

Abstract
Kinesin super family (KIF) genes encode motor kinesins, a family of evolutionary conserved proteins, involved in intracellular trafficking of various cargoes. These proteins are critical for various physiological processes including neuron function and survival, ciliary function and ciliogenesis, and cell‐cycle progression. Recent evidence suggests that alterations in motor kinesin genes can lead to a variety of human diseases, including monogenic disorders. Neuropathies, impaired higher brain functions, structural brain abnormalities and multiple congenital anomalies (i.e., renal, urogenital, and limb anomalies) can result from pathogenic variants in many KIF genes. We expand the phenotype associated with KIF4A variants from developmental delay and intellectual disability with or without epilepsy to a congenital anomaly phenotype with hydrocephalus and various brain anomalies at the more severe end of phenotypic manifestations. Additional anomalies of the kidneys and urinary tract, congenital lymphedema, eye, and dental anomalies seem to be variably associated and overlap with clinical signs observed in other kinesinopathies. Caution still applies to missense variants, but hopefully, future work will further establish genotype–phenotype correlations in a larger number of patients and functional studies may give further insights into the complex function of KIF4A.
Keywords: brain anomalies, hydrocephalus, intellectual disability, KIF4A, kinesinopathies, kinesins
1. INTRODUCTION
Kinesin proteins are molecular motors involved in intracellular trafficking: they are in charge of transporting membranous organelles and protein complexes in a microtubule‐ and ATP‐dependent manner (Miki et al., 2002). Their role in transport is fundamental to cellular logistics and performance not only as effectors of signal transduction cascades but also as modulators of signal transduction molecules function (Hirokawa et al., 2009). Kinesins were first described in 1985 (Brady, 1985; Vale et al., 1985), and since then, 45 different mammalian kinesin genes have been identified in mouse (44 in human genome) and classified into 16 kinesin families (kinesin‐1 to kinesin 14B) by phylogenetic analyses (Hirokawa & Tanaka, 2015). Due to their role in cellular membrane trafficking, kinesins are involved in chromosome segregation, cell division, and cytokinesis, and they are essential for the functioning of many polar cell types such as neurons, epithelial cells, sperm cells, or stem cells during organogenesis. Due to their importance in various physiological processes—such as neuron function and survival, ciliary function, and ciliogenesis—alterations in motor kinesin genes can be genetic contributors to many human diseases by various pathological mechanisms including cancer, multifactorial diseases and monogenic disorders. Pathogenic variants in 18 out of 44 human KIF genes are responsible for hereditary neuropathies, disturbance of higher brain functions, structural brain anomalies and multiple congenital anomalies (in particular, renal, urogenital, and limb anomalies). Kalantari and Filges (2020) reviewed the emerging role of kinesinopathies particularly in birth defects (Kalantari & Filges, 2020).
The central and peripheral nervous systems are often affected in these monogenic disorders, leading to intellectual disability and central system developmental anomalies such as microcephaly or cortical dysplasia and hereditary neuropathies, paraplegia, or recurrent seizures. Anomaly patterns reminiscent of ciliopathies have been described particularly for pathogenic variants in KIF7 (Acrocallosal syndrome/Joubert syndrome 12, OMIM #200990; Hydrolethalus syndrome 2, OMIM #614120; Putoux et al., 2011) and KIF14 (Meckel syndrome 12, OMIM #616258; primary Microcephaly, OMIM# 617914; Filges et al., 2013; Moawia et al., 2017).
KIF4A belongs to the family 4 of kinesins and is composed by a NH2‐terminal globular motor domain (N‐kinesin), a central α‐helical stalk domain, and COOH‐terminal tail domain (Figure 1; Peretti et al., 2002). Its function is important for the cell cycle and for neuronal development. It regulates PARP‐1 activity, a protein crucial for brain development, and neuronal survival (Midorikawa et al., 2006). Kif4A knockout mouse models display an altered balance between inhibitory and excitatory synaptic transmission, highlighting its importance as a regulator of synaptic function (Willemsen et al., 2014). Furthermore, KIF4A is responsible for the translocation of the cytokinesis protein PRC1, thus participating in the organization of central spindle and midzone formation during mitosis (Kurasawa et al., 2004). KIF4A is located on the long arm of the X‐chromosome (Xq13.1).
FIGURE 1.
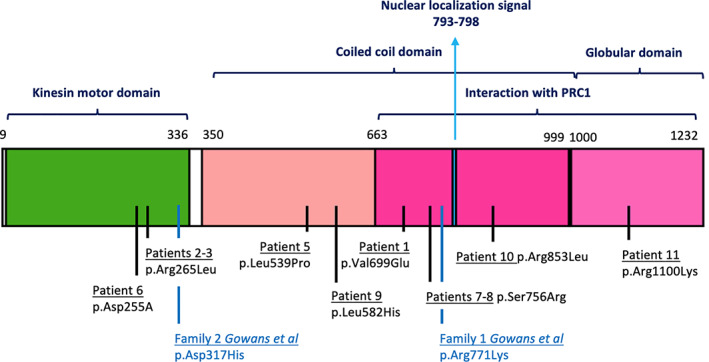
KIF4A protein is characterized by a N‐terminal motor domain (aa 9‐336), a coiled coil domain (aa 350‐999), and a C‐terminal globular domain. A C‐terminal region, overlapping the latter two, is responsible for interaction with PRC1 protein. A nuclear localization signal is found between aa 793 and 798 (The UniProt Consortium, 2019). In the lower part of the image, all the missense variants described in this series and by Gowans et al. (2019) are displayed (Gowans et al., 2019). Patient 4 splicing variant from our cohort (c.1674+1G>A) falls in intron 15, within exons coding for the coiled coil domain. The variant published by Willemsen et al. (2014), c.1489‐8_1490delins10, is a splicing variant falling in the intron–exon junction between intron 14 and exon 15, which was predicted to disrupt the acceptor splice site of exon 15, leading to skipping of exon 15 (within the coiled coil domain; Willemsen et al., 2014)
Only a few patients with likely pathogenic variants in KIF4A have been reported to date, and X‐linked recessive inheritance has been suggested. Willemsen et al. (2014) described four males with X‐linked mild intellectual disability and epilepsy, belonging to the same family and carrying a hemizygous in‐frame variant in KIF4A (Willemsen et al., 2014). Meier et al. (2019) proposed KIF4A as a candidate gene for X‐linked congenital hydrocephalus in a single male fetus (Meier et al., 2019). KIF4A motor activity allows for the anterograde transport of the cell adhesion molecule L1 (L1CAM; Peretti et al., 2002). L1CAM belongs to the immunoglobulin superfamily and has a well‐studied role in neuronal migration, axon growth, guidance, fasciculation, and synaptic plasticity (Itoh & Fushiki, 2015). Pathogenic variants in the L1CAM gene have been already associated with several neurological phenotypes, with or without hydrocephalus (Shaheen et al., 2017) as well as isolated hydrocephalus due to aqueductal stenosis (OMIM 307000).
Based on the suspicion that KIF4A could be associated with a broader range of neurodevelopmental and congenital anomaly phenotypes than previously reported, we collected patients with KIF4A variants through GeneMatcher (Sobreira et al., 2015). We here confirm the initially reported neurodevelopmental phenotype with intellectual disability with or without epilepsy and expand the phenotypic spectrum to include hydrocephalus, either isolated or associated with other congenital anomalies, predominantly of the brain, kidneys and urinary tract.
2. MATERIALS AND METHODS
2.1. Human subjects
We initially identified two unrelated families in which the affected fetus and two affected siblings presented with congenital hydrocephalus due to a suspected deleterious variant in KIF4A. We interrogated GeneMatcher (Sobreira et al., 2015) in order to compare clinical phenotypes of patients with reported KIF4A variants. We included 11 patients, all of whom underwent a comprehensive clinical evaluation by a clinical geneticist and/or a clinician specialist. Clinical data included pregnancy and birth history, the presence of congenital anomalies and dysmorphic signs, brain imaging if done as well as motor and cognitive development. Consent to publish was obtained from all participants. Patient 1 from the case series has previously been reported by Meier et al. (2019).
2.2. Molecular analysis
In all patients, diagnostic exome sequencing (ES) was performed. Due to the genotype‐first recruitment using the GeneMatcher initiative, genetic testing was done in the respective diagnostic laboratories according to the choice of the clinician involved. All molecular diagnostic laboratories are accredited and are performing diagnostic ES according to international standards and according to the locally certified protocols. Interpretation and classification of the identified variants were carried out following the American College of Medical Genetics and Genomics (ACMG) recommendations for variant classification (Richards et al., 2015). In all cases, familial maternal segregation of the variant identified was performed.
3. RESULTS
We collected a total of 11 male patients with variants in KIF4A (NM_012310.5, HGNC 13339, OMIM * 300521) suspected to be causative of the phenotype. Details on genotypes and phenotypes are summarized in Tables 1 and 2. In one patient, the variant occurred de novo (Patient 5), and in the 10 others, the variant is inherited from unaffected mothers, consistent with an X‐linked recessive inheritance. Reported maternal history regarding developmental milestones, school education, and professional achievements did not reveal obvious differences or effects that might be ascribed to the presence of the genetic variants. In all but one patient (Patient 4), variants were missense variants with deleterious in silico predictions (see Table S1 for further details) and affecting the main functional domains of KIF4A. In Patient 4, a splice site variant was identified. Details on variants are summarized in the Table S1.
TABLE 1.
Genotype–phenotype correlations of patients with congenital anomalies
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | Patient 8 | |
|---|---|---|---|---|---|---|---|---|
| Gender | M | M | M | M | M | M | M | M |
| Ethnicities/country of origin | Caucasian | American | American | Egyptian | Japanese | Japanese | Algerian | Algerian |
| Age of examination | TOP at 22 + 3 w GA | Perinatal death (1 h old) | 1 year | 4 years 6 months | 18 years | 2 years 11 months | 4 years | 1 year |
| Prenatal history | + |
+ Oligohydramnios |
+ | No anomalies detected | No anomalies detected | No anomalies detected | No anomalies detected | No anomalies detected |
| Brain anomaly | Hydrocephalus | Brain cysts, relative hydrocephalus, microcephaly | Hydrocephalus, ventriculomegaly, aqueductal stenosis, partial agenesis of CC, small hippocampus, CC and IHEM cysts, few PNH, microcephaly |
Hydranencephaly Corpus callosum agenesis |
Polymicrogyria, heterotopia, hypoplastic pyramidal tracts (Figure 5a) |
Polymicrogyria, high intensity signals in the deep white matter on T2WI (Figure5b) | MRI never performed | MRI never performed |
| Eye anomaly | Not present | Not present | Bilateral optic nerve hypoplasia, right macular coloboma | Bilateral pale optic disc | Not present | Not present | Unilateral Peters anomaly with microphthalmia and central leucoma (LE) | Unilateral Peters anomaly with microcornea and dense leucoma (RE); minimal embryotoxon (LE) |
| Kidney anomalies | Not present | Bilateral CDK | Bilateral CDK, right duplicated renal collecting system, bilateral VUR | Not present | Not present | Not present | Not present | Not present |
| Heart anomalies | Not present | Not present | Not present | Not present | Not present | Not present | Not present | Not present |
| Skeletal/limb anomalies | Not present | Not present | Not present | Short long bones | Not present | Not present | Not present | Not present |
| Development/neurology | Not present | Not present | Not present | Severe to profound developmental delay | Mild psychomotor delay (IQ 57 by WAIS‐III) | Severe psychomotor delay (DQ 22 by Japanese Enjoji); seizures. | Not present | Not present |
| Others | Not present | Hypoplastic bladder | Bilateral severe sensorineural hearing loss | Congenital lymphedema of upper and lower limbs | Not present | Not present | Unilateral cryptorchidism (left) | Unilateral cryptorchidism (right); microdontia, mild and nonspecific dysmorphisms |
| Variant description | c.2096T>A, p.(Val699Glu) Hemizygous | c.794G>T, p.(Arg265Leu) Hemizygous | c.794G>T, p.(Arg265Leu) Hemizygous | c.1674+1G>A Hemizygous | c.1616T>C, p.(Leu539Pro) Hemizygous | c.763G>A, p.(Asp255Asn) Hemizygous | c.2266A>C, p.(Ser756Arg) Hemizygous | c.2266A>C, p.(Ser756Arg) Hemizygous |
| Inheritance | Maternal | Maternal | Maternal | Maternal | De novo | Maternal | Maternal | Maternal |
| Other genetic findings | None | None | None | Normal chromosomal microarray | Normal chromosomal G‐banding | Normal chromosomal G‐banding | None | None |
| Family history | Unremarkable | Brother carrying the same variant in KIF4A (Patient 3) | Brother carrying the same variant in KIF4A (Patient 2) | Similarly affected deceased brother | Unremarkable | Unremarkable | Brother carrying the same variant in KIF4A (Patient 8) | Brother carrying the same variant in KIF4A (Patient 7) |
Abbreviations: CC, corpus callosum; CDK, cystic dysplastic kidneys; DQ, developmental quotient; IHEM, interhemispheric; IQ, intelligent quotient; LE, left eye; PNH, periventricular nodular heterotopia; RE, right eye; TOP, termination of pregnancy; VUR, vesicoureteral reflux; WAIS‐III, Wechsler Adult Intelligence Scale 3rd edition.
TABLE 2.
Genotype–phenotype correlations of patients with developmental delay/intellectual disability without congenital structural anomalies, compared with the family described by Willemsen et al. (2014)
| Patient 9 | Patient 10 | Patient 11 | Willemsen et al. family | |
|---|---|---|---|---|
| Gender | M | M | M | M |
| Ethnicities/Country of origin | Caucasian | Caucasian | Hispanic, Mexican, and Guatemalan ancestry | Caucasian |
| Age of examination | 9 years | 16 years | 2 years 9 months | Five affected males across three generations, specific ages not given |
| Prenatal history | Unremarkable | IUGR and placental insufficiency | Unremarkable | Unremarkable |
| Brain anomaly | Normal MRI at 9 years | Mild ventriculomegaly at birth, normal MRI at 15 years | Normal MRI at 3 years | Central atrophy of lateral hemispheres (III: 7 at age 53) and wide posterior horns (IV: 3 at age 7) |
| Eye anomaly | Not present | Retinopathy related to disease‐causing GPR143 variant (rs137852297) | Not present | Not present |
| Skeletal anomalies | Not present | Delay of osseous age; Perthes disease involving right hip | Not present | Not present |
| Other malformations | Not present | Cryptorchidism; Pelvic left kidney | Not present | Not present |
| Intellectual development | Intellectual disability, speech delay | Borderline intellectual functioning, speech delay, attention deficit, anxiety disturbance, depressive traits. | Intellectual disability, speech delay | Intellectual disability, speech delay |
| Motor development | Not present | Motor delay | Motor delay | ? |
| Global developmental delay | + | + | + | + |
| Autism spectrum disorder | + | Not present | + | Not present |
| Seizures | Not present | Not present | Isolated febrile seizure at 2 years of age | 4/5, developed in late childhood–adolescence, complex partial and generalized, absence and tonic–clonic |
| Other clinical features | Not present | Not present | Dysmorphic features including relative macrocephaly, upslanting palpebral fissures, prominent cupped ears, large forehead, plagiocephaly | Mild, nonspecific facial dysmorphisms |
| Variant description | c.1745T>A, p.(Leu582His) Hemizygous | c.2558G>T, p.(Arg853Leu) Hemizygous | c.3299G>A, p.(Arg1100 Lys) Hemizygous | c.1489‐8_delins10, Hemizygous |
| Inheritance | Maternal | Maternal | Maternal | Maternal |
| Other genetic findings | Chromosomal microarray: 13q31.3 duplication (93,281,466‐94,095,389x3), classified as a CNV of unclear significance by GeneDx, found to be maternally inherited. | Normal karyotype, chromosomal microarray, Methylation at IGF2‐H19, IC1 loci | Normal chromosomal microarray and FMR1 | Normal karyotyping, DNA analysis of FMR1 and ARX genes |
| Family history | Mother has history of anxiety and depression. | Unremarkable | Unremarkable | Five affected males across three generations |
Eight patients (including fetal presentation in Patient 1) had congenital anomalies, including four single cases and two male sibling pairs (Patients 2 and 3 and Patients 7 and 8). Brain anomalies were the predominant clinical finding, with hydrocephalus in 4 (Patients 1–4) and ventriculomegaly in 1 patient (Patient 4), with or without associated microcephaly, partial agenesis of the corpus callosum, small or malrotated hippocampus, polymicrogyria, interhemispheric cysts, periventricular heterotopia, and optic nerve hypoplasia. Minor ventriculomegaly was documented in Patient 11, while, in Patients 7 and 8, MRIs were not performed. In siblings 2 and 3, variable anomalies of the kidney and urinary tract such as bilateral cystic kidney dysplasia, hypoplastic bladder, and unilateral renal collecting system were described. Hearing loss and congenital lymphedema of the upper and lower limbs or short long bones were isolated findings in single patients. Eye anomalies, such as Peter's anomaly, microphthalmia, and leukoma, were the presenting signs in siblings 7 and 8. Furthermore, Patient 3 had a right macular coloboma. The sibling Patients 7 and 8 showed an ocular phenotype together with some dental anomalies (upper lateral incisor agenesis in Patient 7 and microdontia in Patient 8). Minor facial and skeletal anomalies are observed in some of these patients.
Three patients (Table 2: Patients 9–11) presented with intellectual disability, ranging from mild to severe, including speech delay and behavioral disorders, as well as global developmental delay. The detailed history of patients presenting with hydrocephalus and/or congenital anomalies (Patients 1–8) is described in the following paragraphs.
3.1. Patient 1
Patient 1 is a male fetus of a Caucasian mother whose pregnancy was terminated at 22 + 3 weeks gestational age (wGA), after the ultrasound anatomy scan (US) identified severe but likely isolated hydrocephalus internus and a cerebellar size low‐normal (Figure 2). Postmortem autopsy confirmed the sonographic findings and did not identify other organ anomalies neither macroscopically nor histologically. Postmortem tissue changes did not allow examination for aqueductal stenosis, and the vermis cerebelli was described as having irregular borders. ES identified a maternally inherited c.2096T>A, p.(Val699Glu) variant, initially classified as a variant of unknown significance (VUS) based on ACMG criteria (PM1 and PM2 moderate, PP3 and PP5 supporting). Clinical and functional review of genotype–phenotype correlations suggested a disease‐causing role of the variant, and qPCR confirmed a significant reduction of KIF4A mRNA in brain tissue of the affected fetus (12%) compared to FFPE brain tissue of an age‐matched control. The low mRNA levels suggested a loss‐of‐function mechanism as previously proposed (Meier et al., 2019).
FIGURE 2.
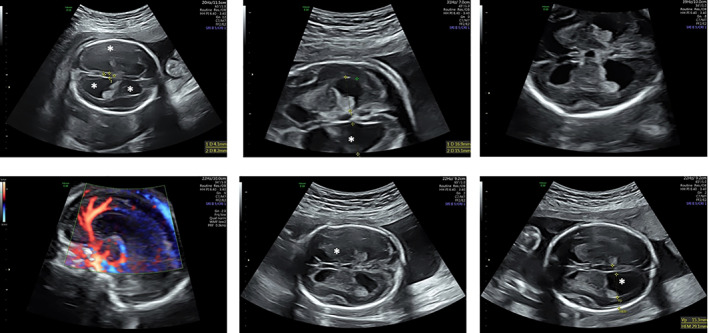
Brain prenatal US imaging of patient 1 at 22 wGA, when hydrocephalus internus was apparent. The enlarged ventricles are highlighted by asterisks. The cerebellar size was described as being at the lower end of normal range, corpus callosum was normal (as shown by the Doppler US scan in the lower left corner)
3.2. Patients 2 and 3
Patients 2 and 3 are male siblings. Patient 2 was born via C‐section at 33 wGA. Prenatal US at 20 weeks GA showed brain and kidney cysts and oligohydramnios developed in the third trimester. The patient died 1 h after birth. At postmortem autopsy, cystic dysplastic kidneys (CDK), hypoplastic bladder, cerebellar hypoplasia, and microcephaly were found. ES identified a c.794G>T, p.(Arg265Leu) maternally inherited variant in KIF4A.
The brother, Patient 3, was born at 34 weeks GA with multiple anomalies as well as pericardial effusion and pulmonary hypertension, which resolved over time. Congenital anomalies included bilateral cerebellar hypoplasia, optic nerve hypoplasia, right macular coloboma, bilateral sensorineural hearing loss (for which he ultimately received cochlear implants), bilateral CDK, a right duplicated renal collecting system, and bilateral vesicoureteral reflux (VUR). His weight was 3.8 kg (−2.1 SD), length 52.5 cm (−2.4 SD), and head circumference (OFC) 36 cm (−4 SD) at 2 months of age (+2 weeks corrected). Microcephaly persisted with OFC measurements falling to −5 SD at 1 year of age. Cerebral MRI confirmed severe ventriculomegaly, aqueductal stenosis, partial agenesis of the corpus callosum, small hippocampus, periventricular nodular heterotopia, and interhemispheric cysts. ES identified the maternally inherited VUS in KIF4A (c.794G>T (p.Arg265Leu)) in the index younger brother, but co‐segregating with the phenotype in the affected sibling. The two siblings are also part of a larger cerebellar malformation cohort (Aldinger et al., 2019).
3.3. Patient 4
Patient 4 is a 4‐year‐old boy of Egyptian descent born to consanguineous parents, whose prenatal history is unknown, and prenatal US was not performed. He presented with congenital lymphedema of upper and lower limbs at birth. Hydranencephaly and corpus callosum agenesis were identified by cerebral MRI imaging at 6 months of age (Figure 3). At 4 years and months of age, he exhibits severe developmental delay, including an inability to sit and absence of speech. Family history includes a similarly affected, previously deceased son as reported by the parents, without additional clinical information available (Figure 4). ES revealed a maternally inherited variant in KIF4A, c.1674+1G>A, which was classified as likely pathogenic based on ACMG criteria (PVS1 very strong, PM2 moderate, PP3 supporting).
FIGURE 3.
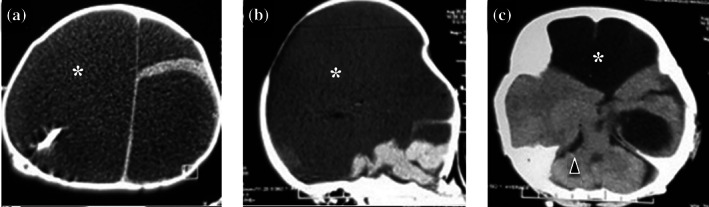
Brain CT scan of patient 4 at 6 months of age, from upper to lower panel: (a) Axial image showing hydranencephaly (white asterisk); (b) midsagittal image showing nonvisualized corpus callosum; (c) lower axial image showing relatively intact cerebellum and brainstem (arrowhead)
FIGURE 4.
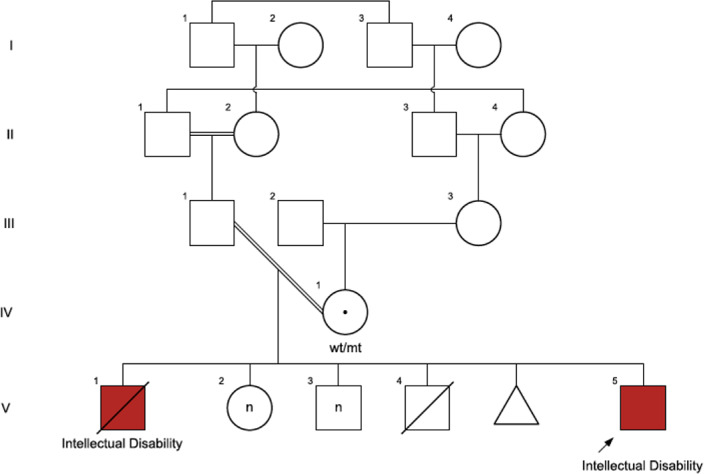
Patient 4 family tree, showing the other likely affected brother and extensive consanguinity in the family
3.4. Patient 5
Patient 5 is a Japanese male born to unrelated healthy parents. His mother had three episodes of miscarriage at 6–8 weeks of gestation. The patient was delivered at 38 wGA, his birth weight was 2808 g (+0.0 SD), length was 49.0 cm (+0.5 SD), and head circumference was 32.0 cm (−0.7 SD). He showed apneic episodes after birth and was admitted to the hospital for 1 month when polymicrogyria was recognized on brain MRI. He showed developmental delay with head control at 5–6 months, sitting unsupported at 8 months, speaking meaningful words at 15 months, walking at 3 years and 2 months, and speaking two‐word sentences at 3 years and 6 months. He showed microcephaly at 5 years with a head circumference at 45.8 cm (−3.1 SD). He had no epileptic seizures, and the findings of electroencephalogram (EEG) at 2 years were normal. Brain MRI at 18 years of age showed dilatation of the lateral ventricles, perisylvian polymicrogyria, heterotopia, and hypoplastic pyramidal tracts (Figure 5a). ES identified a de novo variant in KIF4A, c.1616T>C, p.(Leu539Pro).
FIGURE 5.
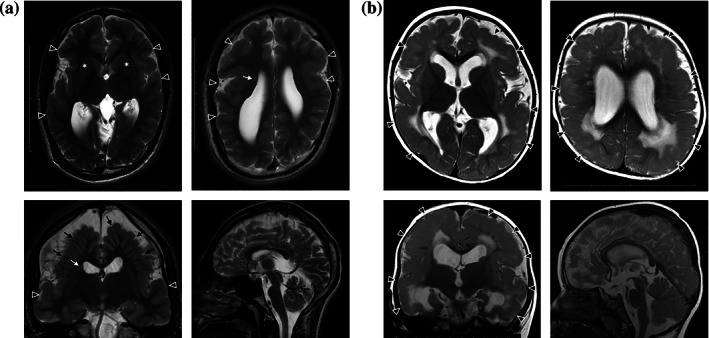
Axial, coronal and sagittal T2‐weighted imaging (T2WI) of Patients 5 and 6 brain MRI. (a) Patient 5 brain MRI at 18 years of age showing dilatation of the lateral ventricles, perisylvian polymicrogyria (arrowheads), heterotopia (white arrows), radially‐oriented gyri (black arrows), and hypoplastic pyramidal tracts (asterisks). (b) Patient 6 brain MRI at 12 months of age, showing dilatation of the lateral ventricles, polymicrogyria (arrowheads) and high intensity signals in the deep white matter on T2WI
3.5. Patient 6
Patient 6 is the second child born to unrelated healthy parents. The pregnancy was uneventful, and he was delivered via C‐section at 38 wGA. His birth weight was 2776 g (−0.6 SD), length was 50.0 cm (+0.9 SD), and head circumference was 31.0 cm (−1.5 SD). He showed eye‐pursuit at 3 months and social smile at 4–5 months. He had unstable head control, frequent opisthotonic posture, and grasping hands at 4 months. Brain MRI revealed polymicrogyria (Figure 5b). He obtained head control at 7 months, but could not sit alone. At 14 months, head circumference was 43.5 cm (−1.8 SD), and developmental quotient (Japanese Enjoji developmental scale) was 45. He showed frequent multifocal epileptic discharges at C3 and C4‐P4 on EEG and then exhibited focal to bilateral tonic–clonic seizures at 23 months. His focal motor seizures originated from the left hand and were refractory to levetiracetam, nitrazepam, and valproic acid. At 2 years and 7 months, he had atypical absences and was diagnosed with epilepsy with continuous spike‐waves during slow wave sleep. ES identified a maternally inherited variant in KIF4A, c.763G>A, p.(Asp255Asn), which was classified as VUS according to ACMG criteria (PM1 moderate, PM2 moderate, PP3 supporting).
3.6. Patients 7 and 8
Patient 7 is a 10‐year‐old boy. He is the first child of healthy parents originating from the same Algerian Village. Family history was unremarkable. He was born at term after an uneventful pregnancy and presented with Peter's anomaly of the left eye with microphthalmia and central leukoma, agenesis of upper lateral incisors, and left cryptorchidism. Echocardiography and renal ultrasound were normal, as were acquisition of psychomotor development milestones. At 3 years and 9 months, his development was within the normal limits for height (−2 SD), weight (3rd percentile), and head circumference (−1.5 SD). At 9 years, he was normally schooled, and his height, weight, and head circumference were −1 SD, −2 SD, and −1.5 SD, respectively.
Patient 8 is the brother of Patient 7. He was born at 37 wGA after a pregnancy marked by religious fasting within the last month and maternal hyperthermia at the first trimester. He displayed unilateral Peter's anomaly, microdontia, and right cryptorchidism. Echocardiography was unremarkable. A failure to thrive, and moderate hypotonia was noted at 9.5 months of age with −2.5 SD for weight and height and −1 SD for head circumference. He walked at 14 months, like his brother. At 6 years, his head circumference and height were −1 SD, his weight was −2 SD, and he was normally schooled. ES revealed a maternally inherited variant in KIF4A, c.2266A>C, p.(Ser756Arg), present in both siblings, and classified as a VUS according to ACMG criteria (PM2 moderate, PP3 supporting).
4. DISCUSSION
Although phenotypic and functional studies remain limited, there is a growing body of evidence, including the clinical and genetic data presented in our patient series, demonstrating the impact of kinesin dysfunction in human disease. Such kinesinopathies, caused by pathogenic variants in various KIF genes, present with recurrent phenotypes including neurodevelopmental differences and congenital anomalies (Kalantari & Filges, 2020). Kinesins are a large superfamily of molecular motors that are of key importance in fundamental cellular processes, using microtubules as rails for directional anterograde intracellular transport, and functioning in regulation and modulation of signal transduction (Hirokawa & Tanaka, 2015). Their complex functional role mirrors a multitude of common as well as heterogeneous clinical signs in conditions caused by variants in this group of genes.
Here, we report a series of 11 male patients, including two sets of siblings, harboring variants in KIF4A compatible with X‐linked recessive inheritance and consistent with the phenotypes of the few previously reported families. Willemsen and collaborators (2014) reported the first family in which a disruptive variant co‐segregated in five affected males over three generations (Willemsen et al., 2014). Functional studies at the level of synaptic function in primary rat hippocampal neurons confirmed that KIF4A is a critical protein in controlling the balance between excitatory and inhibitory inputs during development (Willemsen et al., 2014). The patients described had mild to moderate ID, lived, and worked in sheltered placements, and four had epilepsy including both complex partial and generalized seizures manifesting in later childhood or adolescence (Willemsen et al., 2014). Meier et al. (2019) reported hydrocephalus internus in a fetus, described in detail here as Patient 1, with a maternally inherited hemizygous missense variant in KIF4A and an experimentally confirmed decrease of mRNA in brain tissue compared to age‐matched controls (Meier et al., 2019). The authors proposed KIF4A as a candidate gene for congenital hydrocephalus as an allelic phenotype, also based on the additional functional consideration that KIF4A is a member of the L1CAM recycling pathway and variants in L1CAM are well known to cause X‐linked isolated and syndromic hydrocephalus (Basel‐Vanagaite et al., 2006). A recent study of two families attributed a possible role for KIF4A in dental morphogenesis and identified two missense variants (c.2312G>A, p.(Arg771Lys), and c.304G>A, p.(Asp102Asn)) as causative of taurodontism, microdontia, and dens invaginatus in two patients (Gowans et al., 2019; Figure 1).
In our case series, we provide confirmation of the KIF4A‐associated intellectual disability phenotype and provide additional evidence for an expanded KIF4A phenotype encompassing congenital anomalies including brain malformations. We identified three patients with hydrocephalus, hydranencephaly, and ventriculomegaly, with confirmed aqueductal stenosis in one, similar to L1CAM hydrocephalus phenotypes. Two of the affected patients reported by Willemsen et al. (2014) were described with head circumferences small to low normal and presented with central atrophy of lateral hemispheres and wide posterior horns at brain CT scans. We observed additional brain anomalies in our patients including microcephaly, cerebellar hypoplasia, agenesis of the corpus callosum, hippocampal hypoplasia and malrotation, polymicrogyria, perinodular hyperplasia, and interhemispheric cysts. Furthermore, anomalies of the kidney and urinary tract, such as CDK, were observed in our cohort. These malformations overlap with anomalies described in other kinesinopathies and/or ciliopathies (Kalantari & Filges, 2020). The underlying mechanism is not entirely understood, but likely associated with the various roles of these genes in cell division (Reilly et al., 2019). Congenital lymphedema of upper and lower limbs identified in one patient is also reported in patients with variants in KIF11 (OMIM #152950; Mirzaa et al., 2014). Regarding ophthalmologic phenotypes, optic nerve hypoplasia, chorioretinopathy, coloboma, exudative retinopathy, and congenital fibrosis of the extraocular muscles were reported in other kinesinopathies and ciliopathies, while Peters anomaly and dysgenesis of the anterior segment in two of our patients are novel associations for KIF4A. Pathway convergence may explain these findings, although the prevalence remains unknown until more KIF4A patients can be ascertained. Dental anomalies were present in Patients 7 and 8, further confirming a possible role of the gene in dental morphogenesis as previously suggested (Gowans et al., 2019).
In total, in this cohort, 10 patients harbored missense variants in KIF4A and a single patient harbored a splicing variant (though notably KIF4A is predicted to be loss‐of‐function intolerant with a pLI = 1 in gnomAD, synonymous Z score 0.74, and missense Z score 2.56), making genotype–phenotype correlations challenging. For formal reasons, all missense variants identified had to be classified VUS using strict ACMG criteria. The missense variants, however, are all localized within known functional domains including the kinesin motor domain, responsible for ATP hydrolysis and microtubule binding, and the PRC1 binding and coiled coil domain (Figure 1). KIF4A effects a cell‐cycle‐dependent translocation of PRC1, a cytokinesis protein, to the ends of the spindle molecules during mitosis (Zhu et al., 2005). The KIF4A variants involve the same functional domains in the group of patients with congenital anomalies and the group with primarily ID without major anomalies, as displayed in Figure 1. However, variants affecting the kinesin motor domain are only present in patients with congenital anomalies. The absolute number of patients is, however, too small to draw definite conclusions, and our observations are limited by the retrospective recruiting of patients through GeneMatcher from various clinical settings without an initial standardized collection of genetic and clinical data.
In conclusion, we propose an expanded KIF4A‐associated phenotype, encompassing both developmental delay and intellectual disability with or without epilepsy and a multiple congenital anomaly phenotype with hydrocephalus and various brain anomalies. Additional anomalies of the kidney and urinary tract, congenital lymphedema, dental, and eye anomalies seem to be variably associated and overlap with clinical signs observed in other kinesinopathies. Some caution is advisable since the majority are missense variants lacking individual functional studies. Further genotype–phenotype correlations await the reporting of a larger number of patients, and additional functional studies may give further insights into the mechanism of pathogenicity and the complex biological role of KIF4A.
CONFLICT OF INTEREST
The authors declare no conflict of interests.
AUTHOR CONTRIBUTIONS
Silvia Kalantari, Colleen Carlston and Isabel Filges collected patients. SK and IF wrote the manuscript. IF supervised the work. All authors contributed data and histories from their own patients.
Supporting information
TABLE S1 In silico predictions for all variants reported in the article. Reference transcript: NM_012310.5.
ACKNOWLEDGMENTS
The work was supported by the Swiss National science Foundation (SNSF), Project Grant (320031_160200) and the University of Basel Research Fund to Isabel Filges. Open Access Funding provided by Universitat Basel.
Kalantari, S. , Carlston, C. , Alsaleh, N. , Abdel‐Salam, G. M. H. , Alkuraya, F. , Kato, M. , Matsumoto, N. , Miyatake, S. , Yamamoto, T. , Fares‐Taie, L. , Rozet, J.‐M. , Chassaing, N. , Vincent‐Delorme, C. , Kang‐Bellin, A. , McWalter, K. , Bupp, C. , Palen, E. , Wagner, M. D. , Niceta, M. , … Filges, I. (2021). Expanding the KIF4A ‐associated phenotype. American Journal of Medical Genetics Part A, 185A:3728–3739. 10.1002/ajmg.a.62443
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Aldinger, K. A. , Timms, A. E. , Thomson, Z. , Mirzaa, G. M. , Bennett, J. T. , Rosenberg, A. B. , Roco, C. M. , Hirano, M. , Abidi, F. , Haldipur, P. , Cheng, C. V. , Collins, S. , Park, K. , Zeiger, J. , Overmann, L. M. , Alkuraya, F. S. , Biesecker, L. G. , Braddock, S. R. , Cathey, S. , … Dobyns, W. B. (2019). Redefining the etiologic landscape of cerebellar malformations. American Journal of Human Genetics, 105(3), 606–615. 10.1016/j.ajhg.2019.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basel‐Vanagaite, L. , Straussberg, R. , Friez, M. J. , Inbar, D. , Korenreich, L. , Shohat, M. , & Schwartz, C. E. (2006). Expanding the phenotypic spectrum of L1CAM‐associated disease. Clinical Genetics, 69(5), 414–419. 10.1111/j.1399-0004.2006.00607.x [DOI] [PubMed] [Google Scholar]
- Brady, T. S. (1985). A novel brain ATPase with properties expected for the fast axonal transport motor. Nature, 317(6032), 73–75. [DOI] [PubMed] [Google Scholar]
- Filges, I. , Nosova, E. , Bruder, E. , Tercanli, S. , Townsend, K. , Gibson, W. T. , Röthlisberger, B. , Heinimann, K. , Hall, J. G. , Gregory‐Evans, C. Y. , Wasserman, W. W. , Miny, P. , & Friedman, J. M. (2013). Exome sequencing identifies mutations in KIF14 as a novel cause of an autosomal recessive lethal fetal ciliopathy phenotype. Clinical Genetics, 86(3), 220–228. 10.1111/cge.12301 [DOI] [PubMed] [Google Scholar]
- Gowans, L. J. J. , Cameron‐Christie, S. , Slayton, R. L. , Busch, T. , Romero‐Bustillos, M. , Eliason, S. , Sweat, M. , Sobreira, N. , Yu, W. , Kantaputra, P. N. , Wohler, E. , Adeyemo, W. L. , Lachke, S. A. , Anand, D. , Campbell, C. , Drummond, B. K. , Markie, D. M. , van Vuuren, W. J. , van Vuuren, L. J. , … Butali, A. (2019). Missense pathogenic variants in KIF4A affect dental morphogenesis resulting in X‐linked taurodontism, microdontia and dens‐invaginatus. Frontiers in Genetics, 10(September), 1–8. 10.3389/fgene.2019.00800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa, N. , Noda, Y. , Tanaka, Y. , & Niwa, S. (2009). Kinesin superfamily motor proteins and intracellular transport. Nature Reviews Molecular Cell Biology, 10(10), 682–696. 10.1038/nrm2774 [DOI] [PubMed] [Google Scholar]
- Hirokawa, N. , & Tanaka, Y. (2015). Kinesin superfamily proteins (KIFs): Various functions and their relevance for important phenomena in life and diseases. Experimental Cell Research, 334(1), 16–25. 10.1016/j.yexcr.2015.02.016 [DOI] [PubMed] [Google Scholar]
- Itoh, K. , & Fushiki, S. (2015). The role of L1cam in murine corticogenesis, and the pathogenesis of hydrocephalus. Pathology International, 65(2), 58–66. 10.1111/pin.12245 [DOI] [PubMed] [Google Scholar]
- Kalantari, S. , & Filges, I. (2020). “Kinesinopathies”: Emerging role of the kinesin family member genes in birth defects. Journal of Medical Genetics, 57, 1–11. 10.1136/jmedgenet-2019-106769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurasawa, Y. , Earnshaw, W. C. , Mochizuki, Y. , Dohmae, N. , & Todokoro, K. (2004). Essential roles of KIF4 and its binding partner PRC1 in organized central spindle midzone formation. EMBO Journal, 23(16), 3237–3248. 10.1038/sj.emboj.7600347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier, N. , Bruder, E. , Lapaire, O. , Hoesli, I. , Kang, A. , Hench, J. , Hoeller, S. , de Geyter, J. , Miny, P. , Heinimann, K. , Chaoui, R. , Tercanli, S. , & Filges, I. (2019). Exome sequencing of fetal anomaly syndromes: Novel phenotype–genotype discoveries. European Journal of Human Genetics, 27, 730–737. 10.1038/s41431-018-0324-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midorikawa, R. , Takei, Y. , & Hirokawa, N. (2006). KIF4 motor regulates activity‐dependent neuronal survival by suppressing PARP‐1 enzymatic activity. Cell, 125(2), 371–383. 10.1016/j.cell.2006.02.039 [DOI] [PubMed] [Google Scholar]
- Miki, H. , Setou, M. , Kaneshiro, K. , & Hirokawa, N. (2002). All kinesin superfamily protein, KIF, genes in mouse and human. Proceedings of the National Academy of Sciences of the United States of America, 98(13), 7004–7011. 10.1073/pnas.111145398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzaa, G. M. , Enyedi, L. , Parsons, G. , Collins, S. , Medne, L. , Adams, C. , Ward, T. , Davitt, B. , Bicknese, A. , Zackai, E. , Toriello, H. , Dobyns, W. B. , & Christian, S. (2014). Congenital microcephaly and chorioretinopathy due to de novo heterozygous KIF11 mutations: Five novel mutations and review of the literature. American Journal of Medical Genetics, Part A, 164(11), 2879–2886. 10.1002/ajmg.a.36707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moawia, A. , Shaheen, R. , Rasool, S. , Waseem, S. S. , Ewida, N. , Budde, B. , Kawalia, A. , Motameny, S. , Khan, K. , Fatima, A. , Jameel, M. , Ullah, F. , Akram, T. , Ali, Z. , Abdullah, U. , Irshad, S. , Höhne, W. , Noegel, A. A. , al‐Owain, M. , … Hussain, M. S. (2017). Mutations of KIF14 cause primary microcephaly by impairing cytokinesis. Annals of Neurology, 82(4), 562–577. 10.1002/ana.25044 [DOI] [PubMed] [Google Scholar]
- Peretti, D. , Rosso, S. , Quiroga, S. , Cáceres, A. , & Peris, L. (2002). Evidence for the involvement of Kif4 in the anterograde transport of L1‐containing vesicles. The Journal of Cell Biology, 149(1), 141–152. 10.1083/jcb.149.1.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putoux, A. , Thomas, S. , Coene, K. L. M. , Davis, E. E. , Alanay, Y. , Ogur, G. , Uz, E. , Buzas, D. , Gomes, C. , Patrier, S. , Bennett, C. L. , Elkhartoufi, N. , Frison, M. H. S. , Rigonnot, L. , Joyé, N. , Pruvost, S. , Utine, G. E. , Boduroglu, K. , Nitschke, P. , … Attié‐Bitach, T. (2011). KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nature Genetics, 43(6), 601–606. 10.1038/ng.826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly, M. L. , Stokman, M. F. , Magry, V. , Jeanpierre, C. , Alves, M. , Paydar, M. , Hellinga, J. , Delous, M. , Pouly, D. , Failler, M. , Martinovic, J. , Loeuillet, L. , Leroy, B. , Tantau, J. , Roume, J. , Gregory‐Evans, C. Y. , Shan, X. , Filges, I. , Allingham, J. S. , … Benmerah, A. (2019). Loss‐of‐function mutations in KIF14 cause severe microcephaly and kidney development defects in humans and zebrafish. Human Molecular Genetics, 28(5), 778–795. 10.1093/hmg/ddy381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , & Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen, R. , Sebai, M. A. , Patel, N. , Ewida, N. , Kurdi, W. , Altweijri, I. , Sogaty, S. , Almardawi, E. , Seidahmed, M. Z. , Alnemri, A. , Madirevula, S. , Ibrahim, N. , Abdulwahab, F. , Hashem, M. , al‐Sheddi, T. , Alomar, R. , Alobeid, E. , Sallout, B. , AlBaqawi, B. , … Alkuraya, F. S. (2017). The genetic landscape of familial congenital hydrocephalus. Annals of Neurology, 81(6), 890–897. 10.1002/ana.24964 [DOI] [PubMed] [Google Scholar]
- Sobreira, N. , Schiettecatte, F. , Valle, D. , & Hamosh, A. (2015). GeneMatcher: A matching tool for connecting investigators with an interest in the same gene. Human Mutation, 36(10), 928–930. 10.1002/humu.22844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium . (2019). UniProt: A worldwide hub of protein knowledge. Nucleic Acids Research, 47(D1), D506–D515. 10.1093/nar/gky1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale, R. D. , Reese, T. S. , & Sheetz, M. P. (1985). Identification of a novel force‐generating protein, kinesin, involved in microtubule‐based motility. Cell, 42(1), 39–50. 10.1016/S0092-8674(85)80099-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen, M. H. , Ba, W. , Wissink‐Lindhout, W. M. , de Brouwer, A. P. M. , Haas, S. A. , Bienek, M. , Hu, H. , Vissers, L. E. L. M. , van Bokhoven, H. , Kalscheuer, V. , Nadif Kasri, N. , & Kleefstra, T. (2014). Involvement of the kinesin family members KIF4A and KIF5C in intellectual disability and synaptic function. Journal of Medical Genetics, 51(7), 487–494. 10.1136/jmedgenet-2013-102182 [DOI] [PubMed] [Google Scholar]
- Zhu, C. , Zhao, J. , Bibikova, M. , Leverson, J. D. , Bossy‐Wetzel, E. , Fan, J.‐B. , Abraham, R. T. , & Jiang, W. (2005). Functional analysis of human microtubule‐based motor proteins, the kinesins and dyneins, in mitosis/cytokinesis using RNA interference. Molecular Biology of the Cell, 16(July), 3187–3199. 10.1091/mbc.E05 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 In silico predictions for all variants reported in the article. Reference transcript: NM_012310.5.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.