

ABSTRACT
Recombinant-modified vaccinia virus Ankara (rMVA) is known to elicit potent antitumor immune responses in preclinical models due to its inherent ability to activate the innate immune system and the activation of adaptive responses mediated by the expression of tumor antigens and costimulus-providing molecules, such as CD40L and CD137L. Here, we evaluated different rMVA vectors in preclinical peritoneal carcinomatosis models (ID8.OVA-Vegf/GFP and MC38). We compared rMVA vectors expressing a tumor antigen (OVA or gp70) either alone or co-expressed with CD40L or/and CD137L. In tumor-free mice, the vector coding for the triple combination was only slightly superior, whereas, in tumor-bearing animals, we observed a synergistic induction of T lymphocytes specific against vector-encoded and non-encoded tumor-associated antigens. The enhanced activation of the immune response was associated with improved survival in mice with peritoneal carcinomatosis treated with a rMVA vector encoding both CD40L and CD137L. Thus, the triple transgene combination in vaccinia viral vectors represents a promising strategy for the treatment of peritoneal carcinomatosis.
KEYWORDS: Peritoneal carcinomatosis, CD40L, CD137L, CD8 immune response, cancer immunotherapy
Introduction
Peritoneal carcinomatosis represents an advanced stage of gynecological and gastrointestinal cancer characterized by the invasion of the peritoneal cavity lining by malignant cells.1 The highest incidence of peritoneal carcinomatosis is observed among patients with ovarian and colorectal cancer.2,3 This advanced stage of peritoneal carcinomatosis is associated with a dismal prognosis, and the limited therapeutic options do not significantly impact the survival of the patients.3,4 To address this unmet medical need, novel therapeutic strategies are being evaluated in clinical trials including intracavitary approaches.
Cancer vaccines are intended to expand tumor antigen-specific CD4+ and CD8+ T lymphocytes. Still, the impact on the overall survival of current vaccines has been limited, and the only cancer vaccine approved by the FDA has been sipuleucel-T for the treatment of prostate cancer, which provides a median overall survival advantage of approximately 4 months.5 Several advances in the field hold promise for the next-generation vaccines. First, the advent of the anti-PD-(L)1 monoclonal antibody provides a means to unleash the antitumor potential of the vaccine-expanded T lymphocytes.6 Second, tumor neoantigens represent a pool of epitopes that evade the central tolerance mechanisms, and personalized antitumor vaccine based on tumor neoantigens has yielded promising results in phase I clinical trials.7 Third, the expression of co-stimulatory molecules and the tumor antigen might provide the required signals to ensure the optimal expansion, differentiation, and persistence of tumor-specific T lymphocytes.8–10 Finally, live viral vaccine vectors induce the release of danger- and pathogen-associated molecular patterns that provide adequate context for the activation of antigen-presenting cells at the same time, that they uptake and present tumor antigens expressed by the viral vectors.11 Modified vaccinia virus Ankara (MVA) is a replication-deficient strain in human cells and potent inductor of type I interferon. The large cloning capacity of the vectors allows for several transgenes and, therefore, the simultaneous expression of tumor antigens and co-stimulatory molecules. Safety and immunogenicity have been demonstrated in clinical trials, and the FDA has approved this vector as a non-replicating vaccine against smallpox and monkeypox.12
Recently, the expression of CD40 ligand or CD137 ligand with tumor-associated antigens by MVA vectors has been shown to enhance the antitumor efficacy of these vectors dramatically.13,14 CD40 plays a critical role in the activation of dendritic cells. Ligation of CD40 on the professional antigen-presenting cells triggers the production of IL-12 and licenses them for CD8+ T lymphocyte priming.15,16 CD137 is expressed in activated T lymphocytes, and its ligation promotes survival, expansion, and effector functions of activated T cells.17,18 Thus, CD40 and CD137 are potent immunostimulatory molecules with complementary mechanisms of action. The potential of combining both co-stimulatory pathways has been previously demonstrated in preclinical cancer models.19
In this study, we evaluated rMVA vector encoding tumor-associated antigens alone or co-expressed with CD40L, CD137L, or both (Combo) in two models representing ovarian and colon cancer peritoneal carcinomatosis. In these difficult-to-treat settings, the rMVA vector encoding tumor-associated antigens alone was inefficient, and the best results in terms of immune response activation and antitumor effect were achieved when both CD40L and CD137L were co-expressed along with tumor-associated antigens.
Results and discussion
Characterization of rMVAs encoding tumor antigens alone or in conjunction with CD40L and/or CD137L
In order to study the potential synergistic activity of co-expressing CD40L and CD137L in rMVA vectors for the treatment of peritoneal carcinomatosis, rMVAs encoding different combinations of tumor-associated antigens and co-stimulatory ligands were constructed and produced. These vectors conferred expression as a surrogate (ovalbumin – OVA-) or a bonafide tumor antigen (gp70) either alone or combined with CD40L and/or CD137L (rMVA-Combo) (Figure 1(a)). In vitro, these vectors infected efficiently Flt3L-derived mouse DCs, and the transgene-encoded co-stimulatory ligands were readily detected by flow cytometry. The expression levels of CD40L and CD137L exhibited a clear dose-response relationship (Figure 1(b)). In tumor-free animals, the intraperitoneal administration of all different rMVAs encoding OVA induced a potent adaptive response as measured by the OVA-specific IFN-γ production by T lymphocytes 1 week after vaccination. Interestingly, the co-expression of CD40L or CD137L alone did not enhance the OVA-specific IFN-γ release induced by the rMVA-OVA, illustrating the potent adjuvant activity of the vector. This response was improved by vector encoding both CD40L and CD137L, which significantly increased the OVA-specific T-cell immune response (Figure 1(c)).
Figure 1.
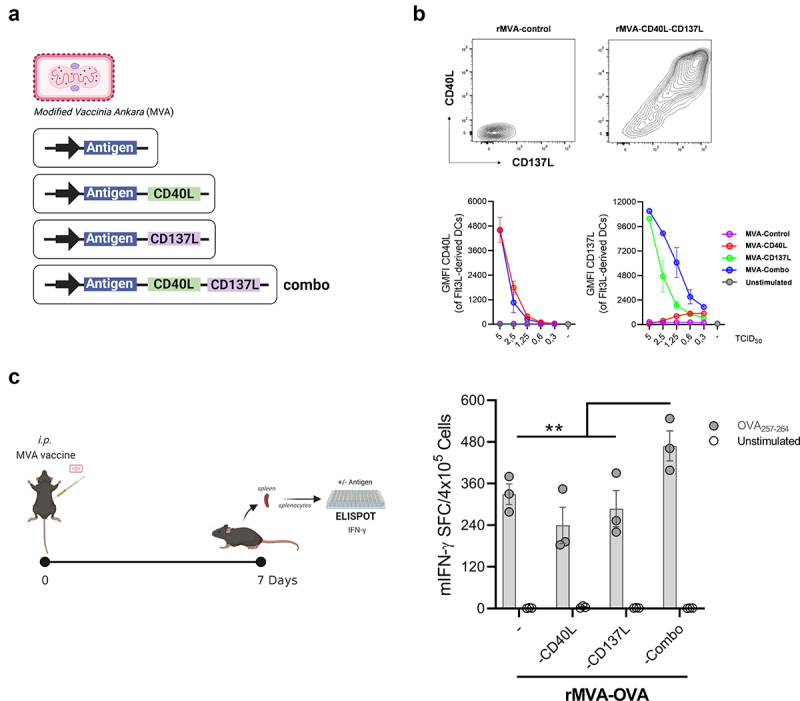
In vitro and in vivo rMVA characterization. (a) Schematic representation of rMVAs constructs. (b) Flow cytometry analysis of Flt3L-derived DCs infected for 18 h with rMVA-Control, rMVA-CD40L, rMVA-CD137L or rMVA-CD40L and rMVA-CD137L (rMVA-Combo). Geometric mean fluorescence intensity (GMFI) of CD40L and CD137L is shown. (c) OVA-specific IFN-γ–producing cells measured by ELISpot seven days after immunization of C57BL/6 mice (n = 3) with rMVA-OVA, rMVA-OVA-CD40L, rMVA-OVA-CD137L or rMVA-OVA-Combo i.p. administration. Data are represented as mean ± SEM. Two-way ANOVA followed by Sidak’s posttest. *p < .05. i.p, intraperitoneal; MVA, modified vaccinia virus Ankara; OVA, ovalbumin.
Loco-regional intracavitary treatment with rMVA-Combo elicits a potent effector T-cell activity and epitope spreading in models of peritoneal carcinomatosis
We intraperitoneally (i.p.) challenged mice with the ovarian cancer cell line ID8.OVA-Vegf/GFP to study the efficacy of rMVA with OVA as antigen in the different constructs. After inoculating ID8.OVA-Vegf/GFP i.p., we treated such mice with PBS (control group), rMVA-OVA, rMVA-OVA-CD40L, rMVA-OVA-CD137L, or rMVA-OVA-Combo. Two vector doses were administered at 7 and 21 days after the inoculation of tumor cells. At 21 days after the first dose (day +28 post-peritoneal carcinomatosis challenge), we collected the spleens to stimulate the splenocytes with OVA257-264 to study the production of IFN-γ by CD8+ T cell (Figure 2(a-b)).
Figure 2.
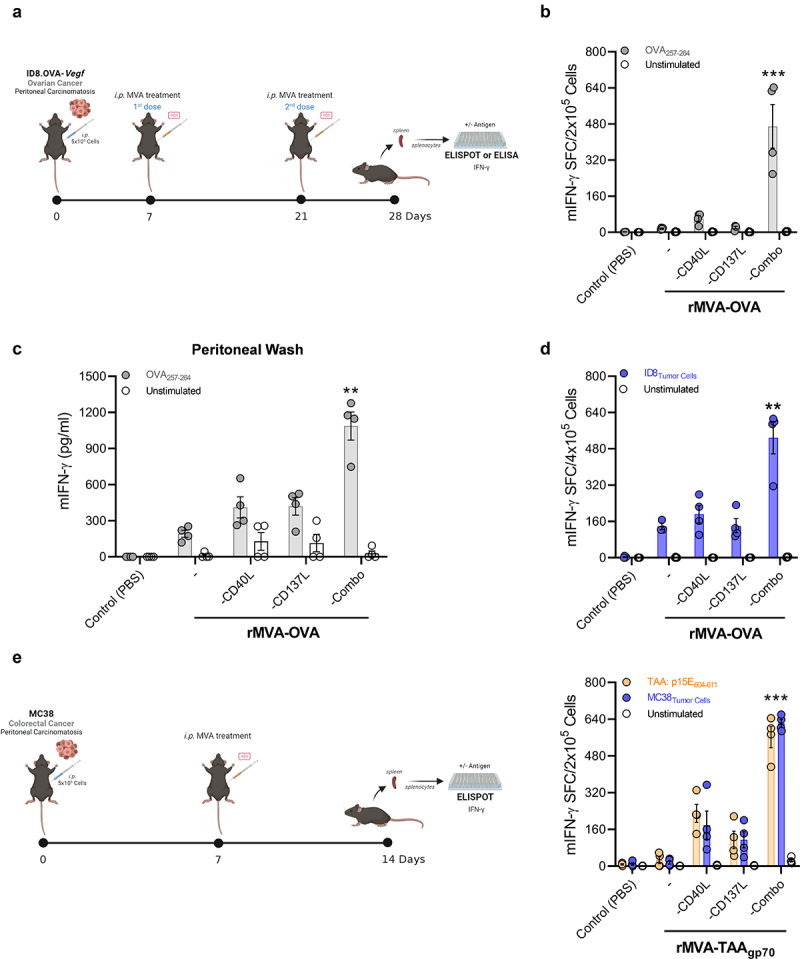
rMVA-Combo induces a potent antitumor-specific immune response in peritoneal carcinomatosis models by intracavitary administration. (a) C57BL/6 mice (n = 4) were challenged i.p. with 5 × 106 ID8.OVA-Vegf/GFP tumor cells. Seven days later, mice were treated by i.p. route with PBS (Control) or 5 × 107 TCID50 of the indicated rMVAs constructs. A second dose (boost) of treatment was administrated twenty-one days after tumor cells inoculation. One week after the boost, splenocytes and peritoneal washes were collected. (b) IFN-γ–producing cells measured by ELISpot in splenocytes stimulated with the OVA257-264 antigen. (c) IFN-γ–production measured by mouse IFN-γ ELISA in peritoneal washes stimulated with the OVA257-264 antigen. (d) IFN-γ–producing cells measured by ELISpot in splenocytes stimulated with 1:10 irradiated (20,000 rads) ID8-Vegf/GFP tumor cells. (e) C57BL/6 mice (n = 4) were challenged i.p. with 5 × 105 MC38 tumor cells. Seven days later, mice were treated by i.p. route with PBS (Control) or 5 × 107 TCID50 of the indicated rMVAs constructs. One week later, mice were euthanized and splenocytes were stimulated with p15E604-611 antigen. IFN-γ–producing cells were measured by ELISpot. Data are expressed as mean ± SEM. Results are representative of two independent experiments. Two-way ANOVA was performed followed by Sidak’s posttest. **p < .01; ***p < .005; i.p, intraperitoneal; MVA, modified vaccinia virus Ankara; OVA, ovalbumin; TCID50, 50% Tissue Culture Infectious Dose.
In contrast to the results obtained in tumor-free mice (Figure 1(c)), rMVA-OVA, rMVA-OVA-CD40L, rMVA-OVA-CD137L failed to activate an IFN-γ-mediated T-cell response. These results might indicate that the immunosuppressive mechanisms deployed by the presence of tumors in the peritoneal cavity abrogated the immunostimulatory effect of these vectors. Remarkably, in this aggressive tumor setting, the rMVA that encoded with both CD40L and CD137L was able to induce a potent effector T cell response with a drastic increase of IFN-γ secretion by antigen-specific CD8+ T cells when compared to rMVA-OVA-CD40L or rMVA-OVA-CD137L (Figure 2(b)). Cells retrieved from peritoneal lavages were also tested to study their specific IFN-γ production. Similarly, the maximum effect was observed in the rMVA-OVA-Combo group, leading to saturation of the signal in the conventional ELISpot assays (Supplementary Fig. 1). Furthermore, the quantification of IFN-γ concentration by ELISA in the supernatant of these in vitro stimulated cells obtained from peritoneal washes showed a clear superiority of the vector co-encoding CD40L and CD137L for the induction of IFN-γ production upon antigen restimulation (Figure 2(c)). Next, we incubated the splenocytes from the different experimental groups with the parental cell line ID8-Vegf/GFP, which did not express OVA, to evaluate the possibility of epitope spreading toward endogenous tumor antigens. Once again, the response in the rMVA-OVA-Combo group was significantly higher than in any of the other treated groups as measured by IFN-γ induction (Figure 2(d)). To further validate this finding, we also evaluated the antigen-specific T-cell response in another peritoneal carcinomatosis model generated by the colorectal cancer cell line (MC38), in this case analyzing the response against the tumor associate antigen (TAA) glycoprotein 70 (gp70) (Figure 2(e)). The synergy of CD40L and CD137L as transgenes in the MVA vectors was confirmed in this model in terms of higher IFN-γ secretion (Figure 2(e)).
Intracavitary rMVA-Combo improves survival in mice bearing peritoneal carcinomatosis and enhances loco-regional antitumor immune responses
Because rMVA-Ag-Combo treatment showed improved ex vivo effector responses in terms of IFN-γ production from antigen-specific T cells, we evaluated the in vivo rMVA-Ag-Combo efficacy challenging mice with ID8.OVA-Vegf/GFP to study the effect on survival. In line with the observed enhanced immune responses, mice treated with rMVA-OVA-Combo showed improved survival compared to the rMVA-OVA group (Figure 3(a)). Of note, rMVA-OVA-CD40L and rMVA-OVA-CD137L did not show significant differences. However, with rMVA-OVA-Combo, a considerable improvement was observed. In addition, at day 76 (when the first mice died in the untreated group), we barely detected any mice with ascites (5%) in the group treated with rMVA-OVA-Combo, whereas a higher proportion of mice showed ascites in the rest of the groups. Ascites were determined as abdominal distention and indicated progression of peritoneal carcinomatosis in ID8 models (Figure 3(a)). The superiority of the rMVA encoding both immunostimulatory molecules was confirmed in the colon carcinoma MC38 model of peritoneal carcinomatosis. In this setting, the retrovirally encoded antigen gp70 was used as a tumor-associated antigen. Treatment of MC38 established in the peritoneal cavity with the vector rMVA-gp70-Combo significantly extended mice survival (Supplementary Figure 2A). Mice that fully eradicated the peritoneal tumors were rechallenged subcutaneously with MC38 cells 90 days after the first MC38 peritoneal inoculation. All naïve mice developed subcutaneous tumors, while mice that had previously eradicated peritoneal tumors were resistant to the rechallenge, indicating the development of immune memory (Supplementary Figure 2B).
Figure 3.
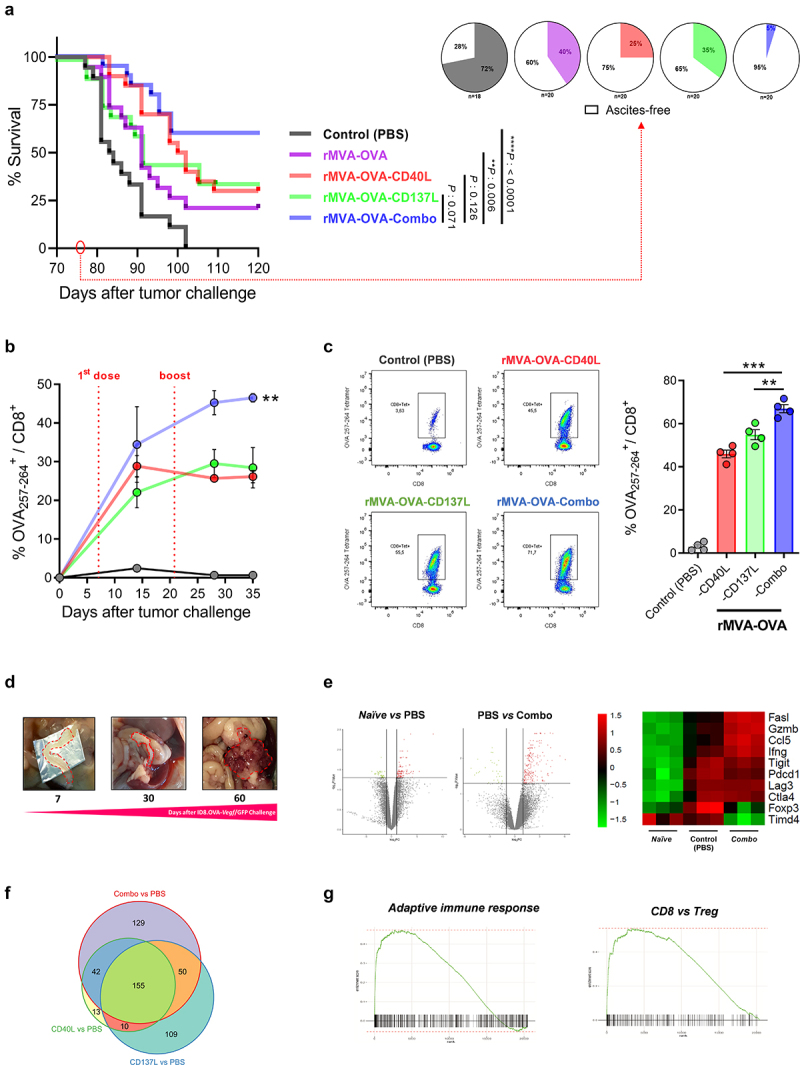
Loco-regional rMVA-Combo increases peritoneal carcinomatosis survival. C57BL/6 mice were challenged i.p. with 5 × 106 ID8.OVA-Vegf/GFP tumor cells. Seven and twenty-one days after tumor challenge, mice were treated by i.p. route with PBS (Control) or 5 × 107 TCID50 of the indicated rMVAs constructs. (a) Kaplan-Meier survival curve and the percentage of ascites development at 76 days after peritoneal carcinomatosis challenge were represented (n = 20). (b) Percentage of OVA257-264 Kb tetramer+/CD8+ cells 15, 28 and 35 days after tumor challenge in blood (n = 3), and (c) in peritoneal washes at 28 days after tumor challenge (n = 4). (d) Peritoneal carcinomatosis ID8.OVA-Vegf/GFP progression in the omentum. (e) Volcano plots and heatmap of z-scored log2CPM expression of relevant genes in the tumor microenvironment identified by RNA-seq analysis in the omentum between the tumor treated with PBS (Control group) vs. non-tumor (Naïve), and rMVA-OVA-Combo vs. PBS. (f) Venn diagram representing the number of genes modulated in each experimental condition. (g) Pathways identified using Gene set enrichment analysis (GSEA). Data are expressed as mean ± SEM. Results are representative of two independent experiments. Two-way ANOVA was performed followed by Sidak’s posttest. **p < .01; ***p < .005; i.p, intraperitoneal; MVA, modified vaccinia virus Ankara; OVA, ovalbumin; TCID50, 50% Tissue Culture Infectious Dose.
In parallel to the survival experiment, we analyzed the kinetics of OVA-specific T lymphocytes in circulation in the groups treated with rMVA-OVA-CD40L, rMVA-OVA-CD137L, and rMVA-OVA-Combo treatments. rMVA-OVA-Combo shows a slight increase of tetramer-positive T cells compared to the other experimental groups after the first dose. Furthermore, differences increased after the second viral administration, indicating a better persistence of the effector immune response in the rMVA-OVA-Combo group (Figure 3(b)). We also analyzed the expansion of tetramer-stained lymphocytes in peritoneal lavages. The results showed a more significant loco-regional increment of tetramer-positive T lymphocytes following treatment with rMVA-OVA-Combo (Figure 3(c)).
One of the most relevant tissues where to study peritoneal carcinomatosis is the omentum. In fact, the omentum is considered to be the origin of peritoneal carcinomatosis, particularly in patients with ovarian cancer. Moreover, the omentum is a critical tissue for the development of immune responses in the peritoneum. In mice challenged with ID8.OVA-Vegf/GFP, the omentum growth correlated with the lethal progression of peritoneal carcinomatosis (Figure 3(d) and Supplementary Figure 3). This was confirmed by quantification of bioluminescence after intraperitoneal administration of ID8.OVA-Vegf/GFP.LUC (Supplementary Fig. 3). Finally, RNA-seq was performed in the omentum to determine the impact of our best therapeutic strategy at a molecular level. Differential gene expression was observed between the non-tumor (Naïve) vs. tumor-untreated or Control (PBS), and Control (PBS) vs. MVA-OVA-Combo group (Figure 3(e) and Supplementary Figure 4).
Interestingly, the RNA expression results showed that tumor inoculation induced an endogenous immune response dampened by several immunosuppressive mechanisms as reflected by high levels of Foxp3, Lag3, Pdcd1, Ctla4, and Tigit, among others (Figure 3(e)). rMVA-OVA-Combo significantly increased the expression of relevant genes that are crucial for the effector antitumor immune responses, such as cytotoxic molecules (Ifng, Gzmb, FasL) and chemokines responsible for T-cell recruitment (Ccl5), while downregulated immunosuppressive genes (FoxP3, Timd4) (Figure 3(e)). Interestingly, 129 genes were modulated exclusively by the rMVA-Combo, indicating a synergistic effect of the immunostimulatory molecules (figure 3(f)). Gene Set Enrichment Analysis (GSEA) also demonstrated that the treatment with rMVA-OVA-Combo upregulated a pathway of genes involved in the adaptive immune response and an inferred increase in the ratio CD8:Tregs (Figure 3(g)).
In conclusion, we have demonstrated that rMVA encoded tumor-associated antigen can be synergistically improved when CD40L and CD137L are co-expressed as molecules promoting T-cell costimulation. The MVA vector with the three transgenes elicited stronger immune responses in mice bearing peritoneal carcinomatosis that clearly improved survival, crucially inducing epitope spreading toward endogenous antigens absent from the vector.
Material and methods
Reagents and cell lines
Peptides OVA257-264 and p15E604-611 were synthesized by GeneCust (Boynes, France) and had a purity above 95% as determined by HPLC and mass spectrometry. Recombinants MVA-BN were developed by Bavarian Nordic and are deposited at the European Collection of Cell Cultures (V00083008). All recombinants were generated from a cloned version of MVA-BN in a bacterial artificial chromosome. Infectious viruses were reconstituted from bacterial artificial chromosomes by transfecting bacterial artificial chromosome DNA into BHK-21 cells and superinfecting them with Shope fibroma virus as a helper virus. After three additional passages of primary embryo fibroblasts, helper virus-free MVA recombinant viruses were obtained. All viruses used in animal experiments were purified twice through a sucrose cushion. ID8.OVA-Vegf/GFP, ID8-Vegf/GFP, and MC38 tumor cell lines were obtained from Dr. George Coukos (Ludwig Cancer Research, Switzerland) and Dr. Karl E. Hellström (University of Washington, Seattle), respectively. ID8.OVA-Vegf/GFP cells were grown in Dulbecco’s modified Eagle’s, high-glucose medium (Invitrogen, Carlsbad, CA) supplemented with 4% fetal bovine serum (Gibco, Thermo Fisher Scientific, Waltham, MA), 100 U/mL penicillin, 100 μg/mL streptomycin (Gibco, Thermo Fisher Scientific, Waltham, MA), 5 μg/mL insulin, 5 μg/mL transferrin, and 5 ng/mL sodium selenite (Roche, Indianapolis, IN). MC38 cells were grown in Roswell Park Memorial Institute 1640 Medium with GlutaMAX supplemented with 10% fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin and 50 µM β-mercaptoethanol (Gibco, Thermo Fisher Scientific, Waltham, MA). All cell lines were grown in a humidified incubator with 5% CO2 at 37°C for at least 7 days before inoculation to mice. All cell lines were routinely tested for mycoplasma contamination using the MycoAlert Mycoplasma Detection Kit (Lonza, Basel, Switzerland).
Analysis of FLT3-Ligand-expanded DC infected with rMVAs
Dendritic cells from FLT3-ligand supplemented bone marrow cultures were generated as described by Minute L., et al.,20 and infected with increasing amounts of rMVA-Control (5PPC), rMVA-CD40L, rMVA-CD137L, or rMVA-Combo (TCID50: 0,3; 0,6; 1,25; 2,5; 5). After 18 h, the infected cells were analyzed for the expression of the co-stimulatory receptors by flow cytometry.
Animal handling
Experiments were performed with 6–8-week-old female C57BL/6 mice with body weights between 18 and 20 g purchased from Harlan Laboratories (Barcelona, Spain). Mice were maintained under pathogen-free conditions and were bred in a temperature-controlled animal facility with a 12 h light–dark cycle. The experimental design was approved by the Ethics Committee for Animal Testing of the University of Navarra (033–20).
Tumor implantation and treatment
Exponentially growing ID8.OVA-Vegf/GFP and MC38 cells were trypsinized and prepared as a single-cell suspension in ice-cold PBS. Animals were injected intraperitoneally with 300 μL of the ID8.OVA-Vegf/GFP and MC38 tumor cell suspension (5 x 106 and 5 × 105 cells, respectively). Seven days after tumor cell inoculation, mice were randomized and treated intraperitoneally with 200 µl of 5 × 107 TCID50 of the respective MVA recombinant or PBS (control group). A second dose (boost) of treatment was administrated 21 days after tumor cell inoculation when indicated. Mice were weighed and closely monitored for ascites development three times a week. Mice were euthanized if their bodyweight reached 28 g or clear signs of pain and distress appeared.
Samples processing
Peritoneal wash, spleens, and omentum were collected after 3 mL of pre-cold PBS (2% FBS) injection in the peritoneal cavity. After a gentle massage of the peritoneum to dislodge any attached cells, a 23 G needle was used to collect all the fluid. Supernatants from peritoneal washes were frozen at −20°C for further analysis. Spleens were mechanically disaggregated and filtered through a 70 μm cell strainer (Thermo Fisher Scientific, Waltham, MA). Splenocytes and peritoneal cells were depleted of erythrocytes, and single-cell suspensions were kept at 4°C until further analysis by ELISpot or flow cytometry. Omentum was weighted and frozen in RNAlater Stabilization Solution at −80°C until RNA isolation.
ELISpot and ELISA
Specific T-cell responses were assessed ex vivo by a mouse IFN-γ Enzyme-linked Immunosorbent Spot (ELISpot) Assay kit (BD-Biosciences). Ninety-six-well Multiscreen IP Plates (Millipore) were coated with 100 µl of assay diluent containing anti–IFN-γ monoclonal antibody and incubated overnight at 4°C. The plates were washed and then blocked with RPMI-1640 medium containing 10% fetal bovine serum (FBS) for 90 min at room temperature. Splenocytes depleted of erythrocytes were added to wells (4 x 105 or 2 × 105 cells) and stimulated with OVA257-264 peptide (1 µg/mL), p15E604-611 peptide (10 µg/mL), 1:10 irradiated (20,000 rads) ID8-Vegf/GFP or MC38 tumor cells in 200 µl/well. Prior to use, ID8-Vegf/GFP tumor cells as a stimulator were treated with 500 IU/mL of IFN-γ for 24 h to increase MHC-I expression. After 24 h of incubation with stimulators, the IFN-producing cells were measured by ELISpot according to manufacturer´s instructions. IFN-γ release levels were measured using BD OptEIA™ Mouse IFN-γ ELISA Set (BD-Biosciences, NJ) following the manufacturer’s recommendations after 48 h of antigen incubation.
Flow cytometry
For peripheral blood MHC-I-tetramer stainings, 100–150 μL of mouse peripheral blood samples collected in 25 μL of Heparine 1% (Mayne Pharma, Raleigh, NC) were directly stained with iTAg Tetramer/PE – H-2Kb OVA257-264 (MBL, Nagoya, Japan) in the presence of FcR-Block (anti-CD16/32 clone 93 BioLegend, San Diego, CA) following the manufacturer’s instructions. For splenocytes and peritoneal cells, single-cell suspensions were first stained with Zombi NIR Fixable viability kit (BioLegend, San Diego, CA) as a live/dead marker and then with iTAg Tetramer/PE – H-2Kb OVA257-264 (MBL, Nagoya, Japan) as described above. Once the samples were stained with the MHC-I -tetramer, co-stainings were performed using the following fluorochrome-labeled antibodies (BioLegend, San Diego, CA): anti-CD3-AF647 (17A2), anti-CD8-BV510 (53–6.7), anti-CD4-BV421 (GK1.5), anti-CD19-BV650 (6D5), and anti-CD45.2-FITC or -PrCPC5 (104). For peripheral blood samples, erythrocytes were lysed with FACS™ Lysis solution (BD-Biosciences, NJ). Flow cytometry was performed using the CytoFLEX S cytometer (Beckman Coulter, Brea, CA). Fluorescence minus one (FMO) or biological comparison controls were used for cell analysis. Data analysis was performed using FlowJo software (TreeStar, Ashland, OR).
mRNA isolation and RNA-seq
Total RNA extraction from omentum was performed using the RNeasy Mini Kit (Qiagen, Hilden, Germany), following the manufacturer’s recommendations. The concentration and RNA integrity of samples were determined using the Qubit RNA HS (High Sensitivity) Assay kit (Invitrogen, Waltham, MA) and the Agilent 2200 TapeStation (Agilent Technologies, Santa Clara, CA). mRNA samples were sent to Macrogen Inc. for library preparation using TruSeq Stranded Total RNA and sequenced to a depth of 80 M reads/sample using NovaSeq6000 (Illumina, San Diego, CA).
All sample processing and subsequent bioinformatics analysis were performed on a workstation equipped with 16x Intel Xeon W-2245 @ 4.7 GHz and 256 GB of RAM in a Linux system (Ubuntu 20.04). Quality control of all samples was performed with the FastQC tool (v.0.11.9) (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc). Before alignment, reads with low quality and adapters were removed using Trimmomatic (v.0.39).21 Alignment was performed with STAR (v.2.7.9a)22 with mm39 assembly as reference. The matrix of raw counts was obtained with featureCounts (v.2.0.0)23 and annotated with Gencode version M27. The analysis of differentially expressed genes was carried out in R/Bioconductor (v.4.1.1) following the bioinformatics workflow provided by edgeR.24 First, genes with less than 5 counts in all the samples (non-expressed genes) were removed from the analysis before normalization. The datasets were normalized using the TMM (trimmed mean of M-values) normalization, then the log2CPM values were calculated and the normalized expression matrix was used for the statistical analysis. We selected the set of genes differentially expressed for each comparison using the criteria of p value < .05%. Gene set enrichment analysis (GSEA) was performed with fgsea (https://doi.org/10.1101/060012) using the M5-GO:BP and C7-IMMNUNESIGDB gene sets from MSigDB v.7.4 database.25 Before running GSEA, the lists of expressed genes from the DEg analysis were pre-ranked by their t-statistic value without any type of filtering. The output of fgsea was filtered keeping those enriched pathways with p value < .05% and re-ranked by normalized enrichment score (NES). Raw data are publicly available on the GEO database with the GSE identifier (GSE196024).
Statistical analysis
GraphPad Prism V.8.2.1 software (GraphPad Software, San Diego, California, USA) was used for statistical analysis. Data were analyzed by two-way ANOVA followed by Sidak’s followed by multiple comparison test. The survival curves of animals treated with different rMVA vectors were plotted according to the Kaplan–Meier method and were compared using log-rank test. Statistical significance was considered when p < .05.
Supplementary Material
Acknowledgments
We are grateful to Paul Miller for English editing. Illustrative Figures 1c, 2a, and 2e were created by BioRender website platform. We would like to express our gratitude to the animal and cytometry facilities from Cima Universidad de Navarra for their support.
Funding Statement
This work was supported by Instituto de Salud Carlos III. (PI20/00002, PI19/01128) cofinanced by Fondos FEDER “A way to make Europe” and Joint Translational Call for Proposals 2015 (JTC 2015), TRANSCAN456 2 (code: TRS-2016-00000371), Gobierno de Navarra Proyecto LINTERNA Ref.: 0011-1411-2020-000075. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 765394. F.A. receives a Miguel Servet I (CP19/00114) contract from ISCIII (Instituto de Salud Carlos III) co-financed by FSE (Fondo Social Europeo) “Investing in your future.” Á.B. is recipients of PFIS fellowship from ISCIII (FI20/00058), and L.A. and M.F.-S. are recipients for a fellowship of the Aid Program Assigned to Projects from the University of Navarra. M.A. is supported by the Spanish Association Against Cancer’s (AECC) Investigator grant (INVES19041ALVA).
Competing interests
M.A. has received research grants from PharmaMar and Highlight Therapeutics. M.H. and H.H are employees of Bavarian Nordic. J.M-E. was employed by Bavarian Nordic when the studies were performed. I.M. reports receiving commercial research grants from BMS, Bioncotech, Alligator, Pfizer, Leadartis, and Roche; has received speakers bureau honoraria from MSD; and is a consultant or advisory board member for BMS, Roche, Genmab, F-Star, Bioncotech, Bayer, Numab, Pieris, Alligator, and Merck Serono. P.B. and F.A. received research grants from Bavarian Nordic.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2022.2098657
References
- 1.Coccolini F, Gheza F, Lotti M, Virzi S, Iusco D, Ghermandi C, et al. Peritoneal carcinomatosis. World J Gastroenterol. 2013;19(41):6979–9. doi: 10.3748/wjg.v19.i41.6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao Y, Chen C, Xu X, Ge X, Ding K, Zheng S, et al . An efficient prognostic immune scoring system for colorectal cancer patients with peritoneal metastasis. Oncoimmunology. 2021;10(1):1901464. doi: 10.1080/2162402X.2021.1901464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fucikova J, Coosemans A, Orsulic S, Cibula D, Vergote I, Galluzzi L, Spisek R.. Immunological configuration of ovarian carcinoma: features and impact on disease outcome. J Immunother Cancer. 2021;9(10):10. doi: 10.1136/jitc-2021-002873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malfroy S, Wallet F, Maucort-Boulch D, Chardonnal L, Sens N, Friggeri A, et al. Complications after cytoreductive surgery with hyperthermic intraperitoneal chemotherapy for treatment of peritoneal carcinomatosis: risk factors for ICU admission and morbidity prognostic score. Surg Oncol. 2016;25(1):6–15. doi: 10.1016/j.suronc.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 5.Handy CE, Antonarakis ES. Sipuleucel-T for the treatment of prostate cancer: novel insights and future directions. Future Oncol. 2018;14(10):907–917. doi: 10.2217/fon-2017-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zalba S, Belsue V, Topp B, de Alwis D, Alvarez M, Troconiz IF, Berraondo P, Garrido MJ. Modulation of intratumoural myeloid cells, the hallmark of the anti-tumour efficacy induced by a triple combination: tumour-associated peptide, TLR-3 ligand and alpha-PD-1. Br J Cancer. 2021;124(7):1275–1285. doi: 10.1038/s41416-020-01239-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ott PA, Hu-Lieskovan S, Chmielowski B, Govindan R, Naing A, Bhardwaj N, Margolin K, Awad MM, Hellmann MD, Lin JJ, et al. A phase Ib trial of personalized neoantigen therapy plus Anti-PD-1 in patients with advanced melanoma, non-small cell lung cancer, or bladder cancer. Cell. 2020;183(2):347–62 e24. doi: 10.1016/j.cell.2020.08.053. [DOI] [PubMed] [Google Scholar]
- 8.Allison JP, Hurwitz AA, Leach DR. Manipulation of costimulatory signals to enhance antitumor T-cell responses. Curr Opin Immunol. 1995;7(5):682–686. doi: 10.1016/0952-7915(95)80077-8. [DOI] [PubMed] [Google Scholar]
- 9.Paijens ST, Vledder A, Loiero D, Duiker EW, Bart J, Hendriks AM, Jalving M, Workel HH, Hollema H, Werner N, et al. Prognostic image-based quantification of CD8CD103 T cell subsets in high-grade serous ovarian cancer patients. Oncoimmunology. 2021;10(1):1935104. doi: 10.1080/2162402X.2021.1935104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petroni G, Buque A, Zitvogel L, Kroemer G, Galluzzi L. Immunomodulation by targeted anticancer agents. Cancer Cell. 2021;39(3):310–345. doi: 10.1016/j.ccell.2020.11.009. [DOI] [PubMed] [Google Scholar]
- 11.Guo ZS, Lu B, Guo Z, Giehl E, Feist M, Dai E, Liu W, Storkus WJ, He Y, Liu Z, et al. Vaccinia virus-mediated cancer immunotherapy: cancer vaccines and oncolytics. J Immunother Cancer. 2019;7(1):6. doi: 10.1186/s40425-018-0495-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pittman PR, Hahn M, Lee HS, Koca C, Samy N, Schmidt D, Hornung J, Weidenthaler H, Heery CR, Meyer TPH, et al. Phase 3 efficacy trial of modified vaccinia Ankara as a vaccine against smallpox. N Engl J Med. 2019;381(20):1897–1908. doi: 10.1056/NEJMoa1817307. [DOI] [PubMed] [Google Scholar]
- 13.Medina-Echeverz J, Hinterberger M, Testori M, Geiger M, Giessel R, Bathke B, Kassub R, Gräbnitz F, Fiore G, Wennier ST, et al. Synergistic cancer immunotherapy combines MVA-CD40L induced innate and adaptive immunity with tumor targeting antibodies. Nat Commun. 2019;10(1):5041. doi: 10.1038/s41467-019-12998-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hinterberger M, Giessel R, Fiore G, Graebnitz F, Bathke B, Wennier S, Chaplin P, Melero I, Suter M, Lauterbach H, et al. Intratumoral virotherapy with 4-1BBL armed modified vaccinia Ankara eradicates solid tumors and promotes protective immune memory. J Immunother Cancer. 2021;9(2):2. doi: 10.1136/jitc-2020-001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tay NQ, Lee DCP, Chua YL, Prabhu N, Gascoigne NRJ, Kemeny DM. CD40L expression allows CD8(+) T cells to promote their own expansion and differentiation through dendritic cells. Front Immunol. 2017;8:1484. doi: 10.3389/fimmu.2017.01484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aranda F, Llopiz D, Diaz-Valdes N, Riezu-Boj JI, Bezunartea J, Ruiz M, Martínez M, Durantez M, Mansilla C, Prieto J, et al. Adjuvant combination and antigen targeting as a strategy to induce polyfunctional and high-avidity T-cell responses against poorly immunogenic tumors. Cancer Res. 2011;71(9):3214–3224. doi: 10.1158/0008-5472.CAN-10-3259. [DOI] [PubMed] [Google Scholar]
- 17.Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellstrom KE, Mittler RS, Chen L. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med. 1997;3(6):682–685. doi: 10.1038/nm0697-682. [DOI] [PubMed] [Google Scholar]
- 18.Weigelin B, Bolanos E, Teijeira A, Martinez-Forero I, Labiano S, Azpilikueta A, Morales-Kastresana A, Quetglas JI, Wagena E, Sánchez-Paulete AR, et al. Focusing and sustaining the antitumor CTL effector killer response by agonist anti-CD137 mAb. Proc Natl Acad Sci U S A. 2015;112(24):7551–7556. doi: 10.1073/pnas.1506357112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eriksson E, Milenova I, Wenthe J, Stahle M, Leja-Jarblad J, Ullenhag G, et al. Shaping the tumor stroma and sparking immune activation by CD40 and 4-1BB signaling induced by an armed oncolytic virus. Clin Cancer Res. 2017;23(19):5846–5857. doi: 10.1158/1078-0432.CCR-17-0285. [DOI] [PubMed] [Google Scholar]
- 20.Minute L, Teijeira A, Sanchez-Paulete AR, Ochoa MC, Alvarez M, Otano I, et al. Cellular cytotoxicity is a form of immunogenic cell death. J Immunother Cancer. 2020;8(1):1. doi: 10.1136/jitc-2019-000325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 24.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.