

ABSTRACT
Endometrial carcinoma (EC) originates from the endometrium and is one of the most common tumors in female patients, and its incidence has continued to increase in recent decades. LncRNAs are involved in the pathogenesis and metastasis of a variety of malignant tumors, which indicates that lncRNAs can be used as tumor diagnostic markers and potential therapeutic targets. In this study, we analyzed the RNA transcripts of EC cells from The Cancer Genome Atlas (TCGA) and first reported a novel lncRNA, BMPR1B-AS1, that was more highly expressed in endometrial cancer tissues than in adjacent tissues, and BMPR1B-AS1 could promote endometrial cancer cell proliferation and metastasis. Bioinformatics prediction and experimental results both suggested that BMPR1B-AS1 could modulate the malignant behaviors of endometrial cancer cell lines by sponging miR-7-2-3p to modulate DCLK1, and a DCLK1 inhibitor blocked the activation of the PI3K/Akt/NF-κB signaling pathway. Collectively, this study suggests that the BMPR1B-AS1/miR-7-2-3p/DCLK1 axis contributes to the proliferation and metastasis of endometrial cancer cells via the PI3K/Akt/NF-κB pathway.
KEYWORDS: Endometrial cancer, proliferation, metastasis, lncRNA BMPR1B-AS1, ceRNA
Introduction
Endometrial carcinoma (EC) originates from the endometrium and is one of the most common tumors in female patients, and its incidence has continued to increase in recent decades [1]. Surgery combined with chemotherapy and radiotherapy can notably prolong the survival of patients with early EC. However, due to the lack of effective treatments for recurrent and metastatic tumors, the relative 5-year survival rate of EC patients has not significantly improved [1]. Therefore, a novel nonsurgical therapeutic option is required [2].
Long noncoding RNAs (lncRNAs) are a class of noncoding RNAs with lengths of greater than 200 nucleotides [3]. LncRNAs are important regulators of gene expression and chromatid modification and are involved in a wide variety of physiological and pathological processes throughout the body [3,4]. Notably, lncRNAs are involved in the pathogenesis and metastasis of a variety of malignant tumors, which indicates that lncRNAs can be used as tumor diagnostic markers and potential therapeutic targets [3,5]. However, the number of disease-related lncRNAs is far lower than the total number of known lncRNAs [6].
MicroRNAs (miRNAs) are endogenous, single-chain, noncoding RNAs that are approximately 22 nucleotides in length; miRNAs play important roles in cell proliferation, differentiation and apoptosis [7]. miRNAs can inhibit and degrade specific genes through cleavage or transcriptional repression [7]. Compared with that in normal cells, lncRNA and miRNA expression in cancer cells may be upregulated or downregulated, and lncRNAs and miRNAs regulate the development and metastasis of cancers [8–10].
LncRNAs can function via various mechanisms; in one such mechanism, lncRNAs may exert their biological effects through their ability to act as competitive endogenous RNAs (ceRNAs) for miRNAs [11]. Typically, miRNAs function as negative regulators of gene expression, and in the presence of ceRNAs, the negative regulatory effects of miRNAs on their targets is impaired [7,12,13]. Various lncRNAs were reported to function as ceRNAs and play crucial roles in the pathological mechanisms of EC. For instance, lncRNA NEAT1 promotes EC cell proliferation and invasion by regulating the miR-144-3p/EZH2 axis [10]. LINC00261 expression is downregulated in endometrial carcinoma cells and increases FOXO1 protein levels by reducing miR-182, miR-183, miR-153, miR-27a, and miR-96 expression to inhibit endometrial carcinoma cell proliferation, migration, and invasion [9]. Despite this knowledge, the key lncRNAs associated with EC remain to be identified and could be potential targets for EC treatment.
In this study, we analyzed the RNA transcripts of EC cells from The Cancer Genome Atlas (TCGA) and first reported a novel lncRNA, BMPR1B-AS1, that was more highly expressed in endometrial cancer tissues than in adjacent tissues. We explored the potential function of BMPR1B-AS1 in endometrial cell lines, and the results indicated that BMPR1B-AS1 could modulate the malignant behaviors of endometrial cell lines via the miR-7-2-3p/DCLK1 axis, and a DCLK1 inhibitor could block the activation of the PI3K/Akt/NF-κB signaling pathway. Collectively, our findings suggested that the BMPR1B-AS1/miR-7-2-3p/DCLK1 axis contributes to the proliferation and metastasis of endometrial cancer cells via the PI3K/Akt/NF-κB pathway, providing new targets for EC diagnosis and treatment.
Materials and methods
Bioinformatics analysis
The RNA transcription data from 405 EC samples were obtained from TCGA. The para-cancerous tissues and carcinoma tissues were distinguished according to the rules for naming TCGA samples, and the differential expression patterns were analyzed with restriction [the mean values of FPKM in the two groups were both greater than 1, |log2 fold change(FC)| >1.0, False Discovery Rate(FDR)<0.05]. MiRanda was used to predict potential target genes, and combined with literature review and test screening, miR-7-2-3p and DCLK1 were selected for further studies.
Patients and specimens
Ten cancer tissues and paired adjacent nontumor tissues (obtained from sites at least 2 cm from the tumor edge) were obtained from patients who were pathologically diagnosed with EC during surgery at the First Affiliated Hospital of Zhengzhou University (Zhengzhou, China) from October 2018 to April 2020. All the fresh specimens were rapidly frozen in liquid nitrogen and then stored in a − 80°C freezer. The protocol of this study was approved by the ethics committee of The First Affiliated Hospital of Zhengzhou University, and all the patients provided informed consent.
Cell culture and transfection
Three human EC cell lines (Ishikawa, Hec-1a and Hec-1b cells) were purchased from the Chinese Academy of Science Cell Bank (Shanghai, China). The Ishikawa, Hec-1a and Hec-1b cells were cultured separately in Roswell Park Memorial Institute (RPMI) 1640 (Solarbio, Beijing, China), DMEM (Solarbio), and MEM (Solarbio) supplemented with 10% fetal bovine serum (FBS, Gibco, CA, USA), 100 μg/mL streptomycin and 100 U/mL penicillin (Solarbio) at 37°C in 5% CO2. Lentivirus vectors (Wanleibio, Shengyang, China) were utilized to establish cell lines stably overexpressing BMPR1B-AS1, and the transfected cells were selected with puromycin (Solarbio) (Ishikawa cells: 3.5 μg/ml; Hec-1b cells: 2 μg/ml) for 1 week. miR-7-2-3p mimics and inhibitors were synthesized and purchased from GenePharma (Shanghai, China). Cell transfection was performed using Lipofectamine 3000 (Invitrogen, Carlsbad, USA) according to the manufacturer’s protocol. All the target sequences are shown in Table 1.
Table 1.
Sequences used in this study.
| ID | Sequences (5’ – 3 ’) |
|---|---|
| BMPR1B-AS1 forward | GTAAGCCACGGAGTCAA |
| BMPR1B-AS1 reverse | CCTTCGCAGTAACAACC |
| DCLK1 forward | TTCTCGGGCAGTGTCTT |
| DCLK1 reverse | TCACCTGTTTCCCATCC |
| β-actin forward | CACTGTGCCCATCTACGAGG |
| β-actin reverse | TAATGTCACGCACGATTTCC |
| miR-7-2-3p forward | CAACAAATCCCAGTCTACCTAA |
| miR-7-2-3p reverse | GCAGGGTCCGAGGTATTC |
| U6 forward | GCTTCGGCAGCACATATACT |
| U6 reverse | GCAGGGTCCGAGGTATTC |
| miR-7-2-3p mimic sense | CAACAAAUCCCAGUCUACCUAA |
| miR-7-2-3p mimic antisense | AGGUAGACUGGGAUUUGUUGUU |
| mimic NC sense | UUCUCCGAACGUGUCACGUTT |
| mimic NC antisense | ACGUGACACGUUCGGAGAATT |
| miR-7-2-3p inhibitor | UUAGGUAGACUGGGAUUUGUUG |
| inhibitor NC | CAGUACUUUUGUGUAGUACAA |
BMPR1B-AS1 WT AAATGAACACATTAAAATTAAGCCTGCAGTTTGAGGTAATGGAGAAGCACTGGAGATTTGTAAGCCACGGAGTCAAATGGTGGACTGGGATTTTCAGGAGATCATTTAGAGAGCAAGATCTTACCAAATCCTTTAGTCATGGTCTATTTCGTTGCACTCATATGGTTGTTACTGCGAAGGTGAAGAACTAATGACTGCAGCAGGAAAAAGAATTGGATGTGTCATGAATTATGGCCCTGCTTATACTTCTACTTCAACCGTAATCATTTGTTTAAACAAAAAGTTCTGCATTTGAATTGT
BMPR1B-AS1 MUT miR-7-2-3p
AAATGAACACATTAAAATTAAGCCTGCAGTTTGAGGTAATGGAGAAGCTGACCACTAAACAAAGCCACGGAGTCAAATGGTGGACTGGGATTTTCAGGAGATCATTTAGAGAGCAAGATCTTACCAAATCCTTTAGTCATGGTCTATTTCGTTGCACTCATATGGTTGTTACTGCGAAGGTGAAGAACTAATGACTGCAGCAGGAAAAAGAATTGGATGTGTCATGAATTATGGCCCTGCTTATACTTCTACTTCAACCGTAATCTAAACAATAAACAAAAAGTTCTGCATTTGAATTGT
Probe of FISH
BMPR1B-AS1 probe 1 TCGCA GAGAT GATAT AATGT CGTGG ATTTC AACTT TTCGG
2 GTCAT GGTCT ATTTC GTTGC ACTCA TATGG TTGTT ACTGC
3 CATAA ACTTA CTAAT TAACA CCTCT ATTCT CATGT ATCAA
18S rRNA probe GGGCAGACGTTCGAATGGGTCGTCGCCGCCACGGG
U6 snRNA probe CGTGTCATCCTTGCGCAGGGGCCATGCTAATCTTCTCTGT
DCLK1 WT TATGGACAACTACTGCTAGAAAGGTCCAATAAATGCTATTTGTTCTAAGTGGTAGATATTGCCAATTGAT
DCLK1 MUT miR-7-2-3p TATGGACAACTACTGCTAGAAAGGTCCAATAAATGCTTAAACAACTAAGTGGTAGATATTGCCAATTGAT
RNA extraction and reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from tissue samples or cultured cells with TRIzol reagent (Takara, Tokyo, Japan) according to the manufacturer’s instructions. The RNA concentration and quality were confirmed using a NanoDrop OneC Microvolume UV-Vis Spectrophotometer (Thermo, Waltham, USA). cDNA was generated using PrimeScript RT Master Mix (Takara). Then, RT-qPCR was conducted with SYBR Green qPCR Mix (BioTeke, Beijing, China). Each sample was standardized to the expression of U6 or β-actin as an internal control gene. Relative gene expression was calculated using the 2–ΔΔCT method. All the primer sequences are listed in Table 1.
Protein extraction and western blotting
Total protein was extracted from the EC cell lines using a radioimmunoprecipitation assay (RIPA) kit (Solarbio). The protein samples were separated by 10% SDS–PAGE (Sangon Biotech, Shanghai, China) and transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, Boston, USA) according to a standard process. After blocking in 5% skim milk (BD Difco, Maryland, USA) or 5% bovine serum albumin(BSA, Biofroxx, Einhausen, Germany) for 2 h, the membranes were incubated overnight at 4 oC with primary antibodies against DCLK1 (JA11-03, HUABIO, Hangzhou, China), E-cadherin (ab40772, Abcam, Cambridge, UK), N-cadherin (ab76011, Abcam), Cyclin D1 (26,939-1-AP, Proteintech, Wuhan, China), CDK4 (#12,790, Cell Signaling Technology, Massachusetts, USA), PI3K p85 (ab191606, Abcam), p-PI3K p85 (phospho Y607) (ab182651, Abcam), Akt (60,203-2-lg, Proteintech), p-Akt (phospho Ser 473) (66,444-1-lg, Proteintech), NF-κB p65 (ab76311, Abcam), or NF-κB p-p65 (phospho S536) (ab76302, Abcam), followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse secondary antibodies(LF101 or LF102, Epizyme, Shanghai, China) for 2 h (1:5000). β-actin (AP0060, Bioworld, Nanjing, China) or GAPDH (AP0063, Bioworld) antibodies were used as the internal controls at a dilution of 1:5000. The protein signals were analyzed using the enhanced chemiluminescence method (Millipore).
Fluorescence in situ hybridization
Digoxigenin (DIG)-labeled BMPR1B-AS1 probes were designed and synthesized by BOSTER (Wuhan, China). Fluorescent in situ hybridization experiments were conducted using a FISH Kit (GenePharma) according to the manufacturer’s instructions. Briefly, Ishikawa cells were fixed in 4% paraformaldehyde (PFA, Biosharp, Hefei, China) at room temperature for 15 min and then washed with PBS. The cells were then permeabilized with Triton X-100 (Beyotime, Shanghai, China) at 4°C for 15 min. Subsequently, the cells were incubated with DIG-labeled BMPR1B-AS1 probes or Control-FISH probes at 55°C for 4 h and washed with 2× PBS for 10 min. The signals were detected with SABC (streptavidin-biotin complex)-FITC (BOSTER). DAPI was used to counterstain the nuclei. Images were obtained by confocal microscopy. The probe sequences are presented in Table 1.
Dual-luciferase reporter assay
A luciferase reporter assay was conducted in 293 T cells. The BMPR1B-AS1 sequence containing the miR-7-2-3p binding site was cloned into the pmirGlo luciferase reporter vector (WT-pmirGlo-BMPR1B-AS1). A pmirGlo luciferase reporter vector containing a corresponding mutant sequence of BMPR1B-AS1 was also constructed (MUT-miR-7-2-3p-pmirGlo-BMPR1B-AS1) (all the sequences are listed in Table 1). WT-pmirGlo-BMPR1B-AS1 and MUT-miR-7-2-3p-pmirGlo-BMPR1B-AS1 were cotransfected with the miR-7-2-3p mimic or negative control (NC) to confirm direct binding between BMPR1B-AS1 and miR-7-2-3p. Additionally, 293 T cells were cotransfected with WT-pmirGlo-BMPR1B-AS1/MUT-miR-548c-3p-pmirGlo-BMPR1B-AS1 and miR-548c-3p mimics/mimics NC. The wild-type and mutant miR-7-2-3p-binding sites in DCLK1 were inserted into pmirGlo luciferase reporter vectors (WT-pmirGlo-DCLK1 and MUT-pmirGlo-DCLK1, respectively) to confirm direct binding between miR-7-2-3p and DCLK1. Luciferase and Renilla signals were measured by a Promega Dual-Luciferase® Reporter Assay System at 24 h and 48 h after transfection. The relative luciferase activity was normalized to the Renilla luciferase activity.
Cell proliferation
Cell proliferation was monitored with a cell counting kit-8 (CCK8, Dojin, Kumamoto, Japan). Briefly, a total of 3 × 103 cells were seeded in each well of a 96-well plate and cultured at 37°C in 5% CO2. At specific time points, 10 μl CCK‑8 solution was added to each well, and the plate was incubated at 37°C for 1 h. Then, cell viability was measured by a microplate reader (Thermo) at 450 nm.
Transwell assay
Cells (5 × 104 cells/well for migration assays or 2 × 105 cells/well for invasion assays) were seeded in 200 μl serum-free media into the upper chambers, which were covered with (invasion assays) or without (migration assays) 40 μl Matrigel (BD, New Jersey, USA). The lower chambers contained 600 μl medium supplemented with antibiotics and 10% FBS. After 24 h, the cells that had invaded or migrated were fixed with 4% PFA (Biosharp) for 20 minutes, and the cells in the lower chambers were stained with 0.4% crystal violet for 20 minutes. Finally, the cells were washed with PBS and counted using an inverted microscope (Olympus Corporation; magnification, 200x) with the image processing program ImageJ software version 1.8 (National Institute of Health). Images from 3 random fields of view were captured for each group for analysis.
Wound healing assay
Cells were seeded into 6-well plates at a density of 5 × 105 cells/well and cultured for 12 h. When the cell monolayer reached 80% confluence, a 200-μl pipette tip was used to create scratch wounds, and the cells were washed twice with serum-free medium to remove the cell debris. The cells were cultured in serum-free medium, and scratch-wound healing was observed by microscopy (Olympus Corporation) and recorded at 0 h, 24 h, and 48 h after scratching.
Cell cycle and apoptosis analysis
Cell cycle assays were performed using a Cell Cycle Detection kit (Beyotime). After digestion and collection, the cells were washed twice with precooled PBS and fixed with 70% ethanol (Aladdin, Shanghai, China) at 4°C overnight. Then, the ethanol was removed, and the cells were washed with PBS. After adding 100 μl RNaseA to the samples, the cells were incubated in a 37°C water bath for 30 minutes. Finally, 500 μl PBS containing 50 μg/ml propidium iodide (PI) was added to the samples and incubated at 4°C in the dark for 30 minutes. A NovoCyteTM Flow Cytometer (ACEA Biosciences, San Diego, USA) was used to analyze the results. An Annexin V-FITC kit (Beyotime) was used to assess cell apoptosis. After collection, the cells were washed twice with PBS, and 500 μl binding buffer was added to resuspend the cells. The samples were mixed well with 5 μl Annexin V-FITC and 10 μl PI and incubated at room temperature in the dark for 15 minutes. A NovoCyteTM Flow Cytometer (ACEA Biosciences) was used to analyze the results.
Tumor xenografts in nude mice
5-week-old female BALB/c nude mice used for this study were purchased from Beijing Huafukang Bioscience Co., ltd., China and maintained in pathogen-free conditions. The animals were divided into 5 groups: Lv-BMPR1B-AS1 group, Lv-BMPR1B-AS1+ agomir-7-2-3p group, Lv-BMPR1B-AS1+ agomir-7-2-3p NC group, Lv-NC group, and blank Hec-1b group. Each mouse was injected subcutaneously in the right flank area with 5 × 106 cells. The agomir-7-2-3p(1 OD, GenePharma) or agomir NC(1 OD) was administrated by intra-tumoral injection once every 3 days after 10 days of inoculation(to allow for tumor growth). The tumor volume was calculated and recorded every 3 days according to the formula: volume(mm3) = length(mm) × width(mm)2 /2. After 25 days, all the mice were sacrificed, and the tumors were removed and weighed. The tumor tissues were subjected to RT-qPCR, and immunohistochemistry.
Immunohistochemistry (IHC)
The tissues were deparaffinized and rehydrated, and then were placed in a retrieval box filled with citric acid antigen retrieval buffer (PH6.0) for antigen retrieval in a microwave oven. DCLK1 antibody (JA11-03, HUABIO) and corresponding secondary antibody were sequentially incubated. Afterward, the freshly prepared DAB (Servicebio, Wuhan, China) solution was applied onto each section, and the nuclei were counterstained with hematoxylin.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, USA). Student’s t-test was used to analyze the differences between two groups. Spearman’s correlation analysis was employed to determine the correlation between two parameters. All the experiments were repeated at least three independent times. The data are presented as the mean ± SD. Statistical significance was defined as p < 0.05 (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Results
The expression of BMPR1B-AS1 was upregulated in endometrial cancer tissues
Analysis of the differential expression of lncRNAs between the paracancerous tissues and carcinoma tissues showed that the expression of 35 lncRNAs was decreased, while the expression of 39 lncRNAs was increased (the mean values of FPKM in the two groups were both greater than 1, |log2 fold change(FC)| >1.0, FDR <.005) (Figure 1a, Sup 1 and 2). Among these lncRNAs with altered expression, the expression of lncRNA BMPR1B-AS1 was most significantly increased. Then, we collected 28 tumor tissues and corresponding paracancerous tissues from EC patients who were pathologically diagnosed, and RT-qPCR was used to detect the expression of BMPR1B-AS1 in these samples. The results showed that the expression of BMPR1B-AS1 in the tumor tissues was significantly higher than that in the adjacent tissues (Figure 1b).
Figure 1.
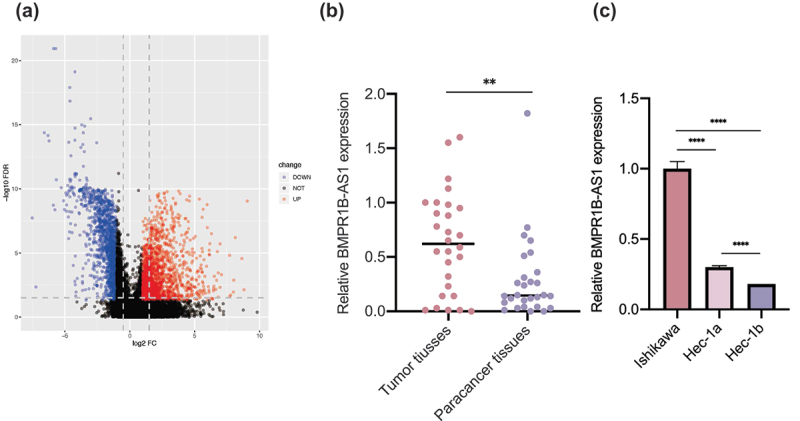
The expression of lncRNA BMPR1B-AS1 in endometrial cancer tissues and cell lines. A, Volcano map of dysregulated lncRNAs between EC tissues and adjacent normal tissues. The results were obtained from TCGA database. B, Expression levels of lncRNA BMPR1B-AS1 in paired EC and adjacent noncancerous tissues (n = 28). C, RT-qPCR analysis of BMPR1B-AS1 expression in human EC cell lines (Ishikawa, Hec-1a and Hec-1b cells). The data are representative of three independent experiments. Bars, ±SD. **p < 0.01, ****p < 0.0001.
Overexpression of BMPR1B-AS1 promotes malignant behaviors of Hec-1b cells
Because of the higher expression of BMPR1B-AS1 in EC tissues, we further explored the oncogenic role of BMPR1B-AS1 in EC cells. First, different expression levels of BMPR1B-AS1 were detected in EC cell lines (Ishikawa, Hec-1a and Hec-1b cells) by RT-qPCR, and the results demonstrated that Ishikawa cells had the highest expression, Hec-1a had intermediate expression, and Hec-1b had the lowest expression (Figure 1c). Then, we used Lv-BMPR1B-AS1 to overexpress BMPR1B-AS1 in Hec-1b cells. The BMPR1B-AS1 overexpression efficiency was confirmed by RT-qPCR (Figure 2a). According to the CCK8 assay, BMPR1B-AS1 overexpression significantly promoted cell growth compared with Lv-NC (Figure 2b). Both wound healing assays and Transwell migration assays revealed that overexpression of BMPR1B-AS1 facilitated the migration of Hec-1b cells (Figure 2c, 2e, 2g, and 2h). Transwell invasion assays showed that the invasion capability of Hec-1b cells was enhanced after BMPR1B-AS1 overexpression (Figure 2d and 2f). Subsequently, apoptosis was inhibited in the Lv-BMPR1B-AS1 group (Figure 2i and 2k). With BMPR1B-AS1 overexpression, we observed increased accumulation of cells in the S-phase and decreased accumulation of cells in the G0/G1 phase, which was consistent with the CCK8 results. (Figure 2j and 2l). In addition, the expression of E-cadherin, N-cadherin, Cyclin D1 and CDK4, which act as key players in tumor cell dissemination and proliferation, was examined by western blotting (Figure 2m-q). The results indicated that overexpression of BMPR1B-AS1 led to decreased expression of E-cadherin but increased expression of N-cadherin, Cyclin D1 and CDK4. These results collectively suggested that overexpression of BMPR1B-AS1 promoted the proliferation, migration and invasion of Hec-1b cells while inhibiting apoptosis.
Figure 2.
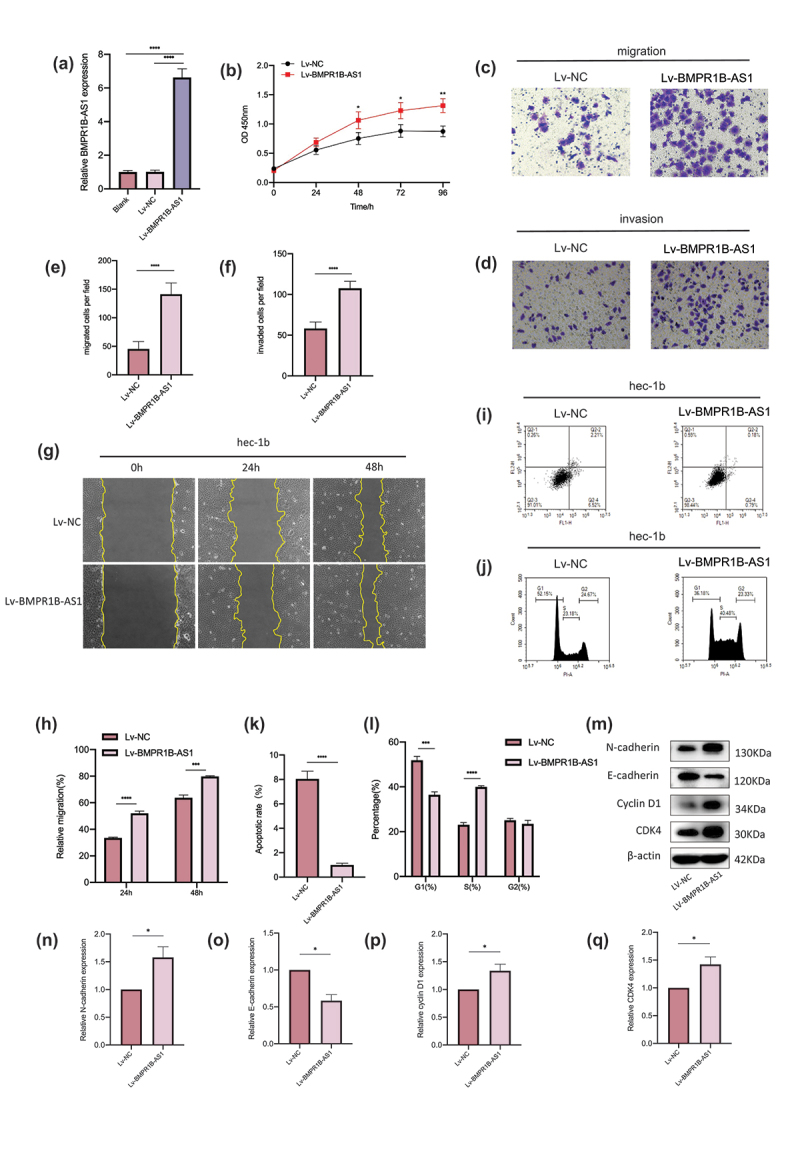
Overexpression of BMPR1B-AS1 promotes the proliferation, migration and invasion of Hec-1b cells while inhibiting apoptosis. A, Overexpression of BMPR1B-AS1 was confirmed by RT-qPCR. B, Overexpression of BMPR1B-AS1 promoted the proliferation of Hec-1b cells. C, E, G, H, Transwell migration assay (magnification, 200×) and wound healing assay (magnification, 100×) showed that overexpression of BMPR1B-AS1 facilitated the migration of Hec-1b cells. D, F, Transwell invasion assay showed that overexpression of BMPR1B-AS1 facilitated the invasion of Hec-1b cells (magnification, 200×). I, K, Flow cytometry results showed that BMPR1B-AS1 overexpression inhibited Hec-1b cell apoptosis. J, L, Flow cytometry results showed that BMPR1B-AS1 overexpression increased the accumulation of cells in the S-phase and decreased the accumulation of cells in the G0/G1 phase. M, N, O, P, Q, Western blot and analysis showed that the protein level of E-cadherin was decreased, but the protein levels of N-cadherin, Cyclin D1 and CDK4 were increased in the BMPR1B-AS1 overexpression group. Lv, lentiviral vector. The data are representative of three independent experiments. Bars, ±SD. *P < .05, **P < .01, ***P < .001, ****P < 0.0001.
Knockdown of BMPR1B-AS1 expression inhibits the malignant behaviors of Ishikawa cells
Next, Ishikawa cells were transfected with Lv-sh-BMPR1B-AS1 to knockdown the expression of BMPR1B-AS1. The knockdown efficiency was determined by RT-qPCR (Figure 3a). According to the CCK8 assay, transfection with Lv-sh-BMPR1B-AS1 significantly inhibited cell growth compared with transfection with Lv-sh-NC (Figure 3b). Both wound healing assays and Transwell migration assays revealed that knockdown of BMPR1B-AS1 expression inhibited the migration of Ishikawa cells (Figure 3c, 3e, 3g, and 3h). Transwell invasion assays showed that the invasion of Ishikawa cells was inhibited after BMPR1B-AS1 knockdown (Figure 3d and 3f). Subsequently, the apoptosis rate was significantly increased in the Lv-sh-BMPR1B-AS1 group (Figure 3i and 3k). With BMPR1B-AS1 knockdown, we observed increased accumulation of cells in the G0/G1 phase and decreased accumulation of cells in the S-phase, which was consistent with the CCK8 results (Figure 3j and 3l). Western blotting assays indicated that knockdown of BMPR1B-AS1 expression led to increased expression of E-cadherin but decreased expression of N-cadherin, Cyclin D1 and CDK4 (Figure 3m-q). These results collectively suggested that knockdown of BMPR1B-AS1 expression inhibited the proliferation, migration and invasion of Ishikawa cells while promoting apoptosis.
Figure 3.
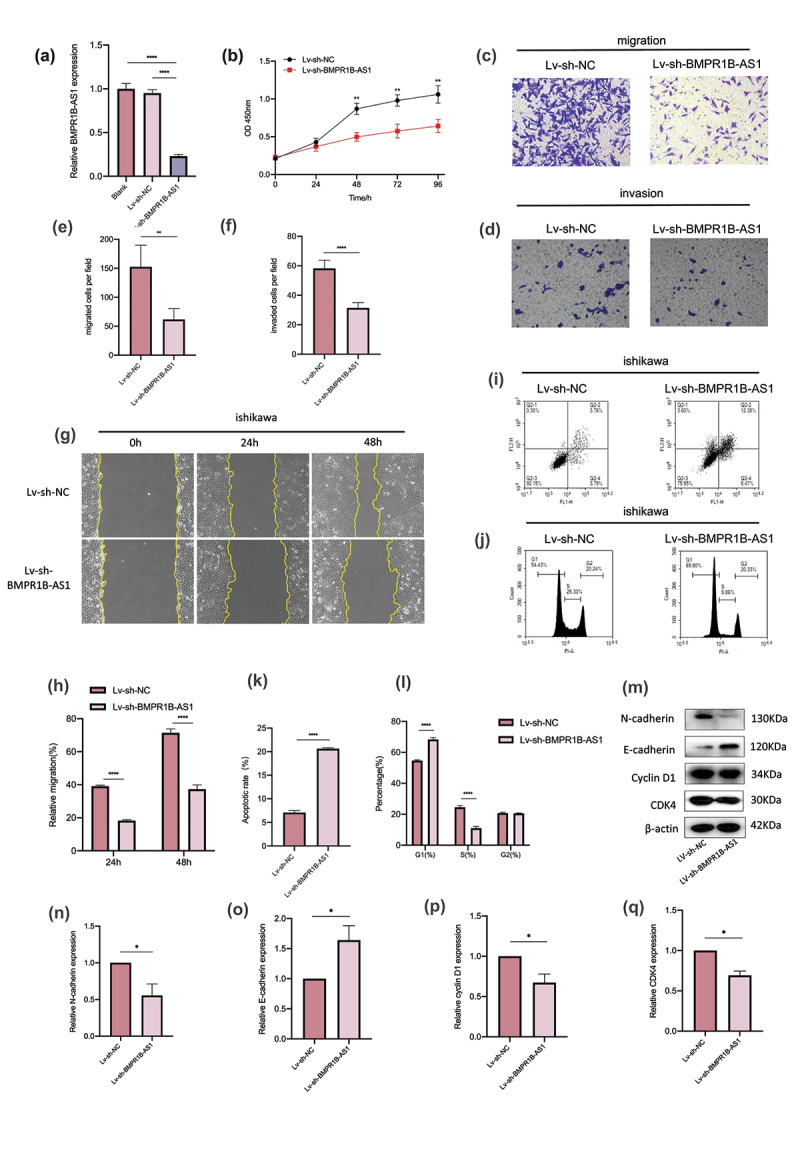
Knockdown of BMPR1B-AS1 expression inhibits the proliferation, migration and invasion of Ishikawa cells while promoting apoptosis. A, BMPR1B-AS1 knockdown efficiency was confirmed by RT-qPCR. B, Knockdown of BMPR1B-AS1 expression inhibited the proliferation of Ishikawa cells. C, E, G, H, Transwell migration assay (magnification, 200×) and wound healing assay (magnification, 100×) showed that knockdown of BMPR1B-AS1 expression decreased the migration of Ishikawa cells. D, F, Transwell invasion assay showed that knockdown of BMPR1B-AS1 expression decreased the invasion of Ishikawa cells (magnification, 200×). I, K, Flow cytometry results showed that BMPR1B-AS1 knockdown facilitated Ishikawa cell apoptosis. J, L, Flow cytometry results showed that BMPR1B-AS1 knockdown increased the accumulation of cells in the G0/G1 phase and decreased the accumulation of cells in the S-phase. M, N, O, P, Q, Western blot and analysis showed that the protein level of E-cadherin was increased, but the protein levels of N-cadherin, Cyclin D1 and CDK4 were decreased in the BMPR1B-AS1 knockdown group. Sh, short hairpin. The data are representative of three independent experiments. Bars, ±SD. **P < .01, ***P < .001, ****P < 0.0001.
BMPR1B-AS1 targets miR-7-2-3p and negatively regulates miR-7-2-3p expression
After BMPR1B-AS1 was shown to play a vital role in EC cell malignant behaviors, we further investigated the mechanism by which BMPR1B-AS1 functions in this process. ceRNA mechanisms are prevalent mechanisms by which lncRNAs exert their oncogenic or anti-oncogenic effects. FISH assays showed that BMPR1B-AS1 was mainly located in the cytoplasm (Figure 4a). Therefore, we hypothesized that BMPR1B-AS1 could bind to miRNAs to regulate EC progression. Then, we used bioinformatics prediction programs (miRdanda) to predict miRNAs that potentially interacted with BMPR1B-AS1 (Sup 3 and 4). Among the 10 identified miRNAs, miR-548c-3p and miR-7-2-3p were confirmed to be tumor suppressor factors in many other cancers. We cotransfected HEK293T cells with a wild-type (WT) or a mutant (MUT) dual-luciferase BMPR1B-AS1 construct and the two miRNAs and performed a dual-luciferase assay. Cotransfection of the miR-7-2-3p mimic and BMPR1B-AS1-WT decreased the luciferase activity, while cotransfection of the miR-7-2-3p mimic and BMPR1B-AS1-MUT and cotransfection of the miR-7-2-3p mimic NC and BMPR1B-AS1-WT did not change the luciferase activity (Figure 4d and 4e). However, cotransfection of the miR-548c-3p mimic and BMPR1B-AS1-WT did not change the luciferase activity compared to cotransfection of the miR-548c-3p mimic and BMPR1B-AS1-MUT and cotransfection of the miR-548c-3p mimic NC and BMPR1B-AS1-WT (Sup 4 and 5). Meanwhile, we also identified that BMPR1B-AS1 expression and miR-7-2-3p expression were significantly negatively correlated in EC tissues (Figure 4c). The expression of miR-7-2-3p was significantly decreased after BMPR1B-AS1 overexpression in Hec-1b cells, and then, an effective miR-7-2-3p mimic was used to rescue the BMPR1B-AS1-overexpressing cell line, causing an increase in miR-7-2-3p expression (figure 4f, 4h and 4i). In contrast, the expression of miR-7-2-3p was significantly increased after BMPR1B-AS1 knockdown in Ishikawa cells, and then, an effective miR-7-2-3p inhibitor was used to rescue the BMPR1B-AS1-knockdown cell line, leading to a continuous decrease in miR-7-2-3p expression (Figure 4g, 4j and 4k). Taken together, these data suggested that BMPR1B-AS1 could function as an efficient miR-7-2-3p sponge in endometrial cancer cells.
Figure 4.
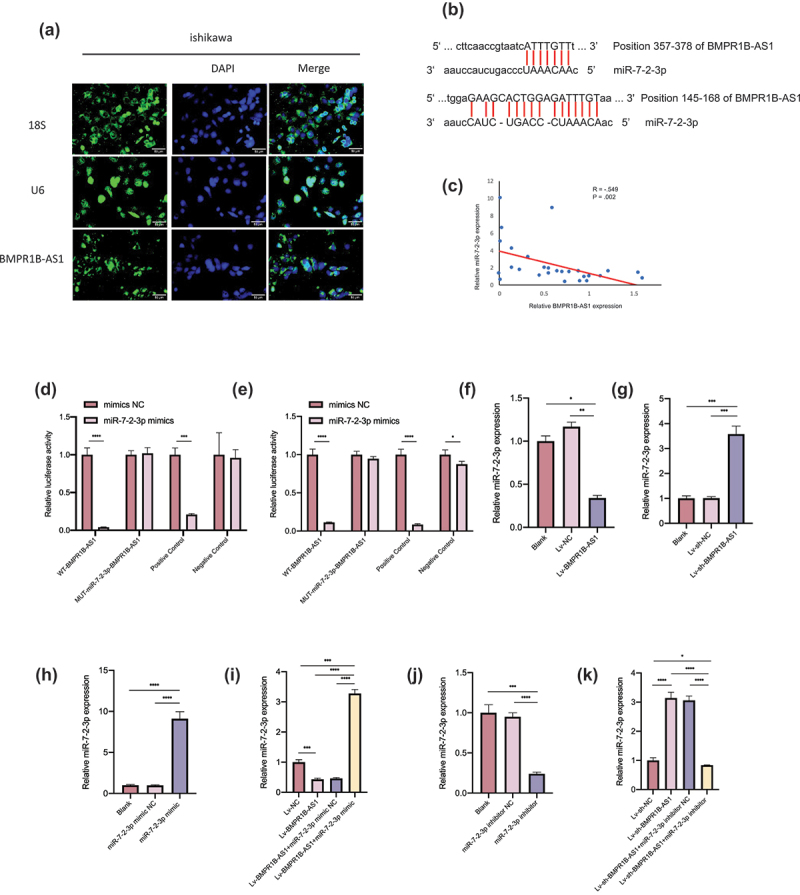
BMPR1B-AS1 functions as an efficient miR-7-2-3p sponge in Hec-1b and Ishikawa cells. A, FISH assay showed that the intracellular localization of BMPR1B-AS1 was mainly in the cytoplasm in Ishikawa cells. 18S and U6 were used as controls. B, Putative binding sites of miR-7-2-3p and BMPR1B-AS1. C, Negative correlation was found between BMPR1B-AS1 expression and miR-7-2-3p expression among the 28 endometrial cancer tissue specimens. R = −.549, P = .002. D, E, The effect of the miR-7-2-3p mimic on the luciferase activities of 293 T cells transfected with WT or MUT BMPR1B-AS1 was detected 24 h and 48 h after transfection, respectively. F, RT-qPCR results showed that miR-7-2-3p expression was downregulated after BMPR1B-AS1 overexpression in Hec-1b cells. G, RT-qPCR results showed that miR-7-2-3p expression was upregulated after BMPR1B-AS1 knockdown in Ishikawa cells. H, miR-7-2-3p expression was upregulated after transfection with the miR-7-2-3p mimic in Hec-1b cells. I, The miR-7-2-3p mimic reversed the BMPR1B-AS1-mediated downregulation of miR-7-2-3p expression. J, miR-7-2-3p expression was downregulated after transfection of Ishikawa cells with the miR-7-2-3p inhibitor. K, The miR-7-2-3p inhibitor attenuated the BMPR1B-AS1-mediated upregulation of miR-7-2-3p expression. The data are representative of three independent experiments. Bars, ±SD. *P < .05, **P < .01, ***P < .001, ****P < 0.0001.
Upregulation of miR-7-2-3p expression rescues the BMPR1B-AS1 overexpression-induced progression of Hec-1b cells.
Rescue experiments were conducted to confirm that BMPR1B-AS1 acts as an oncogene by directly targeting miR-7-2-3p. Cotransfection with the miR-7-2-3p mimic inhibited the proliferation of the BMPR1B-AS1-overexpressing cell lines (Figure 5a). Transwell assays and wound healing assays both indicated that BMPR1B-AS1-overexpressing cell lines cotransfected with the miR-7-3p mimic had lower migration and invasion capabilities than those cotransfected with the miR-7-2-3p mimic NC (Figure 5b-5g). The apoptosis and cell cycle inhibition by Lv-BMPR1B-AS1 was reversed by cotransfection of the miR-7-2-3p mimic (Figure 5h-k).
Figure 5.
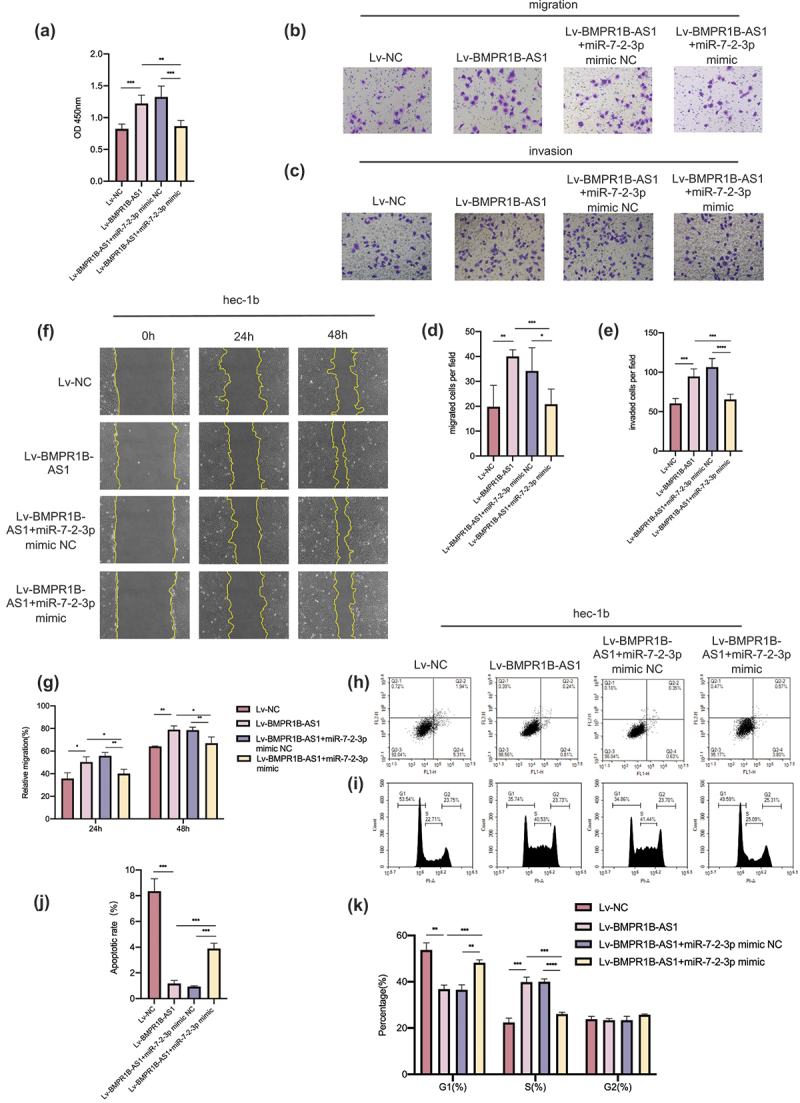
Upregulation of miR-7-2-3p expression reverses the BMPR1B-AS1 overexpression-induced progression of Hec-1b cells. A, CCK8 assay showed that cotransfection with the miR-7-2-3p mimic suppressed the BMPR1B-AS1 overexpression-induced proliferation of Hec-1b cells. B, D, F, G, Transwell migration assay (magnification, 200×) and wound healing assay (magnification, 100×) showed that cotransfection with the miR-7-2-3p mimic suppressed the BMPR1B-AS1 overexpression-induced migration of Hec-1b cells. C, E, Transwell invasion assay (magnification, 200×) showed that cotransfection with the miR-7-2-3p mimic suppressed the BMPR1B-AS1 overexpression-induced invasion of Hec-1b cells. H-K, Flow cytometry showed that cotransfection with the miR-7-2-3p mimic accelerated apoptosis and cell cycle arrest, and these effects were inhibited by BMPR1B-AS1 overexpression in Hec-1b cells. The data are representative of three independent experiments. Bars, ±SD. *P < .05, **P < .01, ***P < .001, ****P < 0.0001.
Downregulation of miR-7-2-3p expression reverses the inhibition of Ishikawa cells mediated by the knockdown of BMPR1B-AS1 expression.
Cotransfection with the miR-7-2-3p inhibitor promoted the proliferation of the BMPR1B-AS1-knockdown cell lines (Figure 6a). Transwell assays and wound healing assays both showed that BMPR1B-AS1 knockdown cell lines cotransfected with the miR-7-3p inhibitor had higher migration and invasion capabilities than those cotransfected with the miR-7-2-3p inhibitor NC (Figure 6b-g). Cotransfection with the miR-7-2-3p inhibitor inhibited apoptosis and cell cycle arrest, and these effects were reversed by knockdown of BMPR1B-AS1 expression in Ishikawa cells (Figure 6h-k).
Figure 6.
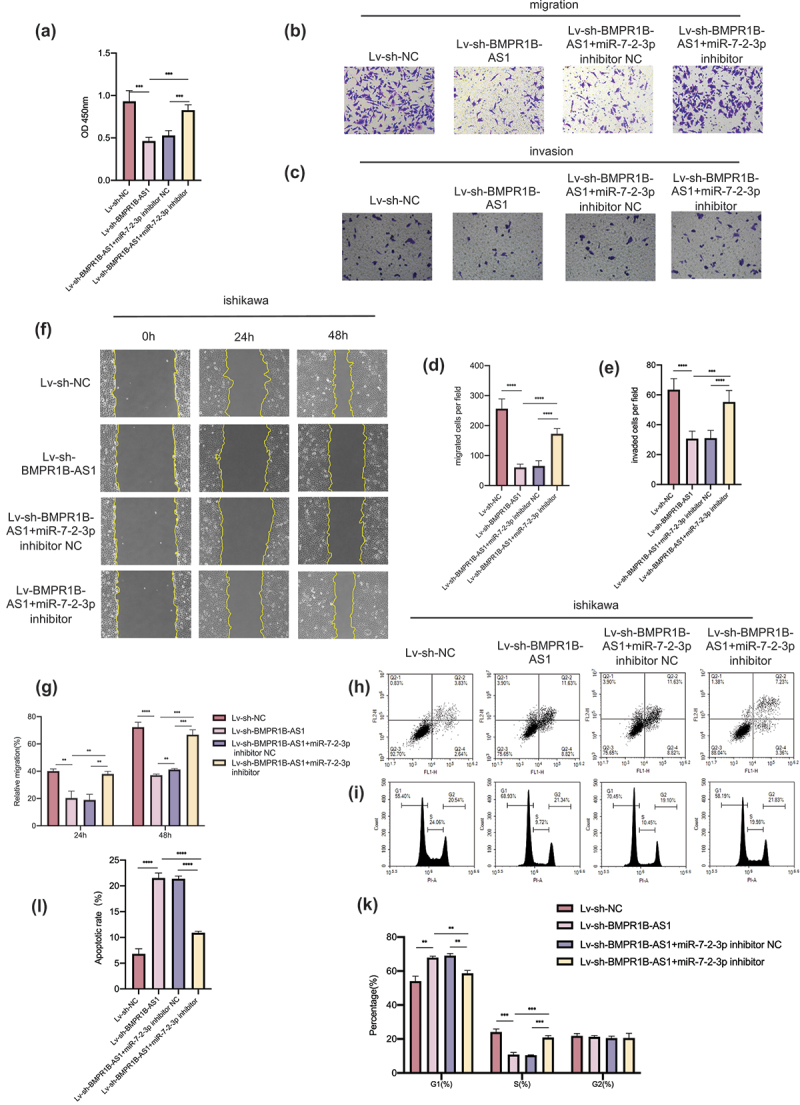
Downregulation of miR-7-2-3p expression reverses the BMPR1B-AS1 knockdown-mediated inhibition of Ishikawa cells. A, CCK8 assay showed that cotransfection with the miR-7-2-3p inhibitor reversed the BMPR1B-AS1 knockdown-mediated inhibition of proliferation of Ishikawa cells. B, D, F, G, Transwell migration assay (magnification, 200×) and wound healing assay (magnification, 100×) showed that cotransfection with the miR-7-2-3p inhibitor reversed the BMPR1B-AS1 knockdown-mediated inhibition of migration of Ishikawa cells. C, E, Transwell invasion assay (magnification, 200×) showed that cotransfection with the miR-7-2-3p inhibitor reversed the BMPR1B-AS1 knockdown-mediated inhibition of invasion of Ishikawa cells. H-K, Flow cytometry showed that cotransfection with the miR-7-2-3p inhibitor inhibited apoptosis and cell cycle arrest, and these effects were enhanced by BMPR1B-AS1 knockdown in Ishikawa cells. The data are representative of three independent experiments. Bars, ±SD. **P < .01, ***P < .001, ****P < 0.0001.
DCLK1 is a target gene of miR-7-2-3p
DCLK1 was predicted to be a target gene of miR-7-2-3p (Figure 7a). A dual-luciferase assay was conducted to elucidate whether miR-7-2-3p can interact with DCLK1. The results showed that cotransfection of DCLK1-WT and the miR-7-2-3p mimic decreased the luciferase activity of the cells, whereas cotransfection of DCLK1-MUT and the miR-7-2-3p mimic and cotransfection of DCLK1-WT and the miR-7-2-3p mimic NC did not change the luciferase activity of the cells (Figure 7b and 7c). Also, we found a negative relationship between miR-7-2-3p expression and DCLK1 mRNA expression in EC tissues (Figure 7d).
Figure 7.
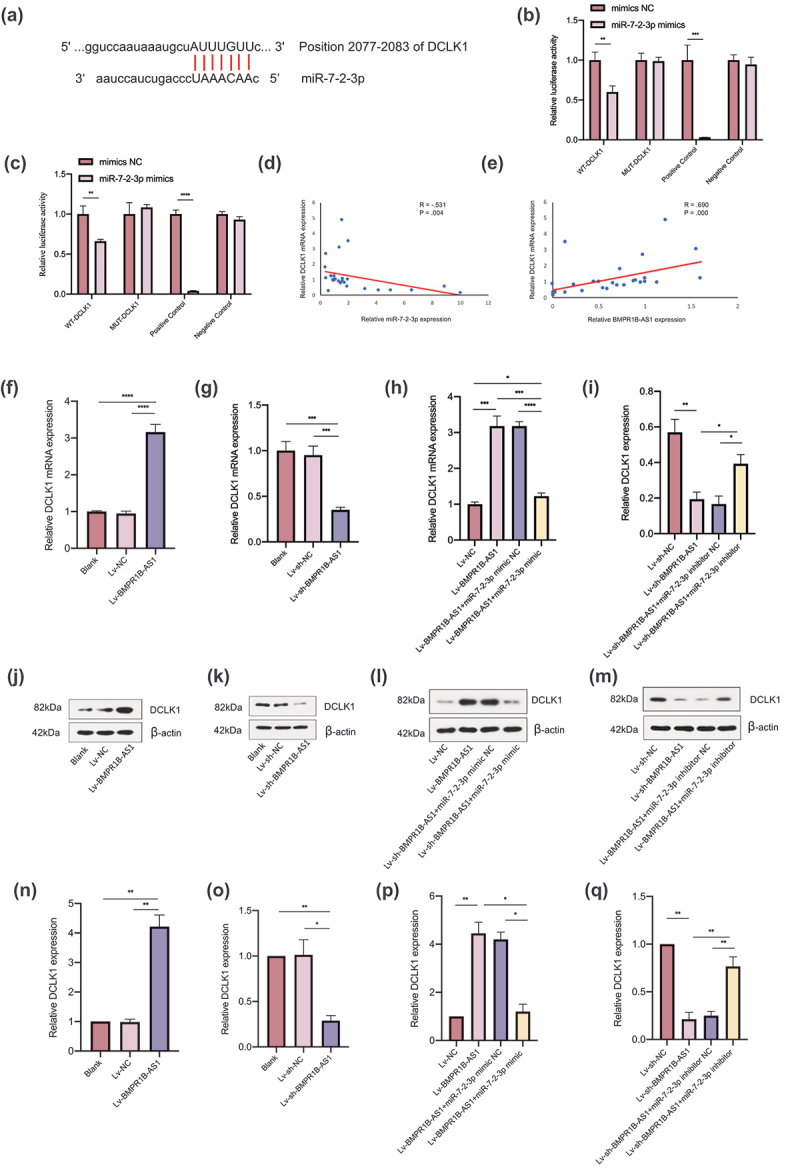
DCLK1 is a target gene of miR-7-2-3p, and BMPR1B-AS1 regulates DCLK1 expression by competitively binding to miR-7-2-3p in EC cell lines. A, Putative binding sites of miR-7-2-3p and DCLK1 mRNA. B, C, The effect of the miR-7-2-3p mimic on the luciferase activities of cells transfected with WT or MUT DCLK1 was detected after 24 h and 48 h, respectively. D, Negative correlation was found between miR-7-2-3p expression and DCLK1 mRNA expression among the 28 endometrial cancer tissue specimens. R = −.531, P = .004. E, Positive correlation was found between BMPR1B-AS1 expression and DCLK1 mRNA expression among the 28 endometrial cancer tissue specimens. R = .690, P = .000. F, J, N, RT-qPCR and western blotting results showed that DCLK1 mRNA and protein expression was upregulated after BMPR1B-AS1 overexpression. G, K, O, RT-qPCR and western blotting results showed that DCLK1 mRNA and protein expression was downregulated after BMPR1B-AS1 knockdown. H, L, P, RT-qPCR and western blotting results showed that the miR-7-2-3p mimic attenuated the BMPR1B-AS1-mediated upregulation of DCLK1 mRNA and protein expression. I, M, Q, RT-qPCR and western blotting results showed that the miR-7-2-3p inhibitor reversed the effects of BMPR1B-AS1 knockdown on DCLK1 mRNA and protein expression. The data are representative of three independent experiments. Bars, ±SD. *P < .05, **P < .01, ***P < .001, ****P < 0.0001.
BMPR1B-AS1 regulates DCLK1 by competitively binding to miR-7-2-3p in EC cell lines
The expression of DCLK1 was significantly increased after BMPR1B-AS1 overexpression (figure 7f, 7j and 7n), while the expression of DCLK1 was significantly decreased after BMPR1B-AS1 knockdown (Figure 7g, 7k and 7o). Moreover, BMPR1B-AS1 level was positively correlated with DCLK1 mRNA level in EC tissues (Figure 7e). Considering the results described above, a rescue assay was performed. The miR-7-2-3p mimic decreased the DCLK1 expression level in the BMPR1B-AS1-overexpressing cell line (Figure 7h, 7l and 7p). The miR-7-2-3p inhibitor rescued the DCLK1 expression level in the BMPR1B-AS1 knockdown cell line (Figure 7i, 7m and 7q). These results demonstrated that BMPR1B-AS1 regulated the expression of DCLK1 in EC cell lines via miR-7-2-3p.
Effects of DCLK1 on the PI3K, Akt, NF-κB expression levels in Ishikawa and Hec-1b cells
We further explored the mechanism by which DCLK1 regulates endometrial cancer cells. It has been reported that DCLK1 promotes cancer progression via the PI3K/Akt/NF-κB axis in other cancers [14]. Therefore, we hypothesized that DCLK1 may regulate the biological behaviors of endometrial cancer cells via the PI3K/Akt/NF-κB pathway. DCLK1-IN-1 is an effective inhibitor of DCLK115. Ishikawa cells and Hec-1b cells were treated with 1 μM DCLK1-IN-1 for 1, 4 and 24 hours. DMSO treatment for the same time points was used as a control. As shown in Figure 8, the levels of p-PI3K p85, p-Akt and p-p65 were decreased in the DCLK1-IN-1 group, but we did not observe any significant changes in the total PI3K p85, Akt or p65 levels. Collectively, our results suggested that the PI3K/Akt/NF-κB pathway may be involved in DCLK1-mediated endometrial cancer progression.
Figure 8.
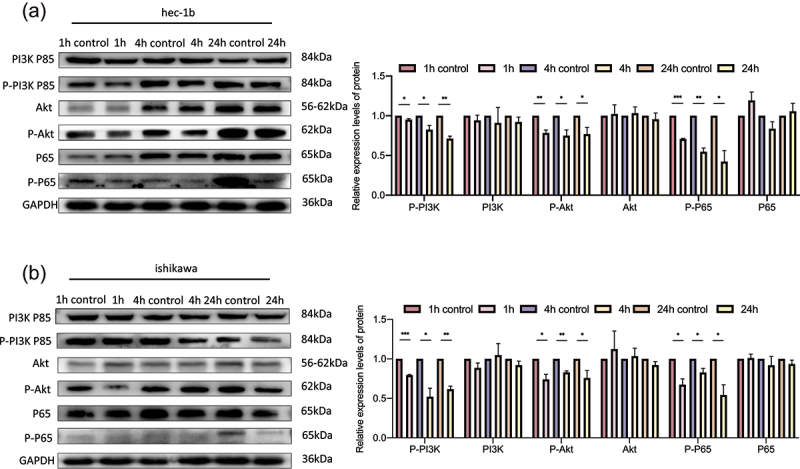
DCLK1 regulates the PI3K/Akt/NF-κB pathway in Hec-1b and Ishikawa cells. A. Western blotting analysis showed that DCLK1-IN-1 downregulated the expression levels of p-P65 via the Akt/NF-κB pathway in Hec-1b cells. B. Western blotting analysis showed that DCLK1-IN-1 downregulated the expression levels of p-P65 via the Akt/NF-κB pathway in Ishikawa cells. 1 h control: DMSO treatment for 1 hour as a control, 1 h: DCLK1-IN-1 treatment for 1 hour, 4 h control: DMSO treatment for 4 hours as a control, 4 h: DCLK1-IN-1 treatment for 4 hours, 24 h control: DMSO treatment for 24 hours as a control, 24 h: DCLK1-IN-1 treatment for 24 hours. The data are representative of three independent experiments. Bars, ±SD. *P < .05, **P < .01, ***P < .001.
BMPR1B-AS1 overexpression promotes the tumor growth of endometrial cancer cells in vivo via miR-7-2-3p/DCLK1
Finally, we carried out a tumor xenograft assay to investigate the function of BMPR1B-AS1 in vivo. As shown in Figure 9a, 9b and 9c, BMPR1B-AS1 significantly promote tumor growth in vivo, which was reversed by treatment with agomir-7-2-3p. As shown in Figure 9d, 9e, 9f and 9g, the overexpression of BMPR1B-AS1 upregulated DCLK1 expression, while administration of agomir-7-2-3p reversed the change. In addition, spearman’s correlation analysis showed that BMPR1B-AS1 expression was negatively correlated with miR-7-2-3p expression (Figure 9h), and miR-7-2-3p (Figure 9i) expression was negatively correlated with DCLK1 mRNA expression, whereas BMPR1B-AS1 was positively correlated with DCLK1 mRNA expression (Figure 9j). Collectively, these results indicated that BMPR1B-AS1 promoted EC tumor growth in vivo through the miR-7-2-3p/DCLK1 axis. A schematic model of the mechanism by which lncRNA BMPR1B-AS1 regulates endometrial cancer cell malignant behaviors is shown in Figure 10.
Figure 9.
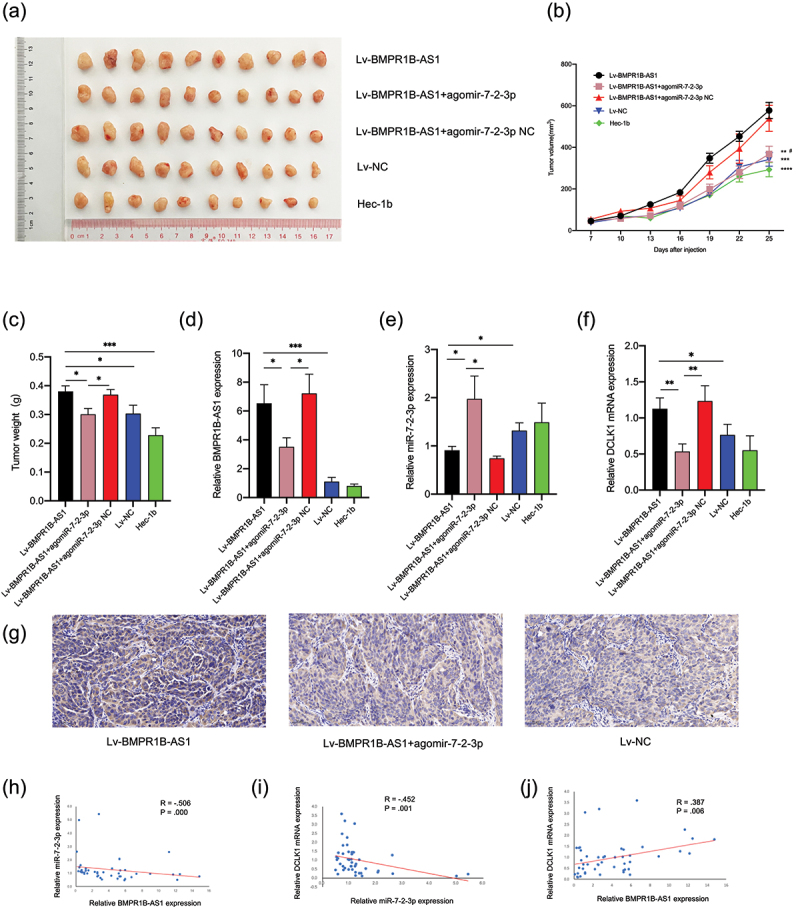
BMPR1B-AS1 promotes endometrial cancer cell growth in vivo by targeting miR-7-2-3p. A, Images of tumor xenografts. B, The growth curves of tumor xenografts demonstrated that BMPR1B-AS1 significantly promote tumor growth in vivo, which was reversed by treatment with agomir-7-2-3p. **P < 0.01 vs. Lv-BMPR1B-AS1, ***P < 0.001 vs. Lv-BMPR1B-AS1, ****P < 0.0001 vs. Lv-BMPR1B-AS1, # P < 0.05 vs. Lv-BMPR1B-AS1+ agomiR-7-2-3p. C, The weights of tumor xenografts derived from five groups. D, The expression levels of BMPR1B-AS1 in five groups were examined by RT-qPCR. E, The expression levels of miR-7-2-3p in five groups were examined by RT-qPCR. F, G, The expression levels of DCLK1 in five groups were examined by RT-qPCR and IHC. H, Negative correlation was found between BMPR1B-AS1 and miR-7-2-3p expression in xenografts tissues. R = −.506, P = .000. I, Negative correlation was found between miR-7-2-3p and DCLK1 mRNA expression in xenografts tissues. R = −.452, P= .001. J. Positive correlation was found between BMPR1B-AS1 and DCLK1 mRNA expression in xenografts tissues. R = .387, P = .006. *P < .05, **P < .01, ***P < .001.
Figure 10.
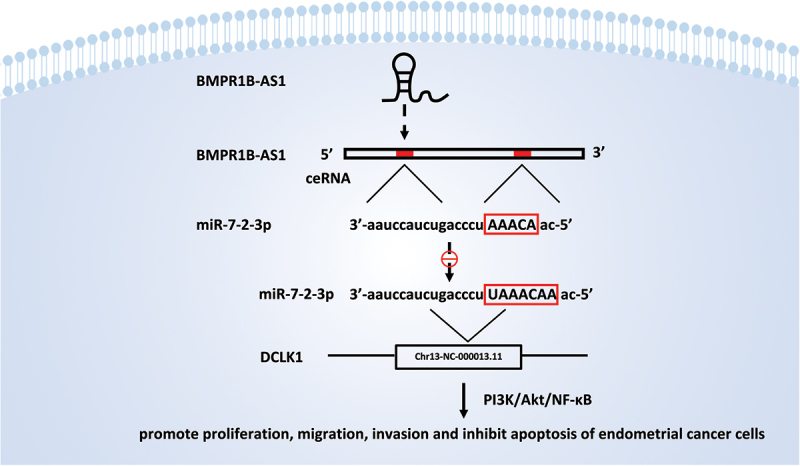
A schematic model of the mechanism by which lncRNA BMPR1B-AS1 regulates endometrial cancer cell malignant behaviors. The BMPR1B-AS1/miR-7-2-3p/DCLK1 axis promotes the progression of endometrial cancer cells via the PI3K/Akt/NF-κB signaling pathway.
Discussion
EC-related lncRNAs have been studied in recent years, and when dysregulated, these lncRNAs act as oncogenes or tumor suppressors. Although an increasing number of studies about lncRNAs have been published in recent years, the study of lncRNAs in EC is still in its infancy [8]. lncRNAs can be divided into different categories, including intronic lncRNAs, bidirectional lncRNAs, intergenic lncRNAs, enhancer RNAs, circular RNAs (circRNAs), pseudogenes, sense lncRNAs, and natural antisense transcripts (NATs), based on their genomic location of origin or their relationship with protein-coding genes [15,16]. NATs are transcribed from the DNA strand opposite to that of the sense transcript and can exert their effects locally, distally, in cis or in trans, and they can also function in multiple subcellular compartments [16–19]. Aberrant expression of NATs is closely related to cancer development [16]. Different mechanisms underlying the biological functions involved in tumorigenesis depend on the subcellular localization of NATs; among these subcellular localizations, cytoplasmic NATs mainly regulate RNA stability and/or mRNA translation [17,20]. IFN-α1 AS is transcribed from the positive strand of chromosome 9, which is the strand opposite the IFNA1 locus (9p22) and overlaps with the conserved secondary structure (CSS) region of IFN-α1 mRNA. This NAT can interact with IFN-α1 mRNA in the cytoplasm by competing for the miR-1270 binding site, preventing the miRNA-induced destabilization of IFN-a1 mRNA [21]. Similarly, BACE1-AS prevents miR-485-5p-induced BACE1 mRNA repression by competing for the same site [22]. LOXL1-AS1 can upregulate the expression of RAP1B, a member of the oncogene family, and then play oncogenic roles in the growth and metastasis of EC cells by sponging miR-28-5p [23]. ZEB1-AS1 expression is significantly upregulated in colon adenocarcinoma (COAD) and can promote PAK2 expression by sponging miR-455-3p, thus facilitating COAD cell growth and metastasis [24]. In the present study, we first defined BMPR1B-AS1 as a cytoplasmic tumor promoter that functions via the miR-7-2-3p/DCLK1 axis through bioinformatics analysis and experiments.
miR-7 is a gene that has attracted substantial attention in the field of tumor research, and its expression is downregulated in a variety of tumor cells. As a tumor suppressor gene, miR-7 participates in tumor growth, development, invasion and metastasis through a variety of signaling pathways [25–27]. miR-7 has three subtypes, namely, miR-7-1, miR-7-2, and miR-7-3, which originate from three different genes. However, these subtypes can be processed into the same mature miR-7 sequence, which comprises 23 nucleotides [28]. miR-7-2-3p is an antisense miRNA star product of miRNA-7-229. Additionally, miR-7-2-3p is generally considered to be a cancer suppressor. miR-7-2-3p expression is downregulated in gallbladder carcinoma (GBC), and upregulation of miR-7-2-3p expression can reverse epithelial-mesenchymal transition (EMT) and decrease gallbladder carcinoma cell metastasis in vitro and in vivo [29]. miR-7-2-3p plays a role in thyroid cancer tumorigenesis by influencing critical pathways, and deficiency in miR-7-2-3p is closely associated with lymph node metastases (LNMs) [30,31]. The correlation of decreased miR-7-2-3p expression with decreased overall survival and progression-free survival in patients with locally advanced esophageal adenocarcinoma (EAC) demonstrated that miR-7-2-3p may serve as a promising prognostic marker in EAC [32]. However, miR-7-2-3p may promote the tumorigenesis process of pancreatic neuroendocrine tumors [33]. This study demonstrated that BMPR1B-AS1 functions as an oncogenic factor by directly sponging miR-7-2-3p and inhibiting its expression, suggesting that miR-7-2-3p is a suppressor of EC.
DCLK1 (doublecortin-like kinase 1) is a member of the microtubule-associated protein (MAP) family and has been identified as a tumor stem cell marker in gastrointestinal tumors [34,35]. DCLK1 is overexpressed in gastric cancer, pancreatic cancer and many other malignant tumors, and DCLK1 overexpression induces the development, metastasis and EMT of tumor cells [14,36–38]. DCLK1 is the direct target gene of miR-7-2-3p in GBC [29]. In this study, the regulatory effect of miR-7-2-3p on DCLK1 was also verified, and DCLK1 expression was positively correlated with the progression of endometrial cancer. Additionally, the DCLK1-positive cell ratio and mean density were both increased in EC tissues (77.5066 ± 15.1461% and 0.3947 ± 0.0577, respectively) compared with paracarcinoma tissues (75.4211 ± 14.0169% and 0.3724 ± 0.0473, respectively), even though there was no significant difference, while the DCLK1 mean density in advanced EC (0.4299 ± 0.0713) was significantly higher than that in early-stage EC (0.3781 ± 0.0431) (p < 0.05) (data not shown). Nevertheless, few studies have examined the role of DCLK1 in the endometrium. DCLK1 expression is upregulated in the cells of the uterine cavity of endometriosis patients before dissemination into the peritoneal cavity, indicating that the eutopic upregulation of DCLK1 expression is crucial for endometriotic cell survival before the establishment of endometriotic lesions [39]. This finding may indicate that DCLK1 is associated with endometrial cell migration capability, which is partially consistent with our findings. It has been reported that DCLK1 plays an oncogenic role via the PI3K/Akt/NF-κB signaling pathway in colorectal cancer, and activation of the PI3K/Akt/NF-κB pathway improves cell proliferation and metastasis, which are key factors in cancer progression [14,40–42]. To elucidate whether the PI3K/Akt/NF-κB pathway is also involved in the DCLK1-mediated progression of endometrial cancer, we treated Hec-1b and Ishikawa cells with a DCLK1 inhibitor and then assessed the levels of activated PI3K, Akt and NF-κB. We observed that the DCLK1 inhibitor continuously blocked the levels of phosphorylated PI3K, phosphorylated Akt and NF-κB subunit p65 in Hec-1b cells and Ishikawa cells.
This is the first study to define BMPR1B-AS1 as an oncogenic factor. We studied the molecular functions that BMPR1B-AS1 performs and the corresponding mechanisms from the perspective of the popular ceRNA theory both in vitro and in vivo. It has been widely thought that the regulation of gene expression by antisense RNAs is a posttranscriptional phenomenon and that antisense RNAs function only as repressors of the translation of specific mRNAs. However, several reports have demonstrated that antisense RNAs may silence their sense genes at the transcriptional level, affecting DNA methylation and histone modifications [17,18,43]. Because BMPR1B expression is significantly decreased in EC samples compared to benign samples [44], it is necessary to further investigate whether BMPR1B-AS1 plays a role in the regulation of BMPR1B transcription.
In conclusion, we first showed that lncRNA BMPR1B-AS1 plays a critical role in the progression of EC by competitively binding to miR-7-2-3p in order to mediate DCLK1-induced PI3K/Akt/NF-κB pathway activation. BMPR1B-AS1 may be a potential therapeutic target for the treatment of EC; however, further investigation is still required to determine the mechanism underlying the role of BMPR1B-AS1 in EC.
Supplementary Material
Acknowledgments
The study was supported by the Joint Funds of the National Science Foundation of China(No. U2004117). We acknowledge assistance with the access of analytic instruments from Translational Medical Center at The First Affiliated Hospital of Zhengzhou University.
Funding Statement
This work was supported by the Joint Funds of the National Science Foundation of China [U2004117].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Siegel RL, Miller KD, Jemal A.. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. [DOI] [PubMed] [Google Scholar]
- [2].Ayakannu T, Taylor A, Willets J, et al. Effect of anandamide on endometrial adenocarcinoma (Ishikawa) cell numbers: implications for endometrial cancer therapy. Lancet. 2015;385(1):S20. [DOI] [PubMed] [Google Scholar]
- [3].Schmitz SU, Grote P, Herrmann BG.. Mechanisms of long noncoding RNA function in development and disease. Cell Mol Life Sci. 2016;73(13):2491–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cesana M, Cacchiarelli D, Legnini I, et al. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011;147(2):358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10(3):155–159. [DOI] [PubMed] [Google Scholar]
- [6].Amândio AR, Necsulea A, Joye E, et al. Hotair is dispensible for mouse development. PLoS Genet. 2016;12(12):e1006232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. [DOI] [PubMed] [Google Scholar]
- [8].Hosseini ES, Meryet-Figuiere M, Sabzalipoor H, et al. Dysregulated expression of long noncoding RNAs in gynecologic cancers. Mol Cancer. 2017;16(1):107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Fang Q, Sang L, Du S. Long noncoding RNA LINC00261 regulates endometrial carcinoma progression by modulating miRNA/FOXO1 expression. Cell Biochem Funct. 2018;36(6):323–330. [DOI] [PubMed] [Google Scholar]
- [10].Wang W, Ge L, X-j X, et al. LncRNA NEAT1 promotes endometrial cancer cell proliferation, migration and invasion by regulating the miR-144-3p/EZH2 axis. Radiol Oncol. 2019;53(4):434–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Salmena L, Poliseno L, Tay Y, et al. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146(3):353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–379. [DOI] [PubMed] [Google Scholar]
- [13].Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505(7483):344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu W, Wang S, Sun Q, et al. DCLK1 promotes epithelial-mesenchymal transition via the PI3K/Akt/NF-κB pathway in colorectal cancer. Int J Cancer. 2018;142(10):2068–2079. [DOI] [PubMed] [Google Scholar]
- [15].Ferguson FM, Nabet B, Raghavan S, et al. Discovery of a selective inhibitor of doublecortin like kinase 1. Nat Chem Biol. 2020;16(6):635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhao S, Zhang X, Chen S, et al. Natural antisense transcripts in the biological hallmarks of cancer: powerful regulators hidden in the dark. J Exp Clin Cancer Res. 2020;39(1):187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pelechano V, Steinmetz LM. Gene regulation by antisense transcription. Nat Rev Genet. 2013;14(12):880–893. [DOI] [PubMed] [Google Scholar]
- [18].Villegas VE, Zaphiropoulos PG. Neighboring gene regulation by antisense long non-coding RNAs. Int J Mol Sci. 2015;16(2):3251–3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gil N, Ulitsky I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat Rev Genet. 2020;21(2):102–117. [DOI] [PubMed] [Google Scholar]
- [20].Schmitt AM, Chang HY. Long Noncoding RNAs in Cancer Pathways. Cancer Cell. 2016;29(4):452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kimura T, Jiang S, Nishizawa M, et al. Stabilization of human interferon-α1 mRNA by its antisense RNA. Cell Mol Life Sci. 2013;70(8):1451–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Faghihi MA, Zhang M, Huang J, et al. Evidence for natural antisense transcript-mediated inhibition of microRNA function. Genome Biol. 2010;11(5):R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yang X, Xing G, Liu S, et al. LncRNA LOXL1-AS1 promotes endometrial cancer progression by sponging miR-28-5p to upregulate RAP1B expression. Biomed Pharmacother. 2020;125:109839. [DOI] [PubMed] [Google Scholar]
- [24].Ni X, Ding Y, Yuan H, et al. Long non-coding RNA ZEB1-AS1 promotes colon adenocarcinoma malignant progression via miR-455-3p/PAK2 axis. Cell Prolif. 2020;53(1):e12723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kong X, Li G, Yuan Y, et al. MicroRNA-7 inhibits epithelial-to-mesenchymal transition and metastasis of breast cancer cells via targeting FAK expression. PLoS One. 2012;7(8):e41523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Okuda H, Xing F, Pandey PR, et al. miR-7 suppresses brain metastasis of breast cancer stem-like cells by modulating KLF4. Cancer Res. 2013;73(4):1434–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li J, Zheng Y, Sun G, et al. Restoration of miR-7 expression suppresses the growth of Lewis lung cancer cells by modulating epidermal growth factor receptor signaling. Oncol Rep. 2014;32(6):2511–2516. [DOI] [PubMed] [Google Scholar]
- [28].Kalinowski FC, Brown RAM, Ganda C, et al. microRNA-7: a tumor suppressor miRNA with therapeutic potential. Int J Biochem Cell Biol. 2014;54:312–317. [DOI] [PubMed] [Google Scholar]
- [29].Lu K, Feng F, Yang Y, et al. High-throughput screening identified miR-7-2-3p and miR-29c-3p as metastasis suppressors in gallbladder carcinoma. J Gastroenterol. 2020;55(1):51–66. [DOI] [PubMed] [Google Scholar]
- [30].H-y W, Wei Y, Pan S-L. Down-regulation and clinical significance of miR-7-2-3p in papillary thyroid carcinoma with multiple detecting methods. IET Syst Biol. 2019;13(5):225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Pan D, Lin P, Wen D, et al. Identification of down-regulated microRNAs in thyroid cancer and their potential functions. Am J Transl Res. 2018;10(8):2264–2276. [PMC free article] [PubMed] [Google Scholar]
- [32].Matsui D, Zaidi AH, Martin SA, et al. Primary tumor microRNA signature predicts recurrence and survival in patients with locally advanced esophageal adenocarcinoma. Oncotarget. 2016;7(49):81281–81291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhou HQ, Chen QC, Qiu ZT, et al. Integrative microRNA-mRNA and protein-protein interaction analysis in pancreatic neuroendocrine tumors. Eur Rev Med Pharmacol Sci. 2016;20(13):2842–2852. [PubMed] [Google Scholar]
- [34].Chandrakesan P, Yao J, Qu D, et al. Dclk1, a tumor stem cell marker, regulates pro-survival signaling and self-renewal of intestinal tumor cells. Mol Cancer. 2017;16(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cao Z, Weygant N, Chandrakesan P, et al. Tuft and cancer stem cell marker Dclk1: a new target to enhance anti-tumor immunity in the tumor microenvironment. Cancers (Basel). 2020;12(12). DOI: 10.3390/cancers12123801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Weygant N, Ge Y, Qu D, et al. Survival of patients with gastrointestinal cancers can be predicted by a surrogate microRNA signature for cancer stem-like cells marked by DCLK1 kinase. Cancer Res. 2016;76(14):4090–4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yan R, Li J, Zhou Y, et al. Inhibition of DCLK1 down-regulates PD-L1 expression through Hippo pathway in human pancreatic cancer. Life Sci. 2020;241:117150. [DOI] [PubMed] [Google Scholar]
- [38].Shan C, Fei F, Li F, et al. miR-448 is a novel prognostic factor of lung squamous cell carcinoma and regulates cells growth and metastasis by targeting DCLK1. Biomed Pharmacother. 2017;89:1227–1234. [DOI] [PubMed] [Google Scholar]
- [39].Proestling K, Birner P, Balendran S, et al. Enhanced expression of the stemness-related factors OCT4, SOX15 and TWIST1 in ectopic endometrium of endometriosis patients. Reprod Biol Endocrinol. 2016;14(1):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ji C, Guo H, Zhang P, et al. AnnexinA5 promote glioma cell invasion and migration via the PI3K/Akt/NF-κB signaling pathway. J Neurooncol. 2018;138(3):469–478. [DOI] [PubMed] [Google Scholar]
- [41].Li W, Du Q, Li X, et al. Eriodictyol inhibits proliferation, metastasis and induces apoptosis of glioma cells PI3K/Akt/NF-κB signaling pathway. Front Pharmacol. 2020;11:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Jiao Y, Li H, Liu Y, et al. Resveratrol inhibits the invasion of glioblastoma-initiating cells via down-regulation of the PI3K/Akt/NF-κB signaling pathway. Nutrients. 2015;7(6):4383–4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Cui I, Cui H. Antisense RNAs and epigenetic regulation. Epigenomics. 2010;2(1):139–150. [DOI] [PubMed] [Google Scholar]
- [44].Richards EG, El-Nashar SA, Schoolmeester JK, et al. Abnormal uterine bleeding is associated with increased BMP7 expression in human endometrium. Reprod Sci. 2017;24(5):671–681. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.