

Abstract
Little information is available concerning the occurrence of natural transformation of bacteria in soil, the frequency of such events, and the actual role of this process on bacterial evolution. This is because few bacteria are known to possess the genes required to develop competence and because the tested bacteria are unable to reach this physiological state in situ. In this study we found that two soil bacteria, Agrobacterium tumefaciens and Pseudomonas fluorescens, can undergo transformation in soil microcosms without any specific physical or chemical treatment. Moreover, P. fluorescens produced transformants in both sterile and nonsterile soil microcosms but failed to do so in the various in vitro conditions we tested. A. tumefaciens could be transformed in vitro and in sterile soil samples. These results indicate that the number of transformable bacteria could be higher than previously thought and that these bacteria could find the conditions necessary for uptake of extracellular DNA in soil.
According to gene sequence analyses and experimental data, gene transfer by natural transformation among microorganisms is involved in bacterial evolution and is likely to occur at present, providing bacteria the flexibility to rapidly adapt to changing environmental conditions (22, 26). However, such transfers remain very difficult to detect under natural conditions, particularly in soil, indicating they occur at very low frequencies (25, 27, 29). According to PCR data, large amounts of extracellular DNA are readily found in most soils and persist for months, indicating that the turnover of naturally released extracellular DNA would be quite slow (10, 19, 23, 24, 31). Extracellular DNA is thought to persist, because soil particles, particularly clay minerals, absorb macromolecules, a process which would not totally inhibit the transforming activity of DNA (7).
The main limitations to natural transformation in soil would be related to the recipient bacteria. Relatively few bacteria have been shown to carry the genes required to develop a natural state of competence; those described belong to phylogenetically distant taxa, including gram-positive and proteobacteria species (16). However, their number could be significantly higher considering that isolates are not systematically tested for this property and that more than 99% of all soil bacteria remain uncultured in vitro. Another limitation to transformation is related to the development under natural conditions of the competence state permitting active uptake of DNA. In soil, bacteria tend to live in a state of dormancy due to prevailing oligotrophic conditions, which would not be particularly favorable for the development of competence (16, 17). For instance, Acinetobacter sp. strain BD413, which exhibits high frequencies of transformation in vitro, is unable to develop a competence state when grown in soil. Moreover, this physiological state is lost very rapidly in situ when in vitro-prepared competent cells are inoculated into soil (18).
In this study, we found that Agrobacterium tumefaciens and Pseudomonas fluorescens, two bacteria present in most soils and used as models for numerous molecular, physiological, and ecological studies, can be transformed without any physical or chemical treatment. Interestingly, the various conditions we tested did not permit P. fluorescens to reach the competence state in vitro, while transformants were detected in situ.
MATERIALS AND METHODS
Bacterial strains, culture media, growth conditions, and cell enumeration.
Bacterial strains and plasmids used in this study are listed in Table 1. Escherichia coli, A. tumefaciens, and P. fluorescens strains were grown overnight at 37°C for E. coli and 28°C for the two other bacteria in liquid Luria-Bertani (LB) medium (containing, per liter, 10 g of Bacto Tryptone, 5 g of yeast extract, and 5 g of NaCl) supplemented with the appropriate antibiotics according to the concentrations given in Table 1. Bacterium counts were determined as the number of CFU on agar-solidified LB medium incubated overnight or for 48 h when targeting E. coli or the other soil bacteria, respectively.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Resistance phenotypea | Use (reference) |
|---|---|---|
| Escherichia coli DH10B | Wild type | Donor cell |
| Pseudomonas fluorescens AK15 | Wild type | Donor cell (32) |
| Escherichia coli 1504 | Tetr (15) Spcr (60) | Recipient cell |
| Pseudomonas fluorescens LP59JG | Rifr (50) Gmr (10) | Recipient cell (11) |
| Agrobacterium tumefaciens GM19023 | Rifr (100) Smr (500) | Recipient cell |
| pKT230 (11.9 kb) | Kanr (50) Smr (100) | Transforming DNA (1) |
| pPZP111 (8.9 kb) | Kanr (25) Cmr (25) | Transforming DNA (13) |
Numbers in parentheses are antibiotic concentrations (in micrograms per milliliter).
Inoculum preparation.
Bacterium inocula were prepared by diluting an overnight culture 20-fold into fresh medium and incubating it for an additional 2-h period (to an optical density of 0.6). These cultures were then centrifuged for 10 min at 6,500 × g before resuspension in the appropriate volume of sterile water to provide a 100-fold bacterial concentration. Using pure sterile water rather than a minimal salt medium avoided modification of competence development and natural transformation.
Soil microcosms.
Microcosms consisted of 50-ml Falcon tubes in which were placed 30 g of air-dried and sieved (2-mm pore size) samples of a sandy loam soil (sand, 50%; silt, 41%; clay, 9%; organic matter, 40.6 g kg of dry soil−1; pH 5.6) collected at La Côte Saint-André (Isère, France). Sterile soil conditions were obtained by gamma-irradiating the microcosms at a dose of 25 kGy from a 60Co source (Conservatome, Dagneux, France). Moisture was adjusted to 3 g of water for each microcosm according to the following protocol: 2.5 g of pure sterile water was added initially to the 30 g of dried soil, which was left to drain naturally for 2 days at room temperature. During the next 3 days, soils were mixed manually from time to time to homogenize moisture. Soils were then seeded with 15 μg of DNA in 0.25 ml of water and 0.25 ml of the concentrated recipient strain in water to provide a soil microcosm with 0.5 μg of DNA and 5 × 109 bacteria g of dry soil−1.
In some experiments, pure plasmid DNA solutions were replaced by donor bacterial suspensions. Donor and recipient bacteria were inoculated at the same population level, with the final concentration remaining at 5 × 109 bacteria g of dry soil−1. Control experiments were systematically conducted with pure water to replace each of the two biological inocula.
Enumeration.
Following a 3-day incubation at 28°C to stabilize the population level (11), soil microcosms were treated with DNase I to degrade any persistent extracellular DNA (incubation for 3 h at 28°C with 1,000 U of DNase I in 90 ml of LB medium). Appropriate dilutions of the soil suspensions were spread on selective (Table 1) and LB media to enumerate transformants and recipient cells, respectively. Colonies were counted following a 2-day incubation at the appropriate temperature. Repetitions were performed as follows. For every experimental combination, six independent microcosms were treated separately, and each one was plated on five petri dishes. Thus, a total of 30 replicates were counted to detect transformants. Moreover, this entire procedure was repeated at least once subsequently.
Recipient cells were selected through their specific resistance to antibiotics (Table 1). The presence of plasmids in the growing colonies was confirmed by using a plasmid extraction kit (Qiagen Inc., Chatsworth, California), according to manufacturer instructions.
Identification of strains.
The PCR amplification method included the use of 1 μl of bacterial cultures as template and a hot start procedure (3 min at 95°C) to release DNA, to avoid initial mispriming and to enhance primer specificity. Primers used, pA and pH (9), were complementary to part of the 16S ribosomal gene amplifying 1.6-kb-long PCR products. Restriction enzymes were from Boehringer Mannheim (Meylan, France) and used according to manufacturer instructions before fragments were separated on agarose gels.
Recovering of transformants from soil.
We put 1, 10, 100, or 1,000 CFU of the plasmid-containing Agrobacterium or Pseudomonas into the sterile soil seeded with the stabilized plasmidless bacteria in the same way as described previously, followed by a 3-h incubation at 28°C in 90 ml of LB medium. Appropriate dilutions of the soil suspensions were spread on selective (Table 1) and LB media to enumerate plasmid-containing strains and plasmidless cells, respectively. Colonies were counted following a 2-day incubation at 28°C. Three independent microcosms were treated separately, and each was repeated subsequently. Each one was plated on five petri dishes.
In vitro transformation protocols.
Plasmids were introduced into E. coli and P. fluorescens strains according to standard electroporation techniques (8). The development of a natural competence state in P. fluorescens was tested by mixing 20 μl of 100-fold-concentrated recipient cells from overnight cultures with 0.5 μg of DNA (20 μl of a 25-μg ml−1 concentrated plasmid solution) or 20 μl of 100-fold-concentrated donor cells and deposited onto a GTTP filter (Millipore, Bedford, Massachusetts) on each of the solid media described below. After drying, the Petri dishes were incubated at 28°C for 24 h before bacteria were resuspended in 2 ml of pure sterile water and plated out on the same media under the appropriate selective conditions. Liquid conditions were also tested by mixing 40 μl of recipient cells from overnight cultures with 950 μl of each of the liquid culture media and 10 μl of a 100-μg ml−1 concentrated plasmid solution. The tubes were incubated for 24 h at 28°C with shaking before the bacteria were plated out on media with appropriate antibiotics. Additional tests included a second 10-μl inoculation of a 100-μg ml−1 concentrated plasmid solution 12 h after the first one and incubation was continued for another 12-h period. The various media we tested included (i) the standard P medium (60 mM lactic acid, 11 mM KH2PO4, 95 mM Na2HPO4, 0.81 mM MgSO4, 37 mM NH4Cl, 68 μM CaCl2, 1.8 μM FeSO4, and 1 ml of trace element liter−1) (21) and derived media, each with a specific deficiency obtained by decreasing the concentration of the corresponding nutrient as follows. To obtain a carbon-limited culture, only 20 mM lactic acid was added. For the nitrogen limitation, only 3 mM NH4Cl was added together with 60 mM lactic acid. The potassium limitation was obtained by adding only 50 μM KH2PO4 and 30 mM Na2HPO4 together with 60 mM lactic acid (20); (ii) the MM medium [containing, per liter, KH2PO4, 3.4 g; (NH4)2SO4, 0.5 g; MgSO4 · 7H2O, 0.05 g; FeSO4 · 7H2O, 0.125 mg; thiamine, 0.25 mg; glucose, 2 g; glycerol, 4 ml]; (iii) two soil-based media: a rough soil medium consisting of 50% (wt vol−1) ground soil in sterile distilled water used as a liquid medium or solidified by adding 15 g of agar liter−1 and a mineral soil medium in which the previous soil suspension in water was centrifuged twice for 15 min at 4,500 × g before the supernatant was filtered (pore size, 0.2 μm; Millipore). The filtered solution was used directly as a liquid medium or supplemented with 15 g of agar liter−1 to be used as a solid medium. Five repetitions were carried out for each sample, repeated at least twice subsequently. Controls consisted of filters with donor or recipient strains alone.
Statistical analysis of data.
Assuming that each petri dish received roughly the same number of bacteria, each petri dish was considered to be a sampling unit, and the presence or absence of any transformant was considered to be the variable. In other words, the result was considered negative only if no transformants appeared. We calculated the probability that the in vitro results were due to a sampling effect and compared this to results of soil experiments. We assumed as a null hypothesis that the frequency of transformation and the probability of observing negative plates were the same in soil and in vitro experiments. We estimated the probability of obtaining negative plates in soil experiments and calculated the probability of observing no positive plates in the in vitro experiments under this hypothesis. We used the zero term of binomial distribution as in the most-probable-number method of Cochran (6), but using only one dilution:
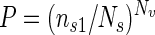 |
1 |
where ns1 is the number of negative plates in the soil experiment, Ns is the total number of plates in the soil experiment, and Nv is the total number of plates in the in vitro experiment.
For example, if we have 20 positive plates among 100 soil replicates and no positive plate among 15 in vitro replicates, the probability that this last observation results from a sampling effect is (80/100)15, or 0.035. Given that a 95% confidence interval is acceptable (P < 0.05), we can thus assume that the two sets of data were not significantly the same.
RESULTS AND DISCUSSION
Transformation of A. tumefaciens and P. fluorescens in soil.
Transformation experiments were conducted directly in soil microcosms by inoculating recipient bacteria and DNA. Detection of clones exhibiting the appropriate antibiotic resistance at frequencies significantly higher than the detection limit (number of recipient cells−1) and possessing the plasmid would indicate the ability of the tested bacteria to be naturally transformed. Because the plasmids we used were lacking the tra function and could not transfer autonomously, they could not be mobilized and could not form cointegrates with additional conjugative plasmids; the only way to transfer the marker genes to the recipient strain was via transformation. We did not detect mutants in the various DNA-less and bacterium-less controls. This would indicate that the spontaneous mutation rate remained below the detection limit. We tested the quantity of transformants (1 to 1,000 CFU) that we could recover in sterile soils containing 5 × 109 plasmidless bacteria under the same conditions as those in transformation experiments. The method we used allowed the detection of 18 ± 15 CFU g of dry soil−1 when 10 CFU was seeded in the 30-g soil microcosms thanks to the enrichment step.
In sterile soil, the total population of A. tumefaciens stabilized at 5.5 × 108 ± 2.4 × 107 cells g of dry soil−1. When the soils were inoculated with a plasmid solution at 0.5 μg of DNA g of dry soil−1, transformants exhibiting resistance to specific antibiotics and carrying plasmid pPZP111 (Fig. 1) were detected at frequencies reaching 1.3 × 10−8 (Table 2), corresponding to 12 ± 15 transformants g of dry soil−1. We also carried out experiments in which the recipient bacteria were coinoculated with donor cells of P. fluorescens AK15 containing the plasmid pPZP111. In such cases, transformants were also detected (Fig. 1) at frequencies reaching 2.7 × 10−9 (Table 2), corresponding to 6.7 ± 13.3 transformants g of dry soil−1.
FIG. 1.

Confirmation of the presence of plasmid pPZP111 in A. tumefaciens GM19023 transformants. Lanes: A, smart ladder (Eurogentec); ND, nondigested profile of extracted plasmids; D, ScaI-digested profile of extracted plasmids; 1, clone from filter experiments with P. fluorescens AK15(pPZP111) as donor; 2, clone from filter experiments with pPZP111 as donor; 3 and 4, clones from experiments with P. fluorescens AK15(pPZP111) as donor in nonsterile soil; 5 and 6, clones from experiments with pPZP111 as donor in sterile soil; 7, A. tumefaciens GMI9023.
TABLE 2.
Transformation rate and detection limit of experiments conducted in sterile soil microcosms
| Donor strain or plasmid | Recipient strain | Transformation ratea | Detection limit |
|---|---|---|---|
| E. coli DH10B (pKT230) | P. fluorescens LP59JG | 5.8 × 10−8 | 9.8 × 10−9 |
| pKT230 | P. fluorescens LP59JG | 8.3 × 10−8 | 2.0 × 10−8 |
| P. fluorescens AK15 (pPZP111) | A. tumefaciens GM19023 | 2.7 × 10−9 | 2.2 × 10−9 |
| pPZP111 | A. tumefaciens GMI9023 | 1.3 × 10−8 | 6.0 × 10−9 |
Efficiencies of transformation were expressed as the number of transformants relative to that of recipient cells.
Similar experiments were conducted with P. fluorescens LP59JG, which stabilized at 2.4 × 108 ± 9.8 × 107 cells g of dry soil−1 after 4 days. When the soils were inoculated with plasmid pKT230, transformants exhibiting resistance to kanamycin and streptomycin and carrying the plasmid were detected (Fig. 2) at frequencies reaching 8.3 × 10−8, corresponding to 20 ± 16.3 transformants g of dry soil−1 (Table 2). We also carried out experiments in which the recipient bacteria were coinoculated with donor cells (E. coli containing the plasmid pKT230). In such cases, transformants were also detected (Fig. 2) at frequencies reaching 5.8 × 10−8 (Table 2), corresponding to 33.3 ± 36.5 transformants g of dry soil−1. E. coli and P. fluorescens strains were identified by comparison of restriction patterns from PCR-restriction fragment length polymorphism analysis conducted on 16S DNA with restriction enzyme AluI (data not shown).
FIG. 2.

Confirmation of the presence of plasmid pKT230 in P. fluorescens LP59JG transformants. Lanes: A, smart ladder (Eurogentec); ND, nondigested profile of extracted plasmids; D, PstI-digested profile of extracted plasmids; 1, E. coli DH10B(pKT230); 2, clone from experiments with E. coli DH10B(pKT230) as donor in sterile soil; 3, clone from experiments with E. coli DH10B(pKT230) as donor in nonsterile soil; 4 and 5, clones from experiments with pKT230 as donor in sterile soil; 6, P. fluorescens LP59JG.
In nonsterile soil samples, the recipient P. fluorescens LP59JG sustained levels up to 2.1 × 107 ± 7.3 × 106 cells g of dry soil−1 after 4 days in soil, while the E. coli donor strain dropped to 3.2 × 105 ± 1.2 × 105 cells g of dry soil−1. Among colonies growing on the selective medium (867 ± 76 cells g of dry soil−1), some were identified as P. fluorescens. This was assessed by the similarity of the 16S ribosomal restriction patterns of PCR products obtained from these clones compared to those of the recipient strain (data not shown). Moreover, these clones harbor an 11.9 kb plasmid, which exhibited the restriction patterns corresponding to the donor plasmid pKT230 (Fig. 2). The presence of indigenous soil bacteria resistant to the four selective antibiotics prevented us from determining the precise transformation rate. However, the transformation rate in nonsterile soil seems to be higher than that in sterile soils. We cannot justify this observation, but we suggest that it may be due to the presence of an organic compound in soil that is in part destroyed by soil sterilization.
Detection of transformants in vitro.
The two bacteria which exhibited evidence of transformation in soil, A. tumefaciens GMI9023 and P. fluorescens LP59JG, as well as E. coli 1504 as the negative control, were tested in vitro for their ability to take up, internalize, and express extracellular DNA.
When transformed with the plasmid pPZP111 harboring the resistance genes to kanamycin and chloramphenicol (Table 1), A. tumefaciens provided recombinant clones at frequencies reaching 6 × 10−9 (14 ± 10 transformants ml−1), while the detection limit remained at 1.6 × 10−10 (Table 3). Moreover, detection of plasmids of the expected size (11.9 kb) (Fig. 1) helped confirm that a transformation event occurred, allowing the recipient strain to take up and replicate the donor plasmid DNA.
TABLE 3.
Transformation rate and detection limit of experiments conducted in vitro
| Donor strain or plasmid | Recipient strain | Transformation ratea | Detection limit |
|---|---|---|---|
| E. coli DH10B (pKT230) | E. coli 1504 | <4.2 × 10−8 | 4.2 × 10−8 |
| pKT230 | E. coli 1504 | <5.0 × 10−8 | 5.0 × 10−8 |
| E. coli DH10B (pKT230) | P. fluorescens LP59JG | <1.3 × 10−7 | 1.3 × 10−7 |
| pKT230 | P. fluorescens LP59JG | <2.6 × 10−9 | 2.6 × 10−9 |
| P. fluorescens AK15 (pPZP111) | A. tumefaciens GMI9023 | NDb | 4.4 × 10−9 |
| pPZP111 | A. tumefaciens GM19023 | 6.0 × 10−9 | 1.6 × 10−10 |
Efficiencies of transformation were expressed as the number of transformants relative to that of recipient cells.
ND, A. tumefaciens GM19023 was not detectable when cultivated concomitantly with P. fluorescens AK15 in vitro.
On the other hand, E. coli 1504 and P. fluorescens LP59JG failed to provide recombinant clones after they were incubated with the donor DNA solution or even with donor cells. As expected, the coculture of the recipient E. coli 1504 with E. coli DH10B(pKT230) or with the pure plasmid solution failed to provide detectable transformants, because E. coli is a nonnaturally competent bacterium for which transformation requires chemical or physical treatments.
More surprisingly, when tested in vitro, P. fluorescens LP59JG also failed to provide detectable transformants. However, two explanations are possible. This could be related to a sampling effect due to the number of replicates. But according to equation 1, we found that this hypothesis was refuted. We observed 76 negative plates and 30 positive plates in the various transformation events we tested with pKT230. The negative probability was thus 76/106. As the in vitro experiment provided 15 negative plates and 0 positive plates with pKT230, P is equal to (76/106)15, or 0.0068. In the same way, transformation experiments conducted with the donor E. coli DH10B(pKT230) provided a P of (41/60)35, or 0.0000016. As P < 0.05, the test is significative, thus proving that the absence of transformant was not due to a sampling effect.
The second hypothesis concerns the way we monitored potential transformations. Conditions tested included liquid and solid LB media and plasmid solutions or living E. coli or P. fluorescens AK15 cells as sources of donor DNA. According to the detection limit, the potential transformation efficiency was thus below 2.6 × 10−9 (Table 3). However, development of competence in naturally transformable bacteria is known to depend upon the chemical composition of the medium, with requirements varying greatly. For some bacteria, specific nutrients are required to induce this physiological state, while in other cases the presence of particular substances inhibits the development of competence. For instance, media with limited carbon and energy sources or deficient in nitrogen and phosphorus have been found to favor the development of competence in Pseudomonas stutzeri (15). On the other hand, bacteria such as Acinetobacter sp. strain BD413 (14, 21) and Ralstonia solanacearum (5) were transformable in media with totally different requirements. In order to cover a wide range of conditions, we tested various media described as promoting competence development in most of the naturally transformable bacteria. This included the use of the P and the MM media favoring competence development in Acinetobacter sp. strain BD413 and R. solanacearum, respectively, but also media characterized by a specific deficiency to test whether inhibition of competence development would be related to the presence of a specific substance. Each medium was tested according to two protocols based on incubation of bacteria on agar-solidified media and in liquid. Finally, we also tested two soil-based media, including a rough medium containing the whole soil sample in water and a medium containing only the soil mineral fraction. None of the conditions tested enabled us to detect transformants, which would have indicated that P. fluorescens can develop competence in vitro. In spite of our efforts and the use of rich, soil-based or deficient media, we were unable to determine whether an essential component for transformation present in the soil would be missing in vitro or whether the media we used contained an inhibiting product. Another hypothesis could involve a rapid degradation of the transforming DNA by excretion of an active nuclease before the cells became competent, as evidenced in Bacillus subtilis (2). However, in spite of a modification of the protocol to include two DNA inoculations separated by a 12-h lag time (see Materials and Methods), transformants remained undetectable. These data confirm that bacterial physiology remains largely unknown with numerous specific limitations. In the case of P. fluorescens, these limitations prevent the development of a competence state in vitro, while for more than 99% of the indigenous microflora it is the growth of colonies on petri dishes which is inhibited.
Transformation mechanism.
A major question regarding these results is related to the mechanism by which bacteria can take up DNA. A first hypothesis deals with the presence of the molecular machinery permitting bacteria to develop a competence state. Tests should be conducted on bacteria belonging to the 99% nonculturable fraction of the soil microflora (28). Another hypothesis to explain the transformants we obtained with A. tumefaciens and P. fluorescens deals with a mechanism that would not be related to the well-defined natural transformation. E. coli transformation has been observed in various environments such as freshwater (4) and even foodstuffs (3). The naturally existing chemical (Ca2+ concentration) or physical (heat shock) parameters could provide E. coli with optimal conditions for a natural process such as transformation. Moreover, it has been hypothesized that induction of competence in E. coli would involve a physiological rather than a physicochemical mechanism (4). From our results, it can be supposed that soils, unlike other media, do not provide the required conditions for physiological or physicochemical transformation of E. coli. This could be due to the fact that E. coli is not a usual inhabitant of soils. When inoculated into nonsterile soil samples, the number of cells decreases rapidly, and so the competitive potential of E. coli becomes much lower than that of indigenous soil bacteria (23). However, the actual status leading to a physical, chemical, physiological, or genetic induction of competence in A. tumefaciens and P. fluorescens, which are natural inhabitants of soils, remains unknown. Additional experiments are necessary to determine whether the currently living strains of these bacteria are fitted with the genes involved in the development of competence, as observed in typical naturally competent bacteria, or whether they can take up DNA by other mechanisms. We can thus assume that the list of bacteria naturally capable of transformation is certainly longer than previously expected. Moreover, the fact that our findings revealed this property in well-known bacteria such as A. tumefaciens and P. fluorescens points out the interest of a more exhaustive study among available isolates, keeping in mind that development of competence was found to be species and even strain specific in bacteria (16).
Whatever the induction mechanisms operating in current bacterial strains, our results, based on experiments simulating natural conditions more closely than previous studies, demonstrate that transformation-mediated gene transfers can occur in soils. Whether such transformation-mediated gene exchange occurs at sufficiently high frequencies to contribute significantly to bacterial genome evolution remains to be investigated. However, factors such as the number of bacteria in the biosphere (5 × 1030 bacterial cells) (30), but also the time scale (3 billion years) will have to be considered carefully when tracking bacterial genome evolution and the related mechanisms.
ACKNOWLEDGMENTS
We are grateful to Stephane Peyrard for technical assistance.
This work was done as part of the Biotechnologies program supported by the Ministère Français de l'Enseignement Supérieur et de la Recherche (MENRT). S.D. and E.K. were funded by a grant from the MENRT.
REFERENCES
- 1.Bagdasarian M, Lurz R, Rückert B, Franklin F C H, Bagdasarian M M, Frey J, Timmis K N. Specific-purpose plasmid cloning vectors. II. Broad host range, high copy number, RSF1010-derived vectors, and a host-vector system for gene cloning in Pseudomonas. Gene. 1981;16:237–247. doi: 10.1016/0378-1119(81)90080-9. [DOI] [PubMed] [Google Scholar]
- 2.Basse G, Lorenz M G, Wackernagel W. A biological assay for the sensitive and quantifiable detection of extracellular microbial DNases. J Microbiol Methods. 1994;20:137–147. [Google Scholar]
- 3.Bauer F, Hertel C, Hammes W P. Transformation of Escherichia coli in foodstuffs. Syst Appl Microbiol. 1999;22:161–168. doi: 10.1016/S0723-2020(99)80061-7. [DOI] [PubMed] [Google Scholar]
- 4.Baur B, Hanselmann K, Schlimme W, Jenni B. Genetic transformation in freshwater: Escherichia coli is able to develop natural competence. Appl Environ Microbiol. 1996;62:3673–3678. doi: 10.1128/aem.62.10.3673-3678.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertolla F, van Gijsegem F, Nesme X, Simonet P. Conditions for natural transformation of Ralstonia solanacearum. Appl Environ Microbiol. 1997;63:4965–4968. doi: 10.1128/aem.63.12.4965-4968.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cochran W G. Estimation of bacterian densities by means of the most probable number. Biometrics. 1950;6:105–116. [PubMed] [Google Scholar]
- 7.Demanèche S, Jocteur-Monrozier L, Quiquampoix H, Simonet P. Evaluation of biological and physical protection against nuclease degradation of clay-bound plasmid DNA. Appl Environ Microbiol. 2001;67:293–299. doi: 10.1128/AEM.67.1.293-299.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drury L. Transformation of bacteria by electroporation. In: Harwood A, editor. Methods in molecular biology. Totowa, N.J: Humana Press Inc.; 1996. [DOI] [PubMed] [Google Scholar]
- 9.Edwards U, Rogall T, Blöcker H, Emde M, Böttger E C. Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding 16S ribosomal RNA. Nucleic Acids Res. 1989;16:7843–7853. doi: 10.1093/nar/17.19.7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gebhard F, Smalla K. Monitoring field releases of genetically modified sugar beets for persistence of transgenic plant DNA and horizontal gene transfer. FEMS Microbiol Ecol. 1999;28:261–272. [Google Scholar]
- 11.Ghiglione J F, Gourbière F, Potier P, Philippot L, Lensi R. Role of respiratory nitrate reductase in ability of Pseudomonas fluorescens YT101 to colonize the rhizosphere of maize. Appl Environ Microbiol. 2000;66:4012–4016. doi: 10.1128/aem.66.9.4012-4016.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghiglione J-F, Philippot L, Normand P, Lensi R, Potier P. Disruption of narG, the gene encoding the catalytic subunit of respiratory nitrate reductase, also affects nitrite respiration in Pseudomonas fluorescens YT101. J Bacteriol. 1999;181:5099–5102. doi: 10.1128/jb.181.16.5099-5102.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hajdukiewicz P, Svab Z, Maliga P. The small, versatile pPZP family of Agrobacterium binary vectors for plant transformation. Plant Mol Biol. 1994;25:989–994. doi: 10.1007/BF00014672. [DOI] [PubMed] [Google Scholar]
- 14.Lorenz M G, Reipschläger K, Wackernagel W. Plasmid transformation of naturally competent Acinetobacter calcoaceticus in non-sterile soil extract and groundwater. Arch Microbiol. 1992;157:355–360. doi: 10.1007/BF00248681. [DOI] [PubMed] [Google Scholar]
- 15.Lorenz M G, Wackernagel W. High frequency of natural genetic transformation of Pseudomonas stutzeri in soil extract supplemented with carbon/energy and phosphorus source. Appl Environ Microbiol. 1991;57:1246–1251. doi: 10.1128/aem.57.4.1246-1251.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lorenz M G, Wackernagel W. Bacterial gene transfer by natural genetic transformation in the environment. Microbiol Rev. 1994;58:563–602. doi: 10.1128/mr.58.3.563-602.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nielsen K M, Bones A M, Van Elsas J D. Induced natural transformation of Acinetobacter calcoaceticus in soil microcosms. Appl Environ Microbiol. 1997;63:3972–3977. doi: 10.1128/aem.63.10.3972-3977.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nielsen K M, van Weerelt M D M, Berg T N, Bones A M, Hagler A N, van Elsas J D. Natural transformation and availability of transforming DNA to Acinetobacter calcoaceticus in soils microcosms. Appl Environ Microbiol. 1997;63:1945–1952. doi: 10.1128/aem.63.5.1945-1952.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paget E, Lebrun M, Freyssinet G, Simonet P. The fate of recombinant plant DNA in soil. Eur J Soil Biol. 1998;34:81–88. [Google Scholar]
- 20.Palmen R, Buijsman P, Hellingwerf K J. Physiological regulation of competence induction for natural transformation in Acinetobacter calcoaceticus. Arch Microbiol. 1994;162:344–351. [Google Scholar]
- 21.Palmen R, Vosman B, Buijsman P, Breek C K D, Hellingwerf K J. Physiological characterization of natural transformation in Acinetobacter calcoaceticus. J Gen Microbiol. 1993;139:295–305. doi: 10.1099/00221287-139-2-295. [DOI] [PubMed] [Google Scholar]
- 22.Paul J H. Microbial gene transfer: an ecological perspective. J Mol Microbiol Biotechnol. 1999;1:45–50. [PubMed] [Google Scholar]
- 23.Recorbet G, Picard C, Normand P, Simonet P. Kinetics of the persistence of chromosomal DNA from genetically engineered Escherichia coli introduced into soil. Appl Environ Microbiol. 1993;59:4289–4294. doi: 10.1128/aem.59.12.4289-4294.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Romanowski G, Lorenz M G, Wackernagel W. Use of polymerase chain reaction and electroporation of Escherichia coli to monitor the persistence of extracellular plasmid DNA introduced into natural soils. Appl Environ Microbiol. 1993;59:3438–3446. doi: 10.1128/aem.59.10.3438-3446.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sikorski J, Graupner S, Lorenz M G, Wackernagel W. Natural genetic transformation of Pseudomonas stutzeri in a non-sterile soil. Microbiology. 1998;144:569–576. doi: 10.1099/00221287-144-2-569. [DOI] [PubMed] [Google Scholar]
- 26.Smith M W, Feng D-F, Doolittle R F. Evolution by acquisition: the case for horizontal gene transfers. Trends Biochem Sci. 1992;17:489–493. doi: 10.1016/0968-0004(92)90335-7. [DOI] [PubMed] [Google Scholar]
- 27.Stotzky G, Gallori E, Khanna M. Transformation in soil. In: Akkermans A D L, van Elsas J D, de Bruijn F J, editors. Molecular microbial ecology manual. Dordrecht, The Netherlands: Kluwer Academic Publishers; 1996. [Google Scholar]
- 28.Strätz M, Mau M, Timmis K N. System to study horizontal gene exchange among microorganisms without cultivation of recipients. Mol Microbiol. 1996;22:207–215. doi: 10.1046/j.1365-2958.1996.00099.x. [DOI] [PubMed] [Google Scholar]
- 29.Trevors J T. DNA in soil: adsorption, genetic transformation, molecular evolution and genetic microchip. Antonie Leeuwenhoek. 1996;70:1–10. doi: 10.1007/BF00393564. [DOI] [PubMed] [Google Scholar]
- 30.Whitman W B, Coleman D C, Wiebe W J. Prokaryotes: the unseen majority. Proc Natl Acad Sci USA. 1998;95:6578–6583. doi: 10.1073/pnas.95.12.6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Widmer F, Seidler R J, Watrud L S. Sensitive detection of transgenic plant marker gene persistence in soil microcosms. Mol Ecol. 1996;5:603–613. [Google Scholar]
- 32.Ye R W, Arunakumari A, Averill B A, Tiedje J M. Mutants of Pseudomonas fluorescens deficient in dissimilatory nitrite reduction are also altered in nitric oxide reduction. J Bacteriol. 1992;174:2560–2564. doi: 10.1128/jb.174.8.2560-2564.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]