

ABSTRACT
Purpose
To assess the incidence of Stargardt disease (STGD1) and to evaluate demographics of incident cases.
Methods
For this retrospective cohort study, demographic, clinical and genetic data of patients with a clinical diagnosis of STGD1 were registered between September 2010 and January 2020 in a nationwide disease registry. Annual incidence (2014‐2018) and point prevalence (2018) were assessed on the basis of this registry.
Results
A total of 800 patients were registered, 56% were female and 83% were of European ancestry. The incidence was 1.67‐1.95:1,000,000 per year and the point prevalence in 2018 was approximately 1:22,000‐1:19,000 (with and without 10% of potentially unregistered cases). Age at onset was associated with sex (p = 0.027, Fisher’s exact); 1.9x more women than men were observed (140 versus 74) amongst patients with an age at onset between 10 and 19 years, while the sex ratio in other age‐at‐onset categories approximated one. Late‐onset STGD1 (≥45 years) constituted 33% of the diagnoses in 2014‐2018 compared to 19% in 2004‐2008. Diagnostic delay (≥2 years between the first documentation of macular abnormalities and diagnosis) was associated with older age of onset (p = 0.001, Mann–Whitney). Misdiagnosis for age‐related macular degeneration (22%) and incidental STGD1 findings (14%) was common in patients with late‐onset STGD1.
Conclusion
The observed prevalence of STGD1 in real‐world data was lower than expected on the basis of population ABCA4 allele frequencies. Late‐onset STGD1 was more frequently diagnosed in recent years, likely due to higher awareness of its phenotype. In this pretherapeutic era, mis‐ and underdiagnosis of especially late‐onset STGD1 and the role of sex in STGD1 should receive special attention.
Keywords: ABCA4, incidence, prevalence, Stargardt Disease, STGD1
Introduction
Stargardt disease (STGD) is an inherited retinal disease (IRD) that was first described comprehensively over a century ago as a rare, progressive, familial macular degeneration that manifested in childhood or adolescence.(Stargardt 1909) In 1997, mutations in the retina‐specific ATP binding cassette transporter (ABCA4) gene were found to cause this disease, from that moment abbreviated as STGD1.(Allikmets et al. 1997) Since then, the phenotypic spectrum of STGD1 has greatly expanded. When presenting in early childhood, STGD1 often evolves to a cone‐rod dystrophy (CRD) that eventually leads to a panretinal degeneration.(Klevering et al. 2004; Lambertus et al. 2015) Late‐onset STGD1 (onset ≥45 years) is hallmarked by sharply demarcated atrophy in a foveal‐sparing pattern.(Westeneng‐van Haaften et al. 2012) Another STGD1 phenotype is the bull’s eye maculopathy in which macular atrophy is often accompanied by exceptionally little lipofuscin.(Nõupuu et al. 2014; Cremers et al. 2020).
STGD1 is widely regarded as the most common Mendelian inherited eye disorder and STGD1 alone accounts for 12% of IRD‐related blindness registrations.(Heath Jeffery et al. 2021) The exact prevalence, however, is unknown. A prevalence of 1:10,000 to 1:8,000 is frequently quoted in the literature, but this estimate was not accompanied by substantive analyses and originates from 1988.(Blacharski 1988) Since then, the disease‐causing gene has been identified, the phenotypic spectrum of ABCA4 disease has been further elucidated, and diagnostic tools, for example imaging and genetic tests, have greatly evolved. Genetic studies have estimated a much higher prevalence of STGD1 of 1:6,578 to even 1:870.(Riveiro‐Alvarez et al. 2009; Hanany et al. 2020) However, these estimates – based on ABCA4 mutation carrier frequencies – do not relate to the observed disease prevalence because the ABCA4 gene holds many variants of unknown significance and because disease expression depends on the severity of the mutation on the other ABCA4 allele.(Cornelis et al. 2017; Zernant et al. 2017; Runhart et al. 2018; Zernant et al. 2018).
Several recent developments require data on the frequency of STGD1. Many strategies to treat IRDs are being tested in preclinical studies or clinical trials.(Vázquez‐Domínguez et al. 2019) Prevalence data are relevant for the planning of clinical trials and are required for pharmacoeconomic evaluation of orphan drugs. Moreover, rare diseases are notably underdiagnosed.(Austin et al. 2018; Auvin et al. 2018) Monitoring of diagnostic trends can provide insight into the degree of underdiagnosis, which would become a critical issue once therapy becomes available. Furthermore, several frequent ABCA4 variants were found to be associated with STGD1 and to have incomplete penetrance.(Zernant et al. 2017; Cremers et al. 2018; Runhart et al. 2018; Zernant et al. 2018; Runhart et al. 2019; Sangermano et al. 2019; Runhart et al. 2020) In the employed penetrance calculations, the actually observed prevalence of disease remained an uncertain variable. A prevalence estimate supported by substantive data would improve research on penetrance of individual ABCA4 variants.
In this nationwide retrospective cohort study, we assessed the annual incidence and the point prevalence of STGD1. To this purpose, we used our collaborative RD5000 disease registry that allows ongoing systematic collection, analysis and interpretation of health data and diagnostic trends.(van Huet et al. 2014).
Methods
Subjects
For this retrospective cohort study, patients with a registered diagnosis of STGD were identified in all referral centres for ophthalmogenetics in the Netherlands. These centres participate in the Dutch registry for Inherited Retinal Dystrophies ‘RD5000’.(van Huet et al. 2014) The diagnosis STGD1 was registered if the inclusion criteria and none of the exclusion criteria in Supplemental Table 1 were met. For this study, records were reviewed to confirm these diagnostic criteria, and patients with a residence outside of the Netherlands were excluded.
Table 1.
Patient and general population characteristics
|
General population Netherlands |
Total STGD1 population | Incident STGD1 cases 2014‐2018 | |
|---|---|---|---|
| Demographics | |||
| Female, n (%) | 8654043 (50%) | 445 (56%) | 78 (56%) |
| Age, mean (SD) | 42 yrs | 47 (19) yrs | 38 (20) yrs |
| Deceased, n (%) | 28 (4%) | 0 (0%) | |
| Age at diagnosis, median (range) | 24 (5‐83) yrs | 36 (6‐83) yrs | |
|
Geographic ancestry European, n (%) North African/Middle Eastern, n (%) Southeast Asian, n (%) Sub‐Saharan African, n (%) Suriname, n (%) Other |
(84%) (6%) (4%) (2%) (2%) (2%) |
603 (83%) 60 (8%) 22 (3%) 15 (2%) 15 (2%) 11 (2%) |
96 (76%) 19 (15%) 2 (2%) 4 (3%) 3 (2%) 2 (2%) |
| Genetics | |||
|
Genetic test in patient or sibling performed, n (%) Genetically confirmed diagnoses in patient or sibling, n (%) |
664 (83%) 615 (77%; 93% of performed tests) |
131 (94%) 125 (89%; 95% of performed tests) |
|
| Patient’s and family history | |||
| Age at onset, median (range) | 20 (1‐82) | 32 (3‐82) | |
| Positive family history STGD1, n (%) | 308 (40%) | 32 (23%) | |
| Consanguinity, n (%) | 61 (10%) | 12 (11%) | |
| Incident cases in 2014‐2018 are all cases who received the diagnosis STGD1 in 2014‐2018. STGD1 = Stargardt disease. | |||
The Medical Research Ethics Committee ‘CMO region Arnhem‐Nijmegen’ ruled that approval was not required for this study. The study is not subject to the Medical Research Involving Human Subjects Act (WMO) because participants were not required to follow procedures or rules of behaviour. The study and data collection were in conformity with all country laws and adhered to the tenets of the Declaration of Helsinki.
Data collection
Data were extracted from medical records between September 2010 and January 2020. Collected variables included demographics, year of STGD1 diagnosis, ABCA4 variants and the family history. Also, the year of initial report of macular abnormalities that retrospectively were considered to be the initial manifestation of STGD1 (i.e. flecks, retinal pigment epithelium atrophy, photoreceptor loss) was recorded. The self‐reported geographic ancestry (first‐ or second‐generation migration) was recorded if these data were available. An age at onset of complaints ≤0 years was considered ‘early‐onset STGD1’, age at onset of complaints ≥45 years was considered ‘late‐onset STGD1,’ based on cut‐offs commonly used in literature.(Westeneng‐van Haaften et al. 2012; Lambertus et al. 2015) The remaining patients were grouped as intermediate‐onset STGD1, age at onset of complaints between 11 to 44 years. Between 1998 and 2018, patients were screened for disease‐causing variants in the coding regions and flanking splice sites and/or non‐coding regions of ABCA4, as part of routine patient care or previous studies. The employed methods are listed in Supplemental Table 2. Sanger sequencing was performed to confirm all identified mutations. Genotypes were classified based on their pathogenicity rating in accordance with the American College of Medical Genetics classification.(Richards et al. 2015).
Table 2.
The most frequent ABCA4 variants in the Dutch patient population
| ABCA4 nucleotide changes | ABCA4 protein changes | Allele count patients | Allele frequency patients | Allele frequency general population a |
|---|---|---|---|---|
| c.5603A>T | p.(Asn1868Ile) | 167 | b | 0.0677 |
| c.5461‐10T>C c | p.[Thr1821Valfs*13, Thr1821Aspfs*6] | 133 | 0.1002 | 0.0003 |
| c.2588G>C d | p.[Gly863Ala, Gly863del] | 115 | 0.0866 | 0.0074 |
| c.768G>T | p.(Leu257Valfs*17) | 103 | 0.0776 | 0.0006 |
| c.5882G>A | p.(Gly1961Glu) | 101 | 0.0761 | 0.0049 |
| c.1822T>A | p.(Phe608Ile) | 47 | 0.0354 | 0.0003 |
| c.3113C>T e | p.(Ala1038Val) | 35 | 0.0264 | 0.0022 |
| c.4539 + 1G>T | p.(?) | 27 | 0.0203 | 0 |
| c.5714 + 5G>A | p.[=; Glu1863Leufs*33] | 23 | 0.0173 | 0.0004 |
| c.4139C>T | p.(Pro1380Leu) | 22 | 0.0166 | 0.0001 |
Not included in this table are deep‐intronic variants c.4253 + 43G>A and c.769‐784C>T, which were identified in 17 and 10 patients. These variants were only recently associated with the disease and therefore not accurately represented in the database.
ABCA4 frequencies in 21,559 control individuals from The Netherlands.(Cremers et al. 2018)
This variant was only recently associated with STGD1 and therefore not accurately represented in the database. It was found as a single variant in 73 alleles.
c.5462‐10T>C is almost always complexed with c.5603A>T.
Only considered penetrant when in cis with c.5603A>T. In the general population of The Netherlands, the allele frequency of c.[2588G>C;5603A>T] is estimated to be 0.0007. (Cremers et al. 2018).
Found in cis with c.1622T>C in 37% of the alleles containing c.3113C>T in patients.
Annual incidence and point prevalence
The annual incidence was assessed for the years 2014 to 2018. This period allowed for the most complete data collection due to digitalization of medical records and improved diagnostic opportunities due to availability of optical coherence tomography (OCT) and short‐wave autofluorescence (SW‐AF) and advances in genetic techniques. The centres participating in this study are the national centres specialized in ophthalmogenetics and cover the vast majority of patient care involving IRDs in The Netherlands. To quantify the smaller proportion of patients with a diagnosis of STGD1 who had never been under the care of an ophthalmologist in any of these centres, we administered an anonymous questionnaire amongst members of a patient association for macular diseases. Patients with a self‐reported diagnosis of STGD1 were asked whether they currently were or ever had been under the care of an ophthalmologist in any of the participating medical centres. The proportion of respondents who had not visited any of the nationally specialized centres, each participating in the registry, was added to the incidence total. To define incidence, the average size of the population of The Netherlands between January 2014 and December 2018 was used, which was 17,042,315 individuals.(Netherlands 2019) The point prevalence on 31 December 2018 was assessed. To define prevalence, the size of the entire population of The Netherlands at the end of the year 2018 was used, which was 17,282,163 individuals.(Netherlands 2019).
Annual incidence and point prevalence were calculated as follows:
Statistical analysis
Descriptive statistics were presented of the complete STGD1 population. The distribution of sex across age–at‐onset categories was evaluated using Fisher’s exact test (two‐sided, α = 0.05). Wilcoxon signed rank test was used to assess whether age at onset was associated with sex in siblings with STGD1 (two‐sided, α = 0.05). Additionally, the Mann–Whitney test and the Fisher’s exact test were performed to assess whether age at onset of complaints, ancestry or sex were associated with diagnostic delay (two‐sided, α = 0.015).
Results
Subjects
A total of 800 patients with STGD1 in accordance with the diagnostic and study criteria were nationally registered until January 2020. Patient characteristics are described in Table 1. Patients had a mean age of 47 ± 19 years, 445 patients (56%) were female and 603 patients (83%) were of European ancestry. Fig. 1 shows the distribution of age at onset plotted by sex, the two of which were associated (p = 0.027, Fisher’s exact); Remarkably, 1.9x more women than men were observed (140 versus 74) amongst patients with an age at onset between 10 and 19 years, while the sex ratio in the other age‐at‐onset categories approximated 1. Cases with a macular dystrophy not further specified who carried only one ABCA4 variant – who represented the less certain STGD1 diagnoses – accounted for only 48 (6%) of the registered patients.
Figure 1.
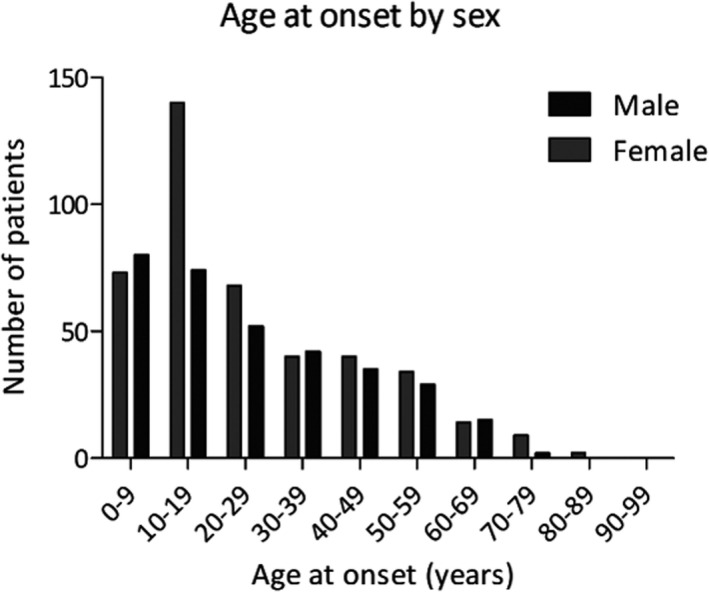
Distribution of the age at onset of Stargardt disease by sex. Age at onset was associated with sex (p = 0.027, Fisher’s exact). Women were overrepresented (1.9:1) amongst patients with an age at onset of 10‐19 years (n = 214)
Annual incidence and point prevalence
Fig. 2 shows an increasing frequency of STGD1 diagnoses over the years 1999‐2018. Between 1 January 2014 and 31 December 2018, 140 incident cases were documented, which corresponds to a mean annual incidence of 28. Based on this registry, annual incidence was 1.64 (95% CI, 1.03‐2.25) per 1,000,000 persons per year. However, this likely is an underestimation of the true incidence, because of potentially unregistered cases.
Figure 2.
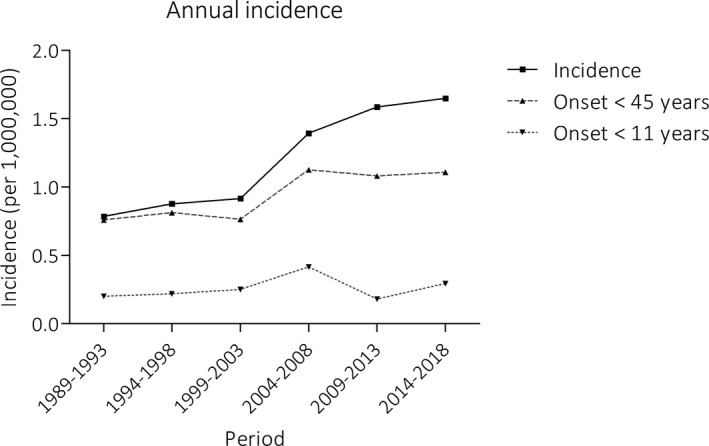
Annual incidence of Stargardt disease based on the national registry. The number of registered STGD1 diagnoses increased over time, which was mostly attributed to an increase in the diagnosis and registration of late‐onset STGD1 (≥45 years). The incidence of early‐onset Stargardt disease (<11 years) remained fairly constant
According to the questionnaire administered in a patient association for macular diseases, five of 49 (10%; 95% CI, 2%‐19%) respondents who had received the diagnosis STGD1 had not visited any of the specialized centres for ophthalmogenetics participating in the registry. Considering the 95% CI of potentially unregistered cases, the annual incidence was between 1.67 and 1.95 per 1,000,000 persons per year.
Until 31 December 2018, a total of 790 patients had been diagnosed of whom 762 patients were alive in 2018. Therefore, the point prevalence in 2018 based solely on the registry was 1 in 22,680 (95% CI, 1:24,413‐1:21,176). Considering potentially unregistered cases (2‐19%), point prevalence was between 1 in 22,295 and 1 in 19,110 individuals.
Diagnostic trends: Increasing frequency of late‐onset STGD1 diagnoses and stable non‐negligible diagnostic delay
Of all patients registered, 193 (24%) experienced initial complaints ≤10 years, 466 (58%) between 11‐44 years and 140 (18%) ≥45 years. Over the years, the proportion of patients diagnosed with late‐onset STGD1 increased gradually as visualized in Fig 2. The median (range) age at onset of patients diagnosed in 2014‐2018 was 32 (3‐82) years, compared to 19 (1‐72) years in 2004‐2008. Prior to the diagnosis of STGD1, 28 (22%) patients with late‐onset STGD1 (≥45 years) had been diagnosed with age‐related macular degeneration (AMD). Another 17 (14%) of late‐onset patients were diagnosed before onset of complaints, during screening visits for diabetic/plaquenil retinopathy or glaucoma (n = 8), regular care for comorbidities (n = 6) or screening because of a positive family history (n = 2) (alternative reason for visit not registered n = 1).
In 58 (8%) of 694 cases, it took more than 2 (2‐21) years between the moment the first macular abnormalities were documented and the moment that STGD1 diagnosis was documented. This diagnostic delay was associated with a higher age at onset of complaints (p = 0.001, Mann–Whitney) and was not associated with sex (p = 0.582, Fisher’s exact) or non‐Western European ancestry (p = 0.584, Fisher’s exact). No trend in the frequency or extent of delay was observed over time. Of patients with a diagnostic delay, 12 patients (21%) had received a diagnosis of AMD prior to STGD1. ABCA4 analysis, performed in 52 patients or a sibling of a patient (n = 1) with a diagnostic delay (91%), confirmed the presence of ≥2 (potentially) pathogenic variants in 50 patients (94%).
ABCA4 genotypes
ABCA4 analysis had been performed in 641 patients (80%). Another 23 patients (3%) had an affected sibling who had undergone ABCA4 testing. A total of 542 of the diagnoses was genetically confirmed by the presence of ≥2 potentially disease‐causing variants in the patient or an affected sibling of the patient (82% of the patients/siblings tested). Another 73 diagnoses (11% of the patients (or siblings) tested) were considered genetically confirmed by the presence of the mild variant c.5603A>T as the second allele. The most frequent variants by far were c.5603A>T, c.5461‐10T>C, c.2588G>C (only considered penetrant when in cis with c.5603A>T), c.768G>T and c.5882G>A (Table 2).
Family history
A total of 623 patients (78%) were considered a ‘proband,’ the first patient in the family. Of these probands, 167 (27%) had family members with STGD1: 137 (22%) had 1 or more affected siblings and 33 (5%) had an affected 2nd or 3rd degree relative. Pseudodominant inheritance was recorded in 9 (1%) families, only 1 of which was known to have consanguinity.
Age at onset could differ many years between siblings, with a median difference of 3 (0‐48) years. Differences of more than 10 years were observed in 18 of 84 families in which multiple siblings were affected (21%) (Table 3). In most of these families with large differences in age at onset (n = 14, 78%), a known mild variant c.2588G>C (n = 5), c.5603A>T (n = 4), c.5882G>A (n = 3), c.[769‐784C>T;5603A>T] (n = 1) or c.4253 + 43G>A (n = 1) had been identified. In 44 families in which the sibling with the highest and lowest age at onset had a different sex, lower age at onset was associated with female sex (p = 0.025, Wilcoxon signed rank): in 24 families, the sister had an earlier onset than the brother, with a median difference of 8 (1‐48) years; in 16 families, the brother was affected earlier, with a median difference of only 2 (1‐7) years.
Table 3.
Sibling pairs with a discordant age at onset
| Family |
ABCA4 nucleotide changes |
ABCA4 protein changes |
Age‐at‐onset difference (yrs) | Sex youngest onset (age onset in yrs) | Sex oldest onset (age onset in yrs) |
|---|---|---|---|---|---|
| 5 | c.5537T>C(;)5603A>T | p.(Ile1846Thr)(;)(Asn1868Ile) | 37 | Female (15) | Male (52) |
| c.5882G>A | p.(Gly1961Glu) | ||||
| 12 | c.5603A>T | p.(Asn1868Ile) | 33 | Female (37) | Male (70) |
| c.5762_5763dup | p.(Ala1922Trpfs*18) | ||||
| 15 | c.3191‐2_3191del | p.(?) | 11 | Male (40) | Male (51) |
| c.5603A>T | p.(Asn1868Ile) | ||||
| 16 | Not tested | 11 | Male (21) | Male (32) | |
| 17 | c.1822T>A | p.(Phe608Ile) | 13 | Male (17) | Male (30) |
| c.2588G>C a | p.[Gly863Ala, Gly863del] | ||||
| 23 | c.2921_3328 + 2del | p.(Ser974_Gly1110delinsCys) | 26 | Male (13) | Male (39) |
| c.5059A>T | p.(Ile1687Phe) | ||||
| 27 | c.768G>T | p.(Leu257Valfs*17) | 22 | Female (49) | Female (71) |
| c.5603A>T | p.(Asn1868Ile) | ||||
| 28 | c.4539 + 2001G>A | p.[=, Arg1514Leufs*36] | 31 | Female (16) | Male (47) |
| c.5882G>A | p.(Gly1961Glu) | ||||
| 35 | c.859‐506G>C | p.[Phe287Thrfs*32,=] | 15 | Female (12) | Male (17) |
| c.5196 + 1137G>A | p.[=, Met1733Glufs*78] | ||||
| 37 | c.768G>T | p.(Leu257Valfs*17) | 16 | Female (14) | Male (30) |
| c.5882G>A | p.(Gly1961Glu) | ||||
| 44 | c.2409_2410del | p.(Phe804Trpfs*3) | 17 | Female (8) | Male (25) |
| c.[2588G>C;5603A>T] (;)2802_2804del | p.[(Gly863Ala, Gly863del); (Asn1868Ile)](;)(Val935del) | ||||
| 48 | c.768G>T | p.(Leu257Valfs*17) | 24 | Female (19) | Male (43) |
| c.5603A>T | p.(Asn1868Ile) | ||||
| 78 | c.656G>C b | p.(Arg219Thr) | 20 | Female (37) | Male (57) |
| c.2588G>C a | p.[Gly863Ala, Gly863del] | ||||
| 88 | c.2588G>C a | p.[Gly863Ala, Gly863del] | 20 | Female (15) | Male (35) |
| c.5461‐10T>C a | p.[Thr1821Valfs*13, Thr1821Aspfs*6] | ||||
| 90 | c.3322C>T | p.(Arg1108Cys) | 14 | Female (38) | Female (52) |
| c.3398T>C | p.(Ile1133Thr) | ||||
| 92 | c.768G>T | p.(Leu257Valfs*17) | 14 | Female (58) | Female (72) |
| c.4253 + 43G>A | p.[=, Ile1377Hisfs*3] | ||||
| 94 | c.[769‐784C>T;5603A>T] | p.[=, Leu257Aspfs*3; Asn1868Ile] | 48 | Female (20) | Male (68) |
| c.4539 + 1G>T | p.(?) | ||||
| 200 | c.768G>T | p.(Leu257Valfs*17) | 18 | Male (17) | Male (35) |
| c.2588G>C a | p.[Gly863Ala, Gly863del] |
Allele 1 in white, allele 2 in grey. Family 5, 35, 37, 44 were previously described by Valkenburg et al. 2019 (Valkenburg et al. 2019)
Variant c.5603A>T is often present on the same allele as c.2588G>C and c.5461‐10T>C. The c.5603A>T was not associated with the disease at the time of genetic analysis in these patients and therefore not reported.
Variants c.656G>C and c. 2588G>C are known to occur on the same allele. Segregation analysis was however not performed.
Discussion
To the best of our knowledge, this is the first study that collected epidemiological data on STGD1 – the full spectrum of ABCA4‐associated disease – supported by genetic data. Point prevalence of STGD1 in the Netherlands in 2018 was approximately 1:22,000‐19,000 individuals, which is far less than the genetically estimated prevalence 1:6,578‐1:870, based on ABCA4 mutation carrier frequencies.(Riveiro‐Alvarez et al. 2009; Hanany et al. 2020) This mismatch is likely due to the recent evidence for reduced prevalence of several mild ABCA4 variants, in addition to the fact that the ABCA4 gene still holds many variants of unknown significance and disease expression depends on the severity of the mutation on the other ABCA4 allele.(Fakin et al. 2016; Cornelis et al. 2017; Zernant et al. 2017; Runhart et al. 2018; Zernant et al. 2018; Heath Jeffery et al. 2021) The point prevalence in this study did also not exceed the crude estimate 1:10,000‐8,000 dating back to 1988, despite huge diagnostic advances in both imaging and genetic techniques since then. In contrast, the annual incidence of STGD1 in the Netherlands (1.67‐1.95:1,000,000) was 1.5x higher than reported in the only previous epidemiological study on STGD1, performed in the United Kingdom.(Spiteri Cornish et al. 2017) This discrepancy might be explained by differences in the definition of STGD1 (bull’s eye maculopathy and CRD were considered other disease entities in that study), study design (a survey amongst ophthalmologists with an inherent incomplete response‐rate), and genetic testing (then rarely performed although it considerably expands the phenotypic range of STGD1).
Late‐onset STGD1 constituted an increasing proportion of the total STGD1 population over the years. This is likely explained by increasing awareness of its existence amongst ophthalmologists since its phenotype was first comprehensively described eight years ago.(Westeneng‐van Haaften et al. 2012) An unknown proportion might still be undiagnosed, for instance due to the absence of symptoms (14% of late‐onset STGD1 was an incidental finding) or misdiagnosis as AMD (22% of late‐onset STGD1). The low age at onset reported in literature suggests that late‐onset STGD1 worldwide is still often not recognized: maximum age at onset being ≤42 years;(Zolnikova et al. 2017) ≤40 years;(Salles et al. 2018) ≤55 years;(Riveiro‐Alvarez et al. 2013) and maximum age at presentation, ≤64 years.(Spiteri Cornish et al. 2017) Considering the current median age at onset of 32 years, and a maximum age at onset of 82 years, the term ‘juvenile’ macular dystrophy is outdated, as previously suggested,(Spiteri Cornish et al. 2017) and misleading to patients and physicians. Indeed, diagnostic delay (≥2 years) was associated with higher age at onset (p = 0.001, Mann–Whitney). In a forthcoming era of therapeutic options for STGD1, the awareness that this illness can manifest at all ages could prevent blindness.
In addition, we will have to focus on factors that impact whether and how disease manifests. Recently, we observed a female predilection amongst patients carrying the mild ABCA4 allele c.5603A>T or c.5882G>A compared to patients who did not carry any known mild allele.(Runhart et al. 2020) Although the current study did not allow a genotype‐specific analysis due to different DNA testing methods employed over time, it did show a remarkable female preponderance amongst patients with age at onset between 10 and 19 years. This female predilection in puberty provides food for thought on the role of sex‐specific disease modifiers. Moreover, a younger age at onset was associated with the female sex when comparing siblings. The fact that the observed prevalence in The Netherlands was not higher than the estimate of 1:10,000, employed in previous penetrance calculations, further corroborates the evidence for incomplete penetrance of several mild ABCA4 variants.(Cremers et al. 2018; Runhart et al. 2018; Runhart et al. 2019) Uncovering the mechanisms underlying sex differences and incomplete penetrance in STGD1 could be of great interest for therapeutic development.
Finally, these data can aid in today’s practice of family counselling. Genetic studies have found a carrier (heterozygous) frequency of approximately 1:10 individuals (10%).(Jaakson et al. 2003; Riveiro‐Alvarez et al. 2009) This knowledge is often used in family counselling, and even in estimating disease prevalence (1:6,578–1:870),(Riveiro‐Alvarez et al. 2009; Hanany et al. 2020) but is difficult to interpret due to uncertain pathogenicity and unknown expression of many ABCA4 alleles. Indeed, we observed a pseudodominant inheritance in only eight reportedly non‐consanguineous families (1.3%), suggesting that at least 1.3% of individuals in the general population carry a pathogenic ABCA4 variant. Although the number of carriers could well be double due to autosomal recessive inheritance, the large gap between this carrier frequency of 1.3‐2.6% derived from the observed pseudodominant inheritance and the aforementioned 10% found in genetic studies exemplifies a large counselling difficulty for ABCA4‐associated disease. A differentiated counselling approach for early‐onset STGD1, caused by severe ABCA4 variants, and late‐onset STGD1, associated with frequent mild variants, could allow for a more accurate prediction of disease risk. The common (20%) discordance between siblings in terms of age at onset (≥10 years) was associated with mild alleles and further highlights the need for developing a differentiated counselling approach.
There are a few limitations to this study. First, the study was not designed to evaluate phenotypic differences between patients: for instance, patients with a CRD caused by ABCA4 mutations often received the diagnosis ‘STGD1’, that is all ABCA4‐associated retinopathy, rather than ‘CRD’. Second, disease registration in the database might be incomplete and diagnosis might not always be correct. The observed prevalence inherently underestimates the true prevalence. The diagnostic trends that are discussed in this study, such as increasing incidence over the years and the increasing frequency of late‐onset STGD1 diagnoses, indeed illustrate this. Ongoing diagnostic advances in combination with continued disease registration would provide data that increasingly reflect the true prevalence and help guide diagnostic and management strategies. Third, ascertainment may have been incomplete due to disease registration in specialized centres only. However, we anticipated that the standard of care in The Netherlands is to at least once send a patient suspected of a retinal dystrophy to one of the participating specialized centres, to allow a full diagnostic work‐up including genetic testing and genetic counselling. This was confirmed by the questionnaire in a patient association: a minority of patients (10%) with a clinical diagnosis of STGD1 had not visited a centre of expertise.
In conclusion, collaborative national disease registries offer a unique opportunity to study longitudinal epidemiological data on rare diseases, giving insight into diagnostic challenges, and providing valuable information for planning of trials and genetic counselling. Mis‐ and underdiagnosis were common in late‐onset STGD1 and should receive special attention in this pretherapeutic era to prevent avoidable blindness in the future.
Acknowledgements
This work was supported by the Foundation Fighting Blindness USA, grant no. PPA‐0517‐0717‐RAD (to FPMC and CBH). The study was also supported by the Oogfonds, Retinafonds, Bartiméus Fonds and ODAS that contributed through UitZicht (2015‐30). The funding organizations had no role in the design or conduct of this research.
References
- Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, Gerrard B, Lupski JR (1997): A photoreceptor cell‐specific ATP‐binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet 15: 236–246. [DOI] [PubMed] [Google Scholar]
- Austin CP, Cutillo CM, Lau LPL, Jonker AH, Rath A, Julkowska D, Thomson D, Terry SF, de Montleau B, Ardigò D, Hivert V, Boycott KM, Baynam G, Kaufmann P, Taruscio D, Lochmüller H, Suematsu M, Incerti C, Draghia‐Akli R, Norstedt I, Wang L & Dawkins HJS (2018): Future of Rare Diseases Research 2017–2027: An IRDiRC Perspective. Clin Transl Sci 11: 21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auvin S, Irwin J, Abi‐Aad P & Battersby A (2018): The Problem of Rarity: Estimation of Prevalence in Rare Disease. Value Health 21: 501–507. [DOI] [PubMed] [Google Scholar]
- Blacharski P (1988): Fundus flavimaculatus. Newsome DA(ed.) Retinal dystrophies and degenerations. New York: Raven Press; 135–159. [Google Scholar]
- Cornelis SS, Bax NM, Zernant J, Allikmets R, Fritsche LG, den Dunnen JT, Ajmal M, Cremers FP (2017): In Silico Functional Meta‐Analysis of 5,962 ABCA4 Variants in 3,928 Retinal Dystrophy Cases. Hum Mutat 38: 400–408. [DOI] [PubMed] [Google Scholar]
- Cremers FPM, Cornelis SS, Runhart EH & Astuti GDN (2018): Author Response: Penetrance of the ABCA4 p.Asn1868Ile allele in Stargardt disease. Invest Ophthalmol Vis Sci 59: 5566–5568. [DOI] [PubMed] [Google Scholar]
- Cremers FPM, Lee W, Collin RWJ & Allikmets R (2020): Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog Retin Eye Res 79: 100861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakin A, Robson AG, Chiang J, Fujinami K, Moore AT, Michaelides M, Holder GE & Webster AR (2016): The Effect on Retinal Structure and Function of 15 Specific ABCA4 Mutations: A Detailed Examination of 82 Hemizygous Patients. Invest Ophthalmol Vis Sci 57: 5963–5973. [DOI] [PubMed] [Google Scholar]
- Hanany M, Rivolta C & Sharon D (2020): Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc Natl Acad Sci U S A 117: 2710–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath Jeffery RC, Mukhtar SA, McAllister IL, Morgan WH, Mackey DA & Chen FK (2021): Inherited retinal diseases are the most common cause of blindness in the working‐age population in Australia. Ophthalmic Genet 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakson K, Zernant J, Külm M, Hutchinson A, Tonisson N, Glavac D, Ravnik‐Glavac M, Allikmets R (2003): Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat 22: 395–403. [DOI] [PubMed] [Google Scholar]
- Klevering BJ, Maugeri A, Wagner A, Go SL, Vink C, Cremers FP & Hoyng CB (2004): Three families displaying the combination of Stargardt's disease with cone‐rod dystrophy or retinitis pigmentosa. Ophthalmology 111: 546–553. [DOI] [PubMed] [Google Scholar]
- Lambertus S, van Huet RAC, Bax NM, Hoefsloot LH, Cremers FPM, Boon CJF, Klevering BJ & Hoyng CB (2015): Early‐Onset Stargardt Disease. Ophthalmology 122(2): 335–344. [DOI] [PubMed] [Google Scholar]
- Netherlands S (2019): Population; key figures. Statistics Netherlands. [Google Scholar]
- Netherlands S (2020): Population; sex, age, migration background and generation, 1 January. Statistics Netherlands. [Google Scholar]
- Nõupuu K, Lee W, Zernant J, Tsang SH & Allikmets R (2014): Structural and genetic assessment of the ABCA4‐associated optical gap phenotype. Invest Ophthalmol Vis Sci 55: 7217–7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz R, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Committee ALQA (2015): Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riveiro‐Alvarez R, Aguirre‐Lamban J, Lopez‐Martinez MA, Trujillo‐Tiebas MJ, Cantalapiedra D, Vallespin E, Avila‐Fernandez A, Ramos C & Ayuso C (2009): Frequency of ABCA4 mutations in 278 Spanish controls: an insight into the prevalence of autosomal recessive Stargardt disease. Br J Ophthalmol 93: 1359–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riveiro‐Alvarez R, Lopez‐Martinez MA, Zernant J, Aguirre‐Lamban J, Cantalapiedra D, Avila‐Fernandez A, Gimenez A, Ayuso C (2013): Outcome of ABCA4 disease‐associated alleles in autosomal recessive retinal dystrophies: retrospective analysis in 420 Spanish families. Ophthalmology 120: 2332–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runhart EH, Khan M, Cornelis SS, Roosing S, Del Pozo‐Valero M, Lamey TM, Liskova P, Dhaenens CM (2020): Association of Sex With Frequent and Mild ABCA4 Alleles in Stargardt Disease. JAMA Ophthalmol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runhart EH, Sangermano R, Cornelis SS, Verheij J, Plomp AS, Boon CJF, Lugtenberg D, Roosing S, Bax NM, Blokland EAW, Jacobs‐Camps MHM, van der Velde‐Visser SD, Pott JWR, Rohrschneider K, Thiadens AAHJ, Klaver CCW, van den Born LI, Hoyng CB, Cremers FPM (2018): The Common ABCA4 Variant p.Asn1868Ile Shows Nonpenetrance and Variable Expression of Stargardt Disease When Present in trans With Severe Variants. Invest Ophthalmol Vis Sci 59: 3220–3231. [DOI] [PubMed] [Google Scholar]
- Runhart EH, Valkenburg D, Cornelis SS, Khan M, Sangermano R, Albert S, Bax NM, Hoyng CB (2019): Late‐Onset Stargardt Disease Due to Mild, Deep‐Intronic ABCA4 Alleles. Invest Ophthalmol Vis Sci 60: 4249–4256. [DOI] [PubMed] [Google Scholar]
- Salles MV, Motta FL, Martin R, Filippelli‐Silva R, Dias da Silva E, Varela P, Costa KA, Sallum JF (2018): Variants in the ABCA4 gene in a Brazilian population with Stargardt disease. Mol Vis 24: 546–559. [PMC free article] [PubMed] [Google Scholar]
- Sangermano R, Garanto A, Khan M, Runhart EH, Bauwens M, Bax NM, van den Born LI, Cremers FPM (2019): Deep‐intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiteri Cornish K, Ho J, Downes S, Scott NW, Bainbridge J & Lois N (2017): The Epidemiology of Stargardt Disease in the United Kingdom. Ophthalmol Retina 1: 508–513. [DOI] [PubMed] [Google Scholar]
- Stargardt K (1909): Über familiäre, progressive Degeneration in der Maculagegend des Auges. Graefes Arch Clin Exp Ophthalmol 71: 534–550. [Google Scholar]
- Valkenburg D, Runhart EH, Bax NM, Liefers B, Lambertus SL, Sánchez CI, Cremers FPM & Hoyng CB (2019): Highly Variable Disease Courses in Siblings with Stargardt Disease. Ophthalmology 126: 1712–1721. [DOI] [PubMed] [Google Scholar]
- van Huet RAC, Oomen CJ, Plomp AS, van Genderen MM, Klevering BJ, Schlingemann RO, Klaver CCW, van den Born LI & Cremers FPM (2014): The RD5000 Database: Facilitating Clinical, Genetic, and Therapeutic Studies on Inherited Retinal Diseases. Investigative Opthalmology & Visual Science 55(11): 7355–7360. [DOI] [PubMed] [Google Scholar]
- Vázquez‐Domínguez I, Garanto A & Collin RWJ (2019): Molecular Therapies for Inherited Retinal Diseases‐Current Standing. Genes (Basel) Opportunities and Challenges, p. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westeneng‐van Haaften SC, Boon CJ, Cremers FP, Hoefsloot LH, den Hollander AI & Hoyng CB (2012): Clinical and genetic characteristics of late‐onset Stargardt's disease. Ophthalmology 119: 1199–1210. [DOI] [PubMed] [Google Scholar]
- Zernant J, Lee W, Collison FT, Fishman GA, Sergeev YV, Schuerch K, Sparrow JR, Allikmets R (2017): Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age‐related macular degeneration. J Med Genet 54: 404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zernant J, Lee W, Nagasaki T, Collison FT, Fishman GA, Bertelsen M, Rosenberg T, Allikmets R (2018): Extremely hypomorphic and severe deep intronic variants in the ABCA4 locus result in varying Stargardt disease phenotypes.Cold Spring Harb Mol Case Stud 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolnikova IV, Strelnikov VV, Skvortsova NA, Tanas AS, Barh D, Rogatina EV, Egorova IV, Ivanova ME (2017): Stargardt disease‐associated mutation spectrum of a Russian Federation cohort. Eur J Med Genet 60: 140–147. [DOI] [PubMed] [Google Scholar]