

Abstract
A selective mono‐N‐arylation strategy of amidines under Chan‐Lam conditions is described. During the reaction optimization phase, the isolation of a mononuclear Cu(II) complex provided unique mechanistic insight into the operation of Chan‐Lam mono‐N‐arylation. The scope of the process is demonstrated, and then applied to access the first mono‐N‐arylated analogues of pentamidine. Sub‐micromolar activity against kinetoplastid parasites was observed for several analogues with no cross‐resistance in pentamidine and diminazene‐resistant trypanosome strains and against Leishmania mexicana. A fluorescent mono‐N‐arylated pentamidine analogue revealed rapid cellular uptake, accumulating in parasite nuclei and the kinetoplasts. The DNA binding capability of the mono‐N‐arylated pentamidine series was confirmed by UV‐melt measurements using AT‐rich DNA. This work highlights the potential to use Chan‐Lam mono‐N‐arylation to develop therapeutic leads against diamidine‐resistant trypanosomiasis and leishmaniasis.
Keywords: antiparasitics, amidine, arylation, copper, medicinal chemistry
Accessing antiparasitic amidines: A selective mono‐N‐arylation strategy of amidines under Chan‐Lam conditions was used to access the first mono‐N‐arylated analogues of pentamidine. Sub‐micromolar activity against kinetoplastid parasites was observed for several analogues with no cross‐resistance in pentamidine and diminazene‐resistant trypanosome strains and against Leishmania mexicana. This work highlights the potential of Chan‐Lam mono‐N‐arylation to develop therapeutic leads against diamidine‐resistant trypanosomiasis and leishmaniasis.

Amidines are essential functional groups used throughout medicinal chemistry. [1] The versatility of this motif arises from their ability to form strong, bifurcated hydrogen bonds and electrostatic interactions with a range of hydrogen bond acceptors and conjugate bases. [2] In addition, modulating the basicity of the amidine group (pK aH∼13–14) alters the overall physicochemical properties, potency, and selectivity of amidine‐containing therapeutics. [3] A prominent exemplar of this approach is the development of the DNA‐binding antiparasitic pentamidine for the treatment of early‐stage Trypanosoma brucei gambiense‐related human African trypanosomiasis (HAT) or sleeping sickness, [4] AIDS‐related pneumocystis pneumonia, and leishmaniasis (Scheme 1a). [5] The broader importance of diamidine antiparasitics is further exemplified by their use as the main treatment for animal African trypanosomiasis (AAT), caused by the related parasite Trypanosoma congolense, which has a billion‐dollar adverse impact on emerging economies. [6]
Scheme 1.

(a) Amidines in antiparasitic agents. (b) Selective amidine mono‐N‐arylation and application to development of pentamidine antiparasitics. MGB=minor groove binder.
Poor oral bioavailability and the emergence of pentamidine‐resistant strains has spurred the exploration of new DNA‐binding analogues (Scheme 1a). [7] Modification of the essential amidine moiety is a nascent strategy to by‐pass resistance mechanisms in these parasites by modulating the affinity of diamidines for key drug transporters in T. brucei [8] [e. g., TbAT1/P2 aminopurine transporters, [9] High Affinity Pentamidine Transporter (HAPT) [10] ]. [11] However, expedient synthetic access to mono‐N‐arylated amidines has been hampered by the lack of robust synthetic methodology suitable for structure‐activity relationship profiling: [12] existing Pinner‐style methodology [13] is unsuited to this challenge due to forcing reaction conditions limiting functional group tolerance, and the lack of chemoselective control leading to competing di/tri‐N‐arylation. [14] By virtue of readily‐available starting materials, Pd‐ [15] and Cu‐catalyzed cross couplings (e. g., Ullmann,[ 3c , 16 ] Chan‐Lam [17] ) provide the opportunity to selectively access mono‐N‐functionalised amidine analogues.
Here we report a mild and chemoselective approach to exemplify amidine mono‐N‐arylation via the optimization of a Chan‐Lam strategy (Scheme 1b). We expand the substrate scope and demonstrate its application to access, for the first time, mono‐N‐arylated pentamidines as anti‐parasitic leads for treatment of HAT, AAT, and leishmaniasis. [18]
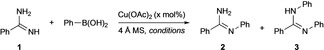
A prominent challenge of amidine functionalization is controlling mono‐ vs. di/tri‐N‐arylation.[ 17b , 19 ] Although selective amidine N‐arylation strategies have been reported, these are embedded within cascade processes alongside C−H activation events.[ 17b , 20 ] As such, the identification of reaction variables which influence mono‐ vs. di/tri‐N‐arylation was first established.
Under standard Chan‐Lam conditions, [21] a model system using benzamidine (1) and PhB(OH)2 gave 73 % overall yield and ca. 4 : 1 selectivity in favor of the mono‐ (2) vs. N,N’‐diarylated product 3 (Entry 1, Table 1). Using an excess of PhB(OH)2 led to 82 % total yield with ca. 9 : 1 selectivity for 3 (Entry 2), which suggested selective mono‐N‐arylation vs. N,N’‐diarylation is possible by controlling reagent stoichiometry. No tri‐N‐functionalization products were observed. Vantourout's B(OH)3‐based conditions were less effective (Entry 3). [22] Li's NaOPiv‐based conditions provided a high conversion and ca. 40 : 1 selectivity for 2 (Entry 4); [17b] however, further exploration of these conditions did not improve the conversion to 2 (Table S5). The observation of a pronounced base effect led to a focused screen and identification of K2CO3 as the optimal additive (Entries 5–9). This screen ultimately provided selective mono‐N‐arylation set of conditions using 20 mol% Cu(OAc)2 giving 2 in 81 % isolated yield and without any observable generation of 3 (Entry 10).
Table 1.
Reaction development.[a]
|
| ||
|---|---|---|
|
Entry |
Conditions |
2/3 (%)[b] |
|
1[c] |
Cu(OAc)2 (100 mol%), Et3N, CH2Cl2, RT, 16 h |
59/14 |
|
2[c,d] |
Cu(OAc)2 (100 mol%), Et3N, CH2Cl2, RT, 16 h |
8/74 |
|
3 |
Cu(OAc)2 (100 mol%), B(OH)3, CH2Cl2, RT, 16 h |
45/11 |
|
4[d] |
Cu(OAc)2 (100 mol%), NaOPiv, DMF, 50 °C, 16 h |
81/2 |
|
5 |
Cu(OAc)2 (100 mol%), K2CO3, CH2Cl2, RT, 16 h |
40/8 |
|
6 |
Cu(OAc)2 (100 mol%), K2CO3, MeOH, RT, 16 h |
59/2 |
|
7[c] |
Cu(OAc)2 (100 mol%), K2CO3, i‐PrOH, RT, 2 h |
83/5 |
|
8 |
Cu(OAc)2 (50 mol%), K2CO3, i‐PrOH, 50 °C, 24 h |
76/2 |
|
9 [c] |
Cu(OAc)2 (20 mol%), K2CO3 , i ‐PrOH, 50 °C, 24 h |
81/0 |
[a] Using 2 : 1 1 : PhB(OH)2 and 2 equiv. of additive unless noted. [b] Determined by HPLC analysis using standard concentration curves of 2 and 3. [c] Isolated yield on 1 mmol scale. [d] 1 : PhB(OH)2=1 : 2. [e] 1 : PhB(OH)2=1 : 1.2.
In contrast to our experience with other Chan‐Lam processes,[ 22 , 23 ] reactions using 1 and K2CO3 produced a purple precipitate within the first 5 minutes of the reaction (Figure S3). X‐ray crystallographic analysis revealed a C2‐symmetric mononuclear Cu(II) complex 4 (Scheme 2a) consisting of a bidentate carbonate ligand and two monodentate benzamidines. This unique complex suggests that K2CO3 plays a dual role in this system, acting both as a ligand and base. A dual ligand/base role has been speculated in some Cu(OAc)2‐based Chan‐Lam reactions; [24] however, structural information has been limited to the observation of a single tetranuclear Cu(II) complex. [22]
Scheme 2.

(a) Structure of Cu(II) complex 4. (b) Chan‐Lam bond formation using complex 4. (c) Proposed mechanistic origin of complex 4. S=solvent.
Exposing 4 to PhB(OH)2 in i‐PrOH afforded 2 in 61 % yield, suggesting 4 is a reaction‐relevant intermediate (Scheme 2a). The role of 4 was further investigated by EPR analysis of reaction mixtures. No EPR signals were observed when [Cu(OAc)2] ⋅ 2H2O was treated with K2CO3 or PhB(OH)2. In contrast, a paramagnetic species was observed upon addition of 1 (Figure S2). This suggests that 1 is responsible for paddlewheel denucleation, consistent with mechanistic proposals for the Chan‐Lam amination.[ 22 , 25 ] Complex 4 could form after paddlewheel denucleation via ligand exchange processes at, for example, putative Cu(II) complexes 5 and 6 (Scheme 2b).
Supplementary to our main aim of using a Chan‐Lam strategy for the late‐stage mono‐N‐arylation of pentamidine, the generality of the approach was assessed using a range of amidines and arylboronic acids, all of which exclusively formed mono‐N‐arylated products (2–29, Scheme 3a, Table S6). The use of 20 mol% Cu(OAc)2 was comparable to stoichiometric Cu(OAc)2; however, for expediency, the latter was used across the series based on the faster reaction time (see Table 1, Entries 7 and 9) with comparable yields afforded for both processes (see yields for catalytic reactions in parentheses).
Scheme 3.

(a) Scope of mono‐N‐arylation. Yields in parentheses for reactions at 50 °C for 24 h using 20 mol% Cu(OAc)2. (b) Library of monoarylated pentamidine derivatives. [a] Isolated as the corresponding TFA salt.
Both mono‐ and bisamidines are known Cu‐chelators, [26] which could hamper the wider utility of this approach to mono‐N‐arylate pentamidine for structure‐activity‐relationship (SAR) profiling. Indeed, standard Chan‐Lam conditions failed to deliver any N‐arylation of pentamidine. Gratifyingly, our reaction conditions were leveraged to directly access a series of exclusively mono‐N‐arylated pentamidines (30–36, Scheme 3b) in a single step from pentamidine.[ 5b , 27 ]
This focused series covered analogues ranging from unsubstituted (30), mono‐substituted (31–33) and disubstituted aromatics (34), a quinolone (35), and the bulky fluorescent analogue (36) suitable for cellular uptake studies.
Compounds (30–36) were then tested for activity against three kinetoplastid parasite species that are commonly treated with pentamidine (T. brucei, L. mexicana) or diminazene aceturate (T. congolense) (Table 2). [28] EC50 values ranged from 0.19 μM to 1.1 μM, with 33 exhibiting the greatest potency followed by 34. Indeed, for T. brucei, the SAR was quite flat, with quinoline (35) and biphenyl (31) analogues displaying similar EC50s to the phenyl analogue (30).
Table 2.
Activity of mono‐N‐aryl pentamidine analogues 30–36 against kinetoplastid parasites.
|
Entry |
Compound |
T. b. brucei |
T. congolense |
L. mexicana |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
EC50 [μM; n=3] |
RF |
t‐test |
EC50 [μM; n=3] |
RF |
t‐test |
EC50 [μM; n=4] |
||||
|
WT |
B48 |
WT |
DA‐Res |
|||||||
|
1 |
30 |
1.10±0.07 |
1.31±0.02 |
1.19 |
0.042 |
2.94±0.10 |
3.26±0.01 |
1.11 |
0.037 |
12.7±0.7 |
|
2 |
31 |
0.91±0.02 |
0.86±0.02 |
0.94 |
0.10 |
n.d. |
n.d. |
– |
– |
2.12±0.07 |
|
3 |
32 |
0.65±0.03 |
0.37±0.006 |
0.57 |
0.0007 |
1.26±0.03 |
1.56±0.06 |
1.23 |
0.012 |
9.71±0.56 |
|
4 |
33 |
0.19±0.003 |
0.28±0.006 |
1.47 |
0.0002 |
0.64±0.02 |
0.75±0.003 |
1.23 |
0.0008 |
0.66±0.04 |
|
5 |
34 |
0.51±0.004 |
0.51±0.02 |
1.01 |
0.82 |
n.d. |
n.d. |
– |
– |
>20 |
|
6 |
35 |
1.09±0.01 |
0.82±0.01 |
0.75 |
0.0001 |
n.d. |
n.d. |
– |
– |
>20 |
|
7 |
36 |
3.04±0.02 |
2.98±0.04 |
0.98 |
0.23 |
6.69±0.11 |
9.29±0.17 |
1.39 |
0.0002 |
7.54±0.59 |
|
8 |
PMD |
0.0024±0.0002 |
0.28±0.002 |
116 |
3.5×10−8 |
1.31±0.02 |
1.42±0.01 |
1.08 |
0.013 |
2.04±0.04 |
|
9 |
DA |
0.082±0.006 |
0.89±0.05 |
10.8 |
0.0001 |
0.27±0.008 |
1.71±0.02 |
6.4 |
2×10−7 |
n.d. |
WT=wild‐type, PMD=pentamidine, DA=diminazene aceturate, DA‐Res=diminazene‐resistant cell line, RF=resistance factor being the ratio of EC50 (resistant line) over EC50(WT). Statistical difference between WT and resistant pairs of cell lines was established using a Student unpaired two‐tailed t‐test.
The compounds were also tested against the related kinetoplastid parasites L. mexicana and T. congolense. Here, 33 displayed >2‐fold and >3‐fold higher activity compared to pentamidine against T. congolense and L. mexicana, respectively. The SAR profile was considerably less flat than for T. brucei, with 33 being 4.6‐fold and 19.2‐fold more active than 30 against T. congolense and L. mexicana, respectively.
The potential for cross‐resistance with diamidines and melaminophenyl arsenicals (MPAs; including melarsoprol, cymelarsan) is particularly problematic in the development of next‐generation amidine antiparasitics. [4] Resistance likely emerges from active drug transport [e. g., TbAT1/P2 aminopurine transporter; [29] High Affinity Pentamidine Transporter (TbAQP2)] into the parasite's interior. [30] Having studied the SAR of the transporter‐diamidine interactions, we hypothesized that mono‐N‐arylation of the pentamidine scaffold would abolish their ability to act as substrates for the T. brucei drug transporters, and therefore would not display cross‐resistance with pentamidine and MPAs. [31] To test this hypothesis, 30–36 were also tested against a T. b. brucei clonal line, B48, [32] lacking both the TbAT1/P2 and HAPT1/AQP2 transporters, and against a diminazene‐resistant T. congolense [11c] clone. Although minor differences in drug sensitivity were observed (±50 % of WT EC50), these variations were trivial compared to the resistance levels to pentamidine (116‐fold) and diminazene (6.4‐fold), respectively. We therefore conclude that mono‐N‐arylated pentamidine analogues are not cross‐resistant with un‐substituted diamidines, arising from differences in their mechanism of uptake.
To gain further insights into the uptake of the mono‐N‐arylated pentamidines, the fluorescent analogue (36) was used to monitor uptake in each of the three parasite species in real‐time (Figure 1). In all three species, 36 was taken up rapidly (3.3 μM and 10 μM), consistent with an EC50≈3–7 μM against the kinetoplastid species (Table 2). The rate of uptake of 36 was dose‐dependent and showed an approximately 2.3–10‐fold higher rate at 10 μM [36] than for 3.3 μM after 30 min (Figure 1). This shows that the uptake mechanism was not saturable in the lower μM range, unlike TbAT1/P2 and HAPT/TbAQP2, which display Km values of 0.26±0.03 and 0.036±0.06 μM, respectively, for pentamidine. [30] Moreover, the rate of uptake of 36 was not substantially different in the presence of 10 μM pentamidine (within 10 % in all cases), clearly indicating that uptake did not involve a high affinity pentamidine transporter. Taken collectively, these data show that mono‐N‐arylated analogues such as 36 evade known pentamidine transporters TbAT1 and TbAQP2 in T. brucei.
Figure 1.

Real time fluorescence development of 36 with bloodstream forms of WT T. b. brucei (A), bloodstream forms of WT T. congolense, (B) and promastigotes of L. mexicana (C). Cells were incubated for 3 h in the presence of 36 at 0, 1, 3.3 or 10 μM, in the presence or absence of 10 μM pentamidine. Measurements were taken at 2‐minute intervals. A.U., artificial units of fluorescence intensity.
One of the putative mechanisms of action of pentamidine is the inhibition of replication via binding to A/T‐rich sequences of duplex DNA. [33] Indeed, selective accumulation of 36 to the nucleus and kinetoplast of T. brucei was observed, as confirmed by co‐localization with Hoechst 33342 (Figure 2), consistent with DNA binding. UV melt analyses were undertaken to explore the ability of 33 and 36 to stabilize DNA duplexes relative to pentamidine (Table S7, Figure S4). A 7.0 °C stabilization of an A‐tract duplex was observed for 33, relative to a 6.0 °C stabilization for pentamidine. Although a reduced level of duplex stabilization was observed using 36 for the same sequence (ΔTm 4.6 °C), both analogues exhibited a similar binding bias for an A‐tract duplex relative to a duplex containing an alternating A‐T sequence. [34] These data indicate that the likely mechanism of action of these analogues is via binding to DNA duplexes with a selectivity profile similar to that observed for pentamidine. The stronger DNA binding of 33 correlated with its stronger anti‐kinetoplastid activity compared to 36.
Figure 2.

Selected immunofluorescence images of T. brucei labelled with a nuclear marker (Hoechst 33342), a dye for the mitochondrion (MitoTracker Green), and with 36 (30 min exposure, [10 μM]) or not (control). Charts show the intensity levels of the DAPI and red filter channels measured in the nuclei and kinetoplast. Scale bars are 5 μm.
In summary, we have developed a general approach to prepare mono‐N‐arylated amidines from mono‐ and bisamidine substrates based on Chan‐Lam cross‐coupling methodology. During our optimization phase, key mechanistic insights into Cu intermediates pertinent to the Chan‐Lam reaction were identified, and the procedure was broadly applicable to the formation of mono‐N‐arylated substrates. This methodology was used to directly prepare mono‐N‐arylated analogues of pentamidine, which displayed promising in vitro activity against three species of kinetoplast parasites of clinical and veterinary importance. Most importantly, the mono‐N‐arylated analogues were not cross‐resistant with pentamidine and diminazene, bypassing known drug transporters. In addition, 36 accumulated rapidly in all three kinetoplastid species and localized to the parasite nucleus and kinetoplast, consistent with a putative mechanism of action being a DNA minor groove binder. These findings highlight that the potential utility of mono‐N‐arylation of diamidines, and potentially their expansion to guanidine analogues, [35] as an emerging class of therapeutic agents against neglected parasitic diseases.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
J.R. and G.A.B. thank GlaxoSmithKline (GSK) and the Engineering and Physical Sciences Research Council (EPSRC) for an industrial CASE studentship (EP/P51066X/1). MMA is supported by a studentship from the government of Libya, and MAU by a studentship from the Petroleum Technology Development Fund (PTDF), Abuja, Nigeria. We thank Dr Alan Kennedy (University of Strathclyde) and Dr Nicola Bell (University of Glasgow) for assistance in the analysis of crystal structures.
J. Robertson, M. A. Ungogo, M. M. Aldfer, L. Lemgruber, F. S. McWhinnie, B. E. Bode, K. L. Jones, A. J. B. Watson, H. P. de Koning, G. A. Burley, ChemMedChem 2021, 16, 3396.
Contributor Information
Dr. Katherine L. Jones, Email: katherine.jones@crl.com.
Prof. Allan J. B. Watson, Email: aw260@st-andrews.ac.uk.
Prof. Harry P. de Koning, Email: harry.de-koning@glasgow.ac.uk.
Prof. Glenn A. Burley, Email: glenn.burley@strath.ac.uk.
References
- 1. Meanwell N. A., J. Med. Chem. 2011, 54, 2529–2591. [DOI] [PubMed] [Google Scholar]
- 2. Peterlin-Mašič L., Kikelj D., Tetrahedron 2001, 57, 7073–7105. [Google Scholar]
- 3.
- 3a. Ilaš J., Jakopin Ž., Borštnar T., Stegnar M., Kikelj D., J. Med. Chem. 2008, 51, 5617–5629; [DOI] [PubMed] [Google Scholar]
- 3b. Lane C. A. L., Hay D., Mowbray C. E., Paradowski M., Selby M. D., Swain N. A., Williams D. H., Bioorg. Med. Chem. Lett. 2012, 22, 1156–1159; [DOI] [PubMed] [Google Scholar]
- 3c. Lawson C. P. A. T., Slawin A. M. Z., Westwood N. J., Chem. Commun. 2011, 47, 1057–1059; [DOI] [PubMed] [Google Scholar]
- 3d. Renton P., Green B., Maddaford S., Rakhit S., Andrews J. S., ACS Med. Chem. Lett. 2012, 3, 227–231; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3e. Ríos Martínez C. H., Nué Martínez J. J., Ebiloma G. U., de Koning H. P., Alkorta I., Dardonville C., Eur. J. Med. Chem. 2015, 101, 806–817. [DOI] [PubMed] [Google Scholar]
- 4. Baker N., de Koning H. P., Mäser P., Horn D., Trends Parasitol. 2013, 29, 110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Brun R., Don R., Jacobs R. T., Wang M. Z., Barrett M. P., Future Microbiol. 2011, 6, 677–691; [DOI] [PubMed] [Google Scholar]
- 5b. Paine M. F., Wang M. Z., Generaux C. N., Boykin D. W., Wilson W. D., De Koning H. P., Olson C. A., Pohlig G., Burri C., Brun R., Murilla G. A., Thuita J. K., Barrett M. P., Tidwell R. R., Curr. Opin. Invest. Drugs 2010, 11, 876–883. [PubMed] [Google Scholar]
- 6. Giordani F., Morrison L. J., Rowan T. G., De Koning H. P., Barrett M. P., Parasitology 2016, 143, 1862–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Soeiro M. N. C., Werbovetz K., Boykin D. W., Wilson W. D., Wang M. Z., Hemphill A., Parasitology 2013, 140, 929–951; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Giordani F., Khalaf A. I., Gillingwater K., Munday J. C., de Koning H. P., Suckling C. J., Barrett M. P., Scott F. J., J. Med. Chem. 2019, 62, 3021–3035. [DOI] [PubMed] [Google Scholar]
- 8. Alghamdi A. H., Munday J. C., Campagnaro G. D., Gurvic D., Svensson F., Okpara C. E., Kumar A., Quintana J., Abril M. E. M., Milic P., Watson L., Paape D., Settimo L., Dimitriou A., Wielinska J., Smart G., Anderson L. F., Woodley C. M., Kelly S. P. Y., Ibrahim H. M. S., Hulpia F., Al-Salabi M. I., Eze A. A., Sprenger T., Teka I. A., Gudin S., Weyand S., Field M., Dardonville C., Tidwell R. R., Carrington M., O′Neill P., Boykin D. W., Zachariae U., De Koning H. P., eLife 2020, 9, e56416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maser P., Sutterlin C., Kralli A., Kaminsky R., Science 1999, 285, 242–244. [DOI] [PubMed] [Google Scholar]
- 10. Munday J. C., Eze A. A., Baker N., Glover L., Clucas C., Andres D. A., Natto M. J., Teka I. A., McDonald J., Lee R. S., Graf F. E., Ludin P., Burchmore R. J. S., Turner C. M. R., Tait A., MacLeod A., Maeser P., Barrett M. P., Horn D., De Koning H. P., J. Antimicrob. Chemother. 2014, 69, 651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Graf F. E., Baker N., Munday J. C., de Koning H. P., Horn D., Mäser P., Int. J. Parasitol. Drugs Drug Res. 2015, 5, 65–68; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Munday J. C., Lopez K. E. R., Eze A. A., Delespaux V., Van den Abbeele J., Rowan T., Barrett M. P., Morrison L. J., de Koning H. P., Int. J. Parasitol. Drugs Drug Resist. 2013, 3, 69–76; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Carruthers L. V., Munday J. C., Ebiloma G. U., Steketee P., Jayaraman S., Campagnaro G. D., Ungogo M. A., Lemgruber L., Donachie A.-M., Rowan T. G., Peter R., Morrison L. J., Barrett M. P., De Koning H. P., Mol. Microbiol. 2021, 10.1111/mmi.14733. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Yao T., Tetrahedron Lett. 2015, 56, 4623–4626; [Google Scholar]
- 12b. Kantin G., Krasavin M., Curr. Org. Chem. 2016, 20, 1370–1388. [Google Scholar]
- 13. Bakunova S. M., Bakunov S. A., Wenzler T., Barszcz T., Werbovetz K. A., Brun R., Tidwell R. R., J. Med. Chem. 2009, 52, 4657–4667. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Dunn P. J., in Comprehensive Organic Functional Group Transformations II (Eds.: Katritzky A. R., Taylor R. J. K.), Elsevier, Oxford, 2005, pp. 655–699; [Google Scholar]
- 14b. Forsberg J. H., Spaziano V. T., Balasubramanian T. M., Liu G. K., Kinsley S. A., Duckworth C. A., Poteruca J. J., Brown P. S., Miller J. L., J. Org. Chem. 1987, 52, 1017–1021; [Google Scholar]
- 14c. Wang J., Xu F., Cai T., Shen Q., Org. Lett. 2008, 10, 445–448; [DOI] [PubMed] [Google Scholar]
- 14d. Caron S., Wei L., Douville J., Ghosh A., J. Org. Chem. 2010, 75, 945–947. [DOI] [PubMed] [Google Scholar]
- 15. McGowan M. A., McAvoy C. Z., Buchwald S. L., Org. Lett. 2012, 14, 3800–3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Cortes-Salva M., Nguyen B.-L., Cuevas J., Pennypacker K. R., Antilla J. C., Org. Lett. 2010, 12, 1316–1319; [DOI] [PubMed] [Google Scholar]
- 16b. Cortes-Salva M., Garvin C., Antilla J. C., J. Org. Chem. 2011, 76, 1456–1459. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a.W. Duo, Tianjin Jude Technology Co., Ltd., Peop. Rep. China. 2013, CN103254100 A;
- 17b. Li J., Benard S., Neuville L., Zhu J., Org. Lett. 2012, 14, 5980–5983. [DOI] [PubMed] [Google Scholar]
- 18. Bray P. G., Barrett M. P., Ward S. A., de Koning H. P., Trends Parasitol. 2003, 19, 232–239. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Rautio J., Meanwell N. A., Di L., Hageman M. J., Nat. Rev. Drug Discovery 2018, 17, 559–587; [DOI] [PubMed] [Google Scholar]
- 19b. Rauws T. R. M., Maes B. U. W., Chem. Soc. Rev. 2012, 41, 2463–2497. [DOI] [PubMed] [Google Scholar]
- 20. Guo W., Zhao M. M., Tan W., Zheng L. Y., Tao K. L., Fan X. L., Org. Chem. Front. 2019, 6, 2120–2141. [Google Scholar]
- 21.
- 21a. West M. J., Fyfe J. W. B., Vantourout J. C., Watson A. J. B., Chem. Rev. 2019, 119, 12491–12523; [DOI] [PubMed] [Google Scholar]
- 21b. Chan D. M. T., Monaco K. L., Wang R.-P., Winters M. P., Tetrahedron Lett. 1998, 39, 2933–2936. [Google Scholar]
- 22. Vantourout J. C., Miras H. N., Isidro-Llobet A., Sproules S., Watson A. J. B., J. Am. Chem. Soc. 2017, 139, 4769–4779. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Vantourout J. C., Li L., Bendito-Moll E., Chabbra S., Arrington K., Bode B. E., Isidro-Llobet A., Kowalski J. A., Nilson M. G., Wheelhouse K. M. P., Woodard J. L., Xie S., Leitch D. C., Watson A. J. B., ACS Catal. 2018, 8, 9560–9566; [Google Scholar]
- 23b. West M. J., Thomson B., Vantourout J. C., Watson A. J. B., Asian J. Org. Chem. 2020, 9, 364–367. [Google Scholar]
- 24. Evans D. A., Katz J. L., West T. R., Tetrahedron Lett. 1998, 39, 2937–2940. [Google Scholar]
- 25. King A. E., Brunold T. C., Stahl S. S., J. Am. Chem. Soc. 2009, 131, 5044–5045. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Mohamed A. A., Coord. Chem. Rev. 2010, 254, 1918–1947; [Google Scholar]
- 26b. Kilner M., Pietrzykowski A., Polyhedron 1983, 2, 1379–1388; [Google Scholar]
- 26c. Barker J., Kilner M., Coord. Chem. Rev. 1994, 133, 219–300. [Google Scholar]
- 27. Kotthaus J., Kotthaus J., Schade D., Schwering U., Hungeling H., Mueller-Fielitz H., Raasch W., Clement B., ChemMedChem 2011, 6, 2233–2242. [DOI] [PubMed] [Google Scholar]
- 28. De Koning H. P., Trop. Med. Infect. Dis. 2020, 5, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.
- 29a. Carter N. S., Fairlamb A. H., Nature 1993, 361, 173–176; [DOI] [PubMed] [Google Scholar]
- 29b. Stewart M. L., Burchmore R. J. S., Clucas C., Hertz-Fowler C., Brooks K., Tait A., MacLeod A., Turner C. M. R., De Koning H. P., Wong P. E., Barrett M. P., Eukaryotic Cell 2010, 9, 336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. De Koning H. P., Mol. Pharmacol. 2001, 59, 586–592. [DOI] [PubMed] [Google Scholar]
- 31. Collar C. J., Al-Salabi M. I., Stewart M. L., Barrett M. P., Wilson W. D., de Koning H. P., J. Biol. Chem. 2009, 284, 34028–34035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bridges D. J., Gould M. K., Nerima B., Mäser P., Burchmore R. J. S., de Koning H. P., Mol. Pharmacol. 2007, 71, 1098–1108. [DOI] [PubMed] [Google Scholar]
- 33. Thomas J. A., Baker N., Hutchinson S., Dominicus C., Trenaman A., Glover L., Alsford S., Horn D., PLoS Neglected Trop. Dis. 2018, 12, e0006980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Edwards K. J., Jenkins T. C., Neidle S., Biochemistry 1992, 31, 7104–7109. [DOI] [PubMed] [Google Scholar]
- 35. Blanc R. S., Richard S., Mol. Cell 2017, 65, 8–24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information