

Summary
Understanding processes that generate and maintain large disjunctions within plant species can provide valuable insights into plant diversity and speciation. The butterfly bush Buddleja alternifolia has an unusual disjunct distribution, occurring in the Himalaya, Hengduan Mountains (HDM) and the Loess Plateau (LP) in China.
We generated a high‐quality, chromosome‐level genome assembly of B. alternifolia, the first within the family Scrophulariaceae. Whole‐genome re‐sequencing data from 48 populations plus morphological and petal colour reflectance data covering its full distribution range were collected.
Three distinct genetic lineages of B. alternifolia were uncovered, corresponding to Himalayan, HDM and LP populations, with the last also differentiated morphologically and phenologically, indicating occurrence of allopatric speciation likely to be facilitated by geographic isolation and divergent adaptation to distinct ecological niches. Moreover, speciation with gene flow between populations from either side of a mountain barrier could be under way within LP. The current disjunctions within B. alternifolia might result from vicariance of a once widespread distribution, followed by several past contraction and expansion events, possibly linked to climate fluctuations promoted by the Kunlun–Yellow river tectonic movement. Several adaptive genes are likely to be either uniformly or diversely selected among regions, providing a footprint of local adaptations.
These findings provide new insights into plant biogeography, adaptation and different processes of allopatric speciation.
Keywords: allopatric speciation, demographic history, Kunlun–Yellow river tectonic movement, Loess Plateau, Scrophulariaceae, whole‐genome sequencing
Introduction
Allopatric speciation has been widely viewed as the most common process of speciation (Rieseberg, 2018). Diverging populations accumulate differences by genetic drift and mutations, providing opportunities for differential selection and adaptation to local environments (Schluter, 2000; Losos & Glor, 2003; Wang et al., 2016; Zhang et al., 2016). Although biogeographic studies on this topic are not uncommon, usually only a limited number of molecular markers has been used (Lexer et al., 2013). The increasing availability of whole‐genome sequencing (WGS) offers opportunities to examine this process in more detail. Genomic data in conjunction with a model testing framework have been applied to address this issue in animals (Kreiner et al., 2019), but it has rarely been done for plants (Feng et al., 2020), and few studies have used WGS data to examine the allopatric speciation process and its outcome, especially in plants (Chen et al., 2019).
The Qinghai–Tibetan Plateau, together with the Hengduan Mountains (HDM) and the Himalaya of its eastern and southern parts, harbours more than 12 000 species of vascular plants in 1500 genera, with exceptional species richness and a high level of endemism, especially for alpine species (Wu, 1988; Li & Li, 1993; Wen et al., 2014). By contrast, the Loess Plateau (LP) has low plant diversity with 3224 vascular plants in an area of 4.5 × 105 km2 (Zhang et al., 2002). To date, relatively few plant species are known to be concurrently distributed across both these upland regions, and this is presumed to be due to their contrasting elevations, climates and soil characteristics (Du, 1997). Nonetheless, the contrast in physical environments between the LP and the Himalaya plus HDM could provide an excellent opportunity to examine the mechanisms underlying allopatric speciation and ecological divergence, in taxa common to both.
The genus Buddleja L. (Butterfly bush), is known for its horticultural value (e.g. B. alternifolia, B. davidii, B. globosa) and the invasive characteristic of B. davidii. It comprises > 90 species distributed across Africa, Asia, North America and South America (Chau et al., 2017). However, some Buddleja species might in fact comprise more than one taxon, a case in point being B. alternifolia, a diploid plant that commonly forms large populations on the Himalaya, but also occurs disjunctionly in both the HDM and LP (Fig. 1a,b). Moreover, plants in LP differ in flowering time, leaf lengths and petal colour from Himalaya plants, and occupy different habitats at substantially lower altitudes (> 2500 m; Fig. 1c,d), which might reflect ecological divergence, and perhaps an ongoing speciation process. Therefore the LP group might represent a separate species from the Himalaya group, or at minimum be on a path towards speciation from it. This species therefore provides a perfect test case to examine the genetic mechanisms underlying intraspecific ecological divergence.
Fig. 1.
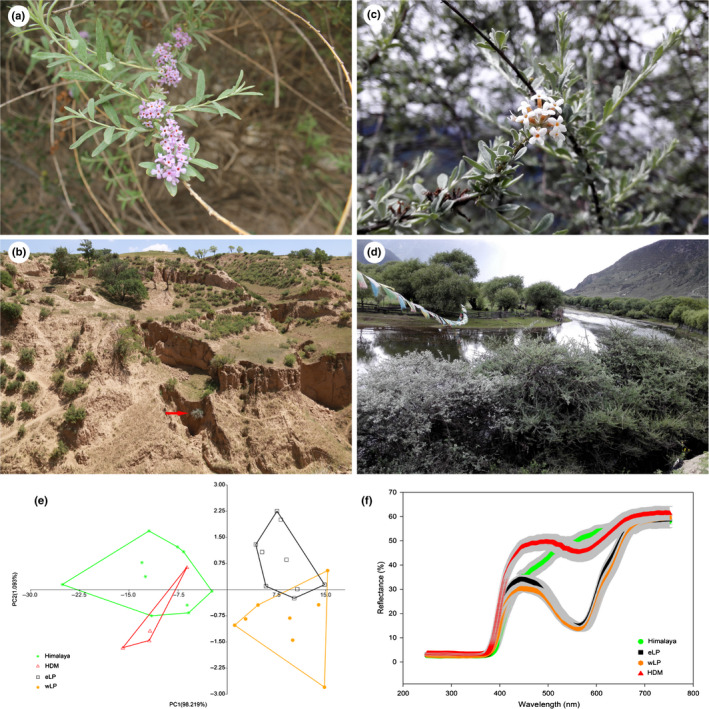
Flower characteristics and contrasting habitats of Buddleja alternifolia in the Loess Plateau (LP) (a, b) and Himalaya (c, d). The red arrow is pointing to individuals of B. alternifolia growing some distance apart in LP in (b), whereas many plants are commonly growing close together alongside the river in the Himalaya (d). (e) PCA result of morphological characteristics in Himalaya, Hengduan Mountains (HDM) and LP, as well as eastern (eLP) and western LP (wLP). (f) Petal colour reflection spectrum result of B. alternifolia in Himalaya, HDM and LP, as well as eastern (eLP) and western LP (wLP).
Here we report a newly produced, high‐quality genome sequence of B. alternifolia, the first for a member of Scrophulariaceae. Using this as a reference, we then examined the population genomics of B. alternifolia to understand its different processes of allopatric speciation in three uplands by re‐sequencing 48 populations covering its full distribution range, that is the Himalaya, LP and the HDM. From this, we characterised the species’ genetic diversity, inferred its population structure and associated demographic history, reconstructed its ancestral areas, and detected isolation by distance (IBD), isolation by adaptation (IBA) and niche divergence. In particular, we sought to capture genomic footprints of adaptation to differential environments.
Materials and Methods
Genome sequencing
Fresh young leaves of B. alternifolia for de novo genome assembly were collected from one individual in Beijing Botanical Garden, Institute of Botany, Chinese Academy of Sciences, Beijing, China. High‐quality genomic DNA was extracted from fresh leaf samples using a cetyltrimethylammonium bromide (CTAB) protocol (Doyle & Doyle, 1987). Long read sequencing using PacBio single molecule real‐time (SMRT) sequencing was performed by the commercial sequencing provider (Beijing Ori‐Gene Science and Technology Co. Ltd, Beijing, China). Three Illumina PCR‐free libraries with insert sizes of 300–500 bp were prepared using 2 μg genomic DNA following the manufacturer's protocol (Illumina). Whole‐genome paired‐end (PE) reads were generated using the Illumina HiSeq X Ten platform. Fastp v.0.19.3 (https://github.com/OpenGene/fastp) was used to filter out low‐quality reads and adaptor sequences. Genomic DNA of high molecular weight (≥ 700 ng) was cross‐linked in situ, extracted and then digested with a restriction enzyme. The biotin‐labelled DNA ligated from the sticky ends of these fragments was enriched and sheared to a fragment size of 300–500 bp to prepare the Hi‐C sequencing library, which was sequenced on a HiSeq X Ten platform.
Genome assembly
PacBio sequencing data were firstly corrected using canu v.1.7 assembler (Koren et al., 2017), and further applied to different assembly strategies including smartdenovo v.1.0 (https://github.com/ruanjue/smartdenovo), wtdbg v.1.2.8 (https://github.com/ruanjue/wtdbg‐1.2.8) and canu v.1.7 to obtain the draft assembly. V0.4 was selected as the best assembly strategy, due to the comprehensive evaluation of genome size, N50, numbers of contigs, and completeness (Supporting Information Table S1). The primary assembly obtained from V0.4 was first polished with Pilon v.1.22 (Walker et al., 2014) based on the high‐quality Illumina sequencing reads, and then piped into the Hi‐C assembly workflow. Hi‐C reads were mapped to the primary assembly with Juicer (Durand et al., 2016), and then a candidate chromosome‐length assembly was generated automatically, using 3d‐dna Pipeline software. Manual review and refinement of the candidate assembly was performed in juicebox Assembly Tools for quality control and interactive correction. To reduce the influence of interactions among chromosomes and to further improve the chromosome‐scale assembly, each chromosome was re‐scaffolded with 3D‐dna separately, and then manually refined with juicebox. Finally, after gap filling with lr_gap closer v.1.1 (Xu et al., 2018), pilon v.1.22 was used to polish the assembly (running for four rounds). We used the long terminal repeat (LTR) Assembly Index (LAI) as the standard for evaluating the assembly of repeat sequences, and to evaluate the assembly continuity (Ou et al., 2018). Additionally, the assembly quality was also evaluated using busco v.4.0.5 (Simao et al., 2015).
Genome annotation
repeatmodeler v.1.0.8 (http://www.repeatmasker.org/RepeatModeler/) was used for de novo identifying repeats within the genome. repeatmasker v.4.0.7 (http://www.repeatmasker.org/) was used to screen for repeats within the assembled genome, using the custom repeat library from RepeatModeler.
Total RNA from the young leaves, stem, flowers and root tissues were extracted using TRIzol reagent (Invitrogen). The PE RNA‐seq libraries were prepared using the NEBNEXT Ultra RNA Library Prep Kit for Illumina (New England Biolabs Inc., Ipswich, MA, USA), and 150‐bp PE sequencing was performed on an Illumina HiSeq X Ten platform. After filtering out low‐quality reads and trimming adapter sequences, RNA‐seq reads were aligned against the genome sequence using hisat2 v.2.1.0 (https://daehwankimlab.github.io/hisat2/). Next, a de novo assembly was constructed using trinity v.2.0.6 (https://github.com/trinityrnaseq/trinityrnaseq), and reference genome‐guided assemblies were generated with stringtie v.1.3.5 (http://ccb.jhu.edu/software/stringtie/) and trinity v.2.0.6.
Transcripts assembled from RNA‐seq data of B. alternifolia as well as protein sequences from Arabidopsis thaliana (Cheng et al., 2017), and two members of the Lamiales (Sesamum indicum and Handroanthus impetiginosus) were used to perform gene annotation (Wang et al., 2014; Silva‐Junior et al., 2018). Based on the repeat‐masked genome, ab initio prediction, transcripts and protein evidence alignments were integrated by the Maker2 gene annotation pipeline to generate a final protein‐coding gene set with annotation edit distance (AED) score calculated for quality control. The Rfam database (http://rfam.xfam.org/) was used for searching noncoding RNAs, while tRNAscan‐SE (Lowe & Eddy, 1997) and RNAmmer (Lagesen et al., 2007) were used to predict tRNAs and rRNAs, respectively.
Identification of orthologous genes and phylogenetic analysis
In total, B. alternifolia and 16 related plant species were sampled considering representativeness of phylogeny within Lamiales (including all species within the order) and other related orders with high‐quality genome (either the genome is chromosome level or scaffold N50 > 10 M). orthofinder2 was used to identify orthologous and paralogous gene clusters (Table S2), with parameters ‘‐M msa’ (Emms and Kelly, 2019). In total, 1227 single‐copy genes identified by orthofinder2 within these 17 species were used to construct a phylogenetic tree. MAFFT (Katoh et al., 2005) was used to perform multiple alignment of protein sequences for each set of single‐copy orthologous genes. All of the 1227 single‐copy genes were concatenated, and IQTree was used to build a maximum‐likelihood (ML) phylogenetic tree with the best‐fit JTT+F+R5 substitution model (Nguyen et al., 2014). In addition, we also inferred a species tree based on the 1227 single‐copy gene trees produced by Astral (Yin et al., 2019), which could provide evidence of gene tree discordance. The divergence time was estimated with r8s v.1.81 (https://github.com/R8S/r8s; Notes S1) and calibrated against crown divergence timing (Magallon et al., 2015) of Monocotyledoneae and Eudicotyledoneae (synchronously 135–130 Ma), Pentapetalae (126–121 Ma), Rosidae (123–115 Ma) and Asteridae (119–110 Ma). All of these dates were assigned as ‘constrained’.
Morphological characteristic measurements and petal colour analysis
In total, 31 populations were sampled for morphological characteristic measurements, comprising 10 populations from the Himalaya, four populations from HDM and 17 populations from LP (eight and nine populations from the western and eastern subgroups, respectively). Within each population in the Himalaya and LP, 30 mature leaves and mature flowers were randomly sampled from each of 30 flowering individuals in the field. For HDM populations, the original field data collected in this way were lost due to a computer failure, therefore measurements were taken from pressed specimens, with between 14 and 18 individuals sampled for per population. Three leaf morphological characters were measured including leaf length, leaf width and length/width ratio. Seven floral characters were measured as follows: (1) corolla tube length; (2) corolla tube width; (3) corolla lobe length; (4) corolla lobe width; (5) anther height; (6) style length; and (7) herkogamy (Dataset S1). past statistical software (v.3.26) (Hammer et al., 2001) was used to perform multivariate analyses of these 10 morphological characters, by a principal component analysis (PCA).
To assess light reflection patterns of B. alternifolia at different wavelengths, we also obtained spectral data from the corolla using a S2000 miniature fibre optic spectrometer with a PX‐2 pulsed xenon lamp (Ocean Optics, Dunedin, FL, USA). All measurements were carried out in the range 250–750 nm, using 0.30 nm increments. Spectral data were collected from the same individuals that were sampled for morphological measurements in the Himalaya and LP. For HDM populations, 30 flowers were sampled from 14 to 18 individuals per population (Dataset S1). Notably, a few successfully cultivated plants in Kunming Botanical Garden exhibited similar flower characteristics to the natural conditions, implying that the morphological differences between areas were partially determined by genetic effect.
Whole‐genome re‐sequencing data filtering and single nucleotide polymorphism (SNP) calling
In total, 96 individuals from 48 populations were re‐sequenced using the Illumina HiSeq X Ten platform (Fig. 2a; Table S13, see later). Raw data were filtered and PE clean reads were mapped to the reference genome. Reads with mapping quality ≤ 30 were removed from all further analysis. In total, 678 659 475 genomic loci were finally obtained. We evaluated the heterozygosity rate among sampled individuals in each of the Himalaya, HDM and LP, and again separately in the eastern and western subregions within LP, testing for significant differences using the Tukey honest significant difference (HSD) post hoc test. Due to differences in the altitudes where B. alternifolia grows in these regions, heterozygosity might also be varied with elevation, and therefore a correlation analysis was used to determine if a relationship exists between heterozygosity and altitudes among all samples. All calculations were performed using Spss v.22.0 for Windows (SPSS, Chicago, IL, USA).
Fig. 2.
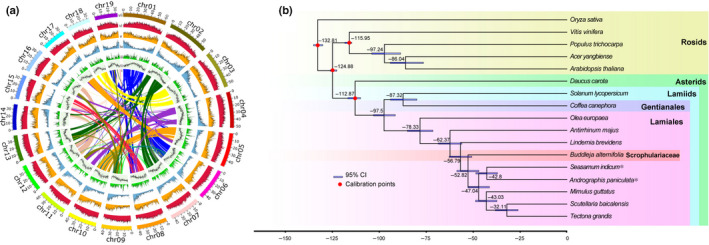
Genome evolution analysis of Buddleja alternifolia. (a) The genome features across 19 chromosomes of B. alternifolia. From the outermost to innermost circles are Class I transposable element (TE) (long and short interspersed nuclear elements) density; Class II TE (DNA and Heliron) density; coding gene (messenger RNA) density, heterozygous (single‐nucleotide polymorphisms, insertions, and deletions) density; GC content and genome colinear blocks. (b) Phylogenetic tree reconstructed using the maximum‐likelihood (ML) method and 1227 single‐copy genes with divergence time estimated with r8s v.1.81 on the basis of three calibration points (red circles). * Indicates conflicting positions between A. paniculata and S. indicum inferred by Astral‐based and ML‐based approaches (see Supporting Information Fig. S1 for details).
SNP calling was then conducted using freebayes (Garrison & Marth, 2012). We performed the following filtering steps to minimise SNP calling bias, and to retain only high‐quality SNPs: (1) removal of SNPs with quality score of < 20; (2) retention of only bi‐allelic SNPs; and (3) treating genotypes with depth < 3 as missing. Finally, we removed SNPs with missing rate > 20%, which resulted in 16 927 185 SNPs. Then all SNPs with maf < 0.05 were also removed, leaving 6373 626 SNPs remaining for downstream analysis.
Linkage disequilibrium decay and calculation of population genetic parameters
Exploring how genetic diversity is distributed among areas could further elucidate the demographic history and potential dispersal routes of B. alternifolia. Here we estimated linkage disequilibrium (LD) decay based on the coefficient of determination (r2 ) between any two loci using poplddecay (https://github.com/ekg/freebayes) (Fig. S2). Genetic diversity parameters including nucleotide diversity π, Watterson's estimator θ W, Tajima's D and F st were calculated at both species and lineage levels from sample allele frequency likelihoods in Angsd (Korneliussen et al., 2014) over 20 kb sliding windows with overlapping 10 kb. Confidently called nonvariant sites were included in this analysis for calculation of these genetic diversity parameters. Furthermore, to see if F st patterns differed between genome regions, we divided the genome into nongenic and genic regions, and further divided the latter into eight small subgroups (codons 1, 2 and 3; intron; utr3; utr5; upstream and downstream).
Population structure analysis
We inferred population structure and admixture among 97 samples using faststructure (Raj et al., 2014) based on 343 459 SNPs after further removing linked loci using Plink (SNP window size: 50 kb; SNPs shifted per step: 10 kb; r2 threshold: 0.2). The most likely number of clusters was computed with 10‐fold cross‐validation (CV), comparing K values from 2 to 10. PCA was conducted to study the genetic relatedness and clustering among populations using Gcta (Yang et al., 2011). Using the same data, a neighbour‐joining phylogeny tree was constructed to observe relationships among samples using Mega7 (Kumar et al., 2016), using the p‐distance method; clade supports were calculated using 1000 bootstrap replicates.
Inference of demographic histories
We used coalescent simulations, applying the composite likelihood method implemented in Fastsimcoal software v.2.6 (Excoffifier et al., 2013), to infer demographic parameters of B. alternifolia using SFS, while excluding individuals with admixture. Two‐dimensional joint SFS (2D‐SFS) was constructed from posterior probabilities of sample allele frequencies by realSFS implemented in Angsd. In total, 563 501 150 sites were retained for analysis. 100 000 coalescent simulations were used for the estimation of the expected 2D‐SFS and log‐likelihood for a set of models that are illustrated in Fig. S3. The best model was identified through the Akaike's information criterion (AIC).
The Stairway plot v.0.2 with 200 bootstrap iterations was also used to infer changes in population size over time, for each of the genetic groups (Liu & Fu, 2015); however individuals were excluded if indicated to have admixture based on site ancestral state, according to SFS as estimated using Angsd based on site ancestral state (Methods S1). We estimated a mutation rate of 1.08 × 7e−9 per site per year (Methods S2) and a generation time of 2 yr in B. alternifolia based on our observation of time period from sowing B. alternifolia seeds to flowering in the Kunming Institute of Botany, when converting estimates to units of years.
Ancestral area reconstruction
We selected 19 representative individuals with high mapping coverage from the neighbour‐joining (NJ) tree (Fig. 3f), comprising five from the Himalaya, three from HDM, five from western LP and six from eastern LP. After removal of missing SNPs, 650 599 SNPs common to all the 19 samples were finally obtained, and B. globosa was defined as an outgroup (Fig. S4). The divergence time of B. alternifolia from B. globosa was set to the range 8.5–25.0 Ma, according to the estimate by Chau (2017).
Fig. 3.
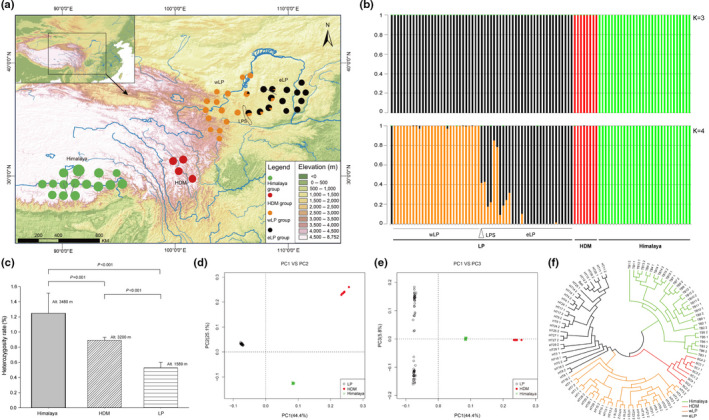
Population genomics of Buddleja alternifolia. (a) Sample collection of 48 populations of B. alternifolia, with colours in the pie charts referring to genetic groups identified by faststructure results, and size to the heterozygosity rate in different areas. Dotted line indicates the location of the Liupanshan mountain (LPS). (b) Population genetic structure of B. alternifolia by faststructure. (c) Significant differences of heterozygosity rates (mean ± SE) among three distribution regions of B. alternifolia. (d, e) PCA plots of genetic variation in B. alternifolia, with the proportion of the variance explained as (d) 44.4% for PC1 and 25.1% for PC2, and (e) 44.4% for PC1 and 5.8% for PC3. (f) A neighbour‐joining (NJ) phylogenetic tree of B. alternifolia based on SNPs from whole‐genome re‐sequencing data.
Divergence time estimation was performed in beast v.2.4.7 based on the SNP matrix (Notes S1). The GTR+ Γ substitution model was selected as the best model, based on AIC testing by iqtree (Nguyen et al., 2014). The clock model was set as the Relaxed Lognormal Clock model with the default settings. For tree prior, the Coalescent Exponential Population model was selected, in accordance to the exponential changes of effective population sizes calculated by Fastimocal 2.6. Analyses were conducted for 100 million generations with sampling every 10 000 generations. Convergence was checked by examining estimated sample sizes > 200, indicating a sufficient level of sampling. The maximum clade credibility (MCC) tree with median node heights was generated in treeannotator v.2.4.1, after discarding the initial 50% as burn‐in. tracer v.1.7.1 (http://tree.bio.ed.ac.uk/software/tracer/) was used to check effective sample sizes.
Four major geographic regions were defined based on current distribution patterns of B. alternifolia, that is the Himalaya, HDM, western LP and eastern LP. We used the R package biogeobears to estimate ancestral distributions under three models: Dec, Divalike (based on dispersal‐vicariance analysis), and Bayarea based on Bayesian inference of historical biogeography for discrete areas, with and without the founder‐event speciation ‘J’ parameter.
Detection of IBD, IBA and niche divergence
We used a simple Mantel test to examine the association of genetic divergence with geographic distance and adaptive divergence. A partial Mantel test may be more informative than a simple Mantel test to gauge the relative importance of the two factors that simultaneously influence genetic structure (Gomez‐Uchida et al., 2011). We therefore further used a partial Mantel test to examine the above associations while controlling for geographic distance and adaptive divergence using the zt‐program (Bonnet & Van de Peer, 2002). Geographic distances between populations are shown in matrix form in Table S3. With reference to Nosil et al. (2008), four out of the above‐mentioned 10 morphological characters examined (length and width of tubes, herkogamy and petal colour) were selected for indicating adaptive divergence, as they were expected to have associations with pollinator preference and differentiation, therefore probably promoting reproductive isolation. Index for petal colour was calculated as the sum of reflectance values from 300 to 700 nm. We calculated Pearson correlation coefficients (r) and their P‐values after 100 000 randomisations.
For ecological divergence, 31 environment variables were collected initially, including 19 bioclimatic variables from the Worldclim database (http://www.worldclim.org/); four measures of global UV‐B radiation from the glUV database (http://www.ufz.de/gluv/); an altitude layer from the EarthEcplorer database (https://earthexplorer.usgs.gov/); three relevant edaphic variables and four other environmental data measures from the FAO database (http://www.fao.org/). In total, 13 environment variables were selected for subsequent analysis using the threshold for Pearson’s correlation coefficients (r) < 0.75 (Dormann et al., 2013; Table S4). In addition, 121 geographical coordinates were obtained from field investigations and specimen resource databases (Table S5). We performed tests of the ecological niche overlap and identity to estimate niche differences using the Schoener's D statistic (Schoener, 1968) and the standardised Hellinger distance (I) in Enmtools. The values for D and I from 0 to 1 represent the similarity of the niches, that is, 0 means completely dissimilar and 1 means identical.
Genomic signals of adaptation
We firstly used a F st‐based approach to investigate the selection signals across the whole genome in populations among the Himalaya, HDM and LP, as well as the western and eastern subgroups within LP. We calculated the F st using the program Angsd in 20‐kb sliding windows along all chromosomes. We further used sweepfinder2 (DeGiorgio et al., 2016), a composite likelihood ratio statistic (CLR) to search for signs of selective sweeps in these populations. The small fraction of SNPs that could not be polarised was excluded from further analysis. As recommended by Huber et al. (2016), we only used sites that were polymorphic or fixed in each group of populations, to scan for sweeps. sweepfinder2 was run with a grid size of 2 kb. We only considered the top 1% of detected regions with significant selection signals in both approaches. CLR scores were merged into sweep regions if neighbouring scores exceeded the top 1% threshold. We performed gene ontology (GO) enrichment analysis for the candidate genes and focused on those genes with significant GO terms (P < 0.05) in the corresponding lineages and sublineages.
Results
Genome sequencing and assembly
In total, c. 90 Gb of data were generated using PacBio SMRT sequencing (11 cells, 10 M reads with 100×); c. 100 Gb (700 M reads, with 120×) with Illumina HiSeq, and c. 110 Gb (740 M reads with 170×) through Hi‐C (Tables S6–S8). Overall, 19 chromosomes (with a total size of 822 846 782 bp, c. 96.4%) were anchored with 676 contigs (with an average size of 43 307 725 bp). In addition, the mitochondrial and chloroplast genomes were assembled into circular DNA of 499 191 bp and 141 138 bp, respectively. The assembled final genome was c. 854 Mb with contig N50 of 1.9 Mb and scaffold N50 of 4.3 Mb (Tables 1, S1). In total, 3919 289 heterozygous sites were detected and the heterozygosity rate was calculated to be c. 0.46%. LAI was estimated to be 17.8, indicating high accuracy and completeness of the assembly.
Table 1.
Statistics of Buddleja alternifolia genome assembly and annotation.
| Characteristics | Size and number |
|---|---|
| Total assembly size | 853 754 563 |
| Total number of contigs | 1096 |
| Maximum contig length | 10 270 997 bp |
| Minimum contig length | 5000 bp |
| Contig N50 length | 1942 762 bp |
| Contig L50 count | 129 |
| Contig N90 length | 514 978 bp |
| Contig L90 count | 431 |
| Total number of scaffolds | 439 |
| Maximum scaffold length | 62 290 481 bp |
| Minimum scaffold length | 5000 bp |
| Scaffold N50 length | 43 104 400 bp |
| Scaffold L50 count | 9 |
| Scaffold N90 length | 33 029 929 bp |
| Scaffold L90 count | 18 |
| Gap number | 657 |
| GC content | 36.10% |
| Gene number | 31 116 |
| Protein‐coding genes | 29 434 |
| Repeat content | 71.70% |
Genome annotations
Of the assembled genome sequence, 71.70% (612.20 Mb) were transposable elements with a dominance of LTRs (Table 1; Fig. 2a). The most abundant repeat element families were Copia (262.90 Mb, 30.8%) and Gypsy (111.22 Mb, 13.02%) (Table S9). We generated 182.81 million raw reads of the RNA‐seq data. In total, 29 434 protein‐coding genes were annotated. The average lengths of transcripts, gene regions and coding DNA sequences were 1447, 4256 and 1283 bp, respectively (Table S10). Moreover, 720 ncRNA, 87 rRNA and 875 tRNA sequences were identified. Based on sequence conservation, 17 281 (c. 58.70%), 27 073 (c. 92.00%), 27 781 (c. 94.20%) and 23 448 (c. 79.70%) genes were annotated. In addition, based on domain conservation, 380 (c. 1.29%), 3809 (c. 12.94%), 22 695 (c. 77.10%), 8110 (c. 27.55%) and 26 207 (c. 89.04%) genes were annotated (Table S11). The annotated proteins were evaluated using busco (embryophyta_odb9 database), and this revealed that 1246 (86.5%) genes were complete and single copy, 122 (8.5%) were complete and duplicated, while 46 (3.2%) genes were missing and 26 (1.8%) were fragmented.
Comparative genomics and phylogenetic analysis
We performed orthogroup analysis and identified 433 576 genes belonging to 18 531 gene families. Of these, 39 gene families and 190 genes were unique to B. alternifolia (Table S12).
As the first sequenced genome representing a member of Scrophulariaceae, our phylogenomic analysis based on both ML and Astral methods (Fig. S1), largely agreed with APG IV (2016), placing B. alternifolia within the Lamiales, except that our analysis suggested that Scrophulariaceae branched off after Linderniaceae (Fig. 2b), whereas APG IV had this reversed. Based on the tree, calibrated using three major divergence events within Angiosperms, Scrophulariaceae diverged from related families (at least among those included) c. 60 Ma (Fig. 2b).
Morphological characters and petal colour analysis
In the PCA analysis of 10 morphological characters, the first principal component (PC1) explained 98.22% of the total variance, and allowed the separation of 31 populations into two groups: populations from the Himalaya and HDM formed one group, whereas LP formed another. Among these characteristics, leaf length and width were mostly related to PC1. The second principal component accounted for 1.09%, and further divided the LP group into western and eastern subgroups (Fig. 1e). Neither component separated Himalaya from HDM, and indeed we found plants from these two regions to be morphologically indistinguishable.
The reflectance spectrum of B. alternifolia petals also showed contrasting patterns between regions, with LP plants consistently showing a marked peak in the reflectance spectrum at 430 nm, whereas no peak of reflectance spectrum occurred in the Himalaya and HDM accessions (Dataset S1; Fig. 1f).
Whole‐genome re‐sequencing, SNP calling and annotation
In total, 1.05 Tb of WGS data (7.15 billion PE raw reads) were generated for 96 B. alternifolia accessions from 48 populations with an average depth of c. 14.9× (Fig. 3a; Table S13). Using the chromosome‐level B. alternifolia reference genome, the mapping rate of these raw reads was 98.83 ± 1.1% (Table S11). On average, a heterozygosity rate of 0.0078 ± 0.0036 was detected among all samples (Table S13). Intriguingly, Himalaya accessions had a significantly higher heterozygosity rate (0.0125 ± 0.0027) and mean elevation (3480 m) than accessions from the other two regions (HDM: 0.0089 ± 0.0004, elevation 3200 m; LP: 0.0053 ± 0.0007, elevation 1594 m; both P < 0.001). Heterozygosity was therefore positively correlated with altitude (r = 0.778, P < 0.001; Fig. 3c).
We finally obtained 6373 626 high‐quality SNPs using a strict filtering standard with an average of 1444 359 per individual. A survey of all these SNPs in the B. alternifolia genome showed that c. 74.4% of SNPs were located in intergenic regions and 4.4% in coding sequences (Table S14).
Population structure of B. alternifolia
We found very similar patterns of genome‐wide LD of B. alternifolia among the three areas, with 10, 12 and 13 kb as half of the maximum r2 in Himalaya, HDM and LP, respectively (Fig. S2). All three lineages of B. alternifolia had high levels of genetic diversity, with an average of 9.6 ± 3.2 × 10−3 for π and 9.16 ± 3.04 × 10−3 for θ w (Table 2). Notably, a significantly higher genetic diversity was observed in the Himalaya lineage (an average π of 13.84 × 10−3; θ w = 13.25 × 10−3) than both the HDM (π = 8.45 × 10−3; θ w = 7.13 × 10−3) and LP (π = 6.51 × 10−3; θ w = 7.11 × 10−3) lineages (π: F = 553.62, P < 0.001; θ w: F = 313.98, P < 0.001), which is in accordance with the patterns of heterozygosity rate observed above.
Table 2.
Sample size and genetic diversity of three lineages of Buddleja alternifolia.
| Region | No. populations/plants | No. of SNPs | π (SD) (10−3) | θ w (SD) (10−3) |
|---|---|---|---|---|
| Himalaya | 15/30 | 4022 382 | 13.84 (0.79) | 13.25 (1.28) |
| LP* | 29/59 | 2539 728 | 6.51 (0.59) | 7.11 (0.57) |
| HDM | 4/8 | 2073 747 | 8.45 (0.72) | 7.13 (0.56) |
| Total | 48/97 | 6373 626 | 9.60 (3.20) | 9.16 (3.04) |
π average number of pairwise nucleotide differences per site, θ W Watterson's estimator of θ per base pair.
The individual for de novo whole‐genome sequencing was included in LP.
faststructure analysis revealed that the three groups corresponding to the Himalaya, LP and HDM were clearly separated when three clusters (K = 3) were assumed (Figs 3b, S5). Two subgroups corresponding to populations located in the east and west of LP were further split when the optimal value (K = 4) was set (Fig. 3b). When K ≥ 5, similar patterns were observed than with K = 4, indicating the stable population structure of B. alternifolia. Notably, in both western and eastern LP, those populations closest to the Liupanshan mountain (LPS) barrier that divides them exhibited an admixture pattern (Fig. 3b), indicating the existence of gene flow across the LPS barrier.
Consistent with the faststructure results, PCA analysis of B. alternifolia revealed three major clusters corresponding to the three uplands (Fig. 3d). Specifically, PC1 (explaining 44.4% of the total genetic variance) separated Himalaya from LP and HDM, suggesting that the latter two lineages shared more genetic background. LP and HDM lineages were separated by PC2, which explained 25.1% of the total genetic variance. East and West LP were further separated by PC3 with 5.8% variation (Fig. 3e). An NJ tree similarly indicated that each of the Himalaya, LP and HDM formed a distinct lineage (Fig. 3f), with LP then subdivided into eastern and western clades, with some admixture in the centre.
Genome‐wide genetic differentiation between populations was F st > 0.5 for any pair of populations, in any of the three regions (Table S15). F st in the gene regions of all these small subgroups are very similar, with F st (LP vs Himalaya) > F st (LP vs HDM) > F st (HDM vs Himalaya). Intriguingly, however, the F st pattern for intergenic regions was different in that F st (LP vs HDM) > F st (LP vs Himalaya) > F st (HDM vs Himalaya) (Table S16). Comparing F st patterns in gene regions to nongene regions, genetic differentiation between LP and HDM was higher in gene regions (0.699 vs 0.641) whereas that between HDM and Himalaya was higher in nongene regions (0.619 vs 0.555).
Divergence and demographic histories of B. alternifolia
We first evaluated three alternative models (Himalaya, LP or HDM diverged first), assuming no subsequent gene flow in each case, based on the faststructure results. The model where the Himalaya diverged first was supported, having the lowest AIC (896 982 610). Then nine models that differed in what types of gene flow occurred were checked and the most likely model (AIC = 887 254 256 and the maximum Akaike's weight = 1; Fig. S3) was selected. This model suggested that the Himalaya group diverged from HDM + LP c. 2.29 Ma, with HDM diverging from LP very soon afterwards. Furthermore, the most likely model also indicated that only a very low level of subsequent gene flow occurred following divergence, presumably due to the geographic isolation between these three areas (Fig. 3a; Table S15). In addition, fastsimcoal analysis revealed that the western and eastern LP groups diverged at c. 0.97 Ma ago, with Ne expanding in the east and contracting in the west (Fig. 3b).
We used the Stairway plot method to infer changes in population size (Ne ) over time for those genetic groups identified by the above structure analysis (Fig. 3c). For each of the three lineages, the earliest bottleneck event was c. 1 Ma, whereas a second occurred at c. 0.2–0.3 Ma in both Himalaya and LP. Both events occurred slightly earlier for Himalaya than for the LP. After the two bottlenecks, the Ne of Himalaya remained fairly constant until 10 000 yr ago, at which point it began to decline. LP underwent a third bottleneck at c. 0.1 Ma and thereafter a stable Ne was maintained until 10 000 yr ago, at which time Ne expanded, then remained stable until the present. For HDM, Ne recovered after the only detected bottleneck but then declined; however these results may be affected by the limited sample sizes.
Treating the western and eastern LP groups separately, the earliest bottleneck event for each occurred at c. 0.8–0.9 Ma, possibly the same demographic event as the 1 Ma bottleneck events for all three lineages mentioned above. All of these were followed by rapid recovery (Fig. 3d). From this point onwards, however, the Ne of western and eastern LP populations followed quite different trajectories. The eastern populations underwent a second population drop at 200–300 ka, then changed little until c. 60 ka, at which time it gradually underwent a recovery towards Ne = 500 000, before a progressive decline towards Ne = 100 000 at c. 2 ka. Conversely, the second population drop in western LP occurred over two stages, between 100 and 200 ka, falling from Ne = c. 800 000 to c. 60 000 then swiftly recovering but only to c. 300 000. There was then a third phase of population decline, this time much slower, between 20 and 5 ka, dropping to Ne = c. 15 000, followed by a steady expansion to Ne = c. 1000 000 by 3 ka (Fig. 4d). This last fall and rise might coincide with a period of local habitat drying and accelerated accumulation of loess populations.
Fig. 4.
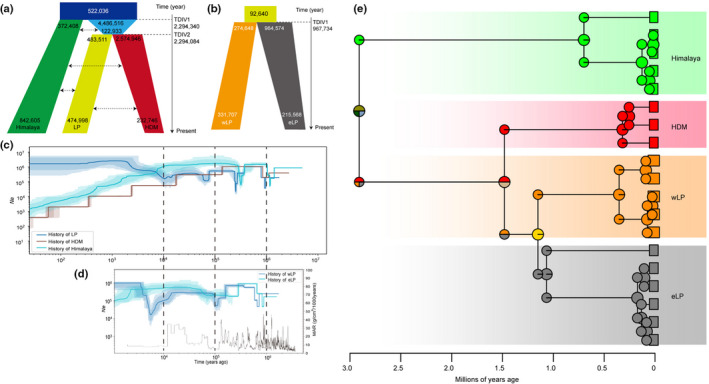
Demography histories and ancestral area reconstruction of Buddleja alternifolia. (a) Schematic of demographic scenarios among the three lineages and (b) eastern and western sublineages within the Loess Plateau (eLP and wLP), modelled using fastsimcoal2, with column width representing the relative effective population size. The numbers on the vertical axis indicate the estimated time of population divergence. (c) Historic changes in Ne for Himalaya, LP and Hengduan Mountains (HDM), and (d) for the two sublineages within LP using stairway plotting, with lighter and darker colours indicating 95% and 75% confident intervals of shaded areas. (e) Maximum clade credibility (MCC) chronogram of ancestral area reconstruction of B. alternifolia inferred from Bayesian analysis in Beast. Pie charts indicate relative probability of ancestral distributions at nodes from BioGeoBears analysis, using the best fitting Divalike model.
Ancestral area reconstruction
Himalaya and (LP + HDM) clades of B. alternifolia diverged at c. 2.91 Ma (confidence interval, CI = 2.14–5.41 Ma), followed by divergence of HDM and LP lineages at c. 1.48 Ma (CI = 1.08–2.75 Ma). Within LP, western and eastern subgroups then diverged at c. 1.15 Ma (CI = 0.84–2.14 Ma). For the models compared in BioGeoBears, the Divalike model had the lowest AIC (12.91) scores (Table S18). This model indicated that the current three lineages are likely to have arisen by vicariance, by contraction of a previously more widespread range for the species and that, during the vicariance process, the Himalaya diverged first (Fig. 3e).
IBD, IBA and niche divergence
Both simple and partial Mantel tests revealed that both geographic distance and adaptive divergence played significant roles influencing genetic distance (all P < 0.05; Table S19). Furthermore, the partial Mantel test suggested that geographic distance played a greater role than adaptive characters, due to the evidently decreased r and increased P‐values when controlling for the latter (Table S19).
Observed measures of niche similarity for both D and I were lower than null distributions for LP vs either Himalaya (observed: D, 0.425; I, 0.571; null distribution: D, 0.691–0.919; I, 0.732–0.929) or HDM (observed: D, 0.392; I, 0.481; null distribution: D, 0.501–0.793; I, 0.602–0.878), suggesting high niche differentiation between LP and both Himalaya and HDM. Alternatively, with regard to the Himalaya vs HDM, both D and I fell within the range of null distributions (observed: D, 0.645; I, 0.750; null distribution: D, 0.541–0.834; I, 0.572–0.866), suggesting a similar niche occupancy.
Genomic signals of adaptation
Selection signals with 65, 59 and 54 genomic regions including 156, 177 and 251 genes were detected in LP and HDM, LP and Himalaya, HDM and Himalaya, respectively, by the F st‐based analysis. In addition, sweepfinder2 detected sweep selection signals in 156, 215 and 152 genomic regions containing 248, 350 and 228 genes in the Himalaya, HDM and LP, respectively. Moreover, 6, 23 and 27 genes were detected by both approaches, and among which 5, 10 and 14 genes had significant overrepresentation with a specific GO term (P < 0.05) in LP, HDM and the Himalaya, respectively (Table S20). Adaptive roles of these genes were functionally annotated, including seed germination and maturation (GO:0010187, GO:0010431 and GO:0009845), response to temperature stimulus (GO:0009266), cold (GO:0070417) and bacterium (GO:0009617) (Table S21). Within LP, 48 and 10 genes were detected by both methods, and among these 26 and 7 genes had significant overrepresentation with a specific GO term (P < 0.05; Table S20) in populations within eLP and wLP respectively, including some of the above‐mentioned GO terms including maintenance of seed germination and dormancy (GO:0010029; GO:0010231), and regulation of defence response to insect and bacterium (GO:2000068, GO:0016045) (Table S21). Notably, both approaches also detected Bualt19G0002200 as having significant overrepresentation with a specific GO term (GO:0009269: response to desiccation) in wLP alone (Table S21), which is the driest area among all distribution ranges of B. alternifolia.
Discussion
Our data strongly indicate that B. alterniflora is monophyletic and sister to B. crispa (Methods S3; Fig. S6), which occupies similar habitats in and around the Himalayas. However, B. alternifolia appears to be heterogeneous. Population structure analysis revealed three genetic groups closely corresponding to the three current distribution areas, and F st was calculated to be > 0.5 between any two of the three areas. Conversely, morphological data clearly divide B. alternifolia into two groups, that is the Himalaya plus HDM and LP, with 98% of morphological data supporting this subdivision in our PCA analysis. LP accessions also differed from the others in flowering time (May in Himalaya vs July in LP; Y. P. Ma, pers. obs.), and in that LP accessions showed a peak in the reflectance spectrum at 430 nm, whereas Himalaya plants exhibited no such peak. As the reflectance spectrum of petal colour could act as an important signal for attracting potential pollinators (Dyer et al., 2012), coupled with the differences in flowering time between these regions, we might expect that the LP linkage would employ different pollinators from the Himalaya linkage. Based on the above evidence, the LP linkage represents, and should be described as, a new species. The HDM lineage, however, was clearly genetically differentiated from both Himalayan and LP linkages, yet appeared to be morphologically indistinguishable from the Himalayan lineage. Therefore, further investigation of the taxonomic status of this group is necessary.
The LP group is divided into western and eastern subgroups by the LPS mountain range, which is 240 km long from north to south and, on average, >2500 m high. The PCA on morphology found a certain amount of subdivision between these subgroups, but our genetic data clearly indicated that some ongoing gene flow persisted between western and eastern LP, although this only involved populations close to the LPS barrier. Therefore the LP populations might be in the early stages of parapatric speciation. However, in the absence of morphological differences, formal taxonomic recognition would be premature for the two groups within the LP.
From the viewpoint of speciation, both IBD and IBA analysis indicated that allopatric speciation would have taken place among three regions promoted by geography isolation and adaptive divergence, probably with the former playing a greater role. The LP range of B. alternifolia differs ecologically from the species’ range in the Himalaya and HDM, in that the region is milder and much drier, and also that LP plants tend to occupy habitats, as opposed to river habitats elsewhere. In line with this, our niche divergence analysis indicated that these ecological differences are likely to have been facilitated the speciation process. Such ecological speciation has been detected many times in plants, especially in the Qinghai–Tibetan Plateau and adjacent areas (e.g. Pinus densata by Mao & Wang, 2011; Salix brachista by Chen et al., 2019).
Most analyses supported the Himalaya as the first‐diverging lineage. Furthermore, higher genetic diversity in the Himalaya is consistent with the other two being derived from the Himalaya. fastsimcoal analysis however, showed that HDM diverged from LP < 1000 yr after the divergence of the Himalaya, c. 2.3 Ma, only 0.6 Ma after the divergence of B. alternifolia itself (Chau, 2017). However, biogeobears indicated that divergence occurred by vicariance, rather than dispersal. With the caveat that this analysis has barely been tested with full genome data before now, it appears to reject a hypothesis that LP and HDM were founded by successive dispersal events. Instead, it supported contraction and separation from a previously continuous range, which could also lead to the relatively short gap between divergence events.
Our WGS‐based population genomic analysis revealed that both genetic diversity and the heterozygosity rate were significantly higher in the Himalaya than in LP or HDM. If the latter two were not founded by dispersal from the Himalaya, then this result could be explained instead by the Himalaya being a refugium that maintains high levels of genetic diversity (Qiu et al., 2011; Zhao et al., 2019). Vicariance followed by population bottlenecks in LP and HDM but not the Himalaya could also produce this effect, but our analysis in fact detects bottlenecks of similar severity in all three. Modern population structure may therefore provide a better explanation: B. alternifolia populations in the Himalaya are often large and intercontinuous, for example along river courses, whereas its population structure is seriously fragmented in LP (Fig. 1c,d; Y. P. Ma, pers. obs.). Therefore genetic drift and inbreeding might have had greater impacts upon genetic diversity and especially heterozygosity in LP than in the Himalaya.
Several episodes of contraction and expansion of effective population sizes of B. alternifolia were revealed by Stairway plot analysis, and these may be linked to geological events. All three lineages experienced a contraction and bottleneck event c. 1 Ma (slightly later if eastern and western LP were examined separately), and these might have been triggered, or at least affected, by the Kunlun–Yellow river tectonic movement which began c. 1.1 Ma and uplifted the eastern Qinghai–Tibetan Plateau to 3000–2500 m in elevation (Cui et al., 2011). This promoted the formation of the modern East‐Asia winter monsoon, triggering major environmental changes, including increased drought in North China, proliferation of deserts, and increases in the rate of sediment loss (Li, 1991; Jiang et al., 2005). Following recovery from the initial bottleneck event, Ne within HDM has decreased steadily that, given that only a few small populations are extant today, is likely to be due to population loss and contraction, and inbreeding among remaining populations.
Both the Himalaya and LP underwent a second rapid population drop and recovery, possibly corresponding to the first known glaciation event in the area, that is the Wangkun Glaciation, c. 0.5–0.7 Ma before present (Cui et al., 2011). Subsequently, the general trend has been for Ne to increase in LP but decrease in the Himalaya. When analysed separately, western LP shows a steady decline from c. 11 to 6 ka, possibly linked to a period of very low rainfall c. 9 ka (Maher, 2016), and followed by recovery to higher Ne than before, which then holds up until the present. Eastern LP seems unaffected during this time, but declines c. 1 ka, indicating that different factors seem to apply on the different sides of the LP.
Genetic footprints of local adaptations to these environments were identified by both approaches. However, genes with significant overrepresentation of a specific GO term did not appear uniformly across all regions, perhaps reflecting differing habitats between lineages. However, selection signals of genes that were functionally involved in seed maturation and dormancy were detected in all four subregions, that is the Himalaya, LP, eLP and wLP (Table S21). All these annotated functions are likely to reflect how seeds of B. alternifolia might adapt to contrasting environments in the different regions. For example, cold and drought are especially strong factors in the Himalaya and LP respectively, but much less so vice versa, because the LP has a milder climate but B. alternifolia typically occupies drier habitats. Further evidence supporting the hypothesis of stronger selection imposed by the drier conditions of LP relative to other areas, comes from the detected gene Bualt19G0002200, having significant overrepresentation with response to desiccation (GO:0009269) in wLP alone, which is the driest area within the distribution ranges of B. alternifolia. Additionally, several genes were functionally annotated involved in response to viral infection (e.g. GO:0060154, GO:0071446, GO:0002230; Table S21) in LP, HDM and eLPS. This result may indicate that pathogen resistance is a strong factor limiting normal growth for B. alternifolia in areas other than the Himalaya, where much stronger UV due to higher elevation could decrease the influence of pathogens. Caveats remain to our GO enrichment analysis, as enrichment of a function might actually indicate that the genes concerned are under selection for an alternate function (Mondragón‐Palomino et al., 2002).
Conclusion
In conclusion, we present a high‐quality and chromosome‐level genome sequence of B. alternifolia, the first such plant genome in the family of Scrophulariaceae. Whole‐genome re‐sequencing‐based population studies uncovered three distinct genetic lineages distributed in the Himalaya, HDM and LP, suggesting that allopatric speciation has taken place. Allopatric speciation with gene flow may also be underway within LP, between populations from either side of a mountain barrier. Niche divergence and GO enrichment analyses indicated an ecological component to the differentiation of LP lineage. Demographic history analysis revealed several contraction and expansion of Ne events in B. alternifolia, with possible causes including climate fluctuations promoted by the Kunlun–Yellow river tectonic movement. Our data are consistent with the current disjunctions resulting from vicariance of a once widespread distribution, and therefore does not support the dispersal‐based ‘out of Tibet’ hypothesis.
Author contributions
Y‐PM, W‐BS, RIM and YVP conceived and designed the experiments; Y‐PM and R‐LL collected samples and made morphological measurements; HMW and R‐GZ analysed the data; Y‐PM, YVP and RIM wrote the paper. YVP, W‐BS, RGO, Q‐ZY and JHC revised the manuscript. All authors read and approved the final manuscript. Y‐PM, H‐MW, R‐LL and R‐GZ contributed equally to this work.
Supporting information
Dataset S1 Morphological measurement and floral colour reflectance data for populations of Buddleja alternifola.
Fig. S1 Phylogenetic trees inferred by ASTRAL‐ and ML‐based approaches.
Fig. S2 Patterns of linkage disequilibrium (LD).
Fig. S3 Models 1–3, during the process of divergence among the three linkages, no gene flows with no changes in effective population size and (Model 1); with changes in effective population sizes starting from the divergence of TB (TDIV1), as well as SC and HT (TDIV2, Model 2); with changes in effective population sizes starting from TDIV1.
Fig. S4 The phylogenomic tree used for time assignment of divergence for ancestral area reconstruction using representative samples of B. alternifolia and three species in the genus are currently available with re‐sequencing data.
Fig. S5 Cross‐validation (CV) error and marginal likelihood values for different model K.
Fig. S6 Reconstructing the phylogenomic relationships for 46 species of Buddleja using single‐copy genes.
Methods S1 Site ancestral state estimation.
Methods S2 Estimating mutation rate of B. alternifolia.
Methods S3 Reconstructing the phylogenomic relationships for 46 species of Buddleja using single‐copy genes.
Notes S1 Reproducibility of analyses for BEAST and r8s files.
Table S1 Statistics of all assemblies.
Table S2 Basic information with regards to genomes of 17 plants that were used to gene family analysis and the phylogenetic tree construction.
Table S3 A matrix information on geographic distances among populations.
Table S4 Environmental parameters used for assessment of ecological niche differentiation in B. alternifolia.
Table S5 Geographical coordinate of B. alternifolia.
Table S6 WGS‐PacBio sequencing statistics.
Table S7 WGS Illumina sequencing statistics.
Table S8 HiC sequencing statistics.
Table S9 Repeat annotations of the Buddleja alternifolia genome assembly.
Table S10 Gene annotation statistics of the Buddleja alternifolia assembly.
Table S11 Functional annotation of predicted genes in the Buddleja alternifolia genome.
Table S12 Summary of the gene family analyses.
Table S13 Basic information on location and genome mapping characteristics of all sampled individuals.
Table S14 Summary of SNP annotations.
Table S15 Global pairwise Fst between areas at the whole‐genome level.
Table S16 Pairwise Fst between areas in the divided nine subgroups of the whole genome, that is, eight in the gene region and one in the intergene region.
Table S17 Results of nine models used in the fastsimocal analysis.
Table S18 Basic parameters of three models compared in BioGeoBears, that is, Dec and Divalike based on dispersal‐vicariance analysis, and Bayarea based on Bayesian inference of historical biogeography for discrete areas, with and without the founder‐event speciation ‘J’ parameter.
Table S19 Results of IBD and IBA analysis using simple and partial Mantel tests.
Table S20 Shared genes detected by both approaches, red colour font indicating the shared genes of a significant overrepresentation with a specific GO term (P < 0.05).
Table S21 Annotation of genes with significant GO terms (P < 0.05) detected by both approaches.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
We thank Li‐dan Tao and Jia‐Ge for field help. This work was equally supported by NSFC (31770240 to WS) and the Second Tibetan Plateau Scientific Expedition and Research Programme (2019QZKK0502 to HS), funded in part by the Yunnan Science and Technology Innovation Team Programme for PSESP Conservation and Utilisation (2019HC015 to WS) and the Youth Innovation Promotion Association, Chinese Academy of Sciences (2018428 to YM).
Contributor Information
Yong‐Peng Ma, Email: mayongpeng@mail.kib.ac.cn.
Richard I. Milne, Email: R.Milne@ed.ac.uk.
Yves Van de Peer, Email: yves.vandepeer@psb.vib-ugent.be.
Wei‐Bang Sun, Email: wbsun@mail.kib.ac.cn.
Data availability
The whole‐genome shotgun project is deposited in DDBJ/ENA/GenBank and under the accessions WHWC00000000. All raw reads generated in this study were deposited in the NCBI database under BioProject accession PRJNA577174.
References
- Bonnet E, Van de Peer Y. 2002. zt: a software tool for simple and partial Mantel tests. Journal of Statistical Software 7: 1–12. [Google Scholar]
- Chau JH. 2017. Systematics of Buddleja (Scrophulariaceae): phylogenetic relationships, historical biogeography, and phylogenomics. PhD thesis, University of Washington, Seattle, WA, USA. [Google Scholar]
- Chau JH, O’Leary N, Sun WB, Olmstead RG. 2017. Phylogenetic relationships in tribe Buddlejeae (Scrophulariaceae) based on multiple nuclear and plastid markers. Botanical Journal of the Linnean Society 184: 137–166. [Google Scholar]
- Chen J‐H, Huang Y, Brachi B, Yun Q‐Z, Zhang W, Lu W, Li H‐n, Li W‐Q, Sun X‐D, Wang G‐Y et al. 2019. Genome‐wide analysis of Cushion willow provides insights into alpine plant divergence in a biodiversity hotspot. Nature Communications 10: 5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CY, Krishnakumar V, Chan AP, Thibaud‐Nissen F, Schobel S, Town CD. 2017. Araport11: a complete reannotation of the Arabidopsis thaliana reference genome. The Plant Journal 89: 789–804. [DOI] [PubMed] [Google Scholar]
- Cui Z, Chen Y, Zhang W, Zhou S, Zhou L, Zhang M, Li C. 2011. Research history, glacial chronology and origins of Quaternary glaciations in China. Quaternary Sciences 31: 749–764. [Google Scholar]
- DeGiorgio M, Huber CD, Hubisz MJ, Hellmann I, Nielsen R. 2016. SweepFinder2: increased sensitivity, robustness and flexibility. Bioinformatics 32: 1895–1897. [DOI] [PubMed] [Google Scholar]
- Dormann CF, Elith J, Bacher S, Buchmann C, Carl G, Carré G, Marquéz JRG, Gruber B, Lafourcade B, Leitão PJ et al. 2013. Collinearity: a review of methods to deal with it and a simulation study evaluating their performance. Ecography 36: 27–46. [Google Scholar]
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin 19: 11–15. [Google Scholar]
- Du Q. 1997. The probe of the boundary between the Qinghai‐Xiang plateau and the Loess plateau regions for vegetation. Acta Biologica Plateau Sinica 13: 83–88. [Google Scholar]
- Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, Aiden EL. 2016. Juicebox provides a visualization system for Hi‐C contact maps with unlimited zoom. Cell Systems 3: 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer AG, Boyd‐Gerny S, McLoughlin S, Rosa MG, Simonov V, Wong B. 2012. Parallel evolution of angiosperm colour signals: common evolutionary pressures linked to hymenopteran vision. Proceedings of the Royal Society B: Biological Sciences 279: 3606–3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms DM, Kelly S. 2019. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biology 20: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffifier L, Dupanloup I, Huerta‐Sanchez E, Sousa VC, Foll M. 2013. Robust demographic inference from genomic and SNP data. PLoS Genetics 9: e1003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Ruhsam M, Wang YH, Li ZH, Wang XM. 2020. Using demographic model selection to untangle allopatric divergence and diversification mechanisms in the Rheum palmatum complex in the Eastern Asiatic Region. Molecular Ecology 29: 1791–1805. [DOI] [PubMed] [Google Scholar]
- Garrison E, Marth G. 2012. Haplotype‐based variant detection from short‐read sequencing. Genomics. [WWW document] URL http://arxiv.org/abs/1207.3907. [Google Scholar]
- Gomez‐Uchida D, Seeb JE, Smith MJ, Habicht C, Quinn TP, Seeb LW. 2011. Single nucleotide polymorphisms unravel hierarchical divergence and signatures of selection among Alaskan sockeye salmon (Oncorhynchus nerka) populations. BMC Evolutionary Biology 11: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer Ø, Harper DA, Ryan PD. 2001. PAST: paleontological statistics software package for education and data analysis. Palaeontologia Electronica 4: 9. [Google Scholar]
- Huber CD, DeGiorgio M, Hellmann I, Nielsen R. 2016. Detecting recent selective sweeps while controlling for mutation rate and background selection. Molecular Ecology 25: 142–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang ZY, Wu YQ, Cui ZJ. 2005. Kunlun‐yellow river tectonic motion and formation of modern physical geography pattern of China. Journal of Beijing Normal University (Natural Sciences) 41: 85–88. [Google Scholar]
- Katoh K, Kuma KI, Toh H, Miyata T. 2005. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Research 33: 511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. 2017. Canu: scalable and accurate long‐read assembly via adaptive k‐mer weighting and repeat separation. Genome Research 27: 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korneliussen TS, Albrechtsen A, Nielsen R. 2014. ANGSD: analysis of next generation sequencing data. BMC Bioinformatics 15: 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiner JM, Giacomini DA, Bemm F, Waithaka B, Regalado J, Lanz C, Hildebrandt J, Sikkema PH, Tranel PJ, Weigel D et al. 2019. Multiple modes of convergent adaptation in the spread of glyphosate‐resistant Amaranthus tuberculatus . Proceedings of the National Academy of Sciences, USA 116: 21076–21084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis v.7.0 for bigger datasets. Molecular Biology and Evolution 33: 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagesen K, Hallin P, Rødland EA, Stærfeldt HH, Rognes T, Ussery DW. 2007. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Research 35: 3100–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lexer C, Mangili S, Bossolini E, Forest F, Stölting KN, Pearman PB, Zimmermann NE, Salamin N. 2013. ‘Next generation’ biogeography: towards understanding the drivers of species diversification and persistence. Journal of Biogeography 40: 1013–1022. [Google Scholar]
- Li J. 1991. The environmental effects of the uplift of the Qinghai‐Xizang Plateau. Quaternary Science Reviews 10: 479–483. [Google Scholar]
- Li XW, Li J. 1993. A preliminary floristic study on the seed plants from the region of Hengduan Mountains. Acta Bototanica Yunnanica 15: 217–231. [Google Scholar]
- Liu X, Fu YX. 2015. Exploring population size changes using SNP frequency spectra. Nature Genetics 47: 555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losos JB, Glor RE. 2003. Phylogenetic comparative methods and the geography of speciation. Trends in Ecology & Evolution 18: 220–227. [Google Scholar]
- Lowe TM, Eddy SR. 1997. tRNAscan‐SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research 25: 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magallon S, Gomez Acevedo S, Sanchez Reyes LL, Hernandez‐Hernandez T. 2015. A metacalibrated time‐tree documents the early rise of flowering plant phylogenetic diversity. New Phytologist 207: 437–453. [DOI] [PubMed] [Google Scholar]
- Maher BA. 2016. Palaeoclimatic records of the loess/palaeosol sequences of the Chinese Loess Plateau. Quaternary Science Reviews 154: 23–84. [Google Scholar]
- Mao JF, Wang XR. 2011. Distinct niche divergence characterizes the homoploid hybrid speciation of Pinus densata on the Tibetan Plateau. American Naturalist 177: 424–439. [DOI] [PubMed] [Google Scholar]
- Mondragón‐Palomino M, Meyers BC, Michelmore RW, Gaut BS. 2002. Patterns of positive selection in the complete NBS‐LRR gene family of Arabidopsis thaliana . Genome Research 12: 1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2014. IQ‐TREE: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Molecular Biology and Evolution 32: 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosil P, Egan SP, Funk DJ. 2008. Heterogeneous genomic differentiation between walking‐stick ecotypes: “Isolation by Adaptation” and multiple roles for divergent selection. Evolution 62: 316–336. [DOI] [PubMed] [Google Scholar]
- Ou S, Chen J, Jiang N. 2018. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Research 46: e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu YX, Fu CX, Comes HP. 2011. Plant molecular phylogeography in China and adjacent regions: tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Molecular Phylogenetics and Evolution 59: 225–244. [DOI] [PubMed] [Google Scholar]
- Raj A, Stephens M, Pritchard JK. 2014. fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics 197: 573–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg LH. 2018. A new model of speciation. National Science Review 6: 289–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter D. 2000. The ecology of adaptive radiation. Oxford, UK: Oxford University Press. [Google Scholar]
- Schoener TW. 1968. The anolis lizards of Bimini: resource partitioning in a complex fauna. Ecology 49: 704–726. [Google Scholar]
- Silva‐Junior OB, Grattapaglia D, Novaes E, Collevatti RG. 2018. Genome assembly of the Pink Ipê (Handroanthusimpetiginosus, Bignoniaceae), a highly valued, ecologically keystone Neotropical timber forest tree. GigaScience 7: gix125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015. BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics 31: 3210–3212. [DOI] [PubMed] [Google Scholar]
- The Angiosperm Phylogeny Group . 2016. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Botanical Journal of the Linnean Society 181: 1–20. [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9: e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wan ZY, Lim HS, Yue GH. 2016. Genetic variability, local selection and demographic history: genomic evidence of evolving towards allopatric speciation in Asian seabass. Molecular Ecology 25: 3605–3621. [DOI] [PubMed] [Google Scholar]
- Wang L, Yu S, Tong C, Zhao Y, Liu Y, Song C, Zhang Y, Zhang X, Wang Y, Hua W et al. 2014. Genome sequencing of the high oil crop sesame provides insight into oil biosynthesis. Genome Biology 15: R39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J, Zhang J, Nie ZL, Zhong Y, Sun H. 2014. Evolutionary diversifications of plants on the Qinghai‐Tibetan Plateau. Frontiers in Genetics 5: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z. 1988. Hengduan mountain flora and her significance. Journal of Japanese Botany 63: 297–311. [Google Scholar]
- Xu GC, Xu TJ, Zhu R, Zhang Y, Li SQ, Wang HW, Li JT. 2018. LR_Gapcloser: a tiling path‐based gap closer that uses long reads to complete genome assembly. GigaScience 8: giy157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Lee SH, Goddard ME, Visscher PM. 2011. GCTA: a tool for genome‐wide complex trait analysis. American Journal of Human Genetics 88: 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J, Zhang C, Mirarab S. 2019. ASTRALMP: scaling ASTRAL to very large datasets using randomization and parallelization. Bioinformatics 35: 3961–3969. [DOI] [PubMed] [Google Scholar]
- Zhang M, Zhou L, Bawa R, Suren H, Holliday JA. 2016. Recombination rate variation, hitchhiking, and demographic history shape deleterious load in Poplar. Molecular Biology and Evolution 33: 2899–2910. [DOI] [PubMed] [Google Scholar]
- Zhang W, Li D, Liu G, Xu X. 2002. The Characteristics of the seed plant flora in Loess Plateau. Bulletin of Botanical Research 22: 373–379. [Google Scholar]
- Zhao Y‐P, Fan G, Yin P‐P, Sun S, Li N, Hong X, Hu G, Zhang He, Zhang F‐M, Han J‐D et al. 2019. Resequencing 545 ginkgo genomes across the world reveals the evolutionary history of the living fossil. Nature Communications 10: 4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dataset S1 Morphological measurement and floral colour reflectance data for populations of Buddleja alternifola.
Fig. S1 Phylogenetic trees inferred by ASTRAL‐ and ML‐based approaches.
Fig. S2 Patterns of linkage disequilibrium (LD).
Fig. S3 Models 1–3, during the process of divergence among the three linkages, no gene flows with no changes in effective population size and (Model 1); with changes in effective population sizes starting from the divergence of TB (TDIV1), as well as SC and HT (TDIV2, Model 2); with changes in effective population sizes starting from TDIV1.
Fig. S4 The phylogenomic tree used for time assignment of divergence for ancestral area reconstruction using representative samples of B. alternifolia and three species in the genus are currently available with re‐sequencing data.
Fig. S5 Cross‐validation (CV) error and marginal likelihood values for different model K.
Fig. S6 Reconstructing the phylogenomic relationships for 46 species of Buddleja using single‐copy genes.
Methods S1 Site ancestral state estimation.
Methods S2 Estimating mutation rate of B. alternifolia.
Methods S3 Reconstructing the phylogenomic relationships for 46 species of Buddleja using single‐copy genes.
Notes S1 Reproducibility of analyses for BEAST and r8s files.
Table S1 Statistics of all assemblies.
Table S2 Basic information with regards to genomes of 17 plants that were used to gene family analysis and the phylogenetic tree construction.
Table S3 A matrix information on geographic distances among populations.
Table S4 Environmental parameters used for assessment of ecological niche differentiation in B. alternifolia.
Table S5 Geographical coordinate of B. alternifolia.
Table S6 WGS‐PacBio sequencing statistics.
Table S7 WGS Illumina sequencing statistics.
Table S8 HiC sequencing statistics.
Table S9 Repeat annotations of the Buddleja alternifolia genome assembly.
Table S10 Gene annotation statistics of the Buddleja alternifolia assembly.
Table S11 Functional annotation of predicted genes in the Buddleja alternifolia genome.
Table S12 Summary of the gene family analyses.
Table S13 Basic information on location and genome mapping characteristics of all sampled individuals.
Table S14 Summary of SNP annotations.
Table S15 Global pairwise Fst between areas at the whole‐genome level.
Table S16 Pairwise Fst between areas in the divided nine subgroups of the whole genome, that is, eight in the gene region and one in the intergene region.
Table S17 Results of nine models used in the fastsimocal analysis.
Table S18 Basic parameters of three models compared in BioGeoBears, that is, Dec and Divalike based on dispersal‐vicariance analysis, and Bayarea based on Bayesian inference of historical biogeography for discrete areas, with and without the founder‐event speciation ‘J’ parameter.
Table S19 Results of IBD and IBA analysis using simple and partial Mantel tests.
Table S20 Shared genes detected by both approaches, red colour font indicating the shared genes of a significant overrepresentation with a specific GO term (P < 0.05).
Table S21 Annotation of genes with significant GO terms (P < 0.05) detected by both approaches.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Data Availability Statement
The whole‐genome shotgun project is deposited in DDBJ/ENA/GenBank and under the accessions WHWC00000000. All raw reads generated in this study were deposited in the NCBI database under BioProject accession PRJNA577174.