

Abstract
RNA interference (RNAi) is a natural biological pathway that inhibits gene expression by targeted degradation or translational inhibition of cytoplasmic mRNA by the RNA induced silencing complex. RNAi has long been exploited in laboratory research to study the biological consequences of the reduced expression of a gene of interest. More recently RNAi has been demonstrated as a therapeutic avenue for rare metabolic diseases. This review presents an overview of the cellular RNAi machinery as well as therapeutic RNAi design and delivery. As a clinical example we present primary hyperoxaluria, an ultrarare inherited disease of increased hepatic oxalate production which leads to recurrent calcium oxalate kidney stones. In the most common form of the disease (Type 1), end‐stage kidney disease occurs in childhood or young adulthood, often necessitating combined kidney and liver transplantation. In this context we discuss nedosiran (Dicerna Pharmaceuticals, Inc.) and lumasiran (Alnylam Pharmaceuticals), which are both novel RNAi therapies for primary hyperoxaluria that selectively reduce hepatic expression of lactate dehydrogenase and glycolate oxidase respectively, reducing hepatic oxalate production and urinary oxalate levels. Finally, we consider future optimizations advances in RNAi therapies.
Keywords: calcium oxalate, end‐stage renal disease, glycolate oxidase, hyperoxaluria, kidney stones, lactate dehydrogenase, micro‐RNA, primary hyperoxaluria, RNA interference, small interfering RNAs
1. BACKGROUND
There is a paucity of disease‐modifying therapies for rare and ultrarare diseases. This has been attributable to a poor understanding of the molecular disease mechanisms and the challenge of recruiting sufficient numbers of patients to perform adequately powered trials of novel therapies. Advances in genomic sequencing have increased the rate of discovery for novel, disease‐associated genotypes and this increases the understanding of disease pathobiology, opening avenues to novel therapeutic targets and mechanisms.
RNA interference (RNAi) describes an innate biological pathway found in almost all eukaryotic cells. This pathway enables homology‐dependent, post‐transcriptional gene silencing by a micro‐RNA (miRNA) or small interfering RNA (siRNA) oligonucleotide complexed with a group of proteins called the RNA‐induced silencing complex (RISC). RNAi has established roles in development, 1 differentiation, 2 , 3 cancer biology 4 , 5 and cellular antiviral defences. 6 , 7
RNAi represents an attractive biological pathway to exploit in human disease therapeutics where the protein product of the gene cannot be easily targeted by small molecules. Indeed, synthetic siRNA molecules have been available for in vitro and in vivo animal model research for many years, reproducibly demonstrating potency for knockdown of gene expression.
The siRNA sequence and structural design critically influence engagement of the administered compound with the endogenous RNAi machinery. Another major challenge of translating RNAi into a clinically viable therapeutic product lies in the targeted delivery of oligonucleotides to the desired cell type, tissue or organ relevant to the disease in question, in doing so minimising off‐target effects. Despite these challenges, several RNAi‐based therapies for rare inherited diseases are now Food and Drug Administration (FDA) approved and/or in later clinical trials. 8
This review serves as a beginner's guide to RNAi therapeutics. We provide an overview of the biology of RNAi as well as the principles of therapeutic siRNA design and pharmacokinetics. We present a summary of primary hyperoxaluria, an ultrarare, inherited enzyme deficiency that can lead to overwhelming oxalate production, recurrent kidney stones, early end‐stage kidney disease (ESKD) and fulminant systemic oxalosis. In this context we discuss two novel RNAi therapies that selectively reduce hepatic expression of key enzymes involved in oxalate metabolism. RNAi therapies stand to significantly alter clinical outcomes in this debilitating disease.
2. BIOLOGY OF RNA SILENCING
First described in 1998, 9 RNAi is an endogenous, highly conserved cellular pathway whereby short strands of RNA guide the RISC to downregulate targeted gene expression, either by direct translational repression, by homology‐dependent mRNA cleavage or deadenylation. 8 , 10 , 11 , 12
There are two types of double‐stranded (ds)RNA molecules that serve as a guide for the RISC (depicted in Figure 1): miRNA or siRNA, which are broadly classified according to their precursor source and mechanism of transcriptional silencing. 13 Primary miRNA transcripts are transcribed from miRNA genes and form large single‐strand RNA molecules with multiple hairpin loops housing the bone fide miRNA within their double‐strand stems. 13 Within the nucleus, a heterotrimeric microprocessor composed of DROSHA (a class 2 ribonuclease III enzyme) and 2 cofactor DGCR8 (also known as Pasha) proteins, binds and cleaves short hairpin loops (called pre‐miRNA). 14 , 15 , 16
FIGURE 1.
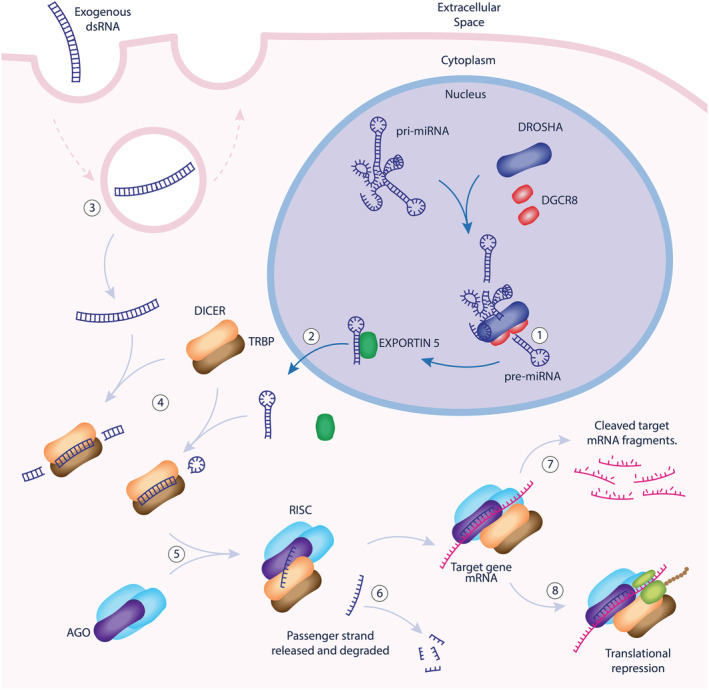
Molecular mechanisms of endogenous cellular RNAi. In the nucleus, pre‐micro RNA (pre‐miRNA) are trimmed from large primary micro‐RNA transcripts (pri‐miRNA) by DROSHA/DGCR8 complexes (1). Pre‐miRNA is exported to the cytoplasm associated with EXPORTIN‐5 (2). Exogenous dsRNA molecules enter the cell by endocytosis and cytoplasmic escape (3). dsRNA in the cytoplasm is further processed by DICER/TRBP complexes (4) before recruitment of Argonaut (AGO) and other proteins to form the RISC (5). The RISC unwinds the dsRNA and incorporates the antisense (or guide) strand, releasing the passenger strand for degradation (6). The antisense strand serves as a guide, selecting mRNA targets according to sequence homology, affecting either target mRNA cleavage and degradation (7) or translational repression (8). dsRNA: double‐stranded RNA; DGCR8: DiGeorge syndrome chromosomal region 8; TRBP: TAR RNA‐binding protein; RISC: RNA‐induced silencing complex, AGO: Argonaut protein
Cytoplasmic transfer of pre‐miRNA occurs by binding to EXPORTIN‐5, a RanGTP dependent dsRNA‐binding protein. 17 Within the cytoplasm, pre‐miRNA binds to TAR RNA binding protein (TRBP) and the terminal loop of the hairpin is cleaved by riboendonuclease DICER leaving approximately 21–23 base pairs of double stranded miRNA. DROSHA processing optimises pre‐miRNA molecules for efficient interaction with DICER's PAZ domain by leaving a 2‐nucleotide 3′overhang and a mono‐phosphorylated 5′ at the end of the hairpin stem. 18 , 19 , 20
An alternative pathway of RNAi more relevant to the field of therapeutic development is initiated when extracellular dsRNA is taken up by endocytosis or from complementary strands of transcribed endogenous mRNA (Figure 1). This dsRNA is cleaved by cytosolic DICER/TRBP in a DROSHA‐independent pathway.
Once the miRNA/siRNA has been processed by DICER, it recruits 1 of 4 Argonaut proteins (AGO1–4), which bind the antisense strand of the miRNA, unwind the dsRNA and release the passenger strand for degradation. 8 , 21 Antisense strand selection is independent of DROSHA and/or DICER cleavage polarity and tends to favour the strand where the 5′ end has a less stable complement pairing. 22 , 23 Whilst AGO1–4 proteins are all capable of loading miRNA/siRNA, only AGO2 retains endonuclease function capable of cleaving mRNA targets directly. 12 DICER‐independent miRNA processing pathways have also been identified for certain miRNA gene products whereby AGO2 performs the cleavage step canonically performed by DICER. 24 , 25
AGO proteins also recruit GW182, which enables translational repression and/or deadenylation of mRNA targets with homology to the 3′ untranslated region of the loaded guide RNA strand. 11 Micro‐RNA‐guided RISCs more often affect gene silencing by translational repression or mRNA degradation and siRNA‐guided RISCs more often affect mRNA cleavage, although crossover is observed. Whilst the efficiency of gene silencing is related to the degree of homology between the guide antisense miRNA/siRNA strand and target mRNA, 26 there may be as few as 7 complementary base pairs with the guide to affect silencing. 27
As more is learned about the components of the RNAi pathways, a more diverse range of functions is being discovered including roles in chromatin modification, chromosome arrangement during the cell cycle and DNA damage responses. These mechanisms are beyond the scope of this review but are discussed in detail in previous reviews. 28 , 29
3. THERAPEUTIC RNAI
The ability of the cell to endocytose RNA from the extracellular environment offers the opportunity to use RNAi as a therapeutic avenue to treat human disease, through the use of specifically designed RNA oligonucleotides targeting genes implicated in human disease pathogenesis. The major design challenges to overcome have included gene‐target specificity and methods for targeting organ‐ or tissue‐specific delivery.
3.1. Design and development
With current technology, short oligonucleotide complexes (21–27 nucleotides) with specific chemical modifications can be synthesized, delivered and evaluated for their activities and drug properties in both in vitro and in vivo models. 30 , 31 , 32 , 33 , 34 , 35 , 36 Therapeutic drug discovery usually starts with a large‐scale screen to identify candidates based on potency of target mRNA or protein inhibition, while computer‐based algorithms developed from cumulative and empirical screening data can be applied to effectively predict potency based on the sequence information in an effort to improve screening efficiency. 33 , 36 Several structural design features have been identified that affect DICER engagement and RNAi potency. Antisense strand selection can be favoured by removing the 2–3‐base overhang from the sense strand. 37 Longer dsRNA molecules are more likely to be cleaved by DICER, appropriately select the antisense strand for RISC integration and ultimately provide a more potent downregulation. 38 Shorter molecules can trigger RNAi with less potency via DICER independent pathways 25 , 39 but this allows greater flexibility in the incorporation of chemical modifications designed to maximize the stability of the molecule. 40
Off‐target effects may include silencing of the target gene in unwanted tissues or silencing of nontarget genes through sequence homology. 41 , 42 This most commonly occurs due to even partial homology between the 3′ untranslated region of the mRNA transcript and the 5′ end of the siRNA guide. 42 , 43 In silico homology analysis of candidate siRNA sequences using human transcriptome libraries is a potential method to predict such effects. Interspecies variation in off‐target transcriptional effects has been observed between mouse and human primary cell cultures; however, there was greater consistency between in vivo mouse liver (delivered by lipid nanoparticles) and in vitro mouse liver tumour cells (delivered by transfection). 44 Intuitively, these data advocate the use of in vitro, organ‐specific human or non‐human primate models as screens for off target effects in human disease.
The innate immune system is capable of responding to foreign RNA, often presented the form of RNA viruses. Immune stimulation by either the siRNA itself or the delivery vehicle can lead to proinflammatory cytokine production (interlekin‐6, tumour necrosis factor‐α) and immune cell activation. 41 Indeed, 1 of the first RNAi‐based therapies aimed at solid tumours (MRX34) closed its early human trials due to immune‐stimulation related adverse events. 45 The downstream effect of immune stimulation can alter transcriptional activity, potentially confounding the therapeutic objectives of the RNAi treatment. Various studies have demonstrated the effect of siRNA length, structure (i.e. single, double strand or hairpin), composition, uridine richness and delivery vehicle (especially cationic liposomes) as factors associated with immune system stimulation. 46 , 47 , 48
Chemical modifications have been widely applied to improve the stability needed for RNA duplexes to escape degradation in RNAse‐rich extracellular or intracellular environments, while simultaneously reducing immunogenicity, improving potency, target‐specificity and enhancing drug‐like properties. 49 , 50 , 51 , 52 , 53 , 54 , 55 These modifications have been extensively applied to all parts of the RNA duplex molecules, including the backbone, the sugars, and the bases on both sense and antisense strands to understand the structure activity relationship and drug stability. 49 , 50 , 51 , 52 , 56 For example, removing the 2–3‐base overhang from both ends of the dsRNA molecule confers increased nuclease resistance and RNAi potency. 57 The 2′ position of the sugar moiety of each nucleoside is often modified from 2′‐hydroxyl into 2′‐deoxy, 2′‐o‐methyl, 2′‐fluoro, 2′‐o‐methoxyethyl and locked nucleic acid to protect the oligonucleotides from RNAse degradation. Chemical modification of guide strand of siRNAs, in particular the 2′‐OMe substitution of position 2 of guide strand, has been demonstrated to significantly reduce off‐target effects. 58 Other backbone modifications include methylphosphonate, phosphorothioate, phosphorodithioate, thioester and other phosphate mimics. 40 , 59 , 60 , 61 The effect of any modification on enhancement or efficiency of RISC loading and AGO2 slicing activities must be validated. Unfortunately, the precise molecular mechanism of how siRNA molecules dynamically load and achieve target silencing is not completely understood even with the availability of crystal structures of complex of siRNA, target mRNA and AGO2. 59 , 62 , 63 , 64 The measurement of in vivo activity in rodents and non‐human primates remains a gold standard to empirically guide the evaluation of structure and activity relationships.
3.2. Advantages and disadvantages of siRNAs
A major advantage of siRNA vs. other antisense‐based approaches for therapeutic application is that its use of an innate cellular machinery allows for highly efficient targeting of complementary transcripts. 9 , 65 , 66 , 67 This reduces production costs and patient doses of siRNA and excipients while achieving sufficient target suppression and reducing toxicity. 68 , 69 Theoretically, siRNAs can be designed for targeting any gene based on its target mRNA sequence alone. RNAi is therefore an appealing therapeutic option for diseases mitigated by pathological gain of gene function or where substrate reduction can be safely implemented without redundancy to alternative metabolic pathways. siRNA is also an option for so‐called undruggable targets, which may not be easily inhibited using conventional small molecule and monoclonal antibody‐based approaches. Additionally, the siRNA drug discovery process can be relatively short compared to conventional small molecule drug approaches. 70 Chemical modifications and oligonucleotide compositions, which provide the main pharmacodynamic properties of siRNA therapeutics, can be readily applied to new sequences to achieve similar effects, whilst inhibiting different gene targets. Chemical modules for tissue‐specific delivery and modifications for activity and stability can be reutilized and applied to other sequences of interest, readying them for in vivo confirmation and subsequent clinical testing.
One major impediment in the development of siRNA therapeutics is focusing the delivery of oligonucleotide duplex to the specific cell types implicated in the disease. The high molecular weight, polyanionic and hydrophobic properties restrict the free diffusion of siRNA across cell membranes to reach cytoplasmic RNAi machinery. 8 , 71 , 72 The development of tissue‐specific delivery vehicles focusing siRNA delivery to the target organ of interest is one strategy to overcome off‐target effects. The development of nano‐carriers (lipid nanoparticle, liposome and polymer‐based) and conjugate‐based delivery has proven fruitful in both preclinical animal models and clinical investigation. 68 , 73 , 74 , 75 However, clinical validation of RNAi is currently only limited to hepatocytes and cancer cells. 8 , 72 Conjugate‐based delivery of siRNA to hepatocytes, utilizing N‐acetylgalactosamine (GalNAc) ligands that bind to the highly abundant hepatocyte cell surface asialoglycoprotein receptor (ASGPR), has revolutionized the therapeutic oligonucleotide field. GalNAc‐conjugated oligonucleotides bind to ASGPR and undergo endocytosis. 74 , 76 By unknown mechanisms, they subsequently escape from endosomes and enter the cytoplasm where they interact with RNAi machinery. In preclinical research, there are reports of delivery using different strategies to hepatic stellate cells, endothelial cells, neurons and other cell types. 77 , 78 , 79 , 80 , 81 , 82
Also, unlike small molecules which can be crudely identified to act either as agonists or antagonists, siRNA can only inhibit targets of interest with currently available technology, although this may change (see below). Finally, the materials and synthetic processes required for oligonucleotide manufacture is higher than for small molecules, representing a greater end‐product cost.
3.3. Predictable pharmacokinetic properties and tolerability profiles
siRNA therapies are currently delivered by subcutaneous or intravenous delivery, and oral delivery of siRNA is yet to be clinically demonstrated. siRNA's long duration of activity has been established in both preclinical and clinical investigation, which make its characteristic short plasma exposure uninformative for predicting duration of action. Nonetheless, investigators are trying to understand the relationship of maximum plasma concentration and direct pharmacodynamics or surrogate markers. 83 , 84 , 85 This long therapeutic effect is partially due to the stable integration of siRNA within RNAi machinery, forming a durable RISC‐capable of catalytic degradation of target mRNA through multiple cycles within targeted cells, even when majority of free siRNA drugs have been removed from metabolism or excretion. 86 , 87 The use of similar siRNA design principles and fixed delivery modules permits a predictability with respect to the pharmacokinetic properties and adverse effects for oligonucleotide duplexes as a therapeutic class, which stands to streamline the development and approval of new siRNA therapeutics, compared to small molecule therapies.
The long duration of siRNA activity has prompted concern from some investigators as to the long period for recovery should any acute toxicity occur. However, conjugate‐based siRNA therapeutics have shown high efficacy and favourable tolerability during clinical development with most common adverse effect as low frequency of mild to moderate injection site reactions, which usually resolve spontaneously. 68 , 69 , 88 Furthermore, concerns of off‐target effect due to sequence‐based complementarity on unrelated genes is yet to be demonstrated in clinical studies.
3.4. Current clinical application of RNAi therapeutics
The strategies described above have led to the advancement of more than two dozen therapeutics in early or late stages of clinical development in cancer, metabolic and chronic viral disease (summarised in 8 , 89 ). Patisiran (ALN‐TTR02 or Onpattro; Alnylam Pharmaeuticals) was the first RNAi therapy to gain FDA approval in August 2018, as a treatment for hereditary transthyretin amyloidosis (OMIM 105210), 85 an autosomal dominant disease where accumulation of misfolded TTR protein throughout the body results in progressive neuropathy, cardiomyopathy and ophthalmic disease among other end‐organ effects. The siRNA in patisiran is packaged in liver specific lipid nanoparticles and silences both wild type and mutant transthyretin genes reducing systemic TTR load. 84 , 85 , 90 Givosiran (Givlaari; Alnylam Pharmaceuticals), an RNAi therapy silencing δ‐aminolevulinic acid synthase 1 for treatment of acute hepatic porphyria, received FDA approval in 2019. 91 Phase 3 trials of givosiran demonstrated significantly lower rates of porphyria attacks, improved pain scores and lower levels of urinary δ‐aminolevulinic acid and porphobilinogen. 92 Many other RNAi based therapeutics are in early phase clinical trials, heralding a new class of therapies with increasing impact on clinical practice for a variety of diseases over the coming decades.
Hypothetically, siRNA therapeutics are well suited for any disease where undesirable disease‐causing proteins can be downregulated at a transcriptional level. It is, of course, imperative to ensure that silencing of the of any targeted gene will not be injurious to the recipient, which may restrict the scope of diseases to which RNAi can be applied. Genetic diseases with dominant negative mutations are another class of diseases that are applicable for siRNA approaches. Additionally, for some metabolic and genetic diseases, siRNA approaches can be designed to remove or reduce substrates of toxic metabolites to prevent or alleviate symptoms of those diseases.
4. PRIMARY HYPEROXALURIA
Primary hyperoxaluria (PH) is an ultra‐rare, autosomal recessive, inherited metabolic condition that leads to an accumulation of glyoxylate in the liver which is metabolised to oxalate by lactate dehydrogenase (LDH; Figure 2). Oxalate is readily filtered by the kidneys and is highly insoluble in urine in the presence of calcium, precipitating as calcium oxalate nephrolithiasis and nephrocalcinosis. 93 In severe cases nephrocalcinosis can progress to chronic kidney disease, at which point the reduced oxalate clearance leads to overwhelming systemic oxalosis affecting blood vessels, bones, retina, myocardium 94 , 95 and skin with a high associated mortality.
FIGURE 2.
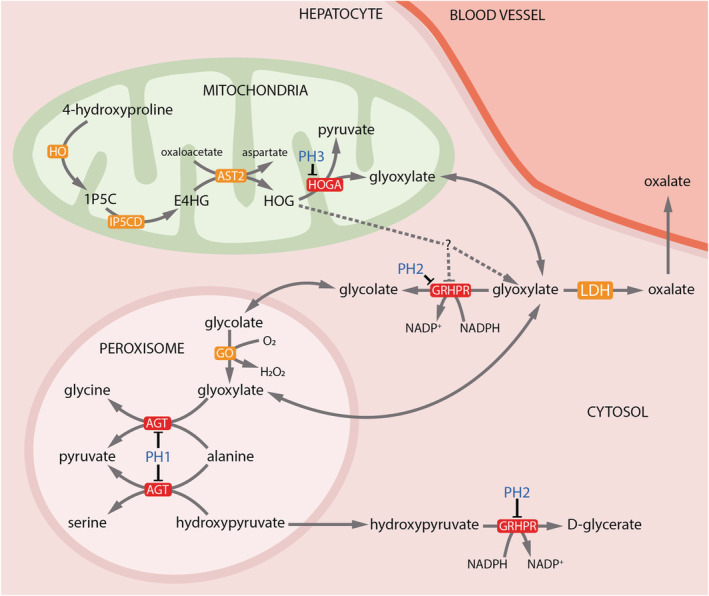
Hepatic metabolism of oxalate. Metabolic pathways involved in oxalate metabolism depicting the enzymes defective in the 3 major types of primary hyperoxaluria (red boxes and blue text). LDH catalyses the final step in the production of oxalate from glyoxalate for all PH types. The precise mechanisms by which HOGA mutations lead to increased oxalate production are not fully understood (depicted by dotted lines). Hypotheses include inhibition of GRHPR by accumulated HOG and metabolism of HOG in the cytoplasm to glyoxylate by an unidentified aldolase enzyme. 1P5C: 1‐pyrroline‐3‐hydroxy‐5‐carboxylate; E4HG: erythrohydroxyglutamate; HOG: 4‐hydroxy‐2‐oxoglutarate; PH: primary hyperoxaluria; AGXT: alanine glyoxylate aminotransferase; GRHPR: glyoxylate reductase/hydroxypyruvate reductase; HOGA1: hydroxyl‐oxoglutarate aldolase 1; LDH: lactate dehydrogenase
PH is sub‐classified into 3 groups based on genotype, each with a distinct phenotype and prognosis (Table 1, Figure 2). The most common phenotype, PH type 1 (PH1, OMIM 259900), accounts for approximately 80% of disease burden and has a population prevalence between 1–3 per million population. PH1 is associated with recessive mutations in alanine‐glyoxylate aminotransferase (AGXT), encoding the enzyme which catalyses the transamination of glyoxylate to glycine within the hepatocyte peroxisome. 96 Tissue expression is specific to the liver. Many PH1 patients will have an elevated urinary glycolate; however, this has also been reported in patients with PH3. 97 , 98 Whilst ESKD in infancy is not uncommon (around 25% of cases), the median age of ESKD is reported at age 10–24 years. 99 , 100 Genotype–phenotype correlations have been observed with Gly170Arg variants demonstrating a median age of ESKD at 47 years for homozygotes in a western European/North African cohort. 99 However, there exists wide variation in severity of renal phenotype even between members of the same family, suggesting that genetic modifiers or potentially kidney‐specific factors may alter phenotype. 101
TABLE 1.
Features of the 3 primary hyperoxaluria genotypes
| Type 1 | Type 2 | Type 3 | |
|---|---|---|---|
| Gene | AGXT | GRHPR | HOGA1 |
| Chromosome | 2q37.3 | 9p13.2 | 10q24.2 |
| Tissue expression | Liver | Liver, kidney, brain, skin, cardiac and skeletal muscle | Liver, kidney, brain |
| Proportion of disease burden | ~80% | ~10% | ~10% |
| Phenotype | Calcium oxalate nephrolithiasis, nephrocalcinosis, childhood CKD | Calcium oxalate nephrolithiasis, nephrocalcinosis, adult CKD | Calcium oxalate nephrolithiasis, nephrocalcinosis, adult CKD |
| Biomarker | Urine glycolate (variable) 97 | Urine L‐glyceric acid (variable) 104 | Dihydroxyglutamate (all patients) 155 |
| Treatment |
Hydration Urinary alkalinisation Pyridoxine (some variants only) Liver +/− kidney transplant |
Hydration Urinary alkalinisation Kidney transplantation (liver transplantation controversial) |
Hydration Urinary alkalinisation Transplantation not required |
| OMIM phenotype number | 259 900 | 260 000 | 613 616 |
CKD, chronic kidney disease
PH2 (OMIM 260000) accounts for roughly 10% of disease burden and is caused by pathogenic variants in the gene encoding glyoxylate reductase and hydroxypyruvate reductase (GRHPR) which metabolises glyoxylate to glycolate and hydroxypyruvate to D‐glycerate (Figure 2). 102 GRHPR is expressed throughout the body but is highest in the liver. 103 GRHPR deficiency leave both substrates vulnerable to LDH metabolism, excreted as oxalate and L‐glycerate respectively, although presence of the either in the urine is variable 102 , 104 necessitating genomic sequencing as gold standard for diagnosis. In the largest reported cohort from the OxalEurope Consortium, almost 10% of patients presented with normal spot urine oxalate:creatinine ratio. 102 Patients develop recurrent kidney stones and nephrocalcinosis, generally progressing to chronic kidney disease or ESKD in mid‐adulthood. 102
PH3 (OMIM 613616) is due to pathogenic variants in hydroxyl‐oxoglutarate aldolase 1 (HOGA1), a mitochondrial enzyme that takes part in hydroxyproline metabolism. 98 PH3 accounts for 10% of PH cases although the population prevalence of the most common variant (c.700 + 5G > T affecting a splice site in intron 5 97 ) is much higher than anticipated for the documented disease frequency, suggesting either incomplete penetrance or underdiagnosis. 97 Given the limitations in access to genomic testing in many settings for patients with mild diseases, patients with PH3 suffering from recurrent but infrequent calcium‐oxalate stones may well be underdiagnosed. The second most common pathogenic variant is p.E315del effecting a 3‐base pair deletion in exon 7, representing 32% of alleles. 97 Patients experience raised urine oxalate excretion with or without recurrent nephrolithiasis and progression to chronic kidney disease is reported but uncommon. 105
Paradoxically, glyoxylate is a product of the HOGA enzyme rather than a substrate and the biological mechanism of oxalate overproduction in PH3 is not well established. 106 The increased oxalate production in HOGA deficiency may be explained by the accumulation of its precursor 4‐hydroxy‐2‐oxoglutarate (HOG). HOG accumulation may inhibit GRHPR; however, increased urinary L‐glycerate is rarely seen, which would argue against this mechanism. 107 , 108 An alternative explanation is the metabolism of HOG to glyoxylate by an as yet uncharacterized cytosolic aldolase. 109
4.1. Current therapies for primary hyperoxaluria are limited in their ability to prevent ESKD
The goals of early conservative therapy for all PH types involves maximizing the solubility of oxalate in the urine in an effort to prevent stone formation and nephrocalcinosis. As for all causes of nephrolithiasis, maintenance of a high intake of water is critical. 110 , 111 Patients are often prescribed to drink up to 2.5 L/m2 water daily. Periods of acute dehydration are avoided and managed aggressively with enteral or parenteral fluid supplementation to avoid acute kidney injury from which recovery can be limited. 112 , 113 Dietary restriction of oxalate rich foods is ineffective 114 ; however, patients are advised to avoid vitamin C supplements as ascorbic acid is metabolised to oxalate. 115 , 116
Supplementation with citrate and orthophosphate serves to complex with urinary calcium preventing calcium‐oxalate crystallization in the urine. 117 , 118 , 119 , 120 Although there is no evidence to support its efficacy, magnesium supplementation is proposed to act via a similar mechanism.
For up to 50% of patients with PH1, high‐dose pyridoxine supplementation has been established to improve enzyme function and reduce urinary oxalate levels. 119 , 121 , 122 , 123 Pyridoxine is metabolised to pyridoxal phosphate, which is an essential catalytic cofactor for the AGXT enzyme as well as being a necessary chaperone for peroxisomal localization. 124 Patients on pyridoxine must be monitored for peripheral neuropathy which is reversible if ceased early. 125 Variants known to respond favourably to pyridoxine include Gly170Arg, Phe152Ile, Ile244Thr and Gly41Arg. 99 , 126 , 127 In particular, the Gly170Arg variant, present in roughly 30% of PH1 cases, is known to be mislocalised to mitochondria but shows both improved activity and peroxisomal localization with pyridoxine treatment. 124 PH1 patients should be trialled on pyridoxine and continued if at least a 30% reduction in urine oxalate can be achieved over 3 months. 112 , 113
4.2. Dialysis and transplantation
Whilst militant adherence to conservative treatments is associated with improved outcomes, the eventual need for dialysis, kidney transplantation and/or liver transplantation in patients with PH1 is almost ubiquitous. 128 Renal clearance is the only avenue to adequately remove such substantial amounts of oxalate in PH, and as a result serum oxalate levels increase exponentially with declining glomerular filtration rate. Systemic deposition of oxalate subsequently accumulates in the eyes, heart, bones, skin, nerves and joints, increasing morbidity. Thus, dialysis is essential not only for renal replacement but for oxalate clearance. Whilst haemodialysis is able to very quickly clear oxalate from the circulating bloodstream, the oxalate production in patients with PH1 is sufficiently high and tissue‐sequestered that patients remain net oxalate positive, even on intensified peritoneal and haemodialysis. 129 PH1 patients on haemodialysis have 2–3 times the mortality rate of nonhyperoxaluria dialysis patients. 130 In undiagnosed PH1 patients and those with pyridoxine‐unresponsive disease, isolated kidney transplantation is associated with high rates of disease recurrence in the graft. 131 Liver transplantation is curative and can be performed in isolation prior to the development of chronic kidney disease or combined with a kidney transplant in ESKD. Sequential liver and kidney transplant can be pursued if there are concerns about immediate risk to the kidney graft 131 , 132 ; however, many patients do well with intraoperative filtration and daily postoperative haemodialysis. 133 Isolated kidney transplant may be considered in pyridoxine‐responsive PH1 and remains the most common approach in ESKD due to PH2 and PH3.
5. NOVEL RNAI THERAPIES FOR PRIMARY HYPEROXALURIA
Whilst the precise mechanisms underpinning the metabolic disturbances giving rise to oxalate overproduction are less well understood in PH3 than for PH1/2, it may be that LDH represents a final enzymatic pathway leading to hyperoxaluria (Figure 2). Hence, an RNAi approach could serve as a potentially effective and safe way to correct the overproduction of oxalate in the liver. In considering a strategy to reduce oxalate production and treat PH, inhibition of glycolate oxidase (GO) is sufficient to reduce the conversion of glycolate to glyoxylate, the proposed main precursor to oxalate 134 , 135 (Figure 2). Inhibition of expression of LDH, the key enzyme responsible for producing oxalate from glyoxylate, has been demonstrated to be effective in treating PH1 and PH2 mice. 136 Lai et al. provided the first in vivo evidence in mammals to support the role of LDH for converting glyoxylate to oxalate, where reduction of hepatic LDH by RNAi achieves efficient reduction in serum oxalate and renal calcium oxalate crystal deposition. 136 Notably, when LDH and GO inhibition was compared in PH1 mice, a disproportionate correlation between GO protein suppression and urinary oxalate reduction was observed. The authors further confirmed that suppression of LDH, but not GO, causes the reduction of urinary oxalate levels in PH2 mouse models. 136 Repression of hepatic LDH in mice and non‐human primates did not cause any acute elevation of circulating liver enzymes, lactate acidosis, or exertional myopathy, suggesting further evaluation of liver‐specific inhibition of LDH as a potential approach for treating PH1 and PH2 was warranted. 136 Subsequently nedosiran (DCR‐PHXC, Dicerna Pharmaceuticals Inc, Table 2) was designed to specially inhibit hepatic expression of LDHA, which encodes the M subtype of LDH, the major isoform of LDH enzyme in the liver. 136 , 137 , 138 Nedosiran is a double‐strand siRNA molecule that is conjugated with GalNAc, which takes advantage of the aforementioned unique ASGPR delivery system in the liver. (Figure 3) Nedosiran is administered by monthly subcutaneous injection. The initial data emerging from a multidose, open‐label trial (PHYOX3, Dicerna Pharmaceuticals, Inc; ClinicalTrials.gov Identifier: NCT0402402) demonstrated sustained, long‐term reduction in urinary oxalate levels as far as the normal range (less than 0.46 mmol/1.73 m2 BSA/24 hours) or near‐normalization in patients with both PH1 and PH2 139 (abstract only). This study advocated that nedosiran was generally well tolerated, which agrees with the observed absence of any liver‐specific adverse effects in natural, systemically LDHA‐deficient patients. 140 , 141 , 142 , 143 , 144 This supports preclinical observations in wild type mice where GalNAc‐conjugated siRNAs designed for liver‐specific knockdown of LDHA had no significant effect on gene expression in muscle, skin or uterine tissue, as well as maintained lactate production and exercise performance. 136 Whilst the definitive pathways by which oxalate production occurs in PH3 are less well established, it is conceivable that LDH silencing could also be of benefit in this disease. Accordingly, clinical testing of nedosiran will soon expand to include PH3 patients (PHYOX4: ClinicalTrials.gov Identifier: NCT04555486) and PH1/2 patients with ESKD (PHYOX7: ClinicalTrials.gov Identifier: NCT04580420).
TABLE 2.
Comparison of the 2 RNAi therapies in development for primary hyperoxaluria
| Nedosiran | Lumasiran | |
|---|---|---|
| Company | Dicerna Pharmaceuticals Inc. | Alnylam Pharmaceuticals |
| Previous names | DCR‐PHXC | ALN‐GO1; Oxlumo |
| Gene product targeted (protein name) |
LDHA (lactate dehydrogenase) |
HAO1 (Glycolate oxidase) |
| Molecule | dsRNA covalently linked to GalNAc residues | dsRNA covalently linked to GalNAc residues |
| Primary hyperoxaluria types targeted |
PH1, PH2 (PH3 trials underway) |
PH1 |
| Administration | Subcutaneous | Subcutaneous |
| Dosing | Monthly |
Loading: Monthly for 3 mo Maintenance: 3 monthly |
| Human trials | PHYOX3 early unpublished results: 6 of 7 patients showed consistent normalization or near normalization of urine oxalate levels after 3 doses (≤0.6 mmol/24 h/1.73m2) 139 |
ILLUMINATE‐A: 53.5% decrease in 24‐h urine oxalate excretion (95% confidence interval 62.3–44.8%) compared to placebo 11.8% reduction. 156 |
| Approvals |
FDA (Nov 2020) EMA (Nov 2020) |
FIGURE 3.
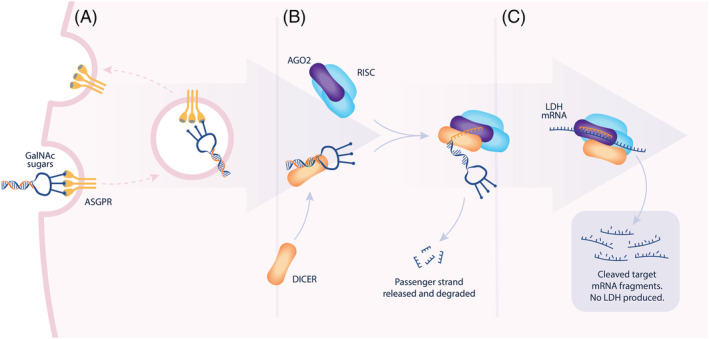
Mechanism of action of nedosiran within hepatocytes. Nedosiran is a GalNAc‐dsRNA conjugate. (A) Bound GalNac sugar residues bind to ASGPR receptors prompting endocytosis of the dsRNA. Via an incompletely understood mechanism, the compound undergoes endocytic escape and enters the cytoplasm. (B) Within the cytoplasm, the dsRNA first interacts with endonuclease DICER (orange). DICER then passes the antisense RNA strand to AGO2 (purple). The dsRNA compound is designed to favour the loading of the antisense RNA strand into the RISC complex. (C) RISC is guided to the target mRNA by homology to the RNA strand, cleaving the target RNA. GalNAc: N‐acetylgalactosamine; ASGPR: asialoglycoprotein receptor; RISC: RNA‐induced silencing complex; dsRNA: double‐stranded ribonucleic acid
Lumasiran (Oxlumo or ALN‐GO1, Alnylam Pharmaceuticals, Table 2) is another RNAi therapy for the treatment of PH1, which gained FDA approval in November 2020. Lumasiran targets GO, reducing the production of glyoxylate, the main oxalate precursor, from glycolate (Figure 2). 145 , 146 Glycolate is excreted by the kidneys but is highly soluble and not associated with a renal phenotype in GO‐deficient (Hao1 knockout) mouse models. 147 Rare human cases of biallelic loss of function variants in HAO1 have also been characterized to have high serum and urine glycolate levels without a disease phenotype, endorsing the safety of an RNAi approach to GO. 148 , 149 In addition, double knockout mice for Agxt and Hao1 demonstrate low levels of urine oxalate excretion. 147 A recently published double‐blind, multinational, placebo‐controlled, randomized controlled trial recruited 39 adult and paediatric patients with PH1 and an estimated glomerular filtration rate of >30 mL/min/1.73 m2. 146 Lumasiran effected a 53.5% reduction in 24 hour urine oxalate levels (95% confidence interval 62.3–44.8%) compared to placebo (mean reduction 11.8%) and 85% of patients achieved a urine oxalate excretion within 150% of the upper limit of the normal range. 146 Furthermore, no serious adverse events were reported.
6. FUTURE PROSPECTS FOR RNAI THERAPIES
Recent regulatory approvals of RNAi‐based drugs include a complex lipid nanoparticle formulation of 1 unconjugated siRNA product and far simpler water‐for‐injection formulations of GalNAc‐conjugated siRNAs and many more are in clinical trials, ranging from first‐in human studies to registration trials.
Beyond synthetic siRNA, there is potential to manipulate RNAi for therapeutic benefit in other ways. As well as gene silencing, dsRNA molecules designed with homology to sequences near target gene promotor regions have been observed to activate gene expression via a non‐endonuclease AGO2‐mediated pathway. 150 , 151 This strategy has been recently applied to the treatment of hepatocellular carcinoma. 152 Anti‐mirs (also known as antagomirs), are antisense oligonucleotides targeting miRNA effectively blocking RISC activity on the host cell transcriptome, 153 a strategy that had been applied to the treatment of hepatitis C (Mirversin, Santaris Pharma); however, this has been superseded by highly efficacious and inexpensive small molecule treatments. 154 Finally, block‐mirs, which anneal with target mRNA, effectively protecting the target transcript from RISC‐mediated degradation, are yet to demonstrate a clinical application.
In principle, good siRNA sequence design to complement the target gene can achieve highly specific target knockdown while avoiding cross‐reactivity to functional protein molecules of same family, which is a challenge for small molecule‐based approaches. This high specificity of the RNAi approach may even allow targeting of disease‐specific alleles and spare the normal allele even when they differ only by 1 or a few nucleotide substitutions. However, it might not be easy to achieve the balance between the activity and specificity due to the requirement of fixed and short length of oligonucleotide duplexes to trigger RNAi activity. Multiple targets can be inhibited simultaneously without changing the therapeutic principles and fundamental physical composition of RNAi‐based therapies, which represents another advantage over small molecule therapeutics.
7. CONCLUSIONS
RNAi involves the processing of endogenous miRNA or exogenous siRNA molecules into RISCs which are capable of downregulating gene expression by homology directed cleavage of target mRNA or direct transcriptional inhibition.
As a platform, synthetic siRNAs are emerging as well tolerated and effective drug candidates as well as approved drugs with high safety margins. Chemical modifications can specify tissue specific delivery of the compound to the tissue of interest. This bodes well for the use of RNAi to treat primary hyperoxaluria with the potential for adaptation of the system for the treatment of other inherited and metabolic diseases.
COMPETING INTERESTS
C.L. and B.D.B. are employees of Dicerna Pharmaceuticals, which is developing siRNAs as therapeutics, including nedosiran. T.A.F. is the Site Principle Investigator at the Royal Children's Hospital in Melbourne, Australia supervising Dicerna (nedosiran) and Alnylam (lumasiran) clinical trials.
Forbes TA, Brown BD, Lai C. Therapeutic RNA interference: A novel approach to the treatment of primary hyperoxaluria. Br J Clin Pharmacol. 2022;88(6):2525–2538. 10.1111/bcp.14925
REFERENCES
- 1. Suh N, Blelloch R. Small RNAs in early mammalian development: from gametes to gastrulation. Development. 2011;138(9):1653‐1661. 10.1242/dev.056234 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Heinrich EM, Dimmeler S. MicroRNAs and stem cells: control of pluripotency, reprogramming, and lineage commitment. Circ Res. 2012;110(7):1014‐1022. 10.1161/circresaha.111.243394 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 3. Ivey KN, Srivastava D. MicroRNAs as Regulators of Differentiation and Cell Fate Decisions. Cell Stem Cell. 2010;7(1):36‐41. 10.1016/j.stem.2010.06.012 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 4. Dhawan A, Scott JG, Harris AL, Buffa FM. Pan‐cancer characterisation of microRNA across cancer hallmarks reveals microRNA‐mediated downregulation of tumour suppressors. Nat Commun. 2018;9(1):5228. 10.1038/s41467-018-07657-1 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nicoloso MS, Spizzo R, Shimizu M, Rossi S, Calin GA. MicroRNAs — the micro steering wheel of tumour metastases. Nat Rev Cancer. 2009;9(4):293‐302. 10.1038/nrc2619 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 6. Malone CD, Hannon GJ. Small RNAs as guardians of the genome. Cell. 2009;136(4):656‐668. 10.1016/j.cell.2009.01.045 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mishra R, Kumar A, Ingle H, Kumar H. The Interplay Between Viral‐Derived miRNAs and Host Immunity During Infection. Front Immunol. 2020;10:3079. 10.3389/fimmu.2019.03079 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Setten RL, Rossi JJ, Han SP. The current state and future directions of RNAi‐based therapeutics. Nat Rev Drug Discov. 2019;18(6):421‐446. 10.1038/s41573-019-0017-4 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 9. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double‐stranded RNA in Caenorhabditis elegans. Nature. 1998;391(6669):806‐811. 10.1038/35888 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 10. Li C, Zamore PD. RNA Interference and Small RNA Analysis. Cold Spring Harb Protoc. 2019;2019(4). 10.1101/pdb.top097436 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 11. Huntzinger E, Kuzuoglu‐Öztürk D, Braun JE, Eulalio A, Wohlbold L, Izaurralde E. The interactions of GW182 proteins with PABP and deadenylases are required for both translational repression and degradation of miRNA targets. Nucleic Acids Res. 2013;41(2):978‐994. 10.1093/nar/gks1078 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, Tuschl T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol Cell. 2004;15(2):185‐197. 10.1016/j.molcel.2004.07.007 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 13. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15(8):509‐524. 10.1038/nrm3838 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 14. Kwon SC, Nguyen Tuan A, Choi Y‐G, et al. Structure of Human DROSHA. Cell. 2016;164(1):81‐90. 10.1016/j.cell.2015.12.019 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 15. Lee Y, Ahn C, Han J, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425(6956):415‐419. 10.1038/nature01957 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 16. Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. The Drosha‐DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18(24):3016‐3027. 10.1101/gad.1262504 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bohnsack MT, Czaplinski K, Gorlich D. Exportin 5 is a RanGTP‐dependent dsRNA‐binding protein that mediates nuclear export of pre‐miRNAs. RNA. 2004;10(2):185‐191. 10.1261/rna.5167604 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang H, Kolb FA, Jaskiewicz L, Westhof E, Filipowicz W. Single processing center models for human Dicer and bacterial RNase III. Cell. 2004;118(1):57‐68. 10.1016/j.cell.2004.06.017 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 19. Vermeulen A, Behlen L, Reynolds A, et al. The contributions of dsRNA structure to Dicer specificity and efficiency. RNA. 2005;11(5):674‐682. 10.1261/rna.7272305 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Park J‐E, Heo I, Tian Y, et al. Dicer recognizes the 5′ end of RNA for efficient and accurate processing. Nature. 2011;475(7355):201‐205. 10.1038/nature10198 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Martinez J, Tuschl T. RISC is a 5′ phosphomonoester‐producing RNA endonuclease. Genes Dev. 2004;18(9):975‐980. 10.1101/gad.1187904 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schwarz DS, Hutvágner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115(2):199‐208. 10.1016/s0092-8674(03)00759-1 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 23. Preall JB, He Z, Gorra JM, Sontheimer EJ. Short interfering RNA strand selection is independent of dsRNA processing polarity during RNAi in Drosophila. Curr Biol. 2006;16(5):530‐535. 10.1016/j.cub.2006.01.061 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 24. Cheloufi S, Dos Santos CO, Chong MM, Hannon GJ. A dicer‐independent miRNA biogenesis pathway that requires Ago catalysis. Nature. 2010;465(7298):584‐589. 10.1038/nature09092 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cifuentes D, Xue H, Taylor DW, et al. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science. 2010;328(5986):1694‐1698. 10.1126/science.1190809 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215‐233. 10.1016/j.cell.2009.01.002 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15‐20. 10.1016/j.cell.2004.12.035 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 28. Gutbrod MJ, Martienssen RA. Conserved chromosomal functions of RNA interference. Nat Rev Genet. 2020;21(5):311‐331. 10.1038/s41576-019-0203-6 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Burger K, Gullerova M. Swiss army knives: non‐canonical functions of nuclear Drosha and Dicer. Nat Rev Mol Cell Biol. 2015;16(7):417‐430. 10.1038/nrm3994 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 30. Aagaard L, Rossi JJ. RNAi therapeutics: principles, prospects and challenges. Adv Drug Deliv Rev. 2007;59(2–3):75‐86. 10.1016/j.addr.2007.03.005 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Castanotto D, Rossi JJ. The promises and pitfalls of RNA‐interference‐based therapeutics. Nature. 2009;457(7228):426‐433. 10.1038/nature07758 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wittrup A, Lieberman J. Knocking down disease: a progress report on siRNA therapeutics. Nat Rev Genet. 2015;16(9):543‐552. 10.1038/nrg3978 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dudek H, Wong DH, Arvan R, et al. Knockdown of beta‐catenin with dicer‐substrate siRNAs reduces liver tumor burden in vivo. Mol Ther. 2014;22(1):92‐101. 10.1038/mt.2013.233 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu Q, Zhou H, Cui J, Cao Z, Xu Y. Reconsideration of in‐silico siRNA design based on feature selection: a cross‐platform data integration perspective. PLoS One. 2012;7(5):e37879. 10.1371/journal.pone.0037879 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thang BN, Ho TB, Kanda T. A semi‐supervised tensor regression model for siRNA efficacy prediction. BMC Bioinformatics. 2015;16:80. 10.1186/s12859-015-0495-2 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vert JP, Foveau N, Lajaunie C, Vandenbrouck Y. An accurate and interpretable model for siRNA efficacy prediction. BMC Bioinformatics. 2006;7:520. 10.1186/1471-2105-7-520 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sano M, Sierant M, Miyagishi M, Nakanishi M, Takagi Y, Sutou S. Effect of asymmetric terminal structures of short RNA duplexes on the RNA interference activity and strand selection. Nucleic Acids Res. 2008;36(18):5812‐5821. 10.1093/nar/gkn584 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Snead NM, Wu X, Li A, et al. Molecular basis for improved gene silencing by Dicer substrate interfering RNA compared with other siRNA variants. Nucleic Acids Res. 2013;41(12):6209‐6221. 10.1093/nar/gkt200 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee HY, Zhou K, Smith AM, Noland CL, Doudna JA. Differential roles of human Dicer‐binding proteins TRBP and PACT in small RNA processing. Nucleic Acids Res. 2013;41(13):6568‐6576. 10.1093/nar/gkt361 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Parmar R, Willoughby JL, Liu J, et al. 5′‐(E)‐Vinylphosphonate: A Stable Phosphate Mimic Can Improve the RNAi Activity of siRNA‐GalNAc Conjugates. Chembiochem. 2016;17(11):985‐989. 10.1002/cbic.201600130 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 41. Jackson AL, Linsley PS. Recognizing and avoiding siRNA off‐target effects for target identification and therapeutic application. Nat Rev Drug Discov. 2010;9(1):57‐67. 10.1038/nrd3010 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 42. Jackson AL, Burchard J, Schelter J, et al. Widespread siRNA "off‐target" transcript silencing mediated by seed region sequence complementarity. RNA (New York, N.Y.). 2006;12(7):1179‐1187. 10.1261/rna.25706 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Birmingham A, Anderson EM, Reynolds A, et al. 3' UTR seed matches, but not overall identity, are associated with RNAi off‐targets. Nat Methods. 2006;3(3):199‐204. 10.1038/nmeth854 [DOI] [PubMed] [Google Scholar]
- 44. Burchard J, Jackson AL, Malkov V, et al. MicroRNA‐like off‐target transcript regulation by siRNAs is species specific. RNA. 2009;15(2):308‐315. 10.1261/rna.1326809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Beg MS, Brenner AJ, Sachdev J, et al. Phase I study of MRX34, a liposomal miR‐34a mimic, administered twice weekly in patients with advanced solid tumors. Invest New Drugs. 2017;35(2):180‐188. 10.1007/s10637-016-0407-y [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Judge AD, Sood V, Shaw JR, Fang D, McClintock K, MacLachlan I. Sequence‐dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat Biotechnol. 2005;23(4):457‐462. 10.1038/nbt1081 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 47. Hornung V, Guenthner‐Biller M, Bourquin C, et al. Sequence‐specific potent induction of IFN‐alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med. 2005;11(3):263‐270. 10.1038/nm1191 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 48. Goodchild A, Nopper N, King A, et al. Sequence determinants of innate immune activation by short interfering RNAs. BMC Immunol. 2009;10:40. 10.1186/1471-2172-10-40 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Burnett JC, Rossi JJ. RNA‐based therapeutics: current progress and future prospects. Chem Biol. 2012;19(1):60‐71. 10.1016/j.chembiol.2011.12.008 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Deleavey GF, Damha MJ. Designing chemically modified oligonucleotides for targeted gene silencing. Chem Biol. 2012;19(8):937‐954. 10.1016/j.chembiol.2012.07.011 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 51. Dirin M, Winkler J. Influence of diverse chemical modifications on the ADME characteristics and toxicology of antisense oligonucleotides. Expert Opin Biol Ther. 2013;13(6):875‐888. 10.1517/14712598.2013.774366 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 52. Rettig GR, Behlke MA. Progress toward in vivo use of siRNAs‐II. Mol Ther. 2012;20(3):483‐512. 10.1038/mt.2011.263 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lundin KE, Gissberg O, Smith CI. Oligonucleotide Therapies: The Past and the Present. Hum Gene Ther. 2015;26(8):475‐485. 10.1089/hum.2015.070 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Janas MM, Zlatev I, Liu J, et al. Safety evaluation of 2′‐deoxy‐2′‐fluoro nucleotides in GalNAc‐siRNA conjugates. Nucleic Acids Res. 2019;47(7):3306‐3320. 10.1093/nar/gkz140 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jackson AL, Burchard J, Leake D, et al. Position‐specific chemical modification of siRNAs reduces "off‐target" transcript silencing. RNA (New York, NY). 2006;12(7):1197‐1205. 10.1261/rna.30706 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Foster DJ, Brown CR, Shaikh S, et al. Advanced siRNA Designs Further Improve In Vivo Performance of GalNAc‐siRNA Conjugates. Mol Ther. 2018;26(3):708‐717. 10.1016/j.ymthe.2017.12.021 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Allerson CR, Sioufi N, Jarres R, et al. Fully 2′‐modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering RNA. J Med Chem. 2005;48(4):901‐904. 10.1021/jm049167j [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 58. Yilmazel B, Hu Y, Sigoillot F, et al. Online GESS: prediction of miRNA‐like off‐target effects in large‐scale RNAi screen data by seed region analysis. BMC Bioinformatics. 2014;15:192. 10.1186/1471-2105-15-192 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8(2):129‐138. 10.1038/nrd2742 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Prakash TP, Kinberger GA, Murray HM, et al. Synergistic effect of phosphorothioate, 5′‐vinylphosphonate and GalNAc modifications for enhancing activity of synthetic siRNA. Bioorg Med Chem Lett. 2016;26(12):2817‐2820. 10.1016/j.bmcl.2016.04.063 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 61. Zlatev I, Foster DJ, Liu J, et al. 5'‐C‐Malonyl RNA: Small Interfering RNAs Modified with 5'‐Monophosphate Bioisostere Demonstrate Gene Silencing Activity. ACS Chem Biol. 2016;11(4):953‐960. 10.1021/acschembio.5b00654 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 62. Swarts DC, Makarova K, Wang Y, et al. The evolutionary journey of Argonaute proteins. Nat Struct Mol Biol. 2014;21(9):743‐753. 10.1038/nsmb.2879 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang Y, Juranek S, Li H, et al. Nucleation, propagation and cleavage of target RNAs in Ago silencing complexes. Nature. 2009;461(7265):754‐761. 10.1038/nature08434 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang G, Reinke V. A C. elegans Piwi, PRG‐1, regulates 21U‐RNAs during spermatogenesis. Curr Biol. 2008;18(12):861‐867. 10.1016/j.cub.2008.05.009 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dykxhoorn DM, Lieberman J. The silent revolution: RNA interference as basic biology, research tool, and therapeutic. Annu Rev Med. 2005;56:401‐423. 10.1146/annurev.med.56.082103.104606 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 66. Perrimon N, Ni JQ, Perkins L. In vivo RNAi: today and tomorrow. Cold Spring Harb Perspect Biol. 2010;2(8):a003640. 10.1101/cshperspect.a003640 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sen GL, Blau HM. A brief history of RNAi: the silence of the genes. FASEB J. 2006;20(9):1293‐1299. 10.1096/fj.06-6014rev [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 68. Fitzgerald K, White S, Borodovsky A, et al. A Highly Durable RNAi Therapeutic Inhibitor of PCSK9. N Engl J Med. 2017;376(1):41‐51. 10.1056/NEJMoa1609243 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sardh E, Harper P, Balwani M, et al. Phase 1 Trial of an RNA Interference Therapy for Acute Intermittent Porphyria. N Engl J Med. 2019;380(6):549‐558. 10.1056/NEJMoa1807838 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 70. Lai C, Martin‐Higueras C, Salido E. siRNA Therapeutics to Treat Liver Disorders. In: Brunetti‐Pierri N, ed. Safety and Efficacy of Gene‐Based Therapeutics for Inherited Disorders. Cham: Springer International Publishing; 2017:159‐190. [Google Scholar]
- 71. Bobbin ML, Rossi JJ. RNA Interference (RNAi)‐Based Therapeutics: Delivering on the Promise? Annu Rev Pharmacol Toxicol. 2016;56:103‐122. 10.1146/annurev-pharmtox-010715-103633 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 72. Springer AD, Dowdy SF. GalNAc‐siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther. 2018;28(3):109‐118. 10.1089/nat.2018.0736 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Coelho T, Adams D, Silva A, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819‐829. 10.1056/NEJMoa1208760 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 74. Matsuda S, Keiser K, Nair JK, et al. siRNA conjugates carrying sequentially assembled trivalent N‐acetylgalactosamine linked through nucleosides elicit robust gene silencing in vivo in hepatocytes. ACS Chem Biol. 2015;10(5):1181‐1187. 10.1021/cb501028c [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 75. Nair JK, Willoughby JL, Chan A, et al. Multivalent N‐acetylgalactosamine‐conjugated siRNA localizes in hepatocytes and elicits robust RNAi‐mediated gene silencing. J Am Chem Soc. 2014;136(49):16958‐16961. 10.1021/ja505986a [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 76. Rajeev KG, Nair JK, Jayaraman M, et al. Hepatocyte‐specific delivery of siRNAs conjugated to novel non‐nucleosidic trivalent N‐acetylgalactosamine elicits robust gene silencing in vivo. Chembiochem. 2015;16(6):903‐908. 10.1002/cbic.201500023 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 77. Tabernero J, Shapiro GI, LoRusso PM, et al. First‐in‐humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013;3(4):406‐417. 10.1158/2159-8290.CD-12-0429 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 78. Sato Y, Murase K, Kato J, et al. Resolution of liver cirrhosis using vitamin A‐coupled liposomes to deliver siRNA against a collagen‐specific chaperone. Nat Biotechnol. 2008;26(4):431‐442. 10.1038/nbt1396 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 79. Khan OF , Zaia EW, Jhunjhunwala S, et al. Dendrimer‐Inspired Nanomaterials for the in Vivo Delivery of siRNA to Lung Vasculature. Nano Lett. 2015;15(5):3008‐3016. 10.1021/nl5048972 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gao Q, Zhang X, Xiang H, Ren G, Zheng Y. Primary breast T‐cell lymphoma, unspecified, treated with autologous peripheral blood stem cell transplantation: A case report and literature review. Oncol Lett. 2014;7(1):156‐158. 10.3892/ol.2013.1676 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Osborn MF, Alterman JF, Nikan M, et al. Guanabenz (Wytensin) selectively enhances uptake and efficacy of hydrophobically modified siRNAs. Nucleic Acids Res. 2015;43(18):8664‐8672. 10.1093/nar/gkv942 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Osborn MF, Khvorova A. Improving siRNA Delivery In Vivo Through Lipid Conjugation. Nucleic Acid Ther. 2018;28(3):128‐136. 10.1089/nat.2018.0725 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nair JK, Attarwala H, Sehgal A, et al. Impact of enhanced metabolic stability on pharmacokinetics and pharmacodynamics of GalNAc‐siRNA conjugates. Nucleic Acids Res. 2017;45(19):10969‐10977. 10.1093/nar/gkx818 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhang X, Goel V, Attarwala H, Sweetser MT, Clausen VA, Robbie GJ. Patisiran Pharmacokinetics, Pharmacodynamics, and Exposure‐Response Analyses in the Phase 3 APOLLO Trial in Patients With Hereditary Transthyretin‐Mediated (hATTR) Amyloidosis. J Clin Pharmacol. 2020;60(1):37‐49. 10.1002/jcph.1480 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhang X, Goel V, Robbie GJ. Pharmacokinetics of Patisiran, the First Approved RNA Interference Therapy in Patients With Hereditary Transthyretin‐Mediated Amyloidosis. J Clin Pharmacol. 2020;60(5):573–585. 10.1002/jcph.1553 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Egli M, Manoharan M. Re‐Engineering RNA Molecules into Therapeutic Agents. Acc Chem Res. 2019;52(4):1036‐1047. 10.1021/acs.accounts.8b00650 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 87. Brown CR, Gupta S, Qin J, et al. Investigating the pharmacodynamic durability of GalNAc‐siRNA conjugates. Nucleic Acids Res. 2020;48(21):11827–11844. 10.1093/nar/gkaa670 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Pasi KJ, Rangarajan S, Georgiev P, et al. Targeting of Antithrombin in Hemophilia A or B with RNAi Therapy. N Engl J Med. 2017;377(9):819‐828. 10.1056/NEJMoa1616569 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 89. Dammes N, Peer D. Paving the Road for RNA Therapeutics. Trends Pharmacol Sci. 2020;41(10):755‐775. 10.1016/j.tips.2020.08.004 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Adams D, Gonzalez‐Duarte A, O'Riordan WD, et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. New England Journal of Medicine. 2018;379(1):11‐21. 10.1056/NEJMoa1716153 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 91. Scott LJ. Givosiran: First Approval. Drugs. 2020;80(3):335‐339. 10.1007/s40265-020-01269-0 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 92. Balwani M, Sardh E, Ventura P, et al. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N Engl J Med. 2020;382(24):2289‐2301. 10.1056/NEJMoa1913147 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 93. Rezvani I, Auerbach V. Primary Hyperoxaluria. N Engl J Med. 2013;369(22):2162‐2163. 10.1056/NEJMc1311606 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 94. Mookadam F, Smith T, Jiamsripong P, et al. Cardiac abnormalities in primary hyperoxaluria. Circ J. 2010;74(11):2403‐2409. 10.1253/circj.cj-10-0107 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias. Kidney Int. 2009;75(12):1264‐1271. 10.1038/ki.2009.32 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Cochat P, Rumsby G. Primary hyperoxaluria. N Engl J Med. 2013;369(7):649‐658. 10.1056/NEJMra1301564 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 97. Hopp K, Cogal AG, Bergstralh EJ, et al. Phenotype‐Genotype Correlations and Estimated Carrier Frequencies of Primary Hyperoxaluria. J Am Soc Nephrol. 2015;26(10):2559‐2570. 10.1681/ASN.2014070698 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Williams EL, Bockenhauer D, van't Hoff WG, et al. The enzyme 4‐hydroxy‐2‐oxoglutarate aldolase is deficient in primary hyperoxaluria type 3. Nephrol Dial Transplant. 2012;27(8):3191‐3195. 10.1093/ndt/gfs039 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 99. Harambat J, Fargue S, Acquaviva C, et al. Genotype‐phenotype correlation in primary hyperoxaluria type 1: the p.Gly170Arg AGXT mutation is associated with a better outcome. Kidney Int. 2010;77(5):443‐449. 10.1038/ki.2009.435 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 100. Mandrile G, van Woerden CS, Berchialla P, et al. Data from a large European study indicate that the outcome of primary hyperoxaluria type 1 correlates with the AGXT mutation type. Kidney Int. 2014;86(6):1197‐1204. 10.1038/ki.2014.222 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 101. Hoppe B, Danpure CJ, Rumsby G, et al. A vertical (pseudodominant) pattern of inheritance in the autosomal recessive disease primary hyperoxaluria type 1: lack of relationship between genotype, enzymic phenotype, and disease severity. Am J Kidney Dis. 1997;29(1):36‐44. 10.1016/s0272-6386(97)90006-8 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 102. Garrelfs SF, Rumsby G, Peters‐Sengers H, et al. Patients with primary hyperoxaluria type 2 have significant morbidity and require careful follow‐up. Kidney Int. 2019;96(6):1389‐1399. 10.1016/j.kint.2019.08.018 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 103. Giafi CF, Rumsby G. Kinetic analysis and tissue distribution of human D‐glycerate dehydrogenase/glyoxylate reductase and its relevance to the diagnosis of primary hyperoxaluria type 2. Ann Clin Biochem. 1998;35(Pt 1):104‐109. 10.1177/000456329803500114 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 104. Rumsby G, Sharma A, Cregeen DP, Solomon LR. Primary hyperoxaluria type 2 without l‐glycericaciduria: is the disease under‐diagnosed? Nephrology Dialysis Transplantation. 2001;16(8):1697‐1699. 10.1093/ndt/16.8.1697 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 105. Allard L, Cochat P, Leclerc A‐L, et al. Renal function can be impaired in children with primary hyperoxaluria type 3. Pediatric Nephrol. 2015;30(10):1807‐1813. 10.1007/s00467-015-3090-x [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 106. Rumsby G, Hulton SA. From pathogenesis to novel therapies in primary hyperoxaluria. Exp Opin Orphan Drugs. 2019;7(2):57‐66. 10.1080/21678707.2019.1571905 [published Online First: Epub Date]. [DOI] [Google Scholar]
- 107. Riedel TJ, Knight J, Murray MS, Milliner DS, Holmes RP, Lowther WT. 4‐Hydroxy‐2‐oxoglutarate aldolase inactivity in primary hyperoxaluria type 3 and glyoxylate reductase inhibition. Biochim Biophys Acta (BBA) ‐ Mol Basis Dis. 2012;1822(10):1544‐1552. 10.1016/j.bbadis.2012.06.014 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Clifford‐Mobley O, Hewitt L, Rumsby G. Simultaneous analysis of urinary metabolites for preliminary identification of primary hyperoxaluria. Ann Clin Biochem. 2016;53(Pt 4):485‐494. 10.1177/0004563215606158 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 109. Belostotsky R, Pitt JJ, Frishberg Y. Primary hyperoxaluria type III‐‐a model for studying perturbations in glyoxylate metabolism. J Mol Med (Berl). 2012;90(12):1497‐1504. 10.1007/s00109-012-0930-z [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 110. Borghi L, Meschi T, Amato F, Briganti A, Novarini A, Giannini A. Urinary volume, water and recurrences in idiopathic calcium nephrolithiasis: a 5‐year randomized prospective study. J Urol. 1996;155(3):839‐843. [PubMed] [Google Scholar]
- 111. Leumann E, Hoppe B. The primary hyperoxalurias. J Am Soc Nephrol. 2001;12(9):1986‐1993. [DOI] [PubMed] [Google Scholar]
- 112. Hoppe B. An update on primary hyperoxaluria. Nat Rev Nephrol. 2012;8(8):467‐475. 10.1038/nrneph.2012.113 [DOI] [PubMed] [Google Scholar]
- 113. Cochat P, Hulton SA, Acquaviva C, et al. Primary hyperoxaluria Type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant. 2012;27(5):1729‐1736. 10.1093/ndt/gfs078 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 114. Sikora P, von Unruh GE, Beck B, et al. [13C2]oxalate absorption in children with idiopathic calcium oxalate urolithiasis or primary hyperoxaluria. Kidney Int. 2008;73(10):1181‐1186. 10.1038/ki.2008.63 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 115. Knight J, Madduma‐Liyanage K, Mobley JA, Assimos DG, Holmes RP. Ascorbic acid intake and oxalate synthesis. Urolithiasis. 2016;44(4):289‐297. 10.1007/s00240-016-0868-7 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Baxmann AC, De OGMC, Heilberg IP. Effect of vitamin C supplements on urinary oxalate and pH in calcium stone‐forming patients. Kidney Int. 2003;63(3):1066‐1071. 10.1046/j.1523-1755.2003.00815.x [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 117. Leumann E, Hoppe B, Neuhaus T. Management of primary hyperoxaluria: efficacy of oral citrate administration. Pediatr Nephrol. 1993;7(2):207‐211. 10.1007/BF00864405 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 118. Marangella M, Bagnis C, Bruno M, Vitale C, Petrarulo M, Ramello A. Crystallization inhibitors in the pathophysiology and treatment of nephrolithiasis. Urol Int. 2004;72(Suppl 1):6‐10. 10.1159/000076583 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 119. Milliner DS, Eickholt JT, Bergstralh EJ, Wilson DM, Smith LH. Results of long‐term treatment with orthophosphate and pyridoxine in patients with primary hyperoxaluria. N Engl J Med. 1994;331(23):1553‐1558. 10.1056/NEJM199412083312304 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 120. Watts RW, Veall N, Purkiss P, Mansell MA, Haywood EF. The effect of pyridoxine on oxalate dynamics in three cases of primary hyperoxaluria (with glycollic aciduria). Clin Sci (Lond). 1985;69(1):87‐90. 10.1042/cs0690087 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 121. Gibbs DA, Watts RW. The action of pyridoxine in primary hyperoxaluria. Clin Sci. 1970;38(2):277‐286. 10.1042/cs0380277 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 122. Hoyer‐Kuhn H, Kohbrok S, Volland R, et al. Vitamin B6 in primary hyperoxaluria I: first prospective trial after 40 years of practice. Clin J am Soc Nephrol. 2014;9(3):468‐477. 10.2215/CJN.06820613 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Yendt ER, Cohanim M. Response to a physiologic dose of pyridoxine in type I primary hyperoxaluria. N Engl J Med. 1985;312(15):953‐957. 10.1056/NEJM198504113121504 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 124. Fargue S, Rumsby G, Danpure CJ. Multiple mechanisms of action of pyridoxine in primary hyperoxaluria type 1. Biochim Biophys Acta (BBA) ‐ Mol Basis Dis. 2013;1832(10):1776‐1783. 10.1016/j.bbadis.2013.04.010 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 125. Hammond N, Wang Y, Dimachkie MM, Barohn RJ. Nutritional neuropathies. Neurol Clin. 2013;31(2):477‐489. 10.1016/j.ncl.2013.02.002 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. van der Hoeven SM, van Woerden CS, Groothoff JW. Primary hyperoxaluria type 1, a too often missed diagnosis and potentially treatable cause of end‐stage renal disease in adults: results of the Dutch cohort. Nephrol Dial Transplant. 2012;27(10):3855‐3862. 10.1093/ndt/gfs320 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 127. Singh P, Chebib FT, Cogal AG, Gavrilov DK, Harris PC, Lieske JC. Pyridoxine Responsiveness in a Type 1 Primary Hyperoxaluria Patient With a Rare (Atypical) AGXT Gene Mutation. Kidney Int Rep. 2020;5(6):955‐958. 10.1016/j.ekir.2020.04.004 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Fargue S, Harambat J, Gagnadoux MF, et al. Effect of conservative treatment on the renal outcome of children with primary hyperoxaluria type 1. Kidney Int. 2009;76(7):767‐773. 10.1038/ki.2009.237 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 129. Illies F, Bonzel KE, Wingen AM, Latta K, Hoyer PF. Clearance and removal of oxalate in children on intensified dialysis for primary hyperoxaluria type 1. Kidney Int. 2006;70(9):1642‐1648. 10.1038/sj.ki.5001806 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 130. Harambat J, van Stralen KJ, Espinosa L, et al. Characteristics and outcomes of children with primary oxalosis requiring renal replacement therapy. Clin J Am Soc Nephrol. 2012;7(3):458‐465. 10.2215/CJN.07430711 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Dhondup T, Lorenz EC, Milliner DS, Lieske JC. Combined Liver‐Kidney Transplantation for Primary Hyperoxaluria Type 2: A Case Report. Am J Transplant. 2018;18(1):253‐257. 10.1111/ajt.14418 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Ellis SR, Hulton SA, McKiernan PJ, de Ville de Goyet J, Kelly DA. Combined liver‐kidney transplantation for primary hyperoxaluria type 1 in young children. Nephrol Dial Transplant. 2001;16(2):348‐354. 10.1093/ndt/16.2.348 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 133. Franssen C, Kema I, Eleveld D, Porte R, Heide J. Intra‐operative continuous renal replacement therapy during combined liver‐kidney transplantation in two patients with primary hyperoxaluria type 1. NDT Plus. 2015;4:113‐116. 10.1093/ndtplus/sfq216 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Dutta C, Avitahl‐Curtis N, Pursell N, et al. Inhibition of Glycolate Oxidase With Dicer‐substrate siRNA Reduces Calcium Oxalate Deposition in a Mouse Model of Primary Hyperoxaluria Type 1. Mol Ther. 2016;24(4):770‐778. 10.1038/mt.2016.4 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Liebow A, Li X, Racie T, et al. An Investigational RNAi Therapeutic Targeting Glycolate Oxidase Reduces Oxalate Production in Models of Primary Hyperoxaluria. J am Soc Nephrol: JASN. 2017;28(2):494‐503. 10.1681/ASN.2016030338 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Lai C, Pursell N, Gierut J, et al. Specific Inhibition of Hepatic Lactate Dehydrogenase Reduces Oxalate Production in Mouse Models of Primary Hyperoxaluria. Mol Ther. 2018;26(8):1983‐1995. 10.1016/j.ymthe.2018.05.016 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Wood KD, Holmes RP, Erbe D, Liebow A, Fargue S, Knight J. Reduction in urinary oxalate excretion in mouse models of Primary Hyperoxaluria by RNA interference inhibition of liver lactate dehydrogenase activity. Biochim Biophys Acta Mol Basis Dis. 2019;1865(9):2203‐2209. 10.1016/j.bbadis.2019.04.017 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Emery AE. The determination of lactate dehydrogenase isoenzymes in normal human muscle and other tissues. Biochem J. 1967;105(2):599‐604. 10.1042/bj1050599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Coenen M, Schalk G, Cochat P, et al. PHYOX3: A Long‐Term, Open‐Label Extension Trial of Nedosiran in Patients with Primary Hyperoxaluria Type 1, 2, or 3 [Abstract]. J Am Soc Nephrol. 2020;31:515. [Google Scholar]
- 140. Kanno T, Sudo K, Maekawa M, Nishimura Y, Ukita M, Fukutake K. Lactate dehydrogenase M‐subunit deficiency: a new type of hereditary exertional myopathy. Clin Chim Acta. 1988;173(1):89‐98. 10.1016/0009-8981(88)90359-2 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 141. Kanno T, Sudo K, Takeuchi I, et al. Hereditary deficiency of lactate dehydrogenase M‐subunit. Clin Chim Acta. 1980;108(2):267‐276. 10.1016/0009-8981(80)90013-3 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 142. Yoshikuni K, Tagami H, Yamada M, Sudo K, Kanno T. Erythematosquamous skin lesions in hereditary lactate dehydrogenase M‐subunit deficiency. Arch Dermatol. 1986;122(12):1420‐1424. [PubMed] [Google Scholar]
- 143. Takahashi Y, Miyajima H, Kaneko E. Genetic analysis of a family of lactate dehydrogenase A subunit deficiency. Intern Med. 1995;34(5):326‐329. 10.2169/internalmedicine.34.326 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 144. Tsujino S, Shanske S, Brownell AK, Haller RG, DiMauro S. Molecular genetic studies of muscle lactate dehydrogenase deficiency in white patients. Ann Neurol. 1994;36(4):661‐665. 10.1002/ana.410360418 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 145. Frishberg Y, Deschenes G, Cochat P, et al. Safety and efficacy study of lumasiran, an investigational RNA interference (RNAi) therapeutic, in adult and pediatric patients with primary hyperoxaluria type 1 (PH1). J Urol. 2019;201(Supplement 4):e174‐e174. [Google Scholar]
- 146. Garrelfs S, Frishberg Y, Hulton S, et al. ILLUMINATE‐A, a phase 3 study of lumasiran, an investigational RNAi therapeutic, in children and adults with primary hyperoxaluria type 1 (PH1). Nephrol Dial Transplant. 2020;35(Supplement_3). [Google Scholar]
- 147. Martin‐Higueras C, Luis‐Lima S, Salido E. Glycolate Oxidase Is a Safe and Efficient Target for Substrate Reduction Therapy in a Mouse Model of Primary Hyperoxaluria Type I. Mol Ther. 2016;24(4):719‐725. 10.1038/mt.2015.224 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. McGregor TL, Hunt KA, Yee E, et al. Characterising a healthy adult with a rare HAO1 knockout to support a therapeutic strategy for primary hyperoxaluria. eLife. 2020;9:e54363. 10.7554/eLife.54363 [published Online First: Epub Date]| [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Frishberg Y, Zeharia A, Lyakhovetsky R, Bargal R, Belostotsky R. Mutations in HAO1 encoding glycolate oxidase cause isolated glycolic aciduria. J Med Genet. 2014;51(8):526‐529. 10.1136/jmedgenet-2014-102529 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 150. Li LC, Okino ST, Zhao H, et al. Small dsRNAs induce transcriptional activation in human cells. Proc Natl Acad Sci U S a. 2006;103(46):17337‐17342. 10.1073/pnas.0607015103 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Janowski BA, Younger ST, Hardy DB, Ram R, Huffman KE, Corey DR. Activating gene expression in mammalian cells with promoter‐targeted duplex RNAs. Nat Chem Biol. 2007;3(3):166‐173. 10.1038/nchembio860 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 152. Setten RL, Lightfoot HL, Habib NA, Rossi JJ. Development of MTL‐CEBPA: Small Activating RNA Drug for Hepatocellular Carcinoma. Curr Pharm Biotechnol. 2018;19(8):611‐621. 10.2174/1389201019666180611093428 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Krutzfeldt J, Rajewsky N, Braich R, et al. Silencing of microRNAs in vivo with 'antagomirs'. Nature. 2005;438(7068):685‐689. 10.1038/nature04303 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 154. Jakobsen JC, Nielsen EE, Feinberg J, et al. Direct‐acting antivirals for chronic hepatitis C. Cochrane Database Syst Rev. 2017;9(9):CD012143‐CD012143. 10.1002/14651858.CD012143.pub3 [published Online First: Epub Date]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Woodward G, Pryke R, Hoppe B, Rumsby G. Rapid liquid chromatography tandem mass‐spectrometry screening method for urinary metabolites of primary hyperoxaluria. Ann Clin Biochem. 2019;56(2):232‐239. 10.1177/0004563218811365 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]
- 156. Garrelfs SF, Frishberg Y, Hulton SA, et al. Lumasiran, an RNAi Therapeutic for Primary Hyperoxaluria Type 1. N Engl J Med. 2021;384(13):1216‐1226. 10.1056/NEJMoa2021712 [published Online First: Epub Date]. [DOI] [PubMed] [Google Scholar]