

Abstract
Hepatitis B immune globulin (HBIG) is a human plasma‐derived immunoglobulin G concentrate that contains a high titer of neutralizing antibodies (anti‐HBs) to the hepatitis B virus (HBV) surface antigen (HBsAg). HBIG is known to be highly effective in treating HBV infections, however, a more systematic characterization of the antibody binding sites on HBsAg and their correlation with emerging “escape” mutations in HBsAg was lacking. By using anti‐HBs antibodies from HBIG lots to screen random peptide phage display libraries, we identified five clusters of peptides that corresponded to five distinct anti‐HBs binding sites on the HBsAg. Three sites, Site II (C121‐C124), Site III (M133‐P135), and Site IV (T140‐G145), were mapped within the “a” determinant, while the two other sites, Site I (Q101‐M103) and Site V (I152‐S154), were outside the “a” determinant. We then tested in binding assays HBsAg peptides containing clinically relevant mutations previously reported within these sites, such as Y134S, P142S, and G145R, and observed a significant reduction in anti‐HBs binding activity to the mutated sites, suggesting a mechanism the virus may use to avoid HBIG‐mediated neutralization. The current HBIG treatment could be improved by supplementing it with site‐specific neutralizing monoclonal antibodies that target these mutations for control of HBV infections.
Keywords: hepatitis B virus, immunoglobulin, mutation
1. INTRODUCTION
Despite increasing vaccination rates and advanced antiviral therapies, infection with hepatitis B virus (HBV) remains a global health problem affecting approximately 257 million people worldwide and is associated with substantial morbidity and mortality, mostly from complications of liver cirrhosis and hepatocellular carcinoma. 1 , 2
HBV is an enveloped virus in the Hepadnaviridae family with a partially double‐stranded DNA genome. 3 HBV strains were originally grouped into four HBsAg serotypes (adr, adw, ayw, and ayr). Genetic analysis has revealed several conserved domains and defined eight genotypes A–H, which are correlated with the four serotypes. 4 , 5
The HBV envelope proteins can be translated from a single open reading frame with at least three distinct in‐frame translation initiation sites: the small hepatitis B surface antigen (HBsAg) comprised only of an “S‐domain” of 226 amino acids (aa), the middle HBsAg with an N‐terminal extension of 55 aa (the preS2‐domain), and the large HBsAg with an additional extension of 108 or 119 aa (the preS1‐domain). 4 , 5 In the HBsAg, a major hydrophilic region between the amino acid positions 121–147 is defined as the “a” determinant, which is common to all HBV subtypes. 6 The “a” determinant contains several loop structures stabilized by a few disulfide bonds, between aa C107‐C138, C121‐C124, C137‐C149, and C139‐C147, resulting in important configurations of HBsAg for HBV infections. 7
Antibody binding to HBsAg, especially to the loops of the “a” determinant, are essential for conveying the broad protection of vaccines against HBV infections. 8 , 9 , 10 In fact, over 90% of human HBsAg‐specific serum antibodies and human B‐cell lines isolated following vaccination are directed towards the “a” determinant loops. 11 , 12 , 13 Conversely, nearly 75% of immunosuppressed subjects with HBV reactivation carry the virus with at least one HBsAg mutation in the “a” determinant. 14 It has also been shown that HBV vaccination, in conjunction with anti‐HBs immunoglobulins, can lead to HBsAg seroclearance in some chronic hepatitis B patients. 15 The inability to produce these anti‐HBs antibodies during acute infection appears to be associated with chronicity, although it remains unknown whether these associations reflect an etiologic role for anti‐HBs antibodies in protecting from or clearing established infection. 16
Hepatitis B immune globulin (HBIG) is derived from the pooled plasma of a large number of vaccinated donors with high levels of polyclonal antibodies that recognize the HBsAg of all HBV serotypes, thus providing an effective treatment option for hepatitis B infections. The level of these anti‐HBs antibodies in each HBIG lot, which is typically determined by an HBsAg immunoassay, represents a measure of the lot's potency or drug activity. A minimum level of anti‐HBs antibodies per lot is usually established as the potency specification for routine product lot release, which is based on clinical trial studies for such indications as providing postexposure prophylaxis, blocking the vertical transmission of infants of HBsAg‐positive mother, and, in combination with potent nucleos(t)ide analog, preventing HBV recurrence in patients after liver transplantation. 17 , 18 However, at the molecular level, the HBsAg binding sites, where these anti‐HBs antibodies act on precisely to exert their neutralizing functions, and the correlation of these binding sites with the emergence of the “escape” mutants have not been fully characterized for HBIG.
Here, we describe multiple conserved epitopes found in the HBsAg that could be targeted by the anti‐HBs antibodies of current HBIG lots. When mutations of these epitopes were generated to mimic those clinically observed “escape” mutations, the binding activity of anti‐HBs antibodies in the HBIG lots was significantly reduced. These observations thus raise the possibility of supplementing the current HBIG treatment with site‐specific neutralizing antibodies that bind to those epitopes where HBsAg mutations could possibly emerge for better treatment outcomes of HBV infections.
2. MATERIALS AND METHODS
2.1. Peptides, proteins, and anti‐HBs antibodies
All peptides were synthesized by GenScript, Inc. The wildtype HBsAg peptides and their corresponding mutants were biotinylated at the N‐terminus with the sequence SGSG as a linker between the peptide (HBsAg 119–125) and the biotin tag, while all other peptides carried the biotin label at their C‐terminus with GGGSAK as a linker between the peptide (HBsAg 101–106; HBsAg 123–137; HBsAg 139–148) and the biotin tag. Recombinant HBsAg proteins of subtypes ad and ay, which were expressed in Saccharomyces cerevisiae, were purchased from Bio‐Rad Laboratories (catalog no. PIP002 and OBT0915, respectively). HBIG (Human) lots made by various FDA‐licensed manufacturers were obtained from the National Institutes of Health (NIH) Clinical Center Pharmacy (Bethesda, MD) and used in this study as the source materials of anti‐HBs antibodies. All these HBIG lots had met the lot release potency specification of ≥312 IU/ml anti‐HBs.
2.2. Partial purification of anti‐HBs antibodies
Before performing the phage display library screening, the anti‐HBs antibodies in the HBIG were purified to create an enriched anti‐HBs working solution by binding with and eluting from HBsAg. Briefly, HBsAg subtype ad or ay, diluted in phosphate‐buffered saline (PBS) at pH of 7.4 to a final concentration of 5 ng/µl, was added to wells in a 96‐well plate at 100 µl/well and kept at 4°C for overnight coating. After washing three times with 200 µl of PBS (pH 7.4) containing 0.05% Tween 20, 1 µg of HBIG, diluted in 200 µl of PBS (pH 7.4), was added to the HBsAg‐coated wells and incubated at room temperature for 30 min. After washing the wells three times, the bound anti‐HBs antibodies were then eluted into 50 µl of 0.1 M glycine‐HCl buffer (pH 2.5) per well. The collected anti‐HBs antibodies were immediately neutralized by adding PBS (pH 7.4). This process was repeated at least three times for partial purification of anti‐HBs antibodies from HBIG.
2.3. Screening of random peptide phage display libraries
The selection of peptides from random peptide phage display libraries (Ph.D.™‐12 and Ph.D.™‐C7C phage display libraries, New England BioLabs) has been described previously. 19 Briefly, 1010 phages were incubated with partially purified anti‐HBs antibodies‐protein G mixtures at room temperature for 20 min. After 6–8 washings with 0.05 M Tris‐HCl buffer (pH 7.5) containing 0.15 M NaCl and 0.05% Tween 20, the phages were eluted from the complexes with 0.1 M HCl and neutralized with 1 M Tris‐HCl buffer (pH 9.0). The eluted phages were then amplified in the Escherichia coli strain ER2738 for 5 h. After 2 or 3 additional rounds of selection by the same preparation of anti‐HBs antibodies, each single‐phage plaque was amplified and then sequenced by automated DNA sequencing at the Core Facility of CBER, FDA (Silver Spring, MD). The corresponding peptide sequences were deduced from the DNA sequences.
2.4. Protein sequence alignment
Sequences of phage‐displayed peptides were aligned and grouped manually into clusters based on the core residues, which appeared to be commonly shared by these peptides to suggest that these were the contact sites of the anti‐HBs antibodies. By further examining the homology of these core residues with the amino acid sequence of HBsAg, the phage‐displayed peptide clusters were mapped to their corresponding sites on the HBsAg, 4 , 5 the phage‐displayed peptides homologous to HBsAg were called HBsAg epitope mimics, while the corresponding sites on the HBsAg were considered to be putative immune epitopes.
2.5. Enzyme‐linked immunosorbent assay (ELISA)
Biotin‐conjugated peptides (500 ng/well) were added to streptavidin‐coated 96‐well Maxisorp plates (Thermo Fisher Scientific), followed by incubation at room temperature for 1 h in SuperBlock blocking buffer (Thermo Fisher Scientific). Blocking buffer was applied to the wells, which were then left to incubate at 37°C for another hour. After the plate was washed four times with PBS, pH 7.4, containing 0.05% Tween 20, to remove unbound peptides, serial dilutions of the test antibodies were added to the plate, followed by incubation at 37°C for 1 h. The plate was then washed four times after which the secondary antibody, a goat anti‐human peroxidase‐conjugated immunoglobulin G (IgG; KPL) at a 1:5000 dilution, was added to the wells, and then incubated at 37°C for 30 min. After four washes, the reactions were developed by adding 100 µl of 1‐Step TMB‐ELISA substrate solution (Thermo Fisher Scientific) and stopped by adding 100 µl of 4 N sulfuric acid. The absorbance of each well was measured at 450 nm using a SpectraMax M2e microplate reader (Molecular Devices). ELISA was performed with an HBIG lot diluted 1:3000, wherein it would typically result in an optical density reading of approximately 0.4 at 450 nm for the binding measurements of wildtype peptide controls. Triplicate readouts of three independent assays were done for each test peptide.
2.6. Statistical analysis
Statistical analysis was performed with GraphPad Prism 8 (GraphPad Software) using an unpaired the Student's t test with a two‐tailed p value (p ≤ 0.01 or p ≤ 0.05). Error bars represent the standard deviation or the SE of the mean.
3. RESULTS
3.1. Identification of anti‐HBs antibody binding sites through screening random peptide phage display libraries with partially purified HBIG
To understand the molecular basis of HBV neutralization by HBIG, we performed a series of experiments of screening random peptide phage display libraries to identify the peptide clusters that could be recognized by anti‐HBs antibodies present in commercially available HBIG lots, and, at the same time, determine the residue‐specificity of these anti‐HBs antibodies.
Before performing the phage display experiments, the anti‐HBs antibodies were partially purified from a mixture of randomly chosen lots of HBIG to reduce any potential cross‐reactivity by the various non‐anti‐HBs polyclonal antibodies contained in these lots. This process involved coating a 96‐well plate first with HBsAg proteins, followed by incubation with the HBIG solution to bind the antigens in the wells. The IgG fractions of HBIG that bound HBsAg were eluted and collected. HBsAg from the two most common HBsAg serotypes, ad and ay, was used for coating the plates to broaden the HBsAg antigenic repertoire and capture the majority of anti‐HBs antibodies. This experimental approach was designed based on the fact that all currently available genetically engineered HBV vaccines use the adw serotype, and that the antibody responses following immunization with recombinant HBV vaccines are largely directed against the common “a” determinant.
When the HBsAg‐selected HBIG antibodies were applied to screen the phage‐displayed libraries, five major clusters of peptides with the common core residues were revealed following sequence alignment (Figure 1A). It should be noted that at aa 122 of HBsAg, it could naturally be an arginine (R) or a lysine (K) (Figure 1B), which is an important site known to be closely associated with subtype d or y expression, wherein the changes are mediated by a shift from K to R. 4 , 5 As HBIG antibodies bound to both types of R‐ or K‐containing peptides displayed on the phages (Figure 1A, Cluster II), this suggested that the HBIG antibodies could interact equally with both ad and ay serotypes. In addition, Cluster IV consisted of multiple phage‐displayed peptides with identical sequences (Figure 1A).
Figure 1.
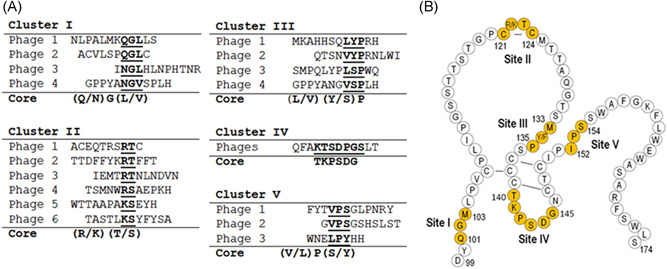
Epitope mapping by screening random peptide phage display libraries. (A) The amino acid sequences of the peptides selected after screening phage‐display libraries with anti‐HBs antibodies partially purified from HBIG lots are aligned to determine the core residues within each cluster. The core residues are indicated in underlined bold font. The letters in the parentheses denote the amino acid residues that are varied at that position. (B) Schematic representation of ant‐HBs antibody binding sites (Sites I–V) that correspond to the core sequences, respectively, as shown in (A). The sequence of HBsAg ayw3 between the amino acid (aa) positions 99–174 is used as the model subtype. The “a” determinant region of HBsAg is located between aa 121–147 with the lines between a pair of cysteines to represent the disulfide bonds. HBsAg, hepatitis B surface antigen
Further alignment of the core residues with the HBsAg sequence, where HBV ayw3 was used as the model subtype in this experiment (Figure 1B), suggested that these identified peptide clusters corresponded, respectively, to five sites on the HBsAg, i.e., Q101‐G102‐M103 (Site I), R122‐T123 (Site II), M133‐Y134‐P135 (Site III), T140‐K141‐P142‐S143‐D144‐G145 (Site IV), and I152‐P153‐S154 (Site V) (Figure 1B). Notably, Cluster IV peptides contained residues that were comparable to those in Site IV of HBsAg, but the linear arrangement of these residues was different (Figure 1B).
To verify if these core residues identified by the phage display screening could represent the contact sites of the anti‐HBs, three peptides from Clusters II, III, and IV (corresponding to Sites II, III, and IV, respectively, within the common “a” determinant of HBsAg), along with their alanine substituted versions, were examined by ELISA for their binding to an HBIG lot. As shown in Figure 2 (left panel), alanine substitutions of two core residues in the mimic peptides of Site II decreased about 25% of the binding level of the wildtype; the reduction was found to be significant (p < 0.05). As the mimic peptide (ACEQT1 R 1 SR 2 T 2C) appeared to contain at least two sets of core residues of RT(S), we interpreted this result to mean that the replacement of one set of RT(S) was not sufficient to severely disrupt the binding.
Figure 2.
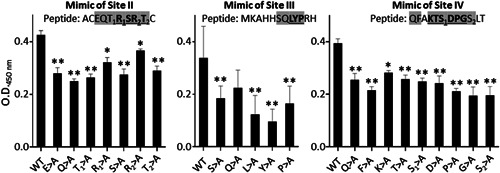
Determination of the core residues of the phage‐displayed peptides needed for the binding of anti‐HBs antibodies present in an HBIG lot. Selected peptides as shown in Figure 1A were chemically synthesized to mimic the epitopes in the HBsAg, and then assayed in an ELISA for binding with an HBIG lot. The core residues are shown in underlined bold font. The shaded letters represent the amino acids that are replaced by alanine in each peptide. A HBIG (5% IgG solution) lot sample prepared at a 1:3000 dilution was used for this assay. X‐axis indicates the substituted amino acids. Y‐axis indicates the binding activity measured by O.D. readings at 450 nm. Data show the mean ± SEM from triplicate readouts of three independent assays. Two asterisks (**) and one asterisk (*) indicate p < 0.01 and p < 0.05, respectively. ELISA, enzyme‐linked immunosorbent assay; HBsAg, hepatitis B surface antigen; IgG, immunoglobulin G; OD, optical density
In contrast, when the mimic peptide (MKAHHSQ LYP RH) of Site III was tested, the substitutions of each of the “core” residues of LYP by alanine led to a significant reduction of the binding activity (p < 0.01) (Figure 2, middle panel).
Similarly with the mimic peptide (QFA KTS 1 DPG S2LT) of Site IV, when each of the six core residues was replaced by alanine (Figure 2, right panel), anti‐HBs antibody binding activity was reduced significantly (p < 0.01) regardless of the difference in the linear sequences between the mimic and Site IV. These results suggested that the core residues in the context of phage‐displayed peptides could be recognized by anti‐HBs antibodies.
3.2. Characterization of residue‐specificity of anti‐HBs antibodies present in HBIG lots
Once the importance of the core residues in the context of phage‐displayed peptides was established for the binding of anti‐HBs antibodies, we tested to see if their corresponding sites in the HBsAg, as depicted in Figure 1B, were critical for the binding of anti‐HBs.
Of all the five corresponding sites on the HBsAg, we focused on Site I, which is located outside the “a” determinant, and the other three sites within the “a” determinant, i.e., Sites II, III, and IV, for their binding to anti‐HBs antibodies. Site V was not included in this experiment as it was not associated with any known “escape” mutants. As shown in Figure 3A, when alanine was introduced into the peptides encompassing Site I, an approximately 50% reduction in anti‐HBs binding activity was observed for residues Q101, G102, and M103 (p < 0.01), while an L104A change showed no detectable effect. This experiment confirmed that the three conserved residues Q101, G102, and M103 in Site I were critical for the binding of anti‐HBs antibodies in the HBIG.
Figure 3.
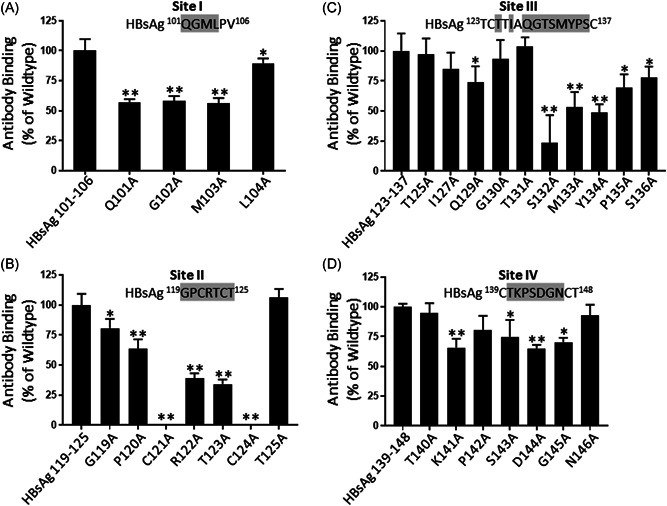
Site‐directed mutational analyses of HBsAg for anti‐HBs antibody binding. Peptides representing the epitopes (Sites I–IV) in the HBsAg and their alanine substitutions were analyzed by ELISA for their binding by anti‐HBs antibodies of an HBIG lot (5% HBIG at 1:3000 dilution). Individual residues substituted by alanine are indicated by the shaded letters. The binding activity of HBIG to the wildtype peptide, as measured by O.D. readings at 450 nm, is expressed as 100% binding, while the binding activity of HBIG to the mutated peptide is expressed as a percentage relative to the wildtype peptide binding. Data show the mean ± SEM from triplicate readouts of three independent assays. Two asterisks (**) and one asterisk (*) indicate p < 0.01 and p < 0.05, respectively. ELISA, enzyme‐linked immunosorbent assay; HBsAg, hepatitis B surface antigen; OD, optical density
Similarly, alanine substitution of C121, R122, T123, or C124 reduced the anti‐HBs binding of the Site II peptides by more than 60% (p < 0.01), whereas a P120A change showed about a 25% reduction (p < 0.01). Neither G119A nor T125A showed any detectable effects that were significant (Figure 3B). This experiment indicated that both R122 and T123, which reside in the tiny loop formed presumably by the disulfide bond between C121‐C124, could be important contact points of anti‐HBs antibodies.
As for Site III shown in Figure 3C, THE most prominent reduction was observed when the peptide contained an S132A, M133A, or Y134A substitution (p < 0.01). Other alanine substitutions at residues between S132‐S136 also decreased the binding activity of anti‐HBs even though the level of reduction varied among them (p < 0.01 or p < 0.05). This result suggested that an anti‐HBs binding site was centered on the residues of S132, M133, and Y134.
It was apparent that certain amino acid substitutions at Site III represented natural variants, whose recognition by anti‐HBs was not expected to be significantly affected. For example, as detected in the phage screening (Figure 1A), Y134 is often replaced by phenylalanine (F) (Table 1), resulting in a sequence of M133‐F134‐P135 which is seen in other serotypes, such as ayw1, adw2, and adw4. Similarly, M133 could also be replaced by an aliphatic amino acid (leucine or valine) without significantly affecting anti‐HBs recognition (Figure 1A and Table 1).
Table 1.
Mutations in the HBsAg observed in clinical studies
| HBsAg | Mutation | Impact | Reference | Epitope |
|---|---|---|---|---|
| Q101 | R | mAb | (28, 32) | Site I |
| G102 | ||||
| M103 | ||||
| L104 | ||||
| G119 | Site II | |||
| P120 | E/S/T | Diagnosis | (20–27) | |
| Vaccine | ||||
| HBIG | ||||
| C121 | ||||
| R122 | ||||
| T123 | N | mAb | (20‐27, 31, 32) | |
| C124 | R/Y | HBIG | ||
| T/M125 | ||||
| T/I126 | A/N/S | Diagnosis | (20–27) | Site III |
| Vaccine | ||||
| HBIG | ||||
| T127 | ||||
| Q129 | H/L | Diagnosis | (20–27) | |
| Vaccine | ||||
| HBIG | ||||
| G130 | D/R | |||
| T131 | ||||
| S132 | ||||
| M133 | L | Diagnosis | (20–27) | |
| Y/F134 | N/R/S | Vaccine | ||
| HBIG | ||||
| P135 | ||||
| S136 | ||||
| T140 | Site IV | |||
| K141 | E/I | mAb | (20–28, 32) | |
| P142 | S | Vaccine | ||
| S/T143 | L | Diagnosis | ||
| D144 | A/E | Vaccine | ||
| G145 | R/A | HBIG | ||
| N146 | ||||
| I152 | Site V | |||
| P153 | Infectivity | (30) | ||
| S154 |
Note: Shaded letter: Critical for HBIG binding; Impact: Amino acid mutations that are known to affect the binding of monoclonal antibodies (mAb), clinical diagnosis, clinical responses to current hepatitis B virus (HBV) vaccines and hepatitis B immune globulins (HBIG), and the in vitro infectivity of HBV.
The critical residues of Site IV for anti‐HBs recognition were also identified by alanine substitution. As shown in Figure 3D, K141A, D144A, and G145A substitutions showed significant reductions of anti‐HBs binding activity (p < 0.01). This experiment confirmed that Site IV is indeed an anti‐HBs binding site, although the linear arrangements of the residues were different between the peptide mimic and Site IV in HBsAg, implying that the phage‐displayed peptides were likely conformational mimics of Site IV.
3.3. Clinical relevance of residue‐specificity of anti‐HBs in 10 HBIG lots
We surveyed previously described HBsAg mutants in HBsAg between residues 101–145 that were related to the treatment and/or diagnosis of HBV infections and correlated the locations of these point mutations with that of the epitopes (Sites I–IV) mapped in the present study (Table 1). A selected set of peptides with or without a specific point mutation was synthesized (Figure 4A,B). Both Y134F in the Site III peptide (HBsAg aa 123–137) and S143T in the Site IV peptide (HBsAg aa 139–148) were chosen as controls because they represented natural HBsAg variants. For testing, Y134S in the Site III peptide, and P142S and G145R in the Site IV peptide were the clinically observed mutants.
Figure 4.
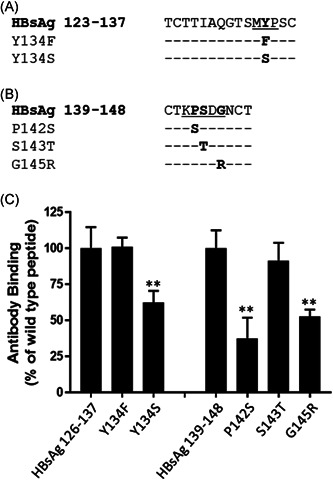
Analysis of clinically relevant HBsAg mutants for their binding to a single HBIG lot. Peptides representing the epitopes in the HBsAg, i.e., Site II (A) and Site III (B), and their clinically observed substitutions were chemically synthesized for ELISA analysis of their binding by anti‐HBs antibodies in an HBIG lot. Residues of interest within the epitopes are underlined. (C) The binding activity of HBIG to the wildtype peptide, as measured by O.D. readings at 450 nm, is expressed as 100% binding, while the binding activity of HBIG to the mutated peptide is expressed as a percentage relative to the wildtype peptide binding. Data show the mean ± SEM from triplicate readouts of three independent assays. Two asterisks (**) and one asterisk (*) indicate p < 0.01 and p < 0.05, respectively. ELISA, enzyme‐linked immunosorbent assay; HBsAg, hepatitis B surface antigen; OD, optical density
As assessed by an ELISA, the presence of Y134F in Site III and S143T in Site IV did not change the levels of anti‐HBs antibody binding activity, while all the other selected mutants (Y134S, P142S, and G145R) significantly reduced the binding activity of an HBIG lot (p < 0.01) (Figure 4C).
To determine whether the observed reduction in anti‐HBs binding of specific mutants was a general effect, we tested 10 additional HBIG lots, which were made by different manufacturers. We found that the wildtype peptide of Site III (HBsAg aa 123–136) and its natural variant peptide (Y134F) showed no detectable difference in their ability to be recognized by the anti‐HBs antibodies (Figure 5A). In contrast, all other mutations including Y134S in Site III, and P142S and G145R in Site IV (except G145R when HBIG lot 1 was used in the experiment), showed reduced anti‐HBs binding activities compared to their wildtype counterpart (p < 0.01 or p < 0.05), although the level of reduction varied among these HBIG lots (Figure 5B,C).
Figure 5.
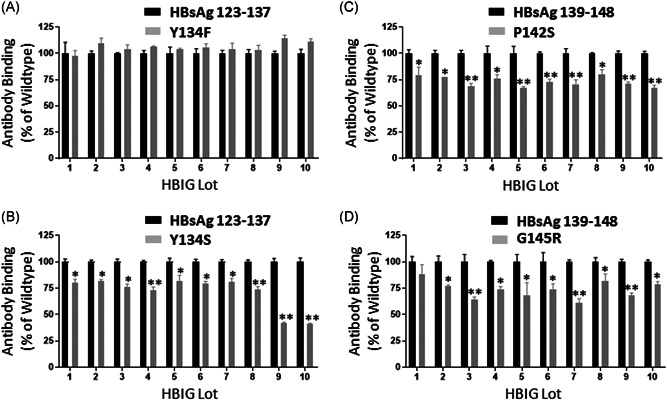
Analysis of clinically relevant HBsAg mutations in two epitopes for their binding to ten additional HBIG lots. Peptides representing specific mutations: (A) Y134F, a natural variant in Site II, (B) Y134S, a potential escape mutant in Site II, (C) P142S, a potential escape mutant in Site IV, and (D) G145R, a well‐recognized escape mutant in Site IV, were analyzed by ELISA for their binding by each of the 1o HBIG lots. The binding activity of HBIG to the wildtype peptide, as measured by O.D. readings at 450 nm, is expressed as 100% binding, while the binding activity of HBIG to the mutated peptide is expressed by as a percentage relative to the wildtype peptide binding. Data show the mean ± SEM from triplicate readouts of three independent assays. Two asterisks (**) and one asterisk (*) indicate p < 0.01 and p < 0.05, respectively. HBsAg, hepatitis B surface antigen; OD, optical density
4. DISCUSSION
By taking advantage of the unique opportunity offered by the very nature of the HBIG itself that it is essentially a highly purified concentrate of polyclonal anti‐HBs neutralizing antibodies from a large number of vaccinated donors, we have identified five immune epitopes in HBsAg, and further established the connection between these immune epitopes and the “escape” mutants of HBsAg that have emerged under various selection pressures.
Of these five epitopes, Sites II, II, and IV were mapped to three regions in the common “a” determinant. Given the importance of the “a” determinant for the antigenicity of HBV vaccines, it is reasonable to assume that the binding of anti‐HBs antibodies to these sites contributes, at least in part, to the overall neutralizing activities associated with the HBIG lots. By incorporating mutations specifically in these epitopes, guided by previously reported “escape” mutants (Table 1), 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 we have further demonstrated that altering a few key residues within these epitopes could significantly disrupt the recognition by anti‐HBs antibodies.
Site IV was mapped to a linear epitope between the two cysteines C139 and C147. The epitope includes amino acids K141, P142, S/T143, D144, and G145, all of which are often mutated during natural infection. Of the abovementioned amino acids, the mutation of G145 to an arginine is among the most frequent human immune escape variations known to alter the projection of the loop structure, thus causing the anti‐HBs antibodies to be unable to recognize it. 26 , 28 The G145R mutant has been reported to occur in the presence of protective levels of anti‐HBs antibodies in infants born to HBV‐infected mothers who received HBIG plus vaccine, in liver transplant patients who received HBIG for prophylaxis, and in HBsAg‐negative chronic carriers of HBV. 17 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 These clinical observations suggest that site‐specific binding by HBIG lots can be a driving force for the selection of escape mutants in these clinical settings. This is consistent with earlier observations that the withdrawal of HBIG treatment could result in a reversion of some mutations back to the original HBsAg sequence. 29 However, this proposal should be made with caution as further studies are needed to exclude the possibility that anti‐HBs antibodies initially present in the unpurified HBIG were able to bind other forms of HBsAg, including those escape mutants, but could have been lost during the process of our experimental purification of the HBIG lots.
Mutation of P142 of Site IV is also able to reduce anti‐HBs antibody binding, but the underlying mechanism might be associated with simply disrupting the stability of the loop structure. 28 In addition, mutation of this proline is known to decrease viral infectivity, 13 implying that compensatory changes in the structure must occur to maintain protein integrity of HBsAg. Therefore, it is likely that anti‐HBs in the HBIG lots, via binding to Site IV, interfere with the virus infectivity resulting in HBV neutralization.
The mutations occurring at Site II and Site III within the “a” determinant, such as T123N and Y134S, have also been documented in patients who have received HBIG treatment, thus supporting the clinical relevance of these sites. Interestingly, a few human monoclonal antibodies with broadly neutralizing activity have been shown to be reactive with these sites. 28 , 30 , 31 The reductions we observed in the HBIG binding to Site II and Site III, as a consequence of the specific amino acid substitutions applied in this study, may provide an explanation of how these mutants can escape from neutralization despite HBIG treatment.
Aside from the three abovementioned epitopes that were mapped within the “a” determinant, the two other epitopes, Site I and Site V, were located outside the “a” determinant. Little is known about the role of Site V in HBV infection, although P153 within this epitope appears to be associated with infectivity in vitro. 13 However, broadly neutralizing monoclonal antibodies that bind specifically to Site I have recently been isolated from memory B cells of HBV vaccinees and controllers. 28 , 31 Knowing that the amino acids within Site I, such as Q101, G102, and M103, are highly conserved among all HBV subtypes, our results appeared to suggest that Site I is one of the major targets for the broadly neutralizing antibodies.
Our study findings essentially filled in several gaps of information by identifying, confirming, and characterizing the critical HBsAg epitope amino acids that are prone to mutation due to selection pressures, including HBIG therapy. It also provided the basis for developing novel residue‐specific neutralizing antibodies that could complement the current HBIG therapy by counteracting future “escape” mutations. Recently, several studies have begun to explore this therapeutic potential. 28 , 30 , 31 , 32 , 33 , 34 , 35 In one promising study, it was shown that HBV infection can be controlled, at least in vitro, by a combination of broadly neutralizing antibodies targeting nonoverlapping epitopes with complementary sensitivity to mutations that commonly emerge during human infection. 28 Future experiments could be carried out in an HBV cell culture system to address if this combinatorial approach will increase the effectiveness of HBIG in targeting specific escape mutants.
Furthermore, for the improvement of current HBIG lots, closer monitoring of the residue‐specificity of anti‐HBs antibodies present in plasma‐derived HBIG may be developed further into additional quality control measures to ensure both the potency and consistency of each HBIG lot.
5. SIGNIFICANCE
The current HBIG lots are manufactured from the pooled plasma of vaccinated donors, thus they contain high titers of anti‐HBs neutralizing antibodies against HBV infections. By taking advantage of this unique feature of HBIG, we have mapped five immunogenic sites in the HBsAg that are recognized by the anti‐HBs antibodies, and, subsequently, established the connection between these immune epitopes and the “escape” mutants of HBsAg that emerged during HBV infections. These findings not only provide the basis for developing residue‐specific neutralizing antibodies that could complement the current HBIG therapy by counteracting future “escape” mutations but also confirm which immune epitopes are important for a quality control‐type of monitoring of anti‐HBs antibodies in HBIG to ensure both the potency and consistency of each lot.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
Pei Zhang designed the study and took responsibility for the integrity of the data and the accuracy of the data analysis. Sreya Tarafdar, Maria Luisa Virata, Hailing Yan, Lilin Zhong, Lu Deng, Yanqun Xu, Yong He, Evi Struble performed the experiments and analyzed the data. Sreya Tarafdar, Maria Luisa Virata, Pei Zhang contributed to the writing of the manuscript. Maria Luisa Virata contributed to the critical revision of the manuscript.
ACKNOWLEDGEMENTS
We thank Drs. Charles Maplethorpe and Mahmood Farshid for their comments on the manuscript. ST was supported by the Research Participation Program at the Center for Biologics Evaluation and Research which was administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and the Food and Drug Administration. This study was supported by the Center for Biologics Evaluation and Research, Food and Drug Administration.
Tarafdar S, Virata ML, Yan H, et al. Multiple Epitopes of Hepatitis B Virus Surface Antigen Targeted by Human Plasma‐derived Immunoglobulins Coincide with Clinically Observed Escape Mutations. J Med Virol. 2022;94:649‐658. 10.1002/jmv.27278
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. World Health Organization . 2017. Global hepatitis report, 2017. (https://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/
- 2. Lim JK, Nguyen MH, Kim WR, Gish R, Perumalswami P, Jacobson IM. Prevalence of chronic hepatitis B virus infection in the United States. Am J Gastroenterol. 2020;115(9):1429‐1438. [DOI] [PubMed] [Google Scholar]
- 3. Magnius L, Mason WS, Taylor J, et al. Ictv report consortium. ICTV virus taxonomy profile: hepadnaviridae. J Gen Virol. 2020;101(6):571‐572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Norder H, Hammas B, Löfdahl S, Couroucé AM, Magnius LO. Comparison of the amino acid sequences of nine different serotypes of hepatitis B surface antigen and genomic classification of the corresponding hepatitis B virus strains. J Gen Virol. 1992;73(Pt 5):1201‐1208. [DOI] [PubMed] [Google Scholar]
- 5. Norder H, Couroucé AM, Coursaget P, et al. Genetic diversity of hepatitis B virus strains derived worldwide: genotypes, subgenotypes, and HBsAg subtypes. Intervirology. 2004;47(6):289‐309. [DOI] [PubMed] [Google Scholar]
- 6. Caligiuri P, Cerruti R, Icardi G, Bruzzone B. Overview of hepatitis B virus mutations and their implications in the management of infection. World J Gastroenterol. 2016;22(1):145‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jaoudé GA, Sureau C. Role of the antigenic loop of the hepatitis B virus envelope proteins in infectivity of hepatitis delta virus. J Virol. 2005;79(16):10460‐10466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chisari FV. Hepatitis B virus biology and pathogenesis. Mol Genet Med. 1992;2:67‐104. [DOI] [PubMed] [Google Scholar]
- 9. Petit MA, Maillard P, Capel F, Pillot J. Immunochemical structure of the hepatitis B surface antigen vaccine–II. Analysis of antibody responses in human sera against the envelope proteins. Mol Immunol. 1986;23(5):511‐523. [DOI] [PubMed] [Google Scholar]
- 10. Lazarevic I. Clinical implications of hepatitis B virus mutations: recent advances. World J Gastroenterol. 2014;20(24):7653‐7664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shokrgozar MA, Shokri F. Subtype specificity of anti‐HBs antibodies produced by human B‐cell lines isolated from normal individuals vaccinated with recombinant hepatitis B vaccine. Vaccine. 2002;20(17‐18):2215‐2220. [DOI] [PubMed] [Google Scholar]
- 12. Gencay M, Hübner K, Gohl P, et al. Ultra‐deep sequencing reveals high prevalence and broad structural diversity of hepatitis B surface antigen mutations in a global population. PLoS One. 2017;12(5):e0172101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Salisse J, Sureau C. A function essential to viral entry underlies the hepatitis B virus “a” determinant. J Virol. 2009;83(18):9321‐9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Salpini R, Colagrossi L, Bellocchi MC, et al. Hepatitis B surface antigen genetic elements critical for immune escape correlate with hepatitis B virus reactivation upon immunosuppression. Hepatology. 2015;61(3):823‐833. [DOI] [PubMed] [Google Scholar]
- 15. Tsuge M, Hiraga N, Uchida T, et al. Antiviral effects of anti‐HBs immunoglobulin and vaccine on HBs antigen seroclearance for chronic hepatitis B infection. J Gastroenterol. 2016;51(11):1073‐1080. [DOI] [PubMed] [Google Scholar]
- 16. Trépo C, Chan HL, Lok A. Hepatitis B virus infection. Lancet. 2014;384(9959):2053‐2063. [DOI] [PubMed] [Google Scholar]
- 17. Terrault NA, Lok ASF, McMahon BJ, et al. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 Hepatitis B Guidance. Clin Liver Dis (Hoboken). 2018;12(1):33‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. European Association for the Study of the Liver . EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J Hepatol. 2017;67(2):370‐398. [DOI] [PubMed] [Google Scholar]
- 19. Zhang P, Yu MY, Venable R, Alter HJ, Shih JW. Neutralization epitope responsible for the hepatitis B virus subtype‐specific protection in chimpanzees. Proc Natl Acad Sci USA. 2006;103(24):9214‐9219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hsu HY, Chang MH, Ni YH, Chiang CL, Wu JF, Chen HL. Universal infant immunization and occult hepatitis B virus infection in children and adolescents: a population‐based study. Hepatology. 2015;61(4):1183‐1191. [DOI] [PubMed] [Google Scholar]
- 21. Ma Q, Wang Y. Comprehensive analysis of the prevalence of hepatitis B virus escape mutations in the major hydrophilic region of surface antigen. J Med Virol. 2012;84(2):198‐206. [DOI] [PubMed] [Google Scholar]
- 22. Gao S, Duan ZP, Coffin CS. Clinical relevance of hepatitis B virus variants. World J Hepatol. 2015;7(8):1086‐1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Araujo NM, Teles SA, Spitz N. Comprehensive analysis of clinically significant hepatitis B virus mutations in relation to genotype, subgenotype and geographic region. Front Microbiol. 2020;11:616023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yamamoto K, Horikita M, Tsuda F, et al. Naturally occurring escape mutants of hepatitis B virus with various mutations in the S gene in carriers seropositive for antibody to hepatitis B surface antigen. J Virol. 1994;68(4):2671‐2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cooreman MP, Leroux‐Roels G, Paulij WP. Vaccine‐ and hepatitis B immune globulin‐induced escape mutations of hepatitis B virus surface antigen. J Biomed Sci. 2001;8(3):237‐247. [DOI] [PubMed] [Google Scholar]
- 26. Alavian SM, Miri SM, Jazayeri SM. Hepatitis B vaccine: prophylactic, therapeutic, and diagnostic dilemma. Minerva Gastroenterol Dietol. 2012;58(2):167‐178. [PubMed] [Google Scholar]
- 27. Lok AS. Clinical significance and molecular characteristics of common hepatitis B virus variants. In: Post T, ed, UpToDate. Waltham, MA: Accessed on May 12 Wolters Kluwer; 2020. [Google Scholar]
- 28. Wang Q, Michailidis E, Yu Y, et al. A combination of human broadly neutralizing antibodies against hepatitis B virus HBsAg with distinct epitopes suppresses escape mutations. Cell Host Microbe. 2020;28(2):335‐349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ghany MG, Ayola B, Villamil FG, et al. Hepatitis B virus S mutants in liver transplant recipients who were reinfected despite hepatitis B immune globulin prophylaxis. Hepatology. 1998;27(1):213‐222. [DOI] [PubMed] [Google Scholar]
- 30. Kucinskaite‐Kodze I, Pleckaityte M, Bremer CM, et al. New broadly reactive neutralizing antibodies against hepatitis B virus surface antigen. Virus Res. 2016;211:209‐221. [DOI] [PubMed] [Google Scholar]
- 31. Hehle V, Beretta M, Bourgine M, et al. Potent human broadly neutralizing antibodies to hepatitis B virus from natural controllers. J Exp Med. 2020;217(10):e20200840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Corti D, Benigni F, Shouval D. Viral envelope‐specific antibodies in chronic hepatitis B virus infection. Curr Opin Virol. 2018;30:48‐57. [DOI] [PubMed] [Google Scholar]
- 33. Golsaz‐Shirazi F, Amiri MM, Farid S, et al. Construction of a hepatitis B virus neutralizing chimeric monoclonal antibody recognizing escape mutants of the viral surface antigen (HBsAg). Antiviral Res. 2017;144:153‐163. [DOI] [PubMed] [Google Scholar]
- 34. Robbiani DF. Neutralizing hepatitis B. J Exp Med. 2020;217(10):e20201261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hong B, Wen Y, Ying T. Recent progress on neutralizing antibodies against hepatitis B virus and its implications. Infect Disord Drug Targets. 2019;19(3):213‐223. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.