

Abstract
DNA hybridization‐capture techniques allow researchers to focus their sequencing efforts on preselected genomic regions. This feature is especially useful when analysing ancient DNA (aDNA) extracts, which are often dominated by exogenous environmental sources. Here, we assessed, for the first time, the performance of hyRAD as an inexpensive and design‐free alternative to commercial capture protocols to obtain authentic aDNA data from osseous remains. HyRAD relies on double enzymatic restriction of fresh DNA extracts to produce RNA probes that cover only a fraction of the genome and can serve as baits for capturing homologous fragments from aDNA libraries. We found that this approach could retrieve sequence data from horse remains coming from a range of preservation environments, including beyond radiocarbon range, yielding up to 146.5‐fold on‐target enrichment for aDNA extracts showing extremely low endogenous content (<1%). Performance was, however, more limited for those samples already characterized by good DNA preservation (>20%–30%), while the fraction of endogenous reads mapping on‐ and off‐target was relatively insensitive to the original endogenous DNA content. Procedures based on two instead of a single round of capture increased on‐target coverage up to 3.6‐fold. Additionally, we used methylation‐sensitive restriction enzymes to produce probes targeting hypomethylated regions, which improved data quality by reducing post‐mortem DNA damage and mapping within multicopy regions. Finally, we developed a fully automated hyRAD protocol utilizing inexpensive robotic platforms to facilitate capture processing. Overall, our work establishes hyRAD as a cost‐effective strategy to recover a set of shared orthologous variants across multiple ancient samples.
1. INTRODUCTION
In the last 15 years, high‐throughput DNA sequencing (HTS) has found many applications in the genetic characterization of present‐day biodiversity. HTS has also been extensively used to characterize past plant and animal communities, utilizing ancient DNA (aDNA) preserved both in sediments and in subfossilized tissues (Leonardi et al., 2016; Orlando & Cooper, 2014). For example, aDNA metabarcoding has helped identify ecological shifts in relation to climatic change (Pedersen et al., 2016; Willerslev et al., 2014) and human activities, including pastoralism (Giguet‐Covex et al., 2014), despite limitations due to the extensively degraded nature of aDNA (Dabney et al., 2013). While massive parallel sequencing of short DNA fragments (Goodwin et al., 2016) has unveiled the full genome sequence of extinct organisms (even beyond the 1 million time range; van der Valk et al., 2021), aDNA libraries are frequently dominated by environmental microbial sources (Prüfer et al., 2010). Therefore, shotgun sequencing often provides a cost‐ineffective strategy for characterizing past sequence variation at the genome scale.
In contrast to shotgun‐sequencing, genome reduction approaches (sensu McCormack et al., 2013), including hybridization‐based target‐enrichment techniques (Kozarewa et al., 2015; Mamanova et al., 2010), are designed to focus sequencing efforts on a fraction of the genome only. These approaches not only reduce analytical costs but also maximize the chances of identifying DNA present even in limited abundance (Slon et al., 2017). Therefore, target‐enrichment provides an increasingly popular strategy for characterizing past biodiversity at both the community (Slon et al., 2017) and the population (Mathieson et al., 2015) levels. The approach is also increasingly used for genetic monitoring of endangered populations for which only noninvasive material can be collected from very limited and/or heavily contaminated sources (Aylward et al., 2018; Fontsere et al., 2020).
One common limitation of hybridization‐based genome reduction techniques pertains to probe design and synthesis. Typically, nucleic acid probes are designed on the basis of existing molecular panels, which are, however, not available across all taxa, especially amongst nonmodel organisms. Probe synthesis also entails significant costs that are generally commensurate with the number of loci targeted. A number of in‐house procedures have been developed to limit probe production costs, through PCR amplification of a number of target loci (Maricic et al., 2010; Peñalba et al., 2014), or in vitro transcription when aiming at characterizing the whole genome (Carpenter et al., 2013). HyRAD technologies have been recently proposed to fill the gap between those two alternatives, allowing users to scale probe production to their research question and sequencing capacity (Schmid et al., 2017; Suchan et al., 2016). The methodology utilizes the versatility of RAD sequencing, which targets genomic regions flanking (Peterson et al., 2012) or encompassing (Baird et al., 2008) user‐selected restriction sites. In hyRAD, enzymatic restriction is first applied to fresh DNA from a set of individuals of the focal or closely related species. Digested DNA fragments are then immortalized to produce target enrichment probes on‐demand for in‐solution aDNA capture (Suchan et al., 2016).
Despite reported success on plant and animal museum specimens (Boucher et al., 2016; Crates et al., 2019; Gauthier et al., 2020; Lang et al., 2020; Linck et al., 2017; Schmid et al., 2018; Toussaint et al., 2021), the full potential of hyRAD to characterize past molecular diversity remains largely unexplored. Currently, only one study retrieved aDNA over the 1,000‐year time scale from ~7,000‐year‐old pine needles that were preserved in the lake sediments (Schmid et al., 2017). The suitability of hyRAD to (i) other preservation conditions, (ii) deeper time ranges and (iii) calcified material, such as bones, teeth and shells, which represent the dominant fraction of the fossil record is, thus, unknown.
In this study, we applied hyRAD for the first time to ancient osseous material going beyond the radiocarbon time range and spanning diverse environmental conditions (from Tunisia, Poland and Russia; Table 1). We benchmarked hyRAD protocols including one or two rounds of capture and different combinations of restriction enzymes. These included methylation‐sensitive enzymes with the aim of targeting hypomethylated genomic regions and limit the impact of post‐mortem DNA damage (Seguin‐Orlando et al., 2015; Smith et al., 2015), while diverting sequencing efforts from the most repetitive, hypermethylated, fraction of the genome (Karam et al., 2015; Larsson et al., 2013). We also developed and validated an automated protocol for targeted capture on Opentrons OT‐2 liquid‐handling robots to minimize hands‐on time and the risk of experimental errors. Overall, the procedures presented in this study successfully retrieved authentic aDNA data from material beyond the radiocarbon time range and could achieve up to 146.46‐fold enrichment of on‐target reads on DNA extracts originally showing 0.34% endogenous DNA. The approach was successful in retrieving orthologous DNA fragments from different samples, even from limited sequencing efforts.
TABLE 1.
Samples used in the study and endogenous DNA content prior to capture
| Sample name | Sample age | Site | Material | Endogenous DNA content (all/unique) |
|---|---|---|---|---|
| PLMie10 | Lusatian culture, Bronze Age/Early Iron Age (1,200–600 bce) | Miechów, Poland | Metatarsus | 0.34/0.34% |
| PLSla2 | Funnelbeaker culture, Eneolithic (3,700–3,000 bce) | Sławęcinek, Poland | Molar | 0.51/0.51% |
| PLMie8 | Found in Funnelbeaker culture feature, Eneolithic (3,700–3,000 bce) | Miechów, Poland | Metacarpus | 6.58/6.55% |
| DIV9 | Upper Palaeolithic, Late Glacial period (13,000–14,500 uncal bp) | Divnogor'ye, Voronezh Region, Russia | Molar | 27.42/1.72% |
| KB217 | Upper Palaeolithic | Medvezhiya cave, upper course of the river Pechora, Urals, Russia | Metapodium | 16.83/11.20% |
| PLMie3 | Przeworsk culture, Roman period (1st–4th century ce) | Miechów, Poland | Femur | 13.09/13.85% |
| LOG3 | Upper Palaeolithic (infinite 14C date, >52,200 uncal bp) | Hyena's Lair, Altai Republic, Russia | Metatarsus | 20.64/20.55% |
| SV2019‐22 | Iron Age, Middle Numidian period (5th–6th century bce) | Althiburos, Tunisia | Tooth | 33.84/33.66% |
| SV2019‐18 | Iron Age, Middle Numidian period (5th–6th century bce) | Althiburos, Tunisia | Tooth | 47.19/46.97% |
| PLKaz4 | Trzciniec culture, Early Bronze Age (1,900–1,200 bce) | Kazimierza Wielka, Poland | Petrous temporal bone | 51.67/51.48% |
| PLKaz1 | Trzciniec culture, Early Bronze Age (1,900–1,200 bce) | Kazimierza Wielka, Poland | Petrous temporal bone | 60.43/60.15% |
| PLKaz2 | Trzciniec culture, Early Bronze Age (1,900–1,200 bce) | Kazimierza Wielka, Poland | Petrous temporal bone | 73.78/73.59% |
bce = Before Common Era. ce = Common Era. bp = Before Present. Uncal = uncalibrated radiocarbon range.
2. METHODS
2.1. DNA extraction and sample library preparation
The samples consisted of 12 bone or tooth samples, ranging from the Late Pleistocene to the 4th century ce (Common Era), from Tunisia, Poland and Russia. They were selected following shallow shotgun sequencing to encompass almost an entire range of endogenous DNA content (0.34%–73.78%; Table 1). DNA extraction followed Gamba et al. (2016) with modifications from Fages et al. (2019). Extracted DNA was treated with USER mix (Uracil‐Specific Excision Reagent, New England Biolabs [NEB]) prior to DNA library construction, following Fages et al. (2019). A total of 14.9 µl of USER‐treated aDNA extract was used for library preparation according to the modified Meyer and Kircher (2010) protocol presented in Fages et al. (2019), including one 6‐bp external index (Orlando et al., 2013) and two 7‐bp internal indices from Rohland et al. (2015) (Figure S1a). To ensure sufficient amounts of DNA templates for hyRAD capture, each of the 12 DNA libraries was amplified in two successive rounds of PCR. First, 3 µl of library was used within a 25‐µl reaction with of 0.2 μm of IS4 primer, 0.2 μm of indexing primer (Meyer & Kircher, 2010), 0.5 µg×µl–1 of BSA (NEB), 1 U of AccuPrime Pfx polymerase (ThermoFisher Scientific) and 1× AccuPrime buffer. PCR consisted of 5 min denaturation at 95°C, followed by 11 cycles of 15 s at 95°C, 30 s at 60°C, 30 s at 68°C, and a final elongation for 5 min at 68°C. The number of PCR cycles for the second reaction was determined using qPCR and the samples were re‐amplified either in two (samples KB2017, PLMie3, LOG3, DIV9, SV2019‐22, SV2019‐18, PLKaz4, PLKaz1; used for the capture with PstI–MseI probes only) or 16 parallel PCRs (samples PLMie10, PLSla2, PLMie8, PLKaz2; used for the capture with all three types of probes and the automated capture test, see below), to obtain the required amount for capture. The second amplification round was carried out using the IS5_reamp.P5 and IS6_reamp.P7 primers (Table S1) from Meyer and Kircher (2010) and the same PCR conditions as above, except that the primer concentration was 0.4 μm and only 1 µl of DNA template was used. For each sample, PCR replicates were pooled, purified and concentrated into 10 µl (for duplicate reactions) or 80 µl (for multiplicate reactions), using MinElute columns (Qiagen).
2.2. Probe library preparation and probe production
Probe production was based on previous hyRAD protocols (Schmid et al., 2017), which rely on in vitro transcription of the probe library molecules containing T7 RNA polymerase promoter. Overall, high‐molecular‐weight DNA extracts (2 µg) from a modern domesticated horse were subject to double enzymatic restriction before ligation to two adapters showing terminal ends complementary to one restriction site (Figure S1b). Different combinations of restriction enzymes were selected to provide different levels of genome reduction. The PstI–MseI combination targeted 6‐bp‐ and 4‐bp‐long restriction sites, respectively, and therefore was expected to produce fewer digested fragments than MspI–MseI and HpaII–MseI combinations, which both build on 4‐bp‐long restriction sites only. The latter two combinations were also selected to target genomic regions showing different DNA methylation background. Both HpaII and MspI enzymes target the same DNA restriction site (C|CGG) but HpaII does not cleave DNA when the central CpG dinucleotide is methylated, in contrast to MspI.
Each digestion reaction used 40 U of PstI‐HF (or MspI/HpaII, NEB) and 20 U of MseI (NEB) in the CutSmart buffer for 3 hr at 37°C in 100 µl. The reaction was purified using AMPure beads (Beckman Coulter), with a bead‐to‐liquid ratio of 2:1 and eluted in 20 µl of Tris 10 mm. The purified DNA was then used in a 3‐hr ligation reaction at 16°C with 800 U of T4 DNA ligase (NEB) and 0.7 μm of each adapter (P1 and P2, see Table S1), with a final volume of 28 µl. Following ligation and purification with AMPure beads (bead‐to‐liquid ratio =1.5:1), DNA templates were selected within the 190–390‐bp size range using the Blue Pippin instrument and 2% agarose cassettes with external marker (Sage Science), which corresponds to inserts of ~100–300 bp. A further purification step allowed the elimination of those probe library constructs that did not incorporate the P2 adapter. This was achieved using the biotin group present in the P2 capture adapter and streptavidin‐coated beads (MyOne C1 Dynabeads, Invitrogen). A total of 40 µl of the beads was first washed and resuspended in 2× TEN buffer (10 mm Tris‐HCl, 1 mm EDTA, 2 m NaCl), then combined with 40 µl of DNA, incubated rotating for 15 min, separated on a magnet, washed three times with 1× TEN buffer and resuspended in 15 µl of water. Finally, the probe library constructs showing both P1 and P2 adapters were PCR‐amplified in a single reaction using KAPA HiFi HotStart ReadyMix (Roche), 0.6 μm of IS4 primer and indexing primer (Meyer & Kircher, 2010), and 7.5 µl of the bead solution obtained in the previous step. The PCR conditions were: 5 min denaturation at 95°C, followed by 14 cycles of 20 s at 98°C, 15 s at 60°C, 30 s at 72°C, and a final elongation for 5 min at 72°C. The probe library was purified using AMPure beads (bead‐to‐liquid ratio =1:1) and eluted in 20 µl of 10 mm Tris. Half of the probe library was kept for sequencing while the rest was digested with 1.5 U of MseI in a 15‐µl reaction for 3 hr at 37°C to remove the P2 adapter prior to probe production through in vitro transcription. Adapter removal was confirmed and concentration estimated by running the sample on a TapeStation 4200 instrument (Agilent). RNA probes were then synthesized following in vitro transcription using the HiScribe T7 High Yield RNA Synthesis Kit (NEB) according to the manufacturer's instructions, with 1/3 of UTP molarity as biotin‐16‐UTP (Roche), and 2 µl of the DNA template in a 40‐µl reaction, for 16 hr at 37°C. Each reaction was then subject to TurboDNAse (Thermo Fisher) treatment at 37°C for 30 min to remove remaining DNA templates and purified on an RNEasy Mini column (Qiagen) using standard procedures, except that 675 µl of ethanol was used with RNA and RTL buffer mix prior to column loading to ensure that RNA probes of all possible lengths were retained. The purified probes were eluted in 50 µl of EB buffer (Qiagen) and supplemented with 2.5 µl of SUPERase‐In (Thermo Fisher).
HyRAD capture requires the use of blocking RNA oligonucleotides that are complementary to the Illumina P5 and P7 adapters present in aDNA libraries (Figure S1a). Blocking RNA was synthesized by annealing synthetic BO.P5 and BO.P7 DNA oligonucleotides with another oligonucleotide consisting of the P7 promoter sequence (Table S1; Carpenter et al., 2013) at 50 μm concentration and using 1 µl of annealed template for in vitro transcription with the HiScribe T7 High Yield RNA Synthesis Kit (NEB). Separate transcription reactions were carried out for each blocking RNA and subject to TurboDNAse treatment and RNEasy Mini column purification, as above.
2.3. Hybridization capture
Hybridization conditions closely followed MyBaits version 3 protocol (MYcroarray; http://www.mycroarray.com/mybaits/manuals.html) and the work from Suchan et al. (2016) and Cruz‐Dávalos et al. (2017). For each reaction, 500 ng of DNA library was mixed with a blocking mix consisting of blocking RNA (0.55 μm), RNA probes (500 ng), human Cot‐1 DNA (2.3 µg) and salmon sperm DNA (2.3 µg), and denatured for 5 min at 95°C. The temperature was then lowered to 55°C for hybridization (Cruz‐Dávalos et al., 2017) and the prewarmed hybridization solution with RNA probes was added. The final hybridization reaction included 5.4× SSPE (equivalent to 0.8 m NaCl), 0.15% SDS, 5.25× Denhardt's solution, 0.9 U of SUPERase‐In (Thermo Fisher) and 13 mm EDTA (including the one present in the SSPE buffer). The 55°C incubation step was carried out for 40 hr during the first round of capture but only for 16 hr during the second round, as the first round already considerably reduced the proportion of nonfocal library material (Figure 1).
FIGURE 1.
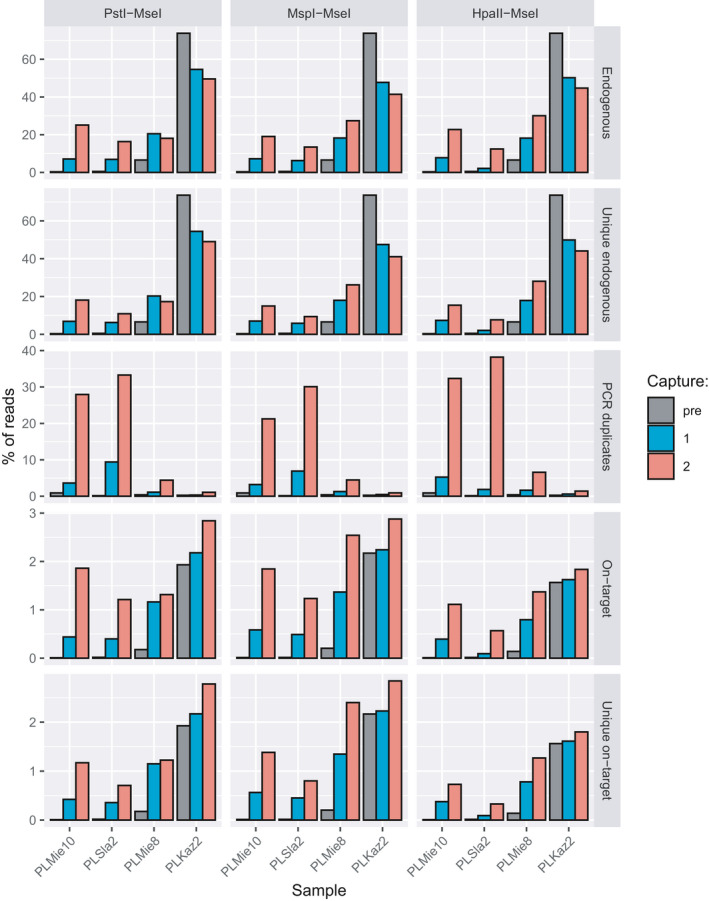
Percentage of endogenous, unique endogenous (after removing PCR duplicates), reads flagged as PCR duplicates, on‐target reads (i.e., showing at least 1 nucleotide overlap with the target regions) and unique on‐target reads. Results are shown for shotgun genomic libraries (pre) and the same libraries with one (capture 1) or two rounds (capture 2) of capture with three types of hyRAD probes, filtering for a minimum mapping quality of 25 (analyses relaxing the mapping quality filter are shown in Figure S10 so as to illustrate the impact of repeated elements in the sequence data)
Next, captured DNA fragments were immobilized for 30 min at 55°C by adding 30 µl of streptavidin‐coated beads (Dynabeads C1, Thermo Fisher) resuspended in 70 µl of TEN buffer (10 mm Tris‐HCl pH 7.5, 1 mm EDTA, 1 m NaCl). Beads were separated on the magnet, resuspended and incubated for 15 min in 180 µl of 1× SSC/0.1% SDS, resuspended and incubated three times for 10 min with 180 µl of 0.1× SSC/0.1% SDS, and finally, resuspended in 30 µl of water.
All samples were subject to capture with PstI–MseI probes. Four samples (PLMie10, PLSla2, PLMie8, PLKaz2) were also captured on two additional sets of probes (MspI–MseI and HpaII–MseI). Since probe libraries contain Illumina sequencing adapters, they could be sequenced prior to in vitro transcription in order to identify the genomic locations (targets) subject to enrichment (Figure S1b). The aDNA libraries were sequenced prior to and following one (samples PLMie10, PLSla2, PLMie8 and PLKaz2) or two rounds of enrichment (all the samples). The four samples sequenced after the first and second round of capture were used to assess the impact of successive rounds of capture. Specifically, the sample libraries obtained following one round of capture were subject to either a second round of capture after re‐amplification for ~500 ng of DNA, or to sequencing following fewer PCR amplification cycles. The number of PCR cycles was determined by qPCR and ranged from 12 to 20 for preparing the second round of capture, five to 15 for sequencing after the first round of capture and three or four for sequencing after the second. PCRs were performed using KAPA HiFi HotStart ReadyMix, 7.5 µl of the beads solution after the capture and wash steps, and 0.5 μm of IS5_reamp.P5 and IS6_reamp.P7 primers (Table S1) from Meyer and Kircher (2010). All resulting sample and probe libraries were sequenced on the Illumina MiniSeq instrument, using a 2 × 80 bp High‐Output Kit.
The detailed protocol is available in the Supporting Information.
2.4. Protocol automation
The automation protocol was implemented using Opentrons OT‐2 robots (opentrons.com; also see May, 2019), equipped with magnetic and PCR modules, and includes all steps underlying hybridization capture, namely: (i) hybridization of aDNA libraries with the blocking mix, prewarming and addition of probes; (ii) incubation in the Opentrons PCR module; and (iii) prewarming of all washing buffers and performing washing by moving the samples to the Opentrons magnetic module for bead separation and buffer change, and back for incubation on the PCR module. The efficacy of the automated capture was assessed by comparison with the performance achieved manually following two‐round captures on four samples (PLMie10, PLSla2, PLMie8 and PLKaz2).
2.5. Data analysis
Paired‐end sequence data obtained from probe libraries were filtered and trimmed using cutadapt version 2.10 (Martin, 2011). We only kept those read pairs showing a fragment of the P1 adapter (consisting of the T7 promoter and the restriction cut‐site) at the beginning of the first read, and the second cut‐site at the beginning of the second read. For the P1 adapter, no more than three errors were allowed, including at best one base indel at read start. For the second adapter, full sequence match to the second cut‐site and full overlap thereof were enforced. The resulting trimmed read pairs were then aligned against the horse EquCab3 reference genome (Kalbfleisch et al., 2018) using paleomix version 1.3.2 (Schubert et al., 2014) and both bwa version 0.7.17 backtrack algorithm (Li et al., 2009) and bowtie2 version 2.3.4.1 (Langmead & Salzberg, 2012). For bowtie2, we used the mapping parameters recommended by Poullet and Orlando (2020), while default parameters were used with bwa. All read alignments were then filtered to a minimal mapping quality of 25. While read alignment was carried out using both bwa and bowtie2 with similar outcomes, we chose the latter for subsequent analyses, based on generally higher numbers of mapped collapsed reads, in line with previous work (Cahill et al., 2018; Poullet & Orlando, 2020; Table S2; Figure S2).
For DNA sequences obtained from aDNA libraries, similar procedures were used, except that (i) adapterremoval version 2.3.1 (Schubert et al., 2016) was used for read trimming and collapsing instead of cutadapt; (ii) seeding was disabled during bwa mapping (Schubert et al. 2012); (iii) rmdup_collapsed from paleomix was used for removing PCR duplicates for collapsed reads and picard tools version 2.18.0 (http://broadinstitute.github.io/picard/) for uncollapsed reads; and (4) post‐mortem DNA damage was assessed using mapdamage version 2.2.1 (Jónsson et al., 2013) and pmdtools version 0.50 (Skoglund et al., 2014).
The expected number and size of DNA fragments obtained following enzymatic digestion were estimated in silico applying fragmatic (Chafin et al., 2018) to the EquCab3.0 reference genome. Here, all three combinations of enzymatic restrictions could be investigated using two analyses only, as HpaII and MspI target the same restriction site. The preseq version 2.0.3 (Daley & Smith, 2013) functions c_curve (step size in extrapolations =10,000) and lc_extrap (maximum extrapolation =50,000,000, step size in extrapolations =10,000) were used to estimate library complexity profiles. This was applied to both probe sequence data and aDNA sequence data following one and two rounds of capture.
Comparison between the different experimental conditions was carried out by random sampling of identical numbers of reads for each given sample for all the treatments (to the lowest number of reads obtained for each sample (range = 88,013–690,062, mean = 464,500).
The following statistics were calculated for the libraries pre‐ and post‐capture, using custom scripts based on samtools version 1.10 (Li et al., 2009) and bedtools version 2.29.2 (Quinlan & Hall, 2010): the fraction of endogenous DNA (number of reads mapping to the reference genome/number of raw reads); the fraction of unique endogenous DNA; the number of PCR duplicates; the fraction of on‐target reads (reads showing at least 1‐bp overlap with probe alignments); the fraction of unique on‐target reads; the average depth‐of‐coverage for unique on‐target reads; the size distribution of the uniquely mapped fragments (based on collapsed read pairs only, as these ensure that full aDNA fragments were sequenced); the GC content of the uniquely mapped reads; and the number of on‐target sites with depth >0 (following PCR duplicate removal). Statistical significance was tested with Wilcoxon's signed‐rank‐sum test in R (R Core Team, 2021), except for the differences between automatic and manual capture and statistics regarding damage rates of reads captured with methylation‐sensitive probes, for which the sample size was too small. These latter analyses should therefore be considered as exploratory only. We have also plotted the size distribution of on‐target reads (based only on collapsed reads for precision), and analysed enrichment folds (number of reads mapped after the procedure/number of reads mapped prior to the procedure) for captures with PstI–MseI probes. These data were further analysed using generalized linear models (GLMs) with inverse Gaussian distribution and the original endogenous content, level of PCR duplicates and age of the samples as independent variables. The relationship between enrichment‐folds and read length was analysed using a GLM with Poisson distribution, stratified by sample, using the lme4 R package (Bates et al., 2015). For probe reads, additional statistics such as the %GC content, CpG content and fragment length were calculated. When probe paired‐end reads were not overlapping, GC and CpG content were calculated based on the underlying genomic region on the reference genome.
To assess the authenticity of the obtained sequences, we quantified nucleotide misincorporations rates using mapdamage (Jónsson et al., 2013) and pmd tools (Skoglund et al., 2014). For data sets obtained using MspI–MseI and HpaII–MseI probes, we also used pmd tools to quantify CpG→TpG misincorporation rates. Such transitions generally derive from post‐mortem cytosine deamination (Briggs et al., 2007). However, methylated CpG dinucleotides that are deaminated post‐mortem are protected from USER‐treatment and sequenced as TpG dinucleotides (Hanghøj et al., 2016), in contrast to unmethylated CpG dinucleotides that are converted into UpG dinucleotides and eliminated following USER‐treatment (Hanghøj et al., 2016). Therefore, CpG→TpG misincorporation rates calculated on sequencing data generated following USER‐treatment offer an opportunity to track the overall methylation levels of the genomic regions effectively enriched. We also assessed the fraction of mappable reads obtained using each probe set, and the proportion of reads flagged as multimapping to determine whether the targeted regions could represent genomic regions with nonaverage numbers of repeated elements.
For all 12 samples captured on PstI–MseI probes, genomic variants were called with freebayes version 1.2.0 (Garrison & Marth, 2012) using sequence data obtained precapture and after second the round of capture, with the following options: ‐‐hwe‐priors‐off ‐‐no‐population‐priors ‐‐genotype‐qualities ‐‐min‐base‐quality 3 ‐‐min‐mapping‐quality 25, and filtered on the genotype level with vcffilter for depth >3 and genotype quality >20 (vcflib 1.0.1; https://github.com/vcflib/vcflib). Data missingness was calculated with vcftools version 0.1.16 (Danecek et al., 2011).
3. RESULTS
3.1. Study design and probe libraries
Over the last few years, our laboratory has undertaken an extensive genomic characterization of ancient horse specimens, which has resulted in an assessment of aDNA preservation levels in over 2,000 osseous remains, spread across the last 100,000 years in North Africa and Eurasia (Fages et al., 2019; Orlando, 2020). This allowed us to select 12 aDNA extracts representing three biomolecular preservation contexts and showing an entire range of endogenous DNA (from 0.34% to 73.78%; Table 1). Target‐enrichment was applied using three panels of hyRAD probes prepared from the genomic DNA extract of a single modern horse mare that was digested by different combinations of restriction enzymes (PstI–MseI, MspI–MseI and HpaII–MseI). The experimental procedures described here allowed for the preparation of extensive amounts of RNA probes. For instance, we obtained 92, 51 and 13.74 µg of RNA from 2 µl of digested PstI–MseI, MspI–MseI and HpaII–MseI probe libraries, respectively. Digesting and using the full volume of probe libraries would, thus, theoretically allow users to perform ~400–2,700 capture reactions.
The size distribution of probe library templates was highly similar to that predicted after in silico digestion of the horse reference genome within the selected size range (Figure S4). From limited sequencing efforts (502,815 read pairs), we could estimate that 1.81% (PstI–MseI), 2.07% (MspI–MseI) and 1.55% (HpaII–MseI) of the horse genome was represented in the probe libraries (Table 2). This equates to ~21%–29% (1.55%/7.16% and 2.07%/7.16%) to 38% (1.81%/4.77%) of the maximal genome coverage expected according to in silico digestion, as sequencing was limited. preseq calculations (Figures S5 and S6) indicated that increasing sequencing efforts to ~5–10 million read pairs would be sufficient to reveal the full probe library content.
TABLE 2.
Sequence characteristics for the sequenced probes libraries (*in‐silico calculations for HpaII–MseI probes do not take methylation‐sensitivity into account)
| Enzyme combination | Raw reads | Properly paired reads | Number of multimapping properly paired reads | Number of targets | Number of targets after merging overlapping targets | Percentage of the genome targeted | Percentage of the genome predicted in silico |
|---|---|---|---|---|---|---|---|
| PstI–MseI | 502,815 | 380,495 | 43,713 | 247,716 | 246,973 | 1.81% | 4.77% |
| MspI–MseI | 502,815 | 381,803 | 55,672 | 260,579 | 257,588 | 2.07% | 7.16% |
| HpaII–MseI | 502,815 | 387,275 | 32,569 | 234,924 | 223,789 | 1.55% | 7.16%* |
HyRAD probes were found evenly distributed across all autosomes and the X chromosome (Figures S7 and S8), while no alignments were retrieved against Y‐chromosomal contigs, in line with probes originating from a mare. Additionally, mitochondrial DNA sequences were extremely rare within the probe libraries (nine for HpaII, six for MspI and absent for PstI; Table S3), as expected from the (nearly) absence of in silico‐predicted digested fragments (absent for PstI–MseI and 10 for MspI/HpaII–MseI).
3.2. Enrichment efficacy
Sequence alignments showed the hallmark of post‐mortem DNA degradation following USER‐treatment, including mild C→T misincorporation rates towards read termini (Figure S9) and fragmentation at cytosine residues (i.e., those that are deaminated into uracil after death and cleaved by USER‐treatment; Rohland et al., 2015). This indicated that the experimental procedure followed in this study succeeded in retrieving authentic aDNA sequence data.
We first assessed hyRAD performance on four specimens by comparing the sequence data obtained following shotgun sequencing and after one or two successive rounds of capture (Figure 1). HyRAD capture resulted in increased overall levels of endogenous DNA present in the three aDNA libraries characterized by low starting endogenous DNA (0.34%–6.55% precapture). This was true for all three probe types, even though these only covered 4.77%–7.16% of the horse genome. For these three samples, the number of horse DNA fragments increased dramatically following one round of enrichment (2.73–21.63‐fold for unique reads; 2.27–22.62‐fold considering PCR duplicates). Endogenous DNA content increased even further following a second round of enrichment for the two samples (on PstI–MseI probes) or three samples (for the rest of the probes) with the lowest endogenous DNA content (4.00‐ to 53.13‐fold and 4.17‐ to 73.07‐fold, for unique reads and considering PCR duplicates, respectively). Endogenous DNA content was reduced after the second capture for the PLMie8 sample on PstI–MseI probes (6.55% original endogenous DNA content), from 3.10‐ to 2.64‐fold for unique reads and 3.12‐ to 2.75‐fold considering PCR duplicates. However, using the robotic protocol, the values after the second capture were higher (4.74‐ and 4.95‐fold, respectively), pointing to the possible variability of the procedure outcome and benefits of automation (see below). Sample PLKaz2, which was characterized by the highest endogenous DNA content precapture (73.59%) stands as an exception as endogenous DNA content was reduced after one round (0.65%–0.74%) or two rounds of capture (0.56%–0.67%). Therefore, the overall benefit of the capture on endogenous DNA content was statistically not significant for the first round of capture (Wilcoxon signed‐rank‐sum test, p = .6772 for both unique and all mapped reads), nor when comparing the first and the second round of capture (p = .1099 and .2036 for all and uniquely mapped reads, respectively). This reduction was not present when reads were not filtered for mapping quality (Figure S10) and was accompanied by a dramatic decrease in mean mapping quality of the aligned reads after the first round of capture (Figure S11, Wilcoxon signed‐rank‐sum test, p = .0005), but not between the first and the second round of capture (Wilcoxon signed‐rank‐sum test, p = .6221). This suggests that capture increased the sequencing of genomic regions found in multiple copies across the genome.
HyRAD‐enriched horse DNA sequences did not map randomly across the genome but were instead preferentially located on probe regions (Figure 1), resulting in on‐target enrichment factors for unique reads of 1.03‐ to 52.88‐fold (median = 6.56‐fold; Wilcoxon signed‐rank‐sum test, p =.0005). Performing two rounds of hyRAD capture increased on‐target coverage further (1.15‐ to 146.46‐fold; median = 16.90‐fold; Wilcoxon signed‐rank‐sum test, p = .0005), despite proportions of PCR duplicates increasing (Wilcoxon signed‐rank‐sum test, p =.0010 and .0005 for the first capture round and between the first and second capture rounds, respectively). Overall, performing one capture round resulted in a 1.23‐ to 67.42‐fold increase in genomic sites covered at least once uniquely (Figure 2; Wilcoxon signed‐rank‐sum test, p = .0005). Performing two capture rounds resulted in a 1.52‐ to 173.80‐fold increase in genomic sites covered at least once uniquely (Figure 2; Wilcoxon signed‐rank‐sum test, p = .0005 both after first capture and between the first and second). This demonstrates the capacity of hyRAD capture to significantly reduce sequencing costs pertaining to the characterization of a preselected fraction of the genome in ancient samples. The expected reduction in experimental costs is two‐fold. First, sequencing costs are directly proportional to the enrichment‐fold, which is rapidly cost‐effective relative to the cost incurred per capture reaction (~15 EUR). Second, the protocol presented here for in‐house probe production costs around 120 EUR for 400–2,700 capture reactions, which outperforms commercial production for target spaces representing substantial genome fractions.
FIGURE 2.
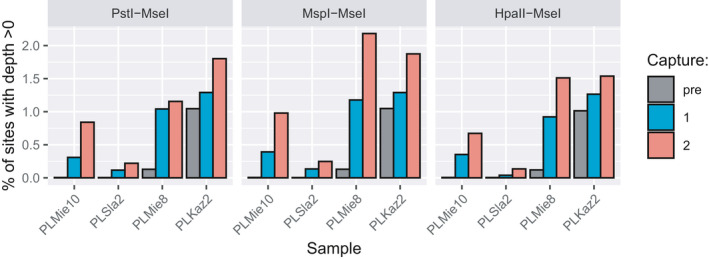
Percentage of sites with non‐null coverage, considering unique on‐target reads. Results are shown for shotgun genomic libraries (pre) and the same libraries with one (capture 1) or two rounds (capture 2) of capture with the three types of hyRAD probes
Both the size and the %GC content of the endogenous DNA molecules sequenced increased after the first round of capture (Figure 3; Wilcoxon signed‐rank‐sum test, p = .0005 and .0010, respectively) and even more after the second round (p =.0015 and .0005, respectively). This is on par with the previously reported performance of target‐enrichment using in‐solution synthetic RNA (Cruz‐Dávalos et al., 2017), with longer and %GC‐richer templates favouring probe‐to‐target annealing.
FIGURE 3.
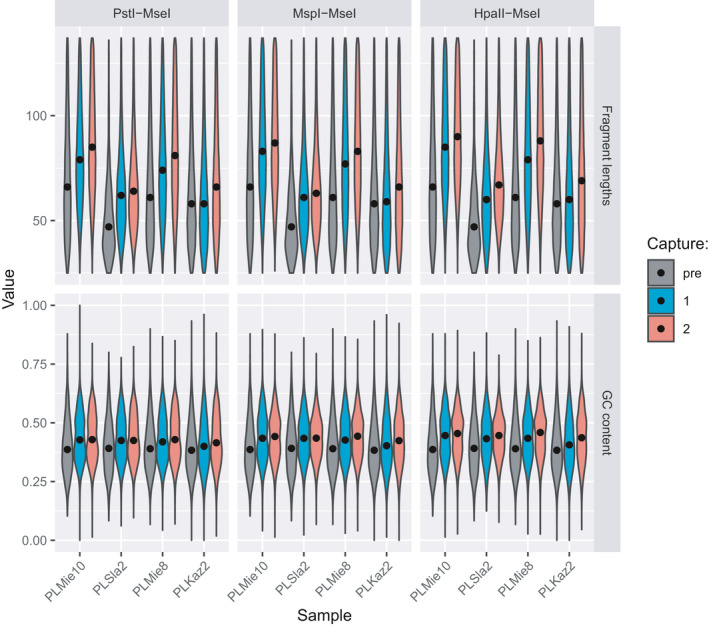
The effect of one (capture 1) or two rounds (capture 2) of capture with three types of hyRAD probes on the %GC content and sequenced fragment lengths, as compared with the shotgun libraries (pre)
We next enriched eight additional samples on PstI–MseI probes to investigate hyRAD performance across an almost continuous range of endogenous DNA preservation levels following two capture rounds (Figure 4). This experiment illustrated the trade‐off between on‐target enrichment rates and the original endogenous DNA library content (Figure 4; Table S5). It showed that hyRAD capture performs best when initial endogenous DNA content (<30%; GLM, inverse Gaussian distribution, p < .0001 for both unique mapped reads and unique on‐target reads) and initial PCR duplicate levels are low (<5.63% in our case; p = .0002 for unique mapped and .0001 for unique on‐target reads). No significant effect of sample age was supported in our model (p = .2268 for unique mapped and .2253 for unique on‐target reads). Additionally, the proportion of unique on‐target/endogenous reads was insensitive to initial endogenous DNA content precapture and sample age (GLM, inverse Gaussian distribution, p = .1327 and .8810). It was, however, sensitive to the duplication rate (p = .0367). Importantly, enrichment‐fold levels increased with the fraction of large (>50‐bp) endogenous fragments available precapture (Figure 5; GLM, Poisson distribution, stratified by sample, p < .0001). This indicates that the prominent limiting factor for aDNA hyRAD capture is the extent of DNA fragmentation ongoing after death, and hence the availability of DNA templates of sufficient size during enrichment.
FIGURE 4.
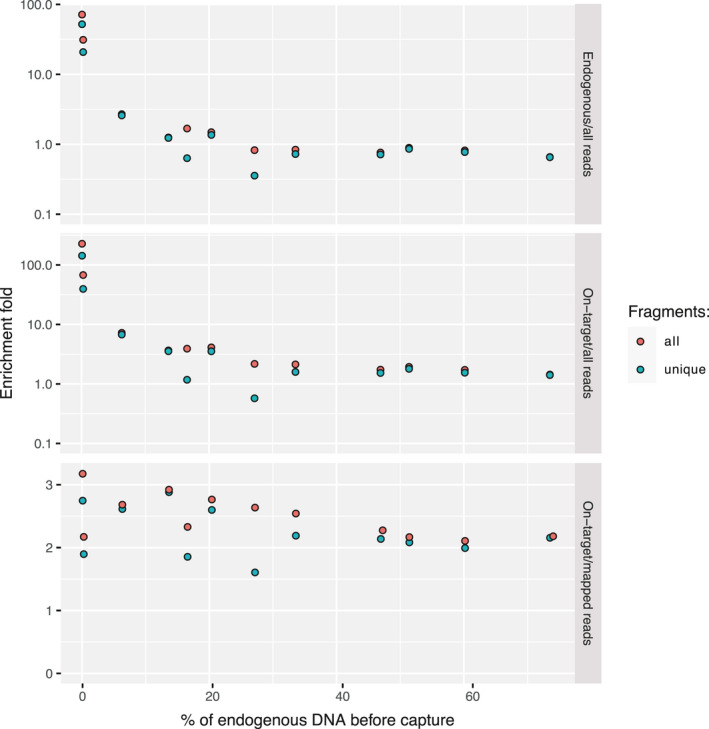
Enrichment‐fold values for 12 samples enriched in two consecutive rounds on PstI–MseI probes, for all and unique fragments (i.e., after PCR duplicate removal). The enrichment is calculated as the proportion of mapped‐to‐all reads, on‐target‐to‐all reads, and on‐target‐to‐mapped reads. Note that the two top panels are shown on log‐scale
FIGURE 5.
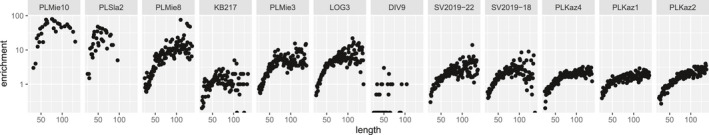
Enrichment‐fold values as a function of the fragment length for 12 samples enriched in two consecutive rounds on PstI–MseI probes
The increased on‐target coverage achieved using hyRAD translated into a higher proportion of shared genomic loci amongst samples (Figure 6a). With our limited sequencing efforts, ~100,000 sites were common to four samples following shotgun sequencing and no sites were shared across all samples. With equivalent sequencing efforts, ~100,000 sites were common to 11 samples and 2,846 were shared across all samples. This led to the identification of 87 variants, 44 of which were present in at least two samples. In contrast, prior to capture, we only detected three variants that were private to two individual samples (Figure 6b,c). The total number of variants identified per sample also increased up to 52 following two rounds of hyRAD capture (Figure 6c). This demonstrates the capacity of hyRAD capture to provide shared orthologous sequence information across multiple samples. preseq calculations showed that after the second round of capture, the number of unique horse reads present in DNA libraries could be discovered faster than after one unique round of capture (Figure S12). As such reads are also significantly more frequently found on target after the second than the first round of capture (Figure 1), this indicates the second round of capture as enriching more efficiently for those loci of interest. It follows that researchers can achieve the sequencing of the shared variation within multiple orthologous regions across many samples with lower sequencing efforts.
FIGURE 6.
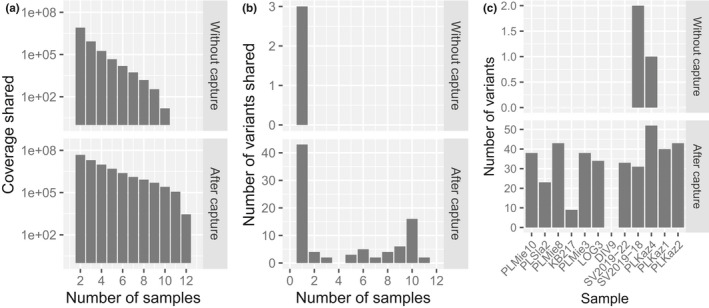
(a) Number of mapped nucleotides shared by n samples without and after two consecutive rounds on PstI–MseI probes. Note the log scale on the y‐axis. (b) Number of shared genomic variants shared by n samples without and after two consecutive rounds of capture on PstI–MseI probes. (c) Number of genomic variants detected in each sample without and after two consecutive rounds of capture on PstI–MseI probes. Note that the upper and lower plots show different scales in both (b) and (c)
Our preseq calculations can also provide guidance for making appropriate decisions on optimal sequencing efforts and, ultimately, scaling experimental costs. In particular, preseq calculations can predict how many reads mapping to the focal genome would be necessary before reaching saturation. Here, this number corresponds to a total of 20–50 million (paired‐end) reads for the vast majority of samples (Figure S12). The only exception was sample DIV9, for which the full library content was discovered when ~10 million reads were sequenced, as the amount of endogenous DNA preserved was extremely limited. We therefore suggest that a conservative starting number of 20–50 million reads are first generated before running preseq calculations and committing to further sequencing costs.
3.3. Scaling and DNA methylation sensitivity
The choice of restriction enzymes allows hyRAD users to scale their experimental design according to the question of interest and sequencing capacity. By choosing restriction enzymes that are sensitive to DNA methylation (e.g., HpaII), they can also target less repetitive hypomethylated regions (Karam et al., 2015; Larsson et al., 2013). This may be especially interesting for aDNA characterization as such regions are less prone to post‐mortem DNA damage (Smith et al. 2014, Seguin‐Orlando et al., 2015). To demonstrate this, we compared on‐target CpG→TpG misincorporation rates as a measure of post‐mortem cytosine deamination, following one and two rounds of hyRAD capture with HpaII–MseI probes (methylation‐sensitive) and MspI–MseI probes (methylation‐insensitive). These rates were indeed lower when the methylation‐sensitive combination was used (Figure 7), but not when including off‐target reads (Figure S13), confirming that the genomic fraction effectively enriched was associated with lower post‐mortem DNA damage. The number of on‐target reads flagged as multimapping was also considerably smaller when the methylation‐sensitive combination was used (Figure S14; Table 2). This was not true for off‐target reads, confirming that capturing hypomethylated regions can indeed help focus on genomic regions with reduced repetitive content.
FIGURE 7.
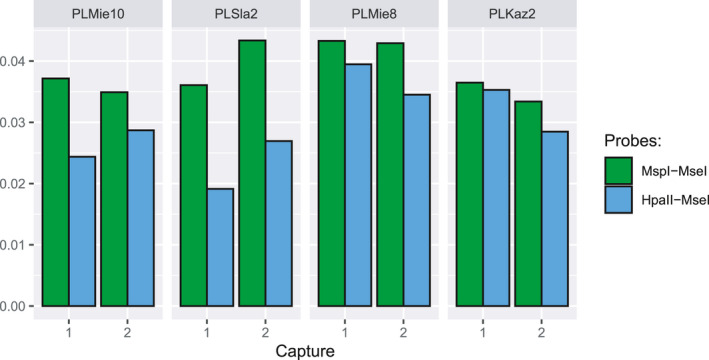
Post‐mortem DNA damage estimates of the unique on‐target reads after one and two rounds of capture on MspI–MseI vs HpaII–MseI probes. The plot shows 5′‐end cytosine deamination rates in CpG context as estimated by pmdtools and cumulated across the first 10 read positions
3.4. Protocol automation
Automated solutions to aDNA analyses have gained increasing interest over the last few years (Rohland et al., 2015; Slon et al., 2017), with the benefit of increased efficiency and reproducibility (Holland & Davies, 2020). However, investment costs for acquiring liquid‐handling robots can be extremely prohibitive and not available outside of large‐scale facilities. To provide an automation solution for most laboratories, we developed an automated hyRAD protocol that can be run on the inexpensive liquid‐handling devices produced by Opentrons. Our automated implementation reduced hands‐on time to 30 min per capture session, vs. 3 hr when carried out manually by skilled laboratory staff, while achieving similar performance (Figure 8). The developed protocol is available at https://github.com/TomaszSuchan/opentrons‐hyRAD.
FIGURE 8.
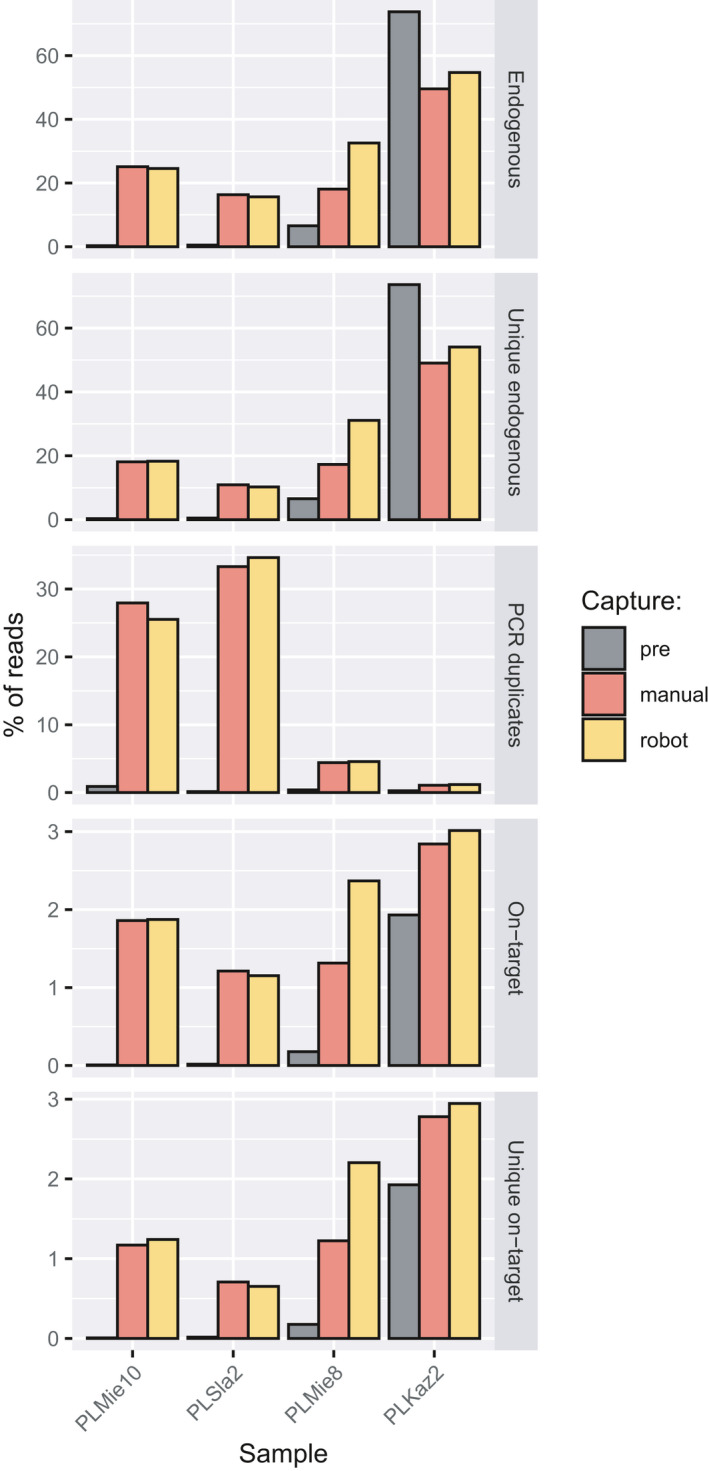
Percentage of endogenous, unique endogenous reads, reads flagged as PCR duplicates, on‐target reads, and unique on‐target reads as compared between manual and robotic capture procedures, using PstI–MseI probes
4. DISCUSSION
In this study, we investigated for the first time the potential of hyRAD target‐enrichment for aDNA extracted from osseous remains. First, we modified the original hyRAD protocol in order to produce RNA probes from the ddRAD‐seq templates, by including T7 polymerase promoter in the library adapter's sequence, as in Schmid et al. (2017). This modification has several benefits over the original approach (Suchan et al., 2016). First, using RNA probes reduces the risk of contamination of aDNA libraries by the probes during capture. Second, RNA–DNA heteroduplexes show higher affinity than DNA–DNA homoduplexes (Lesnik & Freier, 1995), thus improving capture efficacy (Furtwängler et al., 2020). Finally, the T7 polymerase reaction allowed us to obtain sufficient amounts of RNA probes to carry out thousands of capture reactions from relatively modest amounts of starting DNA. We thus solved the main limitation of the original hyRAD protocol, which required many subsequent PCR amplifications to obtain sufficient amounts of capture probes (Suchan et al., 2016).
We assessed hyRAD performance using three types of probes and one single or two rounds of enrichment. We found that the second round of enrichment consistently improved on‐target recovery of unique reads, despite also dramatically increasing the proportion of PCR duplicates. This was especially the case for samples of low endogenous DNA content, consistent with previous research (Fontsere et al., 2020; Hernandez‐Rodriguez et al., 2018). We also confirmed previous reports of limited enrichment for aDNA libraries showing high starting endogenous DNA content (Cruz‐Dávalos et al., 2017). We show that this was mainly driven by the dramatic drop of mean mapping qualities for the captured reads, probably resulting from increased proportions of repeated elements post‐capture.
On‐target enrichment reached up to 146‐fold for the sample with the lowest endogenous DNA content (0.34%) and around 3.5‐fold for samples with 13.90% and 20.64% of endogenous DNA content (not counting samples with high initial PCR duplication rates), which reduces sequencing costs proportionally. Enrichment‐fold values were, however, limited to around 1.4–1.8 for samples with >30% endogenous DNA (Figure 4; Table S5), which is lower than reported for commercial capture protocols (Cruz‐Dávalos et al., 2017). Commercial protocols, however, are based on synthetic probes, and can be carefully designed to represent optimal probe molecular features that cannot be controlled with hyRAD. Our experimental conditions were also less stringent (55°C incubation for 40 and 16 hr during the first and second rounds of enrichment, respectively), in line with the recommendations of Cruz‐Dávalos et al. (2017) on synthetic RNA probes. Further work is necessary to assess whether more stringent annealing conditions could overcome some of the limitations of our current procedure.
Our results confirm that libraries showing high initial rates of PCR duplicates following shotgun‐sequencing (samples DIV9 and KB217 in our case) perform the poorest after enrichment. This demonstrates that library complexity levels have a strong impact on capture outcomes, but that our procedure still succeeds in retrieving substantial amounts of on‐target reads even when library complexity is limited (Figure 4; Table S5).
The size distribution of horse DNA fragments increased following each round of enrichment, as previously shown for aDNA and environmental DNA (Cruz‐Dávalos et al., 2017; Enk et al., 2014). This suggests that more limited enrichment success can be expected for aDNA libraries characterized by extreme fragmentation. Therefore, we suggest that the enrichment potential of a given set of samples can be assessed prior to capture following shallow shotgun sequencing so as to gauge for both complexity levels (i.e., low duplication rates) and the presence of endogenous DNA fragments of a relatively long size (e.g., 80 bp and above).
We also demonstrated that probe preparation with methylation‐sensitive restriction enzymes could help focus sequencing efforts on hypomethylated regions, and limit both the fraction of the repetitive regions sequenced (Karam et al., 2015; Larsson et al., 2013) and the amounts of nucleotide misincorporations pertaining to post‐mortem DNA damage (Smith et al. 2014, Seguin‐Orlando et al., 2015). Therefore, the hyRAD procedure described can not only achieve a cost‐effective characterization of orthologous genomic regions across a set of samples but can also improve the underlying sequence quality. Additionally, automation of the procedure can reduce hands‐on time, and improve experimental reproducibility and traceability. Our approach appears especially appropriate for ancient samples characterized with low endogenous DNA content but not excessively fragmented. It may provide a future avenue for the genetic characterization of environmental (Wilcox et al., 2018) and noninvasive samples (e.g., faeces, Fontsere et al., 2020), which are both characterized by limited DNA amounts of the species of interest.
AUTHOR CONTRIBUTIONS
Designed and conceived the study: T.S. and L.O.. Performed wet‐laboratory work: T.S., M.K., N.K., L.C., L.T.C. and S.S. Provided samples, material and reagents: J.S., M.K., K.K., J.K., A.N.B., A.A.B., A.S.G., S.V.L., J.W., S.P., K.T., M.N., M.M.dH., A.A.T., A.J.E.P., A.K.O. and L.O. Analysed data: T.S. and L.O. Interpreted the data: T.S., L.O. Wrote the article: T.S. and L.O., with input from all co‐authors.
Supporting information
TableS1_FigS1‐S14
Table S2‐S5
ACKNOWLEDGMENTS
We thank all CAGT members for discussion and help, in particular Stefanie Wagner for initial training in the aDNA facilities. This project received funding from: the University Paul Sabatier IDEX Chaire d’Excellence (OURASI); the CNRS Programme de Recherche Conjoint (PRC); the CNRS International Research Project (IRP AMADEUS); the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska‐Curie grant agreement No. 797449; the Russian Foundation for Basic Research, project No. 19‐59‐15001 “Horses and their importance in the life of the ancient population of Altai and adjacent territories: interdisciplinary research and reconstruction”; and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement 681605).
Suchan, T. , Kusliy, M. A. , Khan, N. , Chauvey, L. , Tonasso‐Calvière, L. , Schiavinato, S. , Southon, J. , Keller, M. , Kitagawa, K. , Krause, J. , Bessudnov, A. N. , Bessudnov, A. A. , Graphodatsky, A. S. , Valenzuela‐Lamas, S. , Wilczyński, J. , Pospuła, S. , Tunia, K. , Nowak, M. , Moskal‐delHoyo, M. , … Orlando, L. (2022). Performance and automation of ancient DNA capture with RNA hyRAD probes. Molecular Ecology Resources, 22, 891–907. 10.1111/1755-0998.13518
Contributor Information
Tomasz Suchan, Email: t.suchan@botany.pl.
Ludovic Orlando, Email: ludovic.orlando@univ-tlse3.fr.
DATA AVAILABILITY STATEMENT
The data sets generated for this study can be accessed in the ENA (Accession No. PRJEB43744). The laboratory protocol is available at https://doi.org/10.17504/protocols.io.bywxpxfn. The program for piloting the Opentrons OT‐2 robot can be accessed at https://github.com/TomaszSuchan/opentrons‐hyRAD/tree/v1.0.
REFERENCES
- Aylward, M. L. , Sullivan, A. P. , Perry, G. H. , Johnson, S. E. , & Louis, E. E. (2018). An environmental DNA sampling method for aye‐ayes from their feeding traces. Ecology and Evolution, 8(18), 9229–9240. 10.1002/ece3.4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird, N. A. , Etter, P. D. , Atwood, T. S. , Currey, M. C. , Shiver, A. L. , Lewis, Z. A. , Selker, E. U. , Cresko, W. A. , & Johnson, E. A. (2008). Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One, 3(10), e3376. 10.1371/journal.pone.0003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates, D. , Mächler, M. , Bolker, B. , & Walker, S. (2015). Fitting linear mixed‐effects models using lme4. Journal of Statistical Software, 67(1), 1–48. 10.18637/jss.v067.i01 [DOI] [Google Scholar]
- Boucher, F. C. , Casazza, G. , Szövényi, P. , & Conti, E. (2016). Sequence capture using RAD probes clarifies phylogenetic relationships and species boundaries in Primula sect. Auricula. Molecular Phylogenetics and Evolution, 104, 60–72. 10.1016/j.ympev.2016.08.003. [DOI] [PubMed] [Google Scholar]
- Briggs, A. W. , Stenzel, U. , Johnson, P. L. F. , Green, R. E. , Kelso, J. , Prüfer, K. , Meyer, M. , Krause, J. , Ronan, M. T. , Lachmann, M. , & Pääbo, S. (2007). Patterns of damage in genomic DNA sequences from a Neandertal. Proceedings of the National Academy of Sciences of the United States of America, 104(37), 14616–14621. 10.1073/pnas.0704665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill, J. A. , Heintzman, P. D. , Harris, K. , Teasdale, M. D. , Kapp, J. , Soares, A. E. R. , Stirling, I. , Bradley, D. , Edwards, C. J. , Graim, K. , Kisleika, A. A. , Malev, A. V. , Monaghan, N. , Green, R. E. , & Shapiro, B. (2018). Genomic Evidence of Widespread Admixture from Polar Bears into Brown Bears during the Last Ice Age. Molecular Biology and Evolution, 35(5), 1120–1129. 10.1093/molbev/msy018. [DOI] [PubMed] [Google Scholar]
- Carpenter, M. L. , Buenrostro, J. D. , Valdiosera, C. , Schroeder, H. , Allentoft, M. E. , Sikora, M. , Rasmussen, M. , Gravel, S. , Guillén, S. , Nekhrizov, G. , Leshtakov, K. , Dimitrova, D. , Theodossiev, N. , Pettener, D. , Luiselli, D. , Sandoval, K. , Moreno‐Estrada, A. , Li, Y. , Wang, J. , … Bustamante, C. D. (2013). Pulling out the 1%: Whole‐Genome Capture for the Targeted Enrichment of Ancient DNA Sequencing Libraries. The American Journal of Human Genetics, 93(5), 852–864. 10.1016/j.ajhg.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chafin, T. K. , Martin, B. T. , Mussmann, S. M. , Douglas, M. R. , & Douglas, M. E. (2018). FRAGMATIC: In silico locus prediction and its utility in optimizing ddRADseq projects. Conservation Genetics Resources, 10(3), 325–328. 10.1007/s12686-017-0814-1. [DOI] [Google Scholar]
- Crates, R. , Olah, G. , Adamski, M. , Aitken, N. , Banks, S. , Ingwersen, D. , Ranjard, L. , Rayner, L. , Stojanovic, D. , Suchan, T. , von Takach Dukai, B. , & Heinsohn, R. (2019). Genomic impact of severe population decline in a nomadic songbird. PLoS One, 14(10), e0223953. 10.1371/journal.pone.0223953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz‐Dávalos, D. I. , Llamas, B. , Gaunitz, C. , Fages, A. , Gamba, C. , Soubrier, J. , Librado, P. , Seguin‐Orlando, A. , Pruvost, M. , Alfarhan, A. H. , Alquraishi, S. A. , Al‐Rasheid, K. A. S. , Scheu, A. , Beneke, N. , Ludwig, A. , Cooper, A. , Willerslev, E. , & Orlando, L. (2017). Experimental conditions improving in‐solution target enrichment for ancient DNA. Molecular Ecology Resources, 17(3), 508–522. 10.1111/1755-0998.12595. [DOI] [PubMed] [Google Scholar]
- Dabney, J. , Meyer, M. , & Pääbo, S. (2013). Ancient DNA damage. Cold Spring Harbor Perspectives in Biology, 5(7), a012567. 10.1101/cshperspect.a012567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley, T. , & Smith, A. D. (2013). Predicting the molecular complexity of sequencing libraries. Nature Methods, 10(4), 325–327. 10.1038/nmeth.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C. A. , Banks, E. , DePristo, M. A. , Handsaker, R. E. , Lunter, G. , Marth, G. T. , Sherry, S. T. , McVean, G. , & Durbin, R. , & 1000 Genomes Project Analysis Group (2011). The variant call format and VCFtools. Bioinformatics, 27(15), 2156–2158. 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enk, J. M. , Devault, A. M. , Kuch, M. , Murgha, Y. E. , Rouillard, J.‐M. , & Poinar, H. N. (2014). Ancient whole genome enrichment using baits built from modern DNA. Molecular Biology and Evolution, 31(5), 1292–1294. 10.1093/molbev/msu074. [DOI] [PubMed] [Google Scholar]
- Fages, A. , Hanghøj, K. , Khan, N. , Gaunitz, C. , Seguin‐Orlando, A. , Leonardi, M. , McCrory Constantz, C. , Gamba, C. , Al‐Rasheid, K. A. S. , Albizuri, S. , Alfarhan, A. H. , Allentoft, M. , Alquraishi, S. , Anthony, D. , Baimukhanov, N. , Barrett, J. H. , Bayarsaikhan, J. , Benecke, N. , Bernáldez‐Sánchez, E. , … Orlando, L. (2019). Tracking Five Millennia of Horse Management with Extensive Ancient Genome Time Series. Cell, 177(6), 1419–1435.e31. 10.1016/j.cell.2019.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontsere, C. , Alvarez‐Estape, M. , Lester, J. , Arandjelovic, M. , Kuhlwilm, M. , Dieguez, P. , Agbor, A. , Angedakin, S. , Ayuk Ayimisin, E. , Bessone, M. , Brazzola, G. , Deschner, T. , Eno‐Nku, M. , Granjon, A. , Head, J. , Kadam, P. , Kalan, A. K. , Kambi, M. , Langergraber, K. , … Lizano, E. (2020). Maximizing the acquisition of unique reads in noninvasive capture sequencing experiments. Molecular Ecology Resources, 21(3), 745–761. 10.1111/1755-0998.13300. [DOI] [PubMed] [Google Scholar]
- Furtwängler, A. , Neukamm, J. , Böhme, L. , Reiter, E. , Vollstedt, M. , Arora, N. , Singh, P. , Cole, S. T. , Knauf, S. , Calvignac‐Spencer, S. , Krause‐Kyora, B. , Krause, J. , Schuenemann, V. J. , & Herbig, A. (2020). Comparison of target enrichment strategies for ancient pathogen DNA. BioTechniques, 69(6), 455–459. 10.2144/btn-2020-0100. [DOI] [PubMed] [Google Scholar]
- Gamba, C. , Hanghøj, K. , Gaunitz, C. , Alfarhan, A. H. , Alquraishi, S. A. , Al‐Rasheid, K. A. S. , Bradley, D. G. , & Orlando, L. (2016). Comparing the performance of three ancient DNA extraction methods for high‐throughput sequencing. Molecular Ecology Resources, 16(2), 459–469. 10.1111/1755-0998.12470. [DOI] [PubMed] [Google Scholar]
- Garrison, E. , & Marth, G. (2012). Haplotype‐based variant detection from short‐read sequencing. ArXiv:1207.3907 [q‐Bio]. http://arxiv.org/abs/1207.3907 [Google Scholar]
- Gauthier, J. , Pajkovic, M. , Neuenschwander, S. , Kaila, L. , Schmid, S. , Orlando, L. , & Alvarez, N. (2020). Museomics identifies genetic erosion in two butterfly species across the 20th century in Finland. Molecular Ecology Resources, 20(5), 1191–1205. 10.1111/1755-0998.13167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giguet‐Covex, C. , Pansu, J. , Arnaud, F. , Rey, P.‐J. , Griggo, C. , Gielly, L. , Domaizon, I. , Coissac, E. , David, F. , Choler, P. , Poulenard, J. , & Taberlet, P. (2014). Long livestock farming history and human landscape shaping revealed by lake sediment DNA. Nature Communications, 5(1), 3211. 10.1038/ncomms4211. [DOI] [PubMed] [Google Scholar]
- Goodwin, S. , McPherson, J. D. , & McCombie, W. R. (2016). Coming of age: Ten years of next‐generation sequencing technologies. Nature Reviews Genetics, 17(6), 333–351. 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanghøj, K. , Seguin‐Orlando, A. , Schubert, M. , Madsen, T. , Pedersen, J. S. , Willerslev, E. , & Orlando, L. (2016). Fast, accurate and automatic ancient nucleosome and methylation maps with epiPALEOMIX. Molecular Biology and Evolution, 33(12), 3284–3298. 10.1093/molbev/msw184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez‐Rodriguez, J. , Arandjelovic, M. , Lester, J. , de Filippo, C. , Weihmann, A. , Meyer, M. , Angedakin, S. , Casals, F. , Navarro, A. , Vigilant, L. , Kühl, H. S. , Langergraber, K. , Boesch, C. , Hughes, D. , & Marques‐Bonet, T. (2018). The impact of endogenous content, replicates and pooling on genome capture from faecal samples. Molecular Ecology Resources, 18(2), 319–333. 10.1111/1755-0998.12728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland, I. , & Davies, J. A. (2020). Automation in the Life Science Research Laboratory. Frontiers in Bioengineering and Biotechnology, 8, 571777. 10.3389/fbioe.2020.571777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jónsson, H. , Ginolhac, A. , Schubert, M. , Johnson, P. L. F. , & Orlando, L. (2013). mapDamage2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics, 29(13), 1682–1684. 10.1093/bioinformatics/btt193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalbfleisch, T. S. , Rice, E. S. , DePriest, M. S. , Walenz, B. P. , Hestand, M. S. , Vermeesch, J. R. , O′Connell, B. L. , Fiddes, I. T. , Vershinina, A. O. , Saremi, N. F. , Petersen, J. L. , Finno, C. J. , Bellone, R. R. , McCue, M. E. , Brooks, S. A. , Bailey, E. , Orlando, L. , Green, R. E. , Miller, D. C. , … MacLeod, J. N. (2018). Improved reference genome for the domestic horse increases assembly contiguity and composition. Communications Biology, 1, 197. 10.1038/s42003-018-0199-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karam, M.‐J. , Lefèvre, F. , Dagher‐Kharrat, M. B. , Pinosio, S. , & Vendramin, G. G. (2015). Genomic exploration and molecular marker development in a large and complex conifer genome using RADseq and mRNAseq. Molecular Ecology Resources, 15(3), 601–612. 10.1111/1755-0998.12329. [DOI] [PubMed] [Google Scholar]
- Kozarewa, I. , Armisen, J. , Gardner, A. F. , Slatko, B. E. , & Hendrickson, C. L. (2015). Overview of Target Enrichment Strategies. Current Protocols in Molecular Biology, 112(1), 10.1002/0471142727.mb0721s112. [DOI] [PubMed] [Google Scholar]
- Lang, P. L. M. , Weiß, C. L. , Kersten, S. , Latorre, S. M. , Nagel, S. , Nickel, B. , Meyer, M. , & Burbano, H. A. (2020). Hybridization ddRAD‐sequencing for population genomics of nonmodel plants using highly degraded historical specimen DNA. Molecular Ecology Resources, 20(5), 1228–1247. 10.1111/1755-0998.13168. [DOI] [PubMed] [Google Scholar]
- Langmead, B. , & Salzberg, S. L. (2012). Fast gapped‐read alignment with Bowtie 2. Nature Methods, 9(4), 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson, H. , De Paoli, E. , Morgante, M. , Lascoux, M. , & Gyllenstrand, N. (2013). The Hypomethylated Partial Restriction (HMPR) method reduces the repetitive content of genomic libraries in Norway spruce (Picea abies). Tree Genetics & Genomes, 9(2), 601–612. 10.1007/s11295-012-0582-8. [DOI] [Google Scholar]
- Leonardi, M. , Librado, P. , Der Sarkissian, C. , Schubert, M. , Alfarhan, A. H. , Alquraishi, S. A. , Al‐Rasheid, K. A. S. , Gamba, C. , Willerslev, E. , & Orlando, L. (2016). Evolutionary Patterns and Processes: Lessons from Ancient DNA. Systematic Biology, syw059. 10.1093/sysbio/syw059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesnik, E. A. , & Freier, S. M. (1995). Relative thermodynamic stability of DNA, RNA, and DNA:RNA hybrid duplexes: Relationship with base composition and structure. Biochemistry, 34(34), 10807–10815. 10.1021/bi00034a013. [DOI] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. , & Durbin, R. ., & 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25(16), 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linck, E. B. , Hanna, Z. R. , Sellas, A. , & Dumbacher, J. P. (2017). Evaluating hybridization capture with RAD probes as a tool for museum genomics with historical bird specimens. Ecology and Evolution, 7(13), 4755–4767. 10.1002/ece3.3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamanova, L. , Coffey, A. J. , Scott, C. E. , Kozarewa, I. , Turner, E. H. , Kumar, A. , Howard, E. , Shendure, J. , & Turner, D. J. (2010). Target‐enrichment strategies for next‐generation sequencing. Nature Methods, 7(2), 111–118. 10.1038/nmeth.1419. [DOI] [PubMed] [Google Scholar]
- Maricic, T. , Whitten, M. , & Pääbo, S. (2010). Multiplexed DNA Sequence Capture of Mitochondrial Genomes Using PCR Products. PLoS One, 5(11), e14004. 10.1371/journal.pone.0014004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, M. (2011). Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.Journal, 17(1), 10. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- Mathieson, I. , Lazaridis, I. , Rohland, N. , Mallick, S. , Patterson, N. , Roodenberg, S. A. , Harney, E. , Stewardson, K. , Fernandes, D. , Novak, M. , Sirak, K. , Gamba, C. , Jones, E. R. , Llamas, B. , Dryomov, S. , Pickrell, J. , Arsuaga, J. L. , de Castro, J. M. B. , Carbonell, E. , … Reich, D. (2015). Genome‐wide patterns of selection in 230 ancient Eurasians. Nature, 528(7583), 499–503. 10.1038/nature16152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May, M. (2019). A DIY approach to automating your lab. Nature, 569(7757), 587–588. 10.1038/d41586-019-01590-z. [DOI] [PubMed] [Google Scholar]
- McCormack, J. E. , Hird, S. M. , Zellmer, A. J. , Carstens, B. C. , & Brumfield, R. T. (2013). Applications of next‐generation sequencing to phylogeography and phylogenetics. Molecular Phylogenetics and Evolution, 66(2), 526–538. 10.1016/j.ympev.2011.12.007. [DOI] [PubMed] [Google Scholar]
- Meyer, M. , & Kircher, M. (2010). Illumina Sequencing Library Preparation for Highly Multiplexed Target Capture and Sequencing. Cold Spring Harbor Protocols, 2010(6), pdb.prot5448. 10.1101/pdb.prot5448. [DOI] [PubMed] [Google Scholar]
- Orlando, L. (2020). The Evolutionary and Historical Foundation of the Modern Horse: Lessons from Ancient Genomics. Annual Review of Genetics, 54, 563–581. 10.1146/annurev-genet-021920-011805. [DOI] [PubMed] [Google Scholar]
- Orlando, L. , & Cooper, A. (2014). Using Ancient DNA to Understand Evolutionary and Ecological Processes. Annual Review of Ecology, Evolution, and Systematics, 45(1), 573–598. 10.1146/annurev-ecolsys-120213-091712. [DOI] [Google Scholar]
- Orlando, L. , Ginolhac, A. , Zhang, G. , Froese, D. , Albrechtsen, A. , Stiller, M. , Schubert, M. , Cappellini, E. , Petersen, B. , Moltke, I. , Johnson, P. L. F. , Fumagalli, M. , Vilstrup, J. T. , Raghavan, M. , Korneliussen, T. , Malaspinas, A.‐S. , Vogt, J. , Szklarczyk, D. , Kelstrup, C. D. , … Willerslev, E. (2013). Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature, 499(7456), 74–78. 10.1038/nature12323. [DOI] [PubMed] [Google Scholar]
- Pedersen, M. W. , Ruter, A. , Schweger, C. , Friebe, H. , Staff, R. A. , Kjeldsen, K. K. , Mendoza, M. L. Z. , Beaudoin, A. B. , Zutter, C. , Larsen, N. K. , Potter, B. A. , Nielsen, R. , Rainville, R. A. , Orlando, L. , Meltzer, D. J. , Kjær, K. H. , & Willerslev, E. (2016). Postglacial viability and colonization in North America’s ice‐free corridor. Nature, 537(7618), 45–49. 10.1038/nature19085. [DOI] [PubMed] [Google Scholar]
- Peñalba, J. V. , Smith, L. L. , Tonione, M. A. , Sass, C. , Hykin, S. M. , Skipwith, P. L. , McGuire, J. A. , Bowie, R. C. K. , & Moritz, C. (2014). Sequence capture using PCR‐generated probes: A cost‐effective method of targeted high‐throughput sequencing for nonmodel organisms. Molecular Ecology Resources, n/a‐n/a, 10.1111/1755-0998.12249. [DOI] [PubMed] [Google Scholar]
- Peterson, B. K. , Weber, J. N. , Kay, E. H. , Fisher, H. S. , & Hoekstra, H. E. (2012). Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non‐Model Species. PLoS One, 7(5), e37135. 10.1371/journal.pone.0037135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poullet, M. , & Orlando, L. (2020). Assessing DNA Sequence Alignment Methods for Characterizing Ancient Genomes and Methylomes. Frontiers in Ecology and Evolution, 8, 105. 10.3389/fevo.2020.00105. [DOI] [Google Scholar]
- Prüfer, K. , Stenzel, U. , Hofreiter, M. , Pääbo, S. , Kelso, J. , & Green, R. E. (2010). Computational challenges in the analysis of ancient DNA. Genome Biology, 11(5), R47. 10.1186/gb-2010-11-5-r47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan, A. R. , & Hall, I. M. (2010). BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics, 26(6), 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2021). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. https://www.R‐project.org/ [Google Scholar]
- Rohland, N. , Harney, E. , Mallick, S. , Nordenfelt, S. , & Reich, D. (2015). Partial uracil–DNA–glycosylase treatment for screening of ancient DNA. Philosophical Transactions of the Royal Society B: Biological Sciences, 370(1660), 20130624. 10.1098/rstb.2013.0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid, S. , Genevest, R. , Gobet, E. , Suchan, T. , Sperisen, C. , Tinner, W. , & Alvarez, N. (2017). HyRAD‐X, a versatile method combining exome capture and RAD sequencing to extract genomic information from ancient DNA. Methods in Ecology and Evolution, 8(10), 1374–1388. 10.1111/2041-210X.12785. [DOI] [Google Scholar]
- Schmid, S. , Neuenschwander, S. , Pitteloud, C. , Heckel, G. , Pajkovic, M. , Arlettaz, R. , & Alvarez, N. (2018). Spatial and temporal genetic dynamics of the grasshopper Oedaleus decorus revealed by museum genomics. Ecology and Evolution, 8(3), 1480–1495. 10.1002/ece3.3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert, M. , Ermini, L. , Sarkissian, C. D. , Jónsson, H. , Ginolhac, A. , Schaefer, R. , Martin, M. D. , Fernández, R. , Kircher, M. , McCue, M. , Willerslev, E. , & Orlando, L. (2014). Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using PALEOMIX. Nature Protocols, 9(5), 1056–1082. 10.1038/nprot.2014.063. [DOI] [PubMed] [Google Scholar]
- Schubert, M. , Lindgreen, S. , & Orlando, L. (2016). AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Research Notes, 9(1), 88. 10.1186/s13104-016-1900-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seguin‐Orlando, A. , Gamba, C. , Sarkissian, C. D. , Ermini, L. , Louvel, G. , Boulygina, E. , Sokolov, A. , Nedoluzhko, A. , Lorenzen, E. D. , Lopez, P. , McDonald, H. G. , Scott, E. , Tikhonov, A. , Stafford, T. W. , Alfarhan, A. H. , Alquraishi, S. A. , Al‐Rasheid, K. A. S. , Shapiro, B. , Willerslev, E. , … Orlando, L. (2015). Pros and cons of methylation‐based enrichment methods for ancient DNA. Scientific Reports, 5(1), 11826. 10.1038/srep11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoglund, P. , Northoff, B. H. , Shunkov, M. V. , Derevianko, A. P. , Pääbo, S. , Krause, J. , & Jakobsson, M. (2014). Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal. Proceedings of the National Academy of Sciences, 111(6), 2229–2234. 10.1073/pnas.1318934111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slon, V. , Hopfe, C. , Weiß, C. L. , Mafessoni, F. , de la Rasilla, M. , Lalueza‐Fox, C. , Rosas, A. , Soressi, M. , Knul, M. V. , Miller, R. , Stewart, J. R. , Derevianko, A. P. , Jacobs, Z. , Li, B. , Roberts, R. G. , Shunkov, M. V. , de Lumley, H. , Perrenoud, C. , Gušić, I. , … Meyer, M. (2017). Neandertal and Denisovan DNA from Pleistocene sediments. Science, 356(6338), 605–608. 10.1126/science.aam9695. [DOI] [PubMed] [Google Scholar]
- Smith, O. , Clapham, A. J. , Rose, P. , Liu, Y. , Wang, J. , & Allaby, R. G. (2015). Genomic methylation patterns in archaeological barley show de‐methylation as a time‐dependent diagenetic process. Scientific Reports, 4(1), 5559. 10.1038/srep05559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchan, T. , Pitteloud, C. , Gerasimova, N. S. , Kostikova, A. , Schmid, S. , Arrigo, N. , Pajkovic, M. , Ronikier, M. , & Alvarez, N. (2016). Hybridization Capture Using RAD Probes (hyRAD), a New Tool for Performing Genomic Analyses on Collection Specimens. PLoS One, 11(3), e0151651. 10.1371/journal.pone.0151651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toussaint, E. F. A. , Gauthier, J. , Bilat, J. , Gillett, C. P. D. T. , Gough, H. M. , Lundkvist, H. , Blanc, M. , Muñoz‐Ramírez, C. P. , & Alvarez, N. (2021). HyRAD‐X exome capture museomics unravels giant ground beetle evolution. Genome Biology and Evolution, 13(7),evab112, 10.1093/gbe/evab112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Valk, T. , Pečnerová, P. , Díez‐del‐Molino, D. , Bergström, A. , Oppenheimer, J. , Hartmann, S. , Xenikoudakis, G. , Thomas, J. A. , Dehasque, M. , Sağlıcan, E. , Fidan, F. R. , Barnes, I. , Liu, S. , Somel, M. , Heintzman, P. D. , Nikolskiy, P. , Shapiro, B. , Skoglund, P. , Hofreiter, M. , … Dalén, L. (2021). Million‐year‐old DNA sheds light on the genomic history of mammoths. Nature, 591(7849), 265–269. 10.1038/s41586-021-03224-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox, T. M. , Zarn, K. E. , Piggott, M. P. , Young, M. K. , McKelvey, K. S. , & Schwartz, M. K. (2018). Capture enrichment of aquatic environmental DNA: A first proof of concept. Molecular Ecology Resources, 18(6), 1392–1401. 10.1111/1755-0998.12928. [DOI] [PubMed] [Google Scholar]
- Willerslev, E. , Davison, J. , Moora, M. , Zobel, M. , Coissac, E. , Edwards, M. E. , Lorenzen, E. D. , Vestergård, M. , Gussarova, G. , Haile, J. , Craine, J. , Gielly, L. , Boessenkool, S. , Epp, L. S. , Pearman, P. B. , Cheddadi, R. , Murray, D. , Bråthen, K. A. , Yoccoz, N. , … Taberlet, P. (2014). Fifty thousand years of Arctic vegetation and megafaunal diet. Nature, 506(7486), 47–51. 10.1038/nature12921. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TableS1_FigS1‐S14
Table S2‐S5
Data Availability Statement
The data sets generated for this study can be accessed in the ENA (Accession No. PRJEB43744). The laboratory protocol is available at https://doi.org/10.17504/protocols.io.bywxpxfn. The program for piloting the Opentrons OT‐2 robot can be accessed at https://github.com/TomaszSuchan/opentrons‐hyRAD/tree/v1.0.