 convertases, which have higher affinity for C5. Thus, these high‐affinity C5 convertases may better compete with eculizumab for free C5, possibly displacing C5 from its inhibitor. This eventually enables the terminal pathway of the complement, leading to MAC‐mediated residual intravascular haemolysis. MAC, membrane attack complex; PNH, paroxysmal nocturnal haemoglobinuria.
Biomarkers of complement activity (i.e. CH50) 45 and some pharmacokinetic measurements (free eculizumab and/or free C5 plasma level) 45 , 47 may help to distinguish these patients from those experiencing pharmacodynamic breakthrough, 48 who usually have limited benefit from changes in the eculizumab treatment schedule. 49 However, these sophisticated assessments are not available in routine practice and the most useful laboratory parameter remains LDH, serially monitored before eculizumab dosing and at day 7 from administration. In addition to intravascular haemolysis, extra‐vascular haemolysis emerged as the most relevant factor affecting haematological benefit in PNH patients on eculizumab. 27 , 40 , 50 In PNH patients on eculizumab treatment, extra‐vascular haemolysis is an ineluctable biological phenomenon resulting from the inhibition of the terminal complement (at the level of C5), while PNH erythrocytes remain unable to control early phases of complement activation on their surface (Box 1) 51 , 52
Box 1. Complement biology in PNH.
Eculizumab results in a good control of MAC‐mediated intravascular haemolysis in all PNH patients, with the exception of those carrying rare C5 polymorphisms (mostly of Japanese ethnicity). 31 , 33 , 38
PNH patients on eculizumab (and other anti‐C5 agents) often remain anaemic, despite an adequate control of intravascular haemolysis. 41
Residual intravascular haemolysis due to suboptimal inhibition of C5 may contribute to residual anaemia, but in the majority of patients it does not seem the leading cause.
Residual intravascular haemolysis on eculizumab may be managed by increased doses of eculizumab, or novel anti‐C5 agents.
Underlying bone marrow failure may also contribute to residual anaemia in PNH, but it pertains to a minority of patients who have an indication for anti‐complement therapies (i.e. classic, haemolytic PNH).
C3‐mediated extra‐vascular haemolysis is the main cause of residual anaemia in PNH during anti‐C5 treatment. 40
C3‐mediated extra‐vascular haemolysis is due to the fact that the lack of the endogenous complement regulators CD55 and CD59 on PNH erythrocytes account for the uncontrolled generation of surface C3‐convertase and formation of MAC. Since eculizumab (and other inhibitors intercepting the complement cascade at the level of C5) disables MAC formation, but does not affect early steps of complement activation, surface C3‐convertase continues to generate C3 fragments on PNH erythrocytes, which are spared from MAC‐mediated intravascular haemolysis. These C3 fragments, albeit inactivated (C3b is quickly converted into C3d), 53 serve as opsonins on PNH erythrocytes, eventually promoting their phagocytosis from professional macrophages through their C3 receptors. 54
Inhibitors specifically targeting complement proteins involved in the initial phases of complement activation (upstream C5) and defined proximal complement inhibitors might counteract uncontrolled surface C3 activation occurring on PNH erythrocytes, possibly preventing the emergence of C3‐mediated extra‐vascular haemolysis. 52
Eculizumab targets C5, thereby preventing its C5 cleavage and the generation of the MAC (the effector mechanism of the terminal complement), eventually overcoming the lack of CD59 on PNH erythrocytes. 51 , 52 Nevertheless, PNH erythrocytes also lack CD55 on their surface; as a result, while with eculizumab they are protected from MAC‐mediated haemolysis, PNH erythrocytes continue to undergo chronic, continuous, surface C3 activation. 43 , 51 , 52 This uncontrolled activation leads to progressive deposition of C3 fragments, which may serve as opsonins on the red blood count surface, eventually leading to their removal by tissue macrophages in the spleen and liver. 51 , 52 Both residual intravascular haemolysis and C3‐mediated extra‐vascular haemolysis may benefit from novel therapeutic agents targeting different components of the complement cascade. 41
Specific issues on standard anti‐C5 complement inhibition
PNH, thrombophilia and possible treatments
Thrombophilia of PNH has been defined as “the most vicious acquired thrombophilic state known to medicine”, 51 representing the leading cause of death in PNH. 26 Different pathogenic mechanisms have been proposed to explain this condition, including platelet activation, pro‐thrombotic microparticles generated by haemolysis, impaired nitric oxide bio‐availability, fibrinolytic system impairment and inflammatory mediators. 18
The management of this thrombotic risk using primary prophylaxis with warfarin is still debated, as it may lead to incomplete thromboembolic protection 26 , 55 and a risk of major haemorrhages. 56 On the other hand, eculizumab has been proven effective to prevent thromboembolic complications in PNH 33 ; thus, for patients already on eculizumab, there is no reason to add a specific primary prophylaxis that would increase the risk of complications, without delivering any benefit.
In contrast, there is a broad consensus that any thrombotic event in PNH patients represents an absolute indication for immediate anti‐complement treatment whenever possible, as well as immediate full anti‐coagulation with heparin treatment. In cases of refractoriness to anti‐coagulation and anti‐complement therapies, in these life‐threatening conditions even thrombolytic therapy with tissue plasminogen activator may be considered. 55
Regarding secondary prophylaxis in the case of a thromboembolic event, the most effective treatment strategy nowadays is based on eculizumab 33 ; nevertheless, even in the era of anti‐complement agents, secondary prophylaxis with anti‐coagulants is still recommended. Some biological observations suggest that pathogenic alterations are not fully disabled during eculizumab treatment, eventually supporting this strategy. 57 , 58 Thus, a conservative approach seems conceivable, advising all PNH patients with a prior thromboembolic event to continue anti‐coagulation on top of anti‐complement treatment, unless there are clear contraindications.
Pregnancy and PNH in the era of eculizumab
Historically, the management of pregnancy in PNH women has been challenging, with counselling discouraging maternity in patients. 59 , 60 , 61 In fact, during pregnancy in PNH, intravascular haemolysis tends to become more severe, with more frequent paroxysms, eventually resulting in more severe anaemia and a higher transfusion requirement. 62 , 63 , 64 In addition to the worsening of symptoms, pregnancy in PNH also carries higher morbidity and mortality for both the fetus and the mother, 61 , 63 , 64 and such risk also extends into the post‐partum period. 65
In the eculizumab era, PNH women benefited from anti‐complement treatment, with major clinical improvements and resolution of more severe symptoms, eventually increasing the choice of motherhood. While pregnancy was not allowed within clinical trials, some cases of pregnancy occurred in real life, and taking into account the risk‐to‐benefit for the patients, treating physicians have not necessarily discontinued eculizumab treatment. 66 , 67 , 68
Subsequently, the topic of pregnancy in PNH during eculizumab treatment was addressed in an international study that investigated 75 pregnancies in 61 women occurring in the era of eculizumab treatment. 69 In this large series, there were no fatal events among the mothers, while foetal deaths were three (4%) and miscarriages during the first trimester were six (8%). These rates of morbidity and mortality were comparable with those in non‐PNH healthy women.
Nevertheless, pregnancy in these PNH women remained challenging, because both red blood cell and platelet transfusions were increased while pregnancies progressed. In 54% of pregnancies progressing past the first trimester, breakthrough haemolysis emerged, eventually requiring increased doses and/or reduced intervals of eculizumab. Low‐molecular‐weight heparin was used in 88% of pregnancies, resulting in 10 bleeding events and, during postpartum, two thromboembolic events. 69 Globally, 22 out of the 66 births (29%) were premature; 25 babies were breast‐fed. This study, albeit retrospective, clearly demonstrated that eculizumab leads to remarkable benefits for pregnant PNH women, with a very low rate of maternal complications and high rate of foetal survival. Nevertheless, even in the eculizumab era, pregnancy in PNH women requires the highest attention and a multi‐disciplinary team, including haematologists, gynaecologists and other specialists, is highly recommended.
PNH and haematopoietic stem cell transplantation
Haematopoietic stem cell transplantation (HSCT) remains the only curative treatment for PNH, but it is associated with high morbidity and mortality. 70 In a large retrospective study on 211 PNH patients transplanted between 1978 and 2007, the European Society for Blood and Marrow Transplantation reported 30% overall mortality, with an unacceptable higher risk of mortality in patients with previous thrombosis. 71 Thus, in the era of therapeutic complement inhibition, the role of HSCT seems restricted to a few indications for PNH patients.
Since alternative treatment options are absent, HSCT plays a major role for 5–10% of patients, progressing to a myeloid malignancy (myelodysplastic syndromes or acute myeloid leukaemia), even during anti‐complement treatment, 35 and in 15–20% of patients evolving to bone marrow failure. 34 , 35 In this setting, previous treatment with eculizumab may also affect transplant procedure: the French group has recently reported 21 PNH patients undergoing HSCT between 2007 and 2017, who have previously received eculizumab. 72 In these patients, eculizumab was continued until HSCT, with the recommendation of delivering the last eculizumab dose during the conditioning regimen.
Nevertheless, even in this recent cohort, and despite bridge‐to‐transplant eculizumab treatment, overall mortality was around 30%, mainly due to graft‐versus‐host disease and infectious complications. 72 Based on these data, HSCT should not be considered a reliable treatment option for the rare patients (2–4%) 35 with recurrent thrombosis during eculizumab treatment, and even more so for the 34–51% of patients with clinically meaningful anaemia, despite eculizumab. 27 , 31 , 33 , 39 , 40 , 41 For all these patients there are obvious unmet clinical needs that hopefully may be addressed by the second generation of complement inhibitors.
Iron overload and eculizumab
The spontaneous iron loss through urine, seen as haemoglobinuria or haemosiderinuria, is a peculiar feature of PNH: this is a quite unique circumstance that patients receive large numbers of transfusions without developing iron overload. One of the consequences of eculizumab in PNH is that this somehow protective mechanism is lost due to the inhibition of intravascular haemolysis. In addition, most patients develop a meaningful extra‐vascular haemolysis, which results in substantial changes in iron homeostasis, with possible iron overload, even in the absence of regular transfusions. 73 While the mechanisms of iron overload in PNH on anti‐C5 treatment need to be fully elucidated, regular monitoring of ferritin and transferrin saturation are very important to identify PNH patients who may require therapeutic iron chelation. 74
Second‐generation anti‐complement agents
Several novel anti‐complement agents have been described 75 and are now in clinical development for PNH (Table III). 76 Here we focus on the most advanced programmes, for which phase II and some phase III results are publicly available, as they represent already possible treatment options for the specific PNH patient population, within clinical trials and hopefully even in standard practice. For the purpose of this discussion, novel agents are grouped into terminal complement inhibitors (i.e. C5 inhibitors similar to eculizumab) and proximal complement inhibitors (i.e. those intercepting the complement cascade upstream of C5). Their mechanism of action and their preclinical data are reviewed elsewhere. 41 , 75 , 76
Table III.
Complement inhibitors in clinical development for paroxysmal nocturnal haemoglobinuria.
| Class | Agent | Target | Clinical trial ID | Design | Patient population* | Study treatment | Results |
|---|---|---|---|---|---|---|---|
| Terminal inhibitors | Ravulizumab | C5 | NCT02598583 | Phase I/II, open‐label | Untreated PNH | Intra‐patient DE by IV infusions | Yes 95 |
| NCT02605993 | Phase I/II, open‐label | Untreated PNH | MAD; IV infusions | ||||
| NCT02946463 | Phase III, randomized versus Ecu | Untreated PNH | IV infusions (every eight weeks) | Yes 47 , 48 , 96 | |||
| NCT03056040 | Phase III, randomized versus Ecu | Stable responders PNH | IV infusions (every eight weeks) | Yes 47 , 48 , 79 | |||
| Crovalimab | C5 | NCT03157635 | Phase I/II, multi‐part study | Untreated PNH and stable responders PNH | Intra‐patient DE by IV infusions, followed by SC injections | Yes 83 | |
| LFG316 (tesidolumab) | C5 | NCT02534909 | Phase II, open‐label | Untreated PNH | IV infusions | Pending | |
| REGN3918 (pozelimab) | C5 | NCT03946748 | Phase II, open‐label, POC | Untreated PNH | IV and SC infusions | Pending | |
| NCT04162470 | Phase II, open‐label, extension | Pozelimab‐treated PNH | IV and SC infusions | Ongoing | |||
| ABP959 | C5 | NCT03818607 | Phase III, randomized versus Ecu | Stable responders PNH | IV infusions | Ongoing | |
| Elizaria | C5 | NCT04463056 | Phase III, randomized versus Ecu | Untreated and eculizumab‐treated PNH | IV infusions | Pending | |
| Proximal inhibitors | Pegcetacoplan | C3 | NCT02264639 | Phase Ib, open label, MAD, POC | Poor responders PNH | Daily, SC infusions | Yes 86 |
| NCT02588833 | Phase Ib, open label, MAD, POC | Untreated PNH | Daily, SC infusions | ||||
| NCT03531255 | Phase III, open label, extension | PNH exposed to APL‐2 | Daily, SC infusions | ||||
| NCT03500549 | Phase III, randomized versus Ecu | Poor responders PNH | SC infusions, BIH | Yes 87 | |||
| Danicopan | FD | NCT03053102 | Phase Ib, open label, MD, POC | Untreated PNH | Orally, TID | Yes 89 | |
| NCT03181633 | Phase II, open‐label, extension | PNH exposed to ACH‐4471 | Orally, TID | Ongoing | |||
| NCT03472885 | Phase II, open label, MD, POC | Poor responders PNH | Orally, TID | Yes 90 | |||
| NCT04469465 | Phase III, randomized versus Ecu | Phase III, randomized vs Ecu | Orally, TID | Ongoing | |||
| ACH5020 | FD | NCT04170023 | Phase II, open label, POC | Danicopan‐treated PNH, poor‐responders to anti‐C5 and untreated PNH | Orally, BID | Ongoing | |
| BCX9930 | FD | NCT04330534 | Phase I–II | PNH untreated | Orally, BID | Pending | |
| NCT04702568 | Phase II, open label, extension | PNH, BCX9930‐treated | Orally, BID | Ongoing | |||
| Iptacopan | FB | NCT03439839 | Phase II, open label, POC | Poor responders PNH | Orally, BID | Yes 93 | |
| NCT03896152 | Phase II, open label, POC | Untreated PNH | Orally, BID | Pending | |||
| NCT04558918 | Phase III, randomized versus Ecu | Poor responders PNH | Orally, BID | Ongoing |
BID, bis in die (twice a day); BIH, bis in hebdomade (twice a week); DE, dose escalation; Ecu, eculizumab; IV, intravenous; LDH, lactate dehydrogenase; MAD, multiple ascending doses; MD, multiple doses; PNH, paroxysmal nocturnal haemoglobinuria; POC, proof‐of‐concept; SC, subcutaneous; TID, ter in die (thrice a day).
Stable or poor response is intended to standard eculizumab treatment.
New terminal complement inhibitors
Eculizumab has revolutionized the field but still requires intravenous (IV) injection every two weeks. The need for time‐ and resource‐consuming clinic visits for patients or nurse visits at home for lifelong IV administration are limitations for these mostly young and very active patients. The next generation of terminal complement blockers (Table III) mostly aim to delay clinical visits, with a longer treatment interval or different drug formulation (subcutaneous dosing) allowing treatment independent of the clinical setting.
Ravulizumab (ALXN1210) paved the way for these new long‐acting C5 inhibitors. Four amino acid substitutions in the complementarity‐determining and fragment‐crystallizable (Fc) regions of eculizumab result in ravulizumab’s enhanced endosomal dissociation of C5 and recycling to the vascular compartment through the neonatal Fc receptor pathway. These modifications allow a sustained C5 inhibition with eight‐week dosing intervals. 77
In two large phase III studies in patients who were naive to 78 or previously treated with eculizumab, 79 ravulizumab was shown to be non‐inferior to eculizumab, according to normalization and percentage change in LDH, transfusion avoidance, rates of breakthrough haemolysis and haemoglobin stabilization during the 26‐week primary evaluation period. Improvements in fatigue and quality of life outcomes were also comparable between eculizumab and ravulizumab. 78 , 79 Open‐label extension of phase III studies demonstrated durable efficacy and consistent safety. 80 , 81 Ravulizumab has been approved by the US Food and Drug Administration (FDA) and should also be available soon in most European countries.
Crovalimab (RO7112689 or SKY59; Chugai Pharmaceutical, Tokyo, Japan) is a sequential, highly soluble monoclonal antibody recycling technology (SMART) antibody, allowing for small injection volumes. 82 Crovalimab was engineered for extended self‐administered subcutaneous dosing of small volumes in diseases eligible for C5 inhibition. Moreover, epitope recognition of C5 by crovalimab is different from eculizumab and ravulizumab, which allow binding on a single missense C5 heterozygous mutation (c.2654G→A, single nucleotide polymorphism) present in ∼3% of the Japanese population and associated with a lack of response to eculizumab. 38
A three‐part open‐label adaptive phase I/II trial was conducted to assess safety, pharmacokinetics, pharmacodynamics and exploratory efficacy in healthy volunteers (part 1), as well as in complement blockade‐naive (part 2) and C5 inhibitor‐treated (part 3) PNH patients. Subcutaneous crovalimab was shown to provide complete and sustained terminal complement pathway inhibition in patients with PNH on a monthly injection basis, with no safety concerns, except possible self‐limiting vasculitis‐like symptoms, associated with drug‐target‐drug immune complexes observed in patients switching from eculizumab to crovalimab. 83 Two large phase III studies in patients previously treated with crovalimab (COMMODORE 1; ClinicalTrials.gov Identifier: NCT04432584) and in patients who were naive to standard complement inhibitors (COMMODORE 2; NCT04434092) are ongoing to confirm its efficacy and safety.
Other compounds are in development with no or few data published yet. Pozelimab (REGN3918; Regeneron, Tarrytown, NY, USA), targeting C5, is injected subcutaneously on a weekly basis after an initial IV loading dose. A proof‐of‐concept study to assess the efficacy, safety and pharmacokinetics of tesidolumab (LFG316; Novartis, Basel, Switzerland; NCT02534909), an anti‐C5 antibody in patients with PNH, has never been published. Anti‐C5 biosimilars are also under development, the most mature being elizaria (eculizumab; Generium, Moscow, Russia) approved in Russia.
New proximal complement inhibitors
Proximal complement inhibitors were specifically designed aiming to prevent C3‐mediated extra‐vascular haemolysis; however, depending on their specific mechanism of action, a possible action on residual intravascular haemolysis was also hypothesized (Fig 3). At the moment, three classes of agents are in clinical development, targeting three different components of the complement pathway: (i) complement C3; (ii) complement factor D; (iii) complement factor B. Here we provide a short summary of the recent data from phase II and phase III studies.
Fig 3.
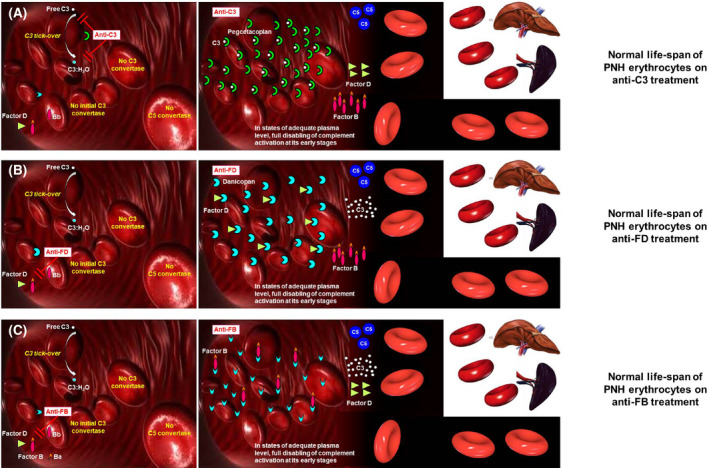
Biology of complement‐mediated haemolysis in PNH on proximal complement inhibitors. (A) Modulation of complement activation on PNH erythrocytes on C3 inhibitors. The C3 inhibitor pegcetacoplan binds C3 in its naive and activated forms, eventually preventing the generation of C3 convertases on the surface of PNH erythrocytes. If the inhibition is pharmacologically sustained, the complement cascade is disabled in its early phases, resulting in inhibition of the MAC‐mediated intravascular haemolysis, and in the prevention of C3 opsonization (and thus of extra‐vascular haemolysis). (B) Modulation of complement activation on PNH erythrocytes on factor D inhibitors. The factor D inhibitor danicopan binds factor D, eventually preventing the cleavage of factor B, which is needed to generate C3 convertases. If the inhibition is pharmacologically sustained, the complement cascade is disabled in its early phases, resulting in inhibition of the MAC‐mediated intravascular haemolysis, and in the prevention of C3 opsonization (and thus of extra‐vascular haemolysis). (C) Modulation of complement activation on PNH erythrocytes on anti‐factor B inhibitors. The factor B inhibitor iptacopan binds factor B, eventually preventing its cleavage needed to generate C3 convertases. If the inhibition is pharmacologically sustained, the complement cascade is disabled in its early phases, resulting in inhibition of the MAC‐mediated intravascular haemolysis, and in the prevention of C3 opsonization (and thus of extra‐vascular haemolysis). MAC, membrane attack complex; PNH, paroxysmal nocturnal haemoglobinuria.
Therapeutic C3 inhibition is based on the small peptide compstatin and its derivatives. Pegcetacoplan is a PEGylated version of compstatin which binds to C3, thereby preventing complement activation along all its pathways. 84 The potential of C3 inhibitors in PNH has been investigated both in vitro 85 and in vivo in a phase II study. 86 The PEGASUS study, a phase III, open‐label, randomized trial, investigated efficacy and safety of pegcetacoplan, versus eculizumab, in adults with PNH and haemoglobin (Hb) < 105 g/l on eculizumab therapy. 87 Patients were randomized to subcutaneous pegcetacoplan monotherapy (n = 41) or intravenous eculizumab (n = 39), after a four‐week run‐in with concomitant pegcetacoplan and eculizumab.
At week 16, pegcetacoplan demonstrated superiority to eculizumab in haemoglobin change from baseline, with an adjusted mean treatment difference of 38·4 g/l (P < 0·0001). Transfusion avoidance was observed in 35/41 (85·4%) pegcetacoplan arms, compared with 6/39 (15·4%) with eculizumab. Non‐inferiority was demonstrated for change in absolute reticulocyte count, but not for LDH. The most common treatment‐emergent adverse events in the pegcetacoplan and eculizumab groups respectively, were injection site reactions (36·6% vs 2·6%), diarrhoea (22·0% vs 2·6%), headache (7·3% vs 23·1%), and fatigue (4·9% vs 15·4%). No cases of meningitis or of severe infection were observed.
These data demonstrate that pegcetacoplan monotherapy was effective in inhibiting intravascular haemolysis of PNH patients and in preventing the emergence of C3‐mediated extra‐vascular haemolysis, eventually resulting in a better haematological response. 87 With follow‐up extended to 48 weeks, patients randomized to pegcetacoplan maintained a high haemoglobin level (mean haemoglobin level was 113 g/l), while patients randomized to eculizumab who switched to pegcetacoplan during the open‐label period demonstrated a significant improvement compared with the baseline (mean haemoglobin increase was 29 g/l). 88
During the extension period, the rate of patients who remained transfusion‐independent was overlapping between the two treatment arms (73% vs 72% in pegcetacoplan–pegcetacoplan and eculizumab–pegcetacoplan arms respectively). No safety alert emerged with this longer follow‐up, even if 12 of the 80 patients discontinued pegcetacoplan, of whom six due to breakthrough haemolysis. Pegcetacoplan is now approved by the FDA for patients with PNH who are either treatment‐naive or switching from anti‐C5 monoclonal antibodies.
The first‐in‐class factor D (FD) inhibitor is danicopan, an oral proximal, complement alternative pathway inhibitor, which was investigated in two open‐label, single‐arm, phase II studies in eculizumab‐poor responders and untreated PNH patients. 89 , 90 In the poor‐responder study, danicopan was given as an add‐on treatment at the dose of 100–200 thrice daily to 12 transfusion dependent PNH patients on eculizumab. 90 Twelve patients received at least one danicopan dose; one discontinued from a serious adverse event, deemed unlikely related to danicopan. In the 11 patients who completed the treatment (one early discontinuation due to a serious adverse event, unlikely related to danicopan), the addition of danicopan resulted in a mean 24 g/l haemoglobin increase at week 24.
The transfusion requirement during the 24 weeks of treatment was one transfusion (two units) in one patient, compared with 10 patients receiving 34 transfusions (58 units) in the 24 weeks prior to danicopan. The most common adverse event was a headache. Thus, the addition of danicopan led to a meaningful improvement in haemoglobin and reduced the transfusion requirement in PNH patients who were transfusion‐dependent on eculizumab. 90
In the untreated PNH study, 10 untreated haemolytic PNH patients received danicopan in monotherapy (100–200 mg thrice daily). 89 All patients reached the primary end‐point and eight completed the treatment (two discontinued: one for a serious adverse event and one for personal reasons unrelated to safety). Danicopan was proven effective in preventing intravascular haemolysis, as demonstrated by mean decreased LDH [5.7 × upper limit of normal (ULN) at baseline vs 1.8 × ULN (day 28) and 2·2 × ULN (day 84); both P < 0·001]. This was associated with meaningful improvement of haemoglobin levels, with increases from baseline (98 g/l) of 11 (day 28) and 17 (day 84) g/l (both P < 0·005). No significant C3 fragment deposition occurred on GPI‐deficient erythrocytes. Most common adverse events were headaches and upper respiratory tract infections.
In this phase II study, danicopan monotherapy resulted in the clinically meaningful inhibition of intravascular haemolysis, without evidence of C3‐mediated extra‐vascular haemolysis, leading to haemoglobin improvement in untreated PNH patients. 89 Danicopan is now under investigation in a phase III study enrolling poor responders to eculizumab; in parallel, a novel analogue with better pharmacokinetic and pharmacodynamic properties is currently being tested in a phase II study, again in poor responders to eculizumab.
Another FD inhibitor in early development is BCX9930, which is given orally twice a day at doses up to 500 mg. Preliminary data show clinical efficacy both as add‐on treatment in PNH patients with inadequate response to eculizumab, 91 as well as in treatment‐naive patients, in monotherapy. 92 Therapeutic inhibition of factor B (FB) exploits iptacopan (previously known as LNP023), an oral, selective and potent first‐in‐class FB inhibitor which was investigated, in a multicentre, open‐label phase II trial enrolling PNH patients with active haemolysis on eculizumab. 93 Iptacopan was initially given as add‐on therapy at the dose of 200 mg twice a day, until the primary end‐point at week 13 was reached; then patients entered into a long‐term extension study, which allowed modification and even discontinuation of eculizumab treatment.
Iptacopan was well tolerated, with no treatment discontinuation or treatment‐related serious adverse event recorded in the 10 patients enrolled. At week 13, iptacopan resulted in a marked reduction of LDH (539 ± 263 vs 245 ± 54 iu/l; P = 0·006), associated with significant improvement of haemoglobin levels (97·7 ± 105 vs 126·3 ± 18·5 g/l; P < 0·001), with 8/10 patients achieving resolution of anaemia. All biomarkers of haemolysis improved on iptacopan treatment, demonstrating substantial efficacy on both intravascular haemolysis and extra‐vascular haemolysis, as confirmed by abrogation of C3 deposition and increase of PNH erythrocyte population.
Observed haematological benefits were maintained longer, throughout the study extension, including seven patients who stopped discontinued eculizumab and continued iptacopan in monotherapy. These data demonstrated that in poor responders to eculizumab, iptacopan, at a chronic dose of 200 mg twice a day, was able to better control intravascular haemolysis and to prevent C3‐mediated extra‐vascular haemolysis, leading to remarkable improvement of haemoglobin levels, even in monotherapy. 93
Additional data on the use of iptacopan in monotherapy come from an ongoing phase II study enrolling treatment‐naive PNH patients. Preliminary data from 13 patients who have been randomized to different doses of iptacopan (25 mg, followed by 100 mg twice a day or 50 mg followed by 200 mg twice a day) demonstrated control of intravascular haemolysis in all treated patients. 94 Looking at biomarkers of haemolysis and to haemoglobin level, the dose of 200 mg twice a day showed the highest efficacy and it has been selected for further studies. Indeed, iptacopan is now be tested in pivotal trials to investigate its haematological benefit, in monotherapy, both in PNH patients, poor responders to anti‐C5 treatment (NCT04558918) or those that are naive to anti‐complement therapy (NCT04820530).
Emerging questions in the era of proximal complement inhibitors
The introduction of eculizumab for the treatment of PNH led to the description of C3‐mediated extra‐vascular haemolysis as a novel mechanism of disease in PNH, unmasked by anti‐C5 therapy (Box 1). 40 Now the clinical use of proximal complement inhibitors is raising different questions about our understanding of complement biology in PNH, eventually identifying possible pitfalls of this novel treatment strategy (Box 2).
Box 2. Pitfalls and caveats of novel anti‐complement therapies in PNH.
Novel anti‐C5 agents seem as effective as eculizumab, with possible advantages related to longer dosing intervals and easier administration routes (i.e. subcutaneous), which result in more convenient treatment options for patients and care providers.
Novel anti‐C5 agents may have better pharmacological properties, possibly allowing for a deeper C5 inhibition; some data suggest that in some cases this may reduce the risk of pharmacokinetic breakthrough haemolysis, while episodes of pharmacodynamic breakthrough haemolysis associated with complement‐amplifying conditions seem unaffected.
The safety profile of novel anti‐C5 agents seems consistent with that of eculizumab, with the exception of transient auto‐immune manifestations, due to drug‐target‐drug immune complexes in patients switching from eculizumab to other anti‐C5 antibodies targeting different C5 epitopes.
Novel anti‐C5 agents, intrinsically, cannot address the unmet clinical need of C3‐mediated extra‐vascular haemolysis.
Proximal complement inhibitors targeting different complement components (Factor B, Factor D and C3) have been safely used in PNH patients; they have easy administration routes (oral or subcutaneous), but a short half‐life, requiring daily or bi‐weekly administration.
The safety profile of proximal complement inhibitors is consistent with that of anti‐C5 agents, even if longer follow‐up is needed to rule out possible impact on the risk of infectious complications, auto‐immune diseases, as well as cancer.
Given the broader impairment of the complement cascade associated with proximal complement inhibitors, anti‐infectious risk mitigation strategies include a broader vaccination schedule, as well as possible pharmacological anti‐microbial prophylaxis.
Once added to anti‐C5 therapies, all proximal complement inhibitors were proven effective in preventing C3‐mediated extra‐vascular haemolysis, eventually leading to a higher haematological benefit in PNH patients.
Proximal complement inhibitors, again added to anti‐C5 therapies, also improved the depth of the control of intravascular haemolysis (likely reducing the activation of the terminal pathway through the inhibition of the complement amplification loop).
The efficacy of proximal complement inhibitors may vary depending on pharmacological properties of the specific agent, likely due to the capability of achieving sustained and full inhibition of their specific molecular targets.
For some proximal complement inhibitors, available data seem to support that they can be used even in monotherapy; however, further data are needed to understand whether in monotherapy the efficacy remains as high as in combination treatment with anti‐C5.
The tremendous efficacy of proximal complement inhibitors is demonstrated not only by improvements in all biomarkers of haemolysis, but also by the large increase of the proportion of erythrocytes with the PNH phenotype.
PNH with such large PNH erythrocyte populations is a novel condition, which might be characterized by a peculiar clinical course and possible complications.
The risk of breakthrough haemolysis emerges as an obvious concern during treatment with proximal complement inhibitors, especially in monotherapy. Such a risk seems highly different among the agents in clinical development, depending on their pharmacological properties, which shape the risk of possible leakage in their inhibition. Current data seem to demonstrate that proximal complement inhibitors may successfully prevent haemolysis, even with such a large PNH erythrocyte population, but breakthrough episodes may develop both in the case of subtherapeutic drug level, or in the presence of complement‐amplifying conditions. Indeed, the inhibitors with the most favourable therapeutic window and with the deepest inhibition of their target, allowing full and sustained inhibition, seem to have the highest chance to be used in monotherapy. Nevertheless, further studies are needed to describe the risk and the mechanisms of breakthrough haemolysis during treatment with proximal complement inhibitors, as well as to establish adequate risk mitigation strategies for its management or prevention.
Recent data have clearly shown that, irrespective of the specific target and of the specific agent, we may improve the control of intravascular haemolysis and may prevent extra‐vascular haemolysis, eventually leading to a greater haematological benefit. Indeed, once clinically meaningful intravascular haemolysis is controlled (as demonstrated by LDH < 1.5 × ULN),an increase of haemoglobin seems the most reasonable goal of PNH treatment.
Most of these data were achieved combining proximal inhibitors with an anti‐C5 agent, but all proximal inhibitors seem very effective, apparently better than anti‐C5 agents, even in monotherapy. Nevertheless, data looking systematically at the difference between combination treatment and monotherapy are limited, and it is not clear whether we lose anything in terms of haematological response by using proximal inhibitors in monotherapy.
Some of the proximal complement inhibitors, such as pegcetacoplan 87 and iptacopan, 93 seem very effective, even in monotherapy, eventually leading to a proportion of PNH erythrocytes as high as >90%, which likely demonstrates a normal life span of PNH erythrocytes. We have to acknowledge that PNH with such large GPI‐deficient erythrocyte populations is a new condition that could not be found in the natural history of the disease, nor during anti‐C5 treatment.
This PNH may represent a novel disease, with possible peculiar presentations that we still have to learn. For instance, such a large mass of erythrocytes susceptible to complement‐mediated lysis raises obvious concerns about the risk of breakthrough haemolysis, even with severe clinical presentation. Available data about breakthrough haemolysis with proximal complement inhibitors are still limited, but we may start by making some observations. For instance, the presence of very high (>95%) PNH erythrocyte populations does not seem a risk for breakthrough haemolysis per se, since many patients harbour such large clones without even laboratory signs of haemolysis.
On the other hand, in this condition, breakthrough haemolysis may appear as a consequence of subtherapeutic doses of the inhibitor, or in the presence of some complement‐amplifying condition, such as infections, which may even overcome therapeutic levels of the inhibitor. When such breakage in complement inhibition occurs, clinical presentation may be severe due to the large mass of erythrocytes susceptible to lysis, and therapeutic interventions are needed, for instance adding, even transiently, a terminal complement inhibitor, or adjusting the dose of the proximal inhibitor, that is in the presence of proven subtherapeutic levels of the inhibitor, or of missed doses.
In contrast, subclinical haemolysis seen as a chronic mild increase of LDH may be associated to some leakage in complement inhibition, and may not require any therapeutic interventions. Future studies will have to address how deep and how sustained complement inhibition is with each of these novel agents to fully understand if they can be safely used in monotherapy.
Conclusions
In conclusion, PNH remains a disease with a pleiotropic clinical presentation and its treatment should be based on the actual clinical presentation: physicians have to assess haemolysis and bone marrow function, as well as the thrombotic complications, before choosing the most appropriate treatment. The hallmark of PNH is complement‐mediated haemolytic anaemia — we should not label any condition as PNH in the absence of meaningful haemolysis. Even if we have to acknowledge that anti‐complement treatment will never be a cure for PNH, it is obvious that anti‐complement treatment is the only aetiologic treatment for PNH. After about 15 years of eculizumab treatment, we know that life expectancy may be almost normal, with major improvement of most disease signs and symptoms.
Nevertheless, very long‐term outcome is required, unmet clinical needs still remain, and room for improvements is vast. We are learning that we can set our bar higher: haemoglobin can be normalized in most PNH patients, even with more convenient treatments that do not require frequent hospitalizations (i.e. orally or subcutaneously available). While clinical trials are on their way to investigate safety and efficacy of individual agents, possibly in head‐to‐head comparison, the message for treating physicians seems obvious: more effective and more convenient treatments will not necessarily mean that everything is solved, since the management of this unique disease will still require specific expertise and continuous efforts to unravel the knowledge of such a mysterious and fascinating illness.
Author contributions
The two authors equally contributed to the preparation of this manuscript drafting the main text, tables and figures.
Conflicts of interest
AMR has received research support from Alexion, Novartis, Alnylam and Rapharma, lecture fees from Alexion, Novartis, Pfizer and Apellis, served as a member of the advisory/investigator board for Alexion, Roche, Achillion, Novartis, Apellis, Biocryst and Samsung, and served as consultant for Amyndas. RPdL has received research funding from Alexion, Amgen, Jazz Pharmaceuticals and Pfizer; consulted for and received honoraria from Alexion, Amgen, Gilead, Jazz Pharmaceuticals, Keocyte, MSD, Novartis, Pfizer, Roche, Samsung and Mallinckrodt.
Acknowledgements
The authors wish to thank the Italian PNH Patient Association (AIEPN), the HPN France/Aplastic Anaemia association, as well as all PNH patients and their families. Open Access Funding provided by Universita degli Studi di Napoli Federico II within the CRUI‐CARE Agreement. [Correction added on 26 May 2022, after first online publication: CRUI funding statement has been added.]
References
- 1. Risitano AM. Paroxysmal nocturnal hemoglobinuria. In: Silverberg DS, editor. Anemia. Rijeka: InTech; 2012. p. 331–74. [Google Scholar]
- 2. Takeda J, Miyata T, Kawagoe K, Iida Y, Endo Y, Fujita T, et al. Deficiency of the GPI anchor caused by a somatic mutation of the PIG‐A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73(4):703–11. [DOI] [PubMed] [Google Scholar]
- 3. Bessler M, Mason PJ, Hillmen P, Miyata T, Yamada N, Takeda J, et al. Paroxysmal nocturnal haemoglobinuria (PNH) is caused by somatic mutations in the PIG‐A gene. EMBO J. 1994;13(1):110–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Miyata T, Yamada N, Iida Y, Nishimura J, Takeda J, Kitani T, et al. Abnormalities of PIG‐A transcripts in granulocytes from patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1994;330(4):249–55. [DOI] [PubMed] [Google Scholar]
- 5. Rotoli B, Luzzatto L. Paroxysmal nocturnal haemoglobinuria. Bailliere's Clin Haematol. 1989;2(1):113–38. [DOI] [PubMed] [Google Scholar]
- 6. Luzzatto L, Notaro R. The, "escape" model: a versatile mechanism for clonal expansion. Br J Haematol 2018;184(3):465–6. [DOI] [PubMed] [Google Scholar]
- 7. Luzzatto L, Risitano AM. Advances in understanding the pathogenesis of acquired aplastic anaemia. Br J Haematol. 2018;182(6):758–76. [DOI] [PubMed] [Google Scholar]
- 8. Mahoney JF, Urakaze M, Hall S, DeGasperi R, Chang HM, Sugiyama E, et al. Defective glycosylphosphatidylinositol anchor synthesis in paroxysmal nocturnal hemoglobinuria granulocytes. Blood. 1992;79(6):1400–3. [PubMed] [Google Scholar]
- 9. Stafford HA, Tykocinski ML, Lublin DM, Holers VM, Rosse WF, Atkinson JP, et al. Normal polymorphic variations and transcription of the decay accelerating factor gene in paroxysmal nocturnal hemoglobinuria cells. Proc Natl Acad Sci USA. 1988;85(3):880–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hirose S, Ravi L, Hazra SV, Medof ME. Assembly and deacetylation of N‐acetylglucosaminyl‐plasmanylinositol in normal and affected paroxysmal nocturnal hemoglobinuria cells. Proc Natl Acad Sci USA. 1991;88(9):3762–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Takahashi M, Takeda J, Hirose S, Hyman R, Inoue N, Miyata T, et al. Deficient biosynthesis of N‐acetylglucosaminyl‐phosphatidylinositol, the first intermediate of glycosyl phosphatidylinositol anchor biosynthesis, in cell lines established from patients with paroxysmal nocturnal hemoglobinuria. J Exp Med. 1993;177(2):517–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nicholson‐Weller A, March JP, Rosenfeld SI, Austen KF. Affected erythrocytes of patients with paroxysmal nocturnal hemoglobinuria are deficient in the complement regulatory protein, decay accelerating factor. Proc Natl Acad Sci USA. 1983;80(16):5066–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nicholson‐Weller A, Burge J, Fearon DT, Weller PF, Austen KF. Isolation of a human erythrocyte membrane glycoprotein with decay‐accelerating activity for C3 convertases of the complement system. J Immunol. 1982;129(1):184–9. [PubMed] [Google Scholar]
- 14. Nicholson‐Weller A. Decay accelerating factor (CD55). Curr Top Microbiol Immunol. 1992;178:7–30. [DOI] [PubMed] [Google Scholar]
- 15. Holguin MH, Fredrick LR, Bernshaw NJ, Wilcox LA, Parker CJ. Isolation and characterization of a membrane protein from normal human erythrocytes that inhibits reactive lysis of the erythrocytes of paroxysmal nocturnal hemoglobinuria. J Clin Investig. 1989;84(1):7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Holguin MH, Wilcox LA, Bernshaw NJ, Rosse WF, Parker CJ. Relationship between the membrane inhibitor of reactive lysis and the erythrocyte phenotypes of paroxysmal nocturnal hemoglobinuria. J Clin Investig. 1989;84(5):1387–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lachmann PJ, Halbwachs L. The influence of C3b inactivator (KAF) concentration on the ability of serum to support complement activation. Clin Exp Immunol. 1975;21(1):109–14. [PMC free article] [PubMed] [Google Scholar]
- 18. Hill A, Kelly RJ, Hillmen P. Thrombosis in paroxysmal nocturnal hemoglobinuria. Blood. 2013;121(25):4985–96, quiz 5105. [DOI] [PubMed] [Google Scholar]
- 19. Rosse WF, Dacie JV. Immune lysis of normal human and paroxysmal nocturnal hemoglobinuria (PNH) red blood cells. I. The sensitivity of PNH red cells to lysis by complement and specific antibody. J Clin Investig. 1966;45(5):736–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ham TH. Chronic hemolytic anemia with paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1937;217:915–7. [Google Scholar]
- 21. Dezern AE, Borowitz MJ. ICCS/ESCCA consensus guidelines to detect GPI‐deficient cells in paroxysmal nocturnal hemoglobinuria (PNH) and related disorders part 1 ‐ clinical utility. Cytometry B Clin Cytom. 2018;94(1):16–22. [DOI] [PubMed] [Google Scholar]
- 22. Sutherland DR, Illingworth A, Marinov I, Ortiz F, Andreasen J, Payne D, et al. ICCS/ESCCA consensus guidelines to detect GPI‐deficient cells in paroxysmal nocturnal hemoglobinuria (PNH) and related disorders part 2 ‐ reagent selection and assay optimization for high‐sensitivity testing. Cytometry B Clin Cytom. 2018;94(1):23–48. [DOI] [PubMed] [Google Scholar]
- 23. Illingworth A, Marinov I, Sutherland DR, Wagner‐Ballon O, DelVecchio L. ICCS/ESCCA consensus guidelines to detect GPI‐deficient cells in paroxysmal nocturnal hemoglobinuria (PNH) and related disorders part 3 ‐ data analysis, reporting and case studies. Cytometry B Clin Cytom. 2018;94(1):49–66. [DOI] [PubMed] [Google Scholar]
- 24. Oldaker T, Whitby L, Saber M, Holden J, Wallace PK, Litwin V. ICCS/ESCCA consensus guidelines to detect GPI‐deficient cells in paroxysmal nocturnal hemoglobinuria (PNH) and related disorders part 4 ‐ assay validation and quality assurance. Cytometry B Clin Cytom. 2018;94(1):67–81. [DOI] [PubMed] [Google Scholar]
- 25. Parker C, Omine M, Richards S, Nishimura J, Bessler M, Ware R, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106(12):3699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Latour RP, Mary JY, Salanoubat C, Terriou L, Etienne G, Mohty M, et al. Paroxysmal nocturnal hemoglobinuria: natural history of disease subcategories. Blood. 2008;112(8):3099–106. [DOI] [PubMed] [Google Scholar]
- 27. Luzzatto L, Gianfaldoni G, Notaro R. Management of paroxysmal nocturnal haemoglobinuria: a personal view. Br J Haematol. 2011;153(6):709–20. [DOI] [PubMed] [Google Scholar]
- 28. Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol. 2007;25(11):1256–64. [DOI] [PubMed] [Google Scholar]
- 29. Hillmen P, Hall C, Marsh JCW, Elebute M, Bombara MP, Petro BE, et al. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2004;350(6):552–9. [DOI] [PubMed] [Google Scholar]
- 30. Hillmen P, Young NS, Schubert J, Brodsky RA, Socié G, Muus P, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355(12):1233–43. [DOI] [PubMed] [Google Scholar]
- 31. Brodsky RA, Young NS, Antonioli E, Risitano AM, Schrezenmeier H, Schubert J, et al. Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood. 2008;111(4):1840–7. [DOI] [PubMed] [Google Scholar]
- 32. Hillmen P, Elebute M, Kelly R, Urbano‐Ispizua A, Hill A, Rother RP, et al. Long‐term effect of the complement inhibitor eculizumab on kidney function in patients with paroxysmal nocturnal hemoglobinuria. Am J Hematol. 2010;85(8):553–9. [DOI] [PubMed] [Google Scholar]
- 33. Hillmen P, Muus P, Dührsen U, Risitano AM, Schubert J, Luzzatto L, et al. Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2007;110(12):4123–8. [DOI] [PubMed] [Google Scholar]
- 34. Kelly RJ, Hill A, Arnold LM, Brooksbank GL, Richards SJ, Cullen M, et al. Long‐term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival. Blood. 2011;117(25):6786–92. [DOI] [PubMed] [Google Scholar]
- 35. Loschi M, Porcher R, Barraco F, Terriou L, Mohty M, de Guibert S, et al. Impact of eculizumab treatment on paroxysmal nocturnal hemoglobinuria: a treatment versus no‐treatment study. Am J Hematol. 2016;91(4):366–70. [DOI] [PubMed] [Google Scholar]
- 36. Hillmen P, Lewis SM, Bessler M, Luzzatto L, Dacie JV. Natural history of paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1995;333(19):1253–8. [DOI] [PubMed] [Google Scholar]
- 37. Socie G, Mary JY, de Gramont A, Rio B, Leporrier M, Rose C, et al. Paroxysmal nocturnal haemoglobinuria: long‐term follow‐up and prognostic factors. French Society of Haematology. Lancet. 1996;348(9027):573–7. [DOI] [PubMed] [Google Scholar]
- 38. Nishimura J‐I, Yamamoto M, Hayashi S, Ohyashiki K, Ando K, Brodsky AL, et al. Genetic variants in C5 and poor response to eculizumab. N Engl J Med. 2014;370(7):632–9. [DOI] [PubMed] [Google Scholar]
- 39. Schubert J, Hillmen P, Rth A, Young NS, Elebute MO, Szer J, et al. Eculizumab, a terminal complement inhibitor, improves anaemia in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2008;142(2):263–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Risitano AM, Notaro R, Marando L, Serio B, Ranaldi D, Seneca E, et al. Complement fraction 3 binding on erythrocytes as additional mechanism of disease in paroxysmal nocturnal hemoglobinuria patients treated by eculizumab. Blood. 2009;113(17):4094–100. [DOI] [PubMed] [Google Scholar]
- 41. Risitano AM, Marotta S, Ricci P, Marano L, Frieri C, Cacace F, et al. Anti‐complement treatment for paroxysmal nocturnal hemoglobinuria: time for proximal complement inhibition? A position paper from the SAAWP of the EBMT. Front Immunol. 2019;10:1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Debureaux P‐E, Cacace F, Sica M, Notaro R, Calado RT, Scheinberg P, et al. Hematological response to eculizumab in paroxysmal nocturnal hemoglobinuria (PNH): application of a novel classification to identify unmet clinical needs and future clinical goals. Blood. 2019;134(Suppl_1):3517. [Google Scholar]
- 43. Hill A, Rother RP, Arnold L, Kelly R, Cullen MJ, Richards SJ, et al. Eculizumab prevents intravascular hemolysis in patients with paroxysmal nocturnal hemoglobinuria and unmasks low‐level extravascular hemolysis occurring through C3 opsonization. Haematologica. 2010;95(4):567–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hillmen P, Muus P, Röth A, Elebute MO, Risitano AM, Schrezenmeier H, et al. Long‐term safety and efficacy of sustained eculizumab treatment in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2013;162(1):62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Peffault de Latour R, Fremeaux‐Bacchi V, Porcher R, Xhaard A, Rosain J, Castaneda DC, et al. Assessing complement blockade in patients with paroxysmal nocturnal hemoglobinuria receiving eculizumab. Blood. 2015;125(5):775–83. [DOI] [PubMed] [Google Scholar]
- 46. Kelly R, Arnold L, Richards S, Hill A, van Bijnen S, Muus P, et al. Modification of the eculizumab dose to successfully manage intravascular breakthrough hemolysis in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2008;112(11):3441. [Google Scholar]
- 47. Peffault de Latour R, Brodsky RA, Ortiz S, Risitano AM, Jang JH, Hillmen P, et al. Pharmacokinetic and pharmacodynamic effects of ravulizumab and eculizumab on complement component 5 in adults with paroxysmal nocturnal haemoglobinuria: results of two phase 3 randomised, multicentre studies. Br J Haematol. 2020;191(3):476–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brodsky RA, Peffault de Latour R, Rottinghaus ST, Roth A, Risitano AM, Weitz IC, et al. Characterization of breakthrough hemolysis events observed in the phase 3 randomized studies of ravulizumab versus eculizumab in adults with paroxysmal nocturnal hemoglobinuria. Haematologica. 2021;106(1):230–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Risitano AM. Dissecting complement blockade for clinic use. Blood. 2015;125(5):742–4. [DOI] [PubMed] [Google Scholar]
- 50. Risitano AM, Hill A, Ricci P, Selleri C, Marando L, Gianfaldoni G, et al. Paroxysmal nocturnal hemoglobinuria (PNH) in the eculizumab era: the bedside and beyond. Haematologica. 2007;92(s3):89. [Google Scholar]
- 51. Luzzatto L, Risitano AM, Notaro R. Paroxysmal nocturnal hemoglobinuria and eculizumab. Haematologica. 2010;95(4):523–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Risitano AM. Paroxysmal nocturnal hemoglobinuria and other complement‐mediated hematological disorders. Immunobiology. 2012;217(11):1080–7. [DOI] [PubMed] [Google Scholar]
- 53. Risitano AM, Notaro R, Pascariello C, Sica M, del Vecchio L, Horvath CJ, et al. The complement receptor 2/factor H fusion protein TT30 protects paroxysmal nocturnal hemoglobinuria erythrocytes from complement‐mediated hemolysis and C3 fragment. Blood. 2012;119(26):6307–16. [DOI] [PubMed] [Google Scholar]
- 54. Lin Z, Schmidt CQ, Koutsogiannaki S, Ricci P, Risitano AM, Lambris JD, et al. Complement C3dg‐mediated erythrophagocytosis: implications for paroxysmal nocturnal hemoglobinuria. Blood. 2015;126(7):891–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Araten DJ, Notaro R, Thaler HT, Kernan N, Boulad F, Castro‐Malaspina H, et al. Thrombolytic therapy is effective in paroxysmal nocturnal hemoglobinuria: a series of nine patients and a review of the literature. Haematologica. 2012;97(3):344–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hall C, Richards S, Hillmen P. Primary prophylaxis with warfarin prevents thrombosis in paroxysmal nocturnal hemoglobinuria (PNH). Blood. 2003;102(10):3587–91. [DOI] [PubMed] [Google Scholar]
- 57. Helley D, de Latour RP, Porcher R, Rodrigues CA, Galy‐Fauroux I, Matheron J, et al. Evaluation of hemostasis and endothelial function in patients with paroxysmal nocturnal hemoglobinuria receiving eculizumab. Haematologica. 2010;95(4):574–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ninomiya H, Kawashima Y, Nagasawa T. Inhibition of complement‐mediated haemolysis in paroxysmal nocturnal haemoglobinuria by heparin or low‐molecular weight heparin. Br J Haematol. 2000;109(4):875–81. [DOI] [PubMed] [Google Scholar]
- 59. Bais J, Pel M, von dem Borne A, van der Lelie H. Pregnancy and paroxysmal nocturnal hemoglobinuria. Eur J Obstet Gynecol Reprod Biol. 1994;53(3):211–4. [DOI] [PubMed] [Google Scholar]
- 60. Bjorge L, Ernst P, Haram KO. Paroxysmal nocturnal hemoglobinuria in pregnancy. Acta Obstet Gynecol Scand. 2003;82(12):1067–71. [DOI] [PubMed] [Google Scholar]
- 61. Fieni S, Bonfanti L, Gramellini D, Benassi L, Delsignore R. Clinical management of paroxysmal nocturnal hemoglobinuria in pregnancy: a case report and updated review. Obstet Gynecol Surv. 2006;61(9):593–601. [DOI] [PubMed] [Google Scholar]
- 62. Spencer JA. Paroxysmal nocturnal haemoglobinuria in pregnancy: case report. Br J Obstet Gynaecol. 1980;87(3):246–8. [DOI] [PubMed] [Google Scholar]
- 63. Ray JG, Burows RF, Ginsberg JS, Burrows EA. Paroxysmal nocturnal hemoglobinuria and the risk of venous thrombosis: review and recommendations for management of the pregnant and nonpregnant patient. Haemostasis. 2000;30(3):103–17. [DOI] [PubMed] [Google Scholar]
- 64. Tichelli A, Socie G, Marsh J, Barge R, Frickhofen N, McCann S, et al. Outcome of pregnancy and disease course among women with aplastic anemia treated with immunosuppression. Ann Intern Med. 2002;137(3):164–72. [DOI] [PubMed] [Google Scholar]
- 65. de Guibert S, Peffault de Latour R, Varoqueaux N, Labussiere H, Rio B, Jaulmes D, et al. Paroxysmal nocturnal hemoglobinuria and pregnancy before the eculizumab era: the French experience. Haematologica. 2011;96(9):1276–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Patriquin C, Leber B. Increased eculizumab requirements during pregnancy in a patient with paroxysmal nocturnal hemoglobinuria: case report and review of the literature. Clin Case Rep. 2015;3(2):88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Danilov AV, Brodsky RA, Craigo S, Smith H, Miller KB. Managing a pregnant patient with paroxysmal nocturnal hemoglobinuria in the era of eculizumab. Leuk Res. 2010;34(5):566–71. [DOI] [PubMed] [Google Scholar]
- 68. Marasca R, Coluccio V, Santachiara R, Leonardi G, Torelli G, Notaro R, et al. Pregnancy in PNH: another eculizumab baby. Br J Haematol. 2010;150(6):707–8. [DOI] [PubMed] [Google Scholar]
- 69. Kelly RJ, Höchsmann B, Szer J, Kulasekararaj A, de Guibert S, Röth A, et al. Eculizumab in pregnant patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2015;373(11):1032–9. [DOI] [PubMed] [Google Scholar]
- 70. Marotta S, Pagliuca S, Risitano AM. Hematopoietic stem cell transplantation for aplastic anemia and paroxysmal nocturnal hemoglobinuria: current evidence and recommendations. Expert Rev Hematol. 2014;7(6):775–89. [DOI] [PubMed] [Google Scholar]
- 71. de Latour RP, Schrezenmeier H, Bacigalupo A, Blaise D, de Souza CA, Vigouroux S, et al. Allogeneic stem cell transplantation in paroxysmal nocturnal hemoglobinuria. Haematologica. 2012;97(11):1666–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Vallet N, de Fontbrune FS, Loschi M, Desmier D, Villate A, Barraco F, et al. Hematopoietic stem cell transplantation for patients with paroxysmal nocturnal hemoglobinuria previously treated with eculizumab: a retrospective study of 21 patients from SFGM‐TC centers. Haematologica. 2018;103(3):e103–e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Risitano AM, Imbriaco M, Marando L, Seneca E, Soscia E, Malcovati L, et al. From perpetual haemosiderinuria to possible iron overload: iron redistribution in paroxysmal nocturnal haemoglobinuria patients on eculizumab by magnetic resonance imaging. Br J Haematol. 2012;158(3):415–8. [DOI] [PubMed] [Google Scholar]
- 74. Hillmen P. The role of complement inhibition in PNH. Hematology. 2008;2008(1):116–23. [DOI] [PubMed] [Google Scholar]
- 75. Risitano AM, Marotta S. Therapeutic complement inhibition in complement‐mediated hemolytic anemias: past, present and future. Semin Immunol. 2016;28(3):223–40. [DOI] [PubMed] [Google Scholar]
- 76. Risitano AM, Marotta S. Toward complement inhibition 2.0: Next generation anti‐complement agents for paroxysmal nocturnal hemoglobinuria. Am J Hematol. 2018;93(4):564–77. [DOI] [PubMed] [Google Scholar]
- 77. Sheridan D, Yu ZX, Zhang Y, Patel R, Sun F, Lasaro MA, et al. Design and preclinical characterization of ALXN1210: a novel anti‐C5 antibody with extended duration of action. PLoS One. 2018;13(4):e0195909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lee JW, Sicre de Fontbrune F, Wong Lee Lee L, Pessoa V, Gualandro S, Fureder W, et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: the 301 study. Blood. 2019;133(6):530–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kulasekararaj AG, Hill A, Rottinghaus ST, Langemeijer S, Wells R, Gonzalez‐Fernandez FA, et al. Ravulizumab (ALXN1210) vs eculizumab in C5‐inhibitor‐experienced adult patients with PNH: the 302 study. Blood. 2019;133(6):540–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Schrezenmeier H, Kulasekararaj A, Mitchell L, Sicre de Fontbrune F, Devos T, Okamoto S, et al. One‐year efficacy and safety of ravulizumab in adults with paroxysmal nocturnal hemoglobinuria naive to complement inhibitor therapy: open‐label extension of a randomized study. Ther Adv Hematol. 2020;11. 10.1177/2040620720966137 (eCollection). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kulasekararaj AG, Hill A, Langemeijer S, Wells R, González Fernández FA, Gaya A, et al. One‐year outcomes from a phase 3 randomized trial of ravulizumab in adults with paroxysmal nocturnal hemoglobinuria who received prior eculizumab. Eur J Haematol. 2021;106(3):389–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fukuzawa T, Sampei Z, Haraya K, Ruike Y, Shida‐Kawazoe M, Shimizu Y, et al. Long lasting neutralization of C5 by SKY59, a novel recycling antibody, is a potential therapy for complement‐mediated diseases. Sci Rep. 2017;7(1):1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Röth A, Nishimura J‐I, Nagy Z, Gaàl‐Weisinger J, Panse J, Yoon S‐S, et al. The complement C5 inhibitor crovalimab in paroxysmal nocturnal hemoglobinuria. Blood. 2020;135(12):912–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Qu H, Magotti P, Ricklin D, Wu EL, Kourtzelis I, Wu Y‐Q, et al. Novel analogues of the therapeutic complement inhibitor compstatin with significantly improved affinity and potency. Mol Immunol. 2011;48(4):481–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Risitano AM, Ricklin D, Huang Y, Reis ES, Chen H, Ricci P, et al. Peptide inhibitors of C3 activation as a novel strategy of complement inhibition for the treatment of paroxysmal nocturnal hemoglobinuria. Blood. 2014;123(13):2094–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. de Castro C, Grossi F, Weitz IC, Maciejewski J, Sharma V, Roman E, et al. C3 inhibition with pegcetacoplan in subjects with paroxysmal nocturnal hemoglobinuria treated with eculizumab. Am J Hematol. 2020;95(11):1334–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hillmen P, Szer J, Weitz IC, Röth A, Höchsmann B, Panse J, et al. Pegcetacoplan versus eculiaumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2021;384:1028–37. [DOI] [PubMed] [Google Scholar]
- 88. Peffault de Latour R, Szer J, Weitz I, Roth A, Hochsmann B, Panse J, et al. Forty‐eight week efficacy and safety of pegcetacoplan in adult patients with paroxysmal nocturnal hemoglobinuria and suboptimal response to prior eculizumab treatment. European Hematology Association 2021; Virtual Congress; 2021.
- 89. Risitano AM, Kulasekararaj AG, Lee JW, Maciejewski JP, Notaro R, Brodsky R, et al. Danicopan: an oral complement factor D inhibitor for paroxysmal nocturnal hemoglobinuria. Haematologica. 2020; [Online ahead of print]. 10.3324/haematol.2020.261826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kulasekararaj A, Risitano AM, Maciejewski JP, Notaro R, Browett P, Lee J‐W, et al. A phase 2 open‐label study of danicopan (ACH‐0144471) in patients with paroxysmal nocturnal hemoglobinuria (PNH) who have an inadequate response to eculizumab monotherapy. Blood. 2019;134(Suppl_1):3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kulasekararaj A, Fureder W, Gandhi S, Cornpropst M, Dobo S, Farmer MK, et al. BCX9930, an oral factor D inhibitor, controls extravascular hemolysis and alleviates associated symptoms in patients with paroxysmal nocturnal hemoglobinuria inadequately controlled on C5 inhibitors. European Hematology Association 2021; Virtual Congress, EHA Library. 325349; EP589. 2021.
- 92. Mc Donald A, Le Roux Malherbe J, Cornpropst M, Dobo S, Farmer MK, Kargl D, et al. BCX9930, a novel oral factor D inhibitor, has positive effects on hemolysis and clinical outcomes in patients with paroxysmal nocturnal hemoglobinuria (PNH) naïve to complement inhibitors. European Hematology Association 2021; Virtual Congress, EHA Library. 325345; EP585. 2021.
- 93. Risitano AM, Röth A, Soret J, Frieri C, Sicre de Fontbrune F, Marano L, et al. Addition of iptacopan, an oral factor B inhibitor, to eculizumab in patients with paroxysmal nocturnal haemoglobinuria and active haemolysis: an open‐label, single‐arm, phase 2, proof‐of‐concept trial. Lancet Haematol. 2021;8:e344–e354. [DOI] [PubMed] [Google Scholar]
- 94. Jang JH, Wong L, Ko BS, Yoon S‐S, Li K, Rozenberg I, et al. First‐line treatment of PNH patients with iptacopan leads to rapid and durable hemoglobin increase by controlling both intra‐ and extravascular hemolysis. European Hematology Association 2021; Virtual Congress, EHA Library, 324581; S173. 2021.
- 95. Roth A, Rottinghaus ST, Hill A, Bachman ES, Kim JS, Schrezenmeier H, et al. Ravulizumab (ALXN1210) in patients with paroxysmal nocturnal hemoglobinuria: results of 2 phase 1b/2 studies. Blood Adv. 2018;2(17):2176–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Lee JW, Peffault de Latour R, Brodsky RA, Jang JH, Hill A, Röth A, et al. Effectiveness of eculizumab in patients with paroxysmal nocturnal hemoglobinuria (PNH) with or without aplastic anemia in the International PNH Registry. Am J Hematol 2019;94(1):E37–E41. [DOI] [PubMed] [Google Scholar]