

ABSTRACT
Immune checkpoint inhibitors (ICIs) have revolutionized the oncology field. However, a significant number of patients do not respond, at least partly due to the lack of preexisting anti-tumor T-cell immunity. Therefore, it is emergent to add an immune-priming step to improve efficacy. Here, we report a combined approach consisting of intratumoral administration of pro-inflammatory allogeneic dendritic cells (AlloDCs) and systemic treatment with αCTLA-4 that can drastically improve the anti-tumor efficacy compared to αCTLA-4 monotherapy. When evaluated in mice with large established CT-26 tumors, monotherapy with αCTLA-4 neither delayed tumor progression nor improved mice survival. However, combination treatment of AlloDCs and αCTLA-4 drastically improved the effectiveness, with 70% of mice being cured. This effect was T cell-dependent, and all survived mice rejected a subsequent tumor re-challenge. Further investigation revealed an immune-inflamed tumor microenvironment (TME) in the combination treatment group characterized by enhanced infiltration of activated antigen-presenting endogenous DCs and CD8+ T cells with a tissue-resident memory (TRM) phenotype (CD49a+CD103+). This correlated with elevated levels of tumor-specific CD39+CD103+CD8+ T cells in the tumor and “tumor-matching” NKG2D+CD39+CX3CR1+CD8+ T cells in peripheral blood. Moreover, splenocytes from mice in the combination treatment group secreted significantly higher IFN-γ upon stimulation with the peptide from the endogenous CT-26 retroviral gp70 (model neoantigen), confirming the induction of a tumor-specific CD8+ T-cell response. Taken together, these data indicate a strong anti-tumor synergy between AlloDCs and αCTLA-4 that warrant further clinical investigation with the corresponding human AlloDC product (ilixadencel) for patients receiving αCTLA-4 therapy.
KEYWORDS: Ilixadencel, pro-inflammatory allogeneic dendritic cells, αCTLA-4 therapy, tissue-residentCD8+ T cells, tumor-reactiveCD8+ T cells, tumor-specificCD8+ T cells, tumor-matching T cells
Introduction
Cancer immunotherapy has revolutionized the field of clinical oncology and has become an important pillar in many oncological treatments, prolonging the survival of patients with otherwise rapidly growing fatal cancer. The success of cancer immunotherapy relies on effector cells such as NK cells and particularly T cells that can specifically recognize and eliminate tumor cells. However, once these cells enter solid tumors, they are faced with a highly immunosuppressive tumor microenvironment (TME) that is a key determinant hampering anti-tumor immunity. For this reason, an effective anti-tumor immune response requires approaches that simultaneously address both the existence of immune inhibitory factors and the lack of appropriate strong activation signals.1,2
CTLA-4 (also known as CD152) is a protein receptor constitutively expressed by Treg and upregulated on conventional T-cells upon activation. It shares two ligands (CD80 and CD86) with a stimulatory receptor, CD28. By binding to CD80 and CD86, CTLA-4 acts as an important immune checkpoint and downregulates immune response.1 Blockade of CTLA-4 can revert the negative signal and re-initiate priming of tumor-specific T cells inside lymph nodes. CTLA-4 blockade can also increase the motility of exhausted effector T cells, and induce tumor infiltration of T cells.2,3 Even though human clinical data have demonstrated that patients with metastatic melanoma can achieve long-term, durable responses and improved overall survival (OS) by CTLA-4 blockade therapy,4 there are still many patients (~80%) who do not respond,4 possibly attributed to a lack of preexisting immunity.5,6 Therefore, combination regimens that can initiate priming of T-cell response might improve the response rate.
Our previous work has demonstrated that intratumoral administration of an immune adjuvant consisting of pro-inflammatory allogeneic dendritic cells (AlloDCs), can break the immunosuppressive effect of the TME and efficiently prime specific anti-tumor T-cell responses.7,8 In contrast to conventional autologous DC vaccine, which relies on injected antigen-loaded autologous DC directly priming the T cell response, the non-loaded AlloDCs approach is through the induction of strong allogeneic reaction to elicit the recruitment, activation, and antigen-loading of “bystander” endogenous DC.8 This concept has also been extensively evaluated in clinical studies (wherein the drug candidate is named ilixadencel) for patients with renal cell carcinoma, hepatocellular carcinoma, and gastrointestinal stromal tumors.9–11 In this study, we aim to combine the potential immune priming effects emerging from AlloDCs with the modulation of effector immune cells’ function/dysfunction elicited by the αCTLA-4 antibody.
We found that intratumoral administration of AlloDCs improved the anti-tumor response of systemic αCTLA-4 treatment. Combining AlloDCs with αCTLA-4 treatment turns non-responsive αCTLA-4 monotherapy into high responsiveness. Effective cancer immunotherapy relies on the induction of specific anti-tumor T-cell responses and maintains the cytotoxicity in the long-term.12 We found that the combination therapy induced infiltration of inflammatory immune cells that created a suitable tumor microenvironment for expansion of CD8+ tissue-resident memory T cells (TRM) (CD49a+CD103+) that was positively correlated with elevated CD39+CD103+ potential tumor-reactive CD8+ T cells in the tumor and “tumor-matching” CD8+ T cells in peripheral blood. These results warrant further investigation of adjuvant administration of the corresponding human AlloDCs (ilixadencel)9,11,13 in patients receiving αCTLA-4 monotherapy.
Materials and methods
Tumor cell culture
For experiments conducted at Charles River (CR) Laboratories, the CT-26 murine colon carcinoma cells were obtained from the American Type Culture Collection (ATCC) and maintained at Charles River Laboratories Discovery Services in RPMI-1640 medium containing 10% (vol/vol) fetal bovine serum (FBS), 2 mM glutamine, 100 units/mL penicillin G sodium, 100 μg/mL streptomycin sulfate (collectively referred as 1% PeSt), and 25 μg/mL gentamicin.
For experiments conducted at Uppsala University, the CT-26 cells were obtained from the American Type Culture Collection (ATCC) and maintained in an RPMI-1640 medium containing 1 mM sodium pyruvate, 1% PeSt, and 10% (vol/vol) heat-inactivated FBS. All components were purchased from Invitrogen (Carlsbad, CA). Tumor cells were cultured in tissue culture flasks in a humidified incubator at 37°C, in an atmosphere of 5% CO2.
Isolation and maturation of mouse bone marrow-derived allogeneic DCs
The Northern Stockholm Research Animal Ethics Committee has approved the animal studies (5.8.18–19434/2019). Bone marrow-derived DCs were generated from the femur and tibia of female 8–10-week-old wild-type (wt) C57BL/6NRj (H-2 Db) mice (The Janvier Labs, France) by exposing the bone marrow and flushing out the cells with a sterile syringe. The harvested cells were cultured in IMDM supplemented with 10% (vol/vol) heat-inactivated FBS, 1% PeSt, 1 mM HEPES, and 50 μM β-mercaptoethanol. All culture medium components were purchased from Invitrogen (Carlsbad, CA). The culture medium was supplemented with 20 ng/mL recombinant murine IL-4 and 20 ng/mL recombinant murine GM-CSF (Nordic BioSite, Sweden). Bone marrow cells were plated on non-tissue culture-treated Petri dishes (Sarstedt, Sweden). The medium was replaced every 3 days supplied with 20 ng/mL recombinant murine IL-4 and 20 ng/mL recombinant murine GM-CSF (Nordic BioSite, Sweden). On day 7 the non-adherent immature dendritic cells (imDCs) were harvested and treated for 18 h with the COMBIG cocktail, consisting of 2.5 μg/mL R848 (InvivoGen, San Diego, CA), 20 μg/mL polyinosinic: polycytidylic acid (polyI:C) (Sigma-Aldrich, St Louis, MO) and 1000 IU/mL IFN-γ (Nordic Biosite)14 to obtain mature DCs, and designated as AlloDCs, as the recipient of these DC is Balb/c (H-2Dd) mice. Cells were cultured in a humidified incubator with a 5% CO2 atmosphere at 37°C.
Animal experiment part 1
The experiment was conducted at Charles River (CR) Laboratories. Charles River Laboratories Discovery Services specifically complies with the recommendations of the Guide for Care and Use of Laboratory Animals concerning restraint, husbandry, surgical procedures, feed and fluid regulation, and veterinary care. The animal care and use program at CR Discovery Services was accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC), which assures compliance with accepted standards for the care and use of laboratory animals.
In vivo therapeutic experiment
The in vivo therapeutic experiment was performed with 9 weeks old female Balb/c mice (H-2Dd) (Charles River Laboratories, United States). Mice were injected subcutaneously (s.c.) on the right hind flank with 3 × 105 CT-26 murine colon cancer cells resuspended in PBS (n = 10/treatment group). Mice received intratumoral (i.t.) vaccinations with AlloDCs generated as mentioned above. AlloDCs were washed and re-suspended in mouse plasma with 10% DMSO to mimic the clinical situation where the cryopreserved and subsequently thawed allogeneic DCs (ilixadencel) are injected without subsequent washing. Mice received two i.t. injections of the vehicle (mouse plasma with 10% DMSO; 25 uL) or 1 × 106 AlloDCs (25 uL) on days 14 and 21 after tumor implantation. Respective groups received αCTLA4 (5 mg/kg BioXcell clone 9H10, United States) on day 14 and 2.5 mg/kg on day 21 intraperitoneally (i.p.). Combination group received i.t. injection of 1 × 106 AlloDCs with i.p. 5 mg/kg αCTLA4 on day 14, and i.t. injection of 1 × 106 AlloDCs with i.p. 2.5 mg/kg αCTLA4 on day 21. Mice were sacrificed when the tumor volume exceeded 1500 mm3 or if bleeding ulcers developed. Tumor volume was calculated by the formula: Volume = length × width2 × π/6. Cured mice (n = 7) and naïve mice (n = 10) were re-challenged with the same amount of CT-26 on day 78, and their tumor growth was followed.
Animal experiment part 2
The experiments were conducted at Uppsala University and the Northern Stockholm Research Animal Ethics Committee (5.8.18–19434/2019) has approved the animal studies.
Depletion
Female Balb/c mice (The Janvier Labs), 6-week-old, were inoculated subcutaneously (s.c.) with 3 × 105 CT-26 tumor cells into the right hind flank in 100 µl of DPBS (Invitrogen) on day 0. Mice received the same treatment as the in vivo therapeutic experiment (n = 6 per treatment group). CD4 depletion antibody (InVivoMab αmouse CD4, clone GK1.5, BioXCell) and CD8 depletion antibody (InVivoMab αmouse CD8a, clone 2.43, BioXCell) were injected (i.p) on day 13, 14, 15, 20, 21 and 22. Mice were sacrificed when the tumor volume exceeded 1500 mm3 or if bleeding ulcers developed. Tumor volume was calculated by the formula: Volume = length × width2 × π/6.
Survival analysis
The animals were monitored individually for tumor growth until humane endpoints were reached or until the tumor volume exceeded the study endpoint volume (EPV, 1500 mm3); tumor size was calculated as the volume length × width2 × π/6. The time to endpoint (TTE) for each mouse was calculated as TTE = [log(EPV)–b]/m, where the constant b is the intercept and m is the slope of the line obtained by linear regression (time vs tumor volume) of a log-transformed tumor growth data set, which comprised of the first measured tumor volume when EPV was exceeded and three consecutive measured tumor volume immediately prior to the attainment of EPV. Any animal determined to have died from treatment-related causes was assigned a TTE value equal to the day of death. The survival curve was generated based on the TTE value using the Kaplan-Meier method and compared using the log-rank (Mantel-Cox) test.
Characterization of tumor and organs
Female Balb/c mice (The Janvier Labs), 6-week-old, were inoculated subcutaneously (s.c.) with 3 ×105 CT-26 tumor cells into the right hind flank in 100 μl of DPBS (Invitrogen) on day 0. Mice received the same treatment as described in the in vivo therapeutic experiment. On day 26, the mice were sacrificed, and tumor tissues, blood, spleen, and lymph nodes were harvested for further analysis.
Analysis of CD45+ tumor-infiltrating cells
NanoString
Tumor samples were harvested as described above. The samples were collected and enzymatically digested (Liberase™, Roche, Solna, Sweden) before total RNA was isolated using the RNeasy® Plus RNA isolation kit (Qiagen AB, Sweden). The gene expression levels were directly measured as mRNA counts using the Mouse-Pan cancer immune-oncology kit (NanoString, Seattle, WA). Gene expression analysis was performed using nSolver Analysis software (NanoString).
Flow cytometry analyses of the tumor microenvironment
In one experiment, tumor samples were harvested as described above, and the samples were collected and enzymatically digested (Liberase™, Roche, Solna, Sweden) into single-cell suspensions. CD45+ cells were sorted using mouse MojoSort™ CD45 isolation beads (Miltenyi Biotec, Germany) and stained with antibodies listed in Supplementary Table S1 for the T-cell panel and myeloid cell panel. In another experiment, tumor samples were harvested as described above and CD45+ cells were analyzed for tissue-residence T cells (antibodies listed in Supplementary Table S1 the tissue-residence T-cell panel). In yet another experiment, T cell exhaustion, as well as T regs and MDSC frequency were analyzed (antibodies listed in Supplementary Table S1 in each corresponding panel).
Stained cells were assessed in CytoFLEX LX flow cytometry (Beckman Coulter Life Sciences, Brea, CA). The data were analyzed and visualized using the software Partek Flow, v10.0 (St. Louis, MO). The CD11c+F4/80− DC population was gated out and exported as .FCS file by Flowjo, and imported into Partek Flow software. Firstly, the FACS data was transformed by Arcsinh value 150. Then transformed data was performed with principal component analysis (PCA). A subset of 12 principal components was identified and non-linear dimensionality reduction with uniform manifold approximation and projection (UMAP) was performed by default setting. The 2D UMAP embedding was used for all subsequent visualization purposes.
Analysis of lymph node and blood
Spleen and lymph node samples were harvested as described above. The samples were sliced, meshed, and passed through a 70 μm cell strainer (VWR International, Sweden) to generate single-cell suspensions. Single cells were stained for the Treg panel and T-cell panel (Supplementary Table S1) and were analyzed in the CytoFLEX LX flow cytometry (Beckman Coulter Life Sciences). Single cells from lymph nodes were also stained for mouse lymph node peripheral TRM cells and DC panel (Supplementary Table S1) and evaluated using BD melody (BD Biosciences) for tissue-resident memory CD8+ T cells. The CD11c+ cells were also sorted for further analysis. The blood was collected as described above and treated with red-blood-cell lysis (BD Biosciences). The cells were stained by a tumor-matching markers panel (Supplementary Table S1) and the stained cells were assessed in CytoFLEX LX flow cytometry (Beckman Coulter Life Sciences). The data were analyzed and visualized by Flowjo (BD Biosciences).
Immunofluorescence analysis of tumor slices
Tumors removed from mice were rapidly deep-frozen using a slush containing dry ice and 2-methylbutane (lab-honeywell Cat.M32631) for 10 min and stored at −80°C. Frozen tumor sections were cut with 8 μm thickness in the cryostat at −25°C and fixed with cold acetone for 15 minutes, air-dried, and kept at −20°C for further use. Sections were rehydrated with 1X PBS for 5 min at RT two times, incubated in blocking buffer (3% BSA in 1× PBS) for 1 h at RT, and stained with FITC-conjugated anti-mouse CD8a (Biolegend, Clone:53–6.7, Cat:100706) and APC-conjugated anti-mouse CD103 (Biolegend, Clone:2E7, Cat:121413) at 1:100 dilution in blocking buffer overnight at +4°C, protected from light. Sections were then washed five times with 1× PBS for 5 min at RT followed by removal of autofluorescence using the Vector®TrueVIEW® Autofluorescence Quenching Kit (Vector Laboratories, Cat: SP-8400). Afterward, the nuclear stain was performed with 5 μg/ml Hoechst (Molecular Probes Invitrogen Hoechst 33342) in 1× PBS for 10 min at RT, slides were washed three times with 1X PBS for 5 min at RT and mounted with VECTASHIELD Vibrance Antifade Mounting Medium and stored at +4°C. All sections were imaged using LSM 700 laser confocal scanning microscope (Zeiss, Germany).
Peptides restimulation assay
H-2Ld-restricted gp70 peptides and beta-galactosidase peptides (as irrelevant control) were purchased (Biosite). Splenocytes were collected after various treatments as described above. Approximately 1 ×105 cells were re-stimulated with each peptide in triplicate, at 20 µg/mL in a 96-well plate for 3 days. The supernatant was collected and analyzed for IFN-γ secretion by ELISA (Mabtech, Nacka Strand, Sweden). The cells were intracellularly stained for IFN-γ (Supplementary Table S1), and the stained cells were assessed in CytoFLEX LX flow cytometry (Beckman Coulter Life Sciences). The data were analyzed and visualized by Flowjo (BD Biosciences).
Statistics
The data are reported as mean ± SEM. Statistical analysis was performed by GraphPad Prism software version 9.0 (La Jolla). The mean values of the 2 groups were compared using a t-test. Non-parametric one-way ANOVA test was used for multiple comparisons between more than 2 groups. Kaplan-Meier survival curves were compared using the log-rank test. Values with P <.05 were considered to be statistically significant.
Results
AlloDCs boost the therapeutic efficacy of αCTLA-4 antibody and the effect is T-cell dependent
Mice bearing subcutaneous syngeneic CT-26 colon tumors were treated with AlloDCs, antagonistic αCTLA-4 antibody, or a combination of both (Figure 1a). AlloDCs treatment did not affect tumor progression or prolonged survival, while αCTLA-4 treatment slightly prolonged the median survival, but no mouse was cured. In striking contrast, combined treatment with AlloDC and αCTLA-4 significantly delayed tumor progression, wherein 70% of mice survived longer than 70 days (7/10 mice) with no detectable tumor (Figure 1b, c). All survived mice (7/10 mice) were re-challenged with CT-26 cells and all mice rejected the newly inoculated tumors compared to control mice (Figure 1d-f) indicating the establishment of an immunological memory response in those mice. Depletion of CD8+ T cells totally abolished the therapeutic benefit of αCTLA-4/AlloDC combination treatment while depletion of CD4+ T cells partially abolished the effect (Figure 1g-i, Supplementary Fig. S1). All these data clearly showed that combining AlloDC with αCTLA-4 enhanced long-term therapeutic efficacy via induction of T cell immunity.
Figure 1.
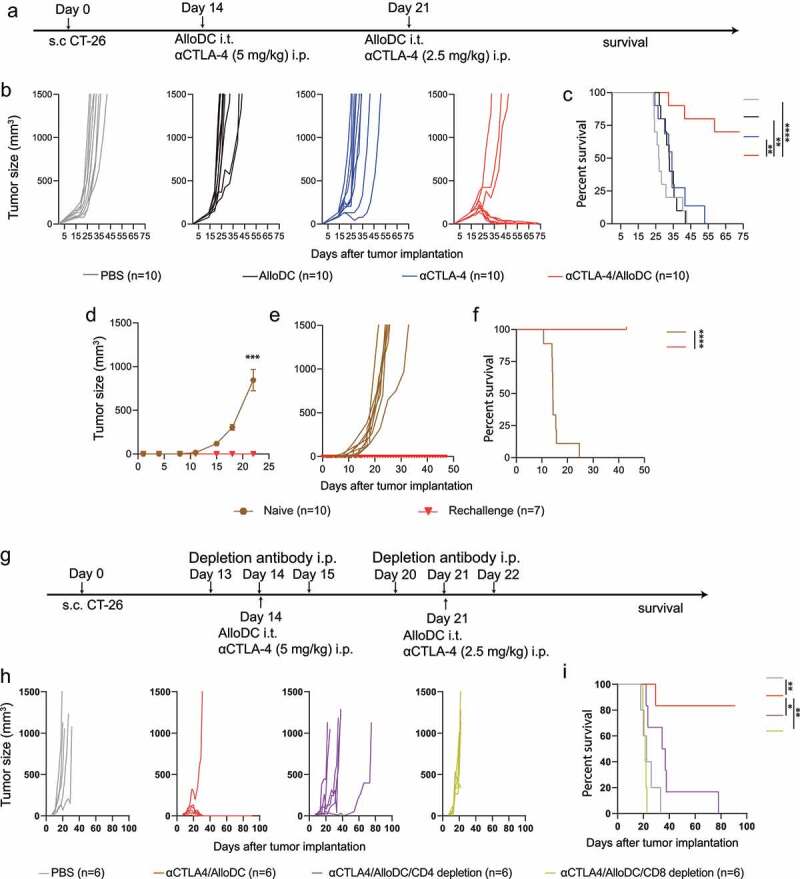
AlloDCs boost the therapeutic efficacy of αCTLA-4 antibody and the effect is dependent on T cells. (a) Schematic illustration of the experiment outline. (b) Tumor size of individual mice and (c) mouse survival (Kaplan-Meier curve) after different treatments (n = 10 per treatment group). (d-f) Seven tumor-free cured mice from the αCTLA-4/AlloDC and control group mice were s.c injected CT-26 tumor cells. The average tumor size (d), individual mice (e), and mouse survival were presented (PBS: n = 10, Rechallenge: n = 7). (g) Treatment schedule of the CD8+ and CD4+ T-cell depletion experiment. (h) Tumor size of individual mice and (i) mouse survival (Kaplan-Meier curve) after treatments (n = 6 per treatment group). The average tumor size from the rechallenge was compared by two-way ANOVA with Tukey correction (***, P < .001). All survival curves were compared using the log-rank test. (*: P < .05, **: P < .01, ****: P < .0001).
Combined αCTLA-4/AlloDC treatment reverts the immunosuppression and boosts the antigen-presentation signature of the tumor microenvironment
We subsequently analyzed the bulk gene expression of the whole tumor after the different treatments (Figure 2a), aiming to elucidate the mechanism of action. Gene set analysis revealed significant pathway signature differences between different treatment groups (Figure 2b, Normalized mRNA counts listed in Supplementary Table S2). Monotherapy with either AlloDC or αCTLA-4 affected the signature scores but did not revert the distribution (Figure 2c). On the other hand, αCTLA-4/AlloDC combination treatment significantly altered the tumor microenvironment, as the pathway signature scores were reverted across the zero-line (Figure 2c). Notably, the top four upregulated pathways in the combination therapy compared to the individual monotherapies (Figure 2d-g) involved genes that classified into myeloid compartment (Figure 2h), antigen presentation (Figure 2i), lymphoid compartment (Figure 2j), and cytokines/chemokine regulation (Figure 2k). Conclusively, these data reflect the changes in the tumor microenvironment after αCTLA-4/AlloDC combination treatment and provide an overview of potential mechanisms that could explain why adding AlloDC to αCTLA-4 drastically improved the therapeutic outcome.
Figure 2.
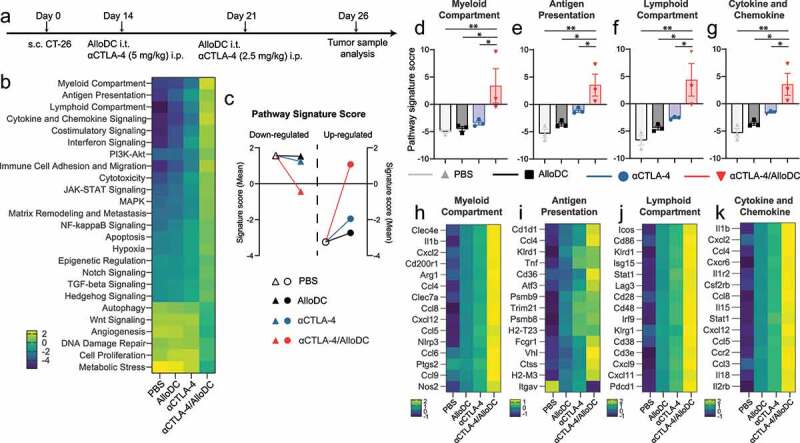
AlloDCs revert the immunosuppressive TME and boost antigen presentation. (a) Schematic illustration of the experimental setup. (b) Pathway signature score (z-scored) of the CT-26 tumor microenvironment after treatment with AlloDC, αCTLA-4 as monotherapy or in combination. (c) The mean of pathway signature scores determined from NanoString mRNA profiling, classified into down-regulated (triangle) and up-regulated (round), were presented as a comparison between the PBS group with either of the treatment groups. (d-f) The pathway score of (d) myeloid compartment, (e) antigen presentation, (f) lymphoid compartment, and (g) cytokine and chemokine. (g-i) Heatmap of the top 15 genes expression from the above-mentioned pathways. Error bars represent SEM and the mean values were compared using nonparametric with multiple comparisons (*: P < .05, **: P < .01).
Combined αCTLA-4/AlloDC treatment enhances infiltration of immune cells with anti-cancer phenotypes
Gene analysis also indicated that there is a tendency for higher tumor-infiltration of immune cells in the combination therapy group, including total CD45+ cells, T cells, NK cells, macrophages, DC, and neutrophils (Figure 3a, Supplementary Fig. S2). We thus further characterized the tumor-infiltrating CD45+ cells by flow cytometry. Four major myeloid cell populations (CD11b+ cells) were identified based on their surface marker expression (Supplementary Fig. S3 A-G), and different treatments altered the cell type compositions (Supplementary Fig. S4). When analyzing the recruited tumor-infiltrating DC population, we observed significantly enhanced infiltration of host DCs in the combination treatment group (Figure 3b) compared to either monotherapy. A detailed characterization of the DCs revealed three top-enriched sub-populations in the αCTLA-4/AlloDC treatment group compared to the rest of the sub-populations (Figure 3c-g Supplementary Fig.S5), named DC-A, DC-B, and DC-C. These DCs showed high IL-12 expression, activation, and maturation (CD86+, IA-IE+) and phenotypic signs of antigen-presenting capacity (CD8a+, CD103+) (Figure 3d), with the difference that Arginase-I was expressed by DC-B but not DC-A, DC-C.
Figure 3.
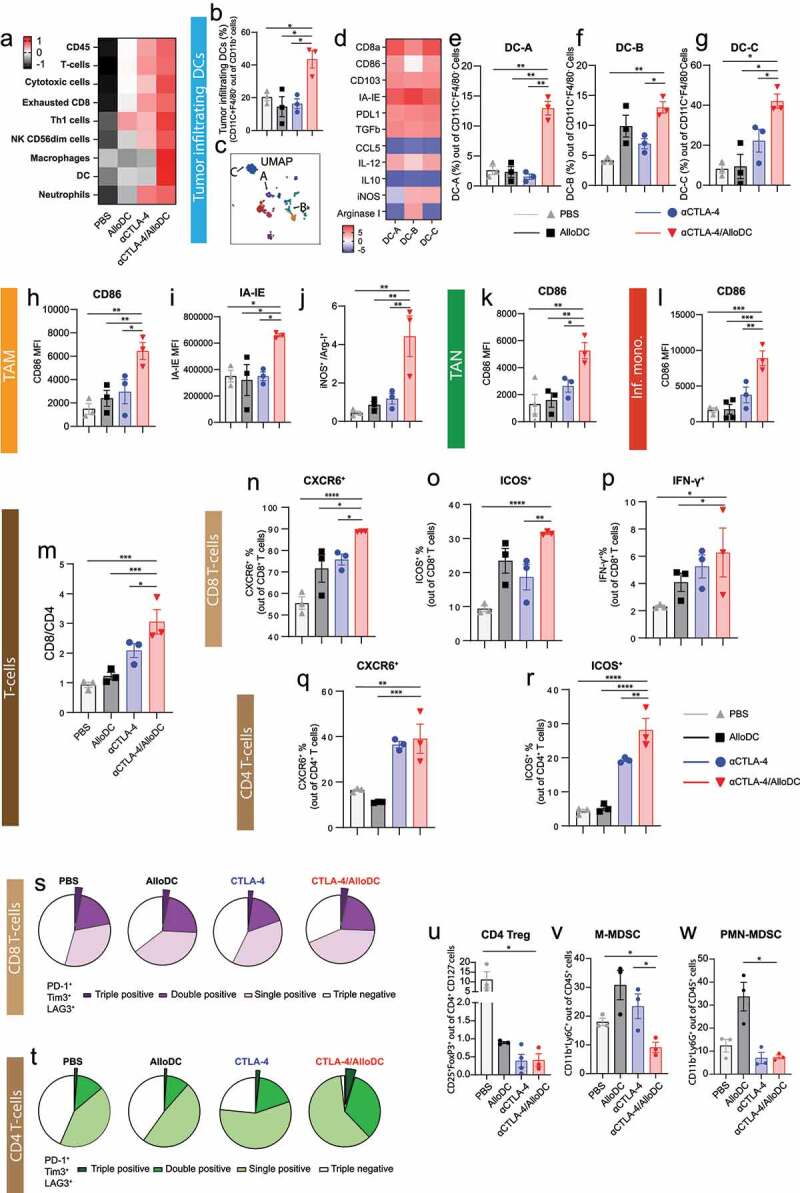
AlloDCs enhance immune cell infiltration and boost the antigen-presentation signature of αCTLA-4 antibody therapy. (a) The abundance of tumor-infiltrating immune cells in each mouse from different treatment groups. Data are presented as cell-type scores from NanoString mRNA profiling. (b) Percentage of the tumor-infiltrating DCs (CD11c+F4/80–) in each treatment group. (c) The U-MAP of clustered tumor-infiltrating DCs. (d) The expression of each marker (z-scored mean value) on tumor-infiltrating DCs (CD11c+F4/80–), clustered (DC-A, DC-B, DC-C) by unsupervised t-SNE analysis. (e-g) Percentage of each clustered in tumor-infiltrating DCs (CD11c+F4/80 – infiltrating DCs). (h, i) Mean fluorescent intensity (MFI) of (h) CD86 and (i) IA-IE on tumor-associated macrophages (TAM, CD11c–F4/80+). (j) The ratio of iNOS+ and Arginase-I+ TAM. (k) MFI of CD86 on tumor-associated neutrophils (TAN, Ly6G+ cells). (l) MFI of CD86 on inflammatory monocytes (CD11c+F4/80+). (m) The ratio of CD8+ and CD4+ T cells in the tumor-infiltrating CD3+ T cells was analyzed by flow cytometry. Percentage of (n) CXCR6+, (o) ICOS+, and (p) IFN-γ+ T cells in the total CD8+ population. Percentage of (q) CXCR6+ and (r) ICOS+ in the total CD4+ T cells. (s-t) Percentage of phenotypic exhaustion CD8+ and CD4+ T cells in the tumor, showing as triple, double, and single positive of PD-1+, Tim3+ and LAG3+ on CD8+ and CD4+ T cells. Percentage of (u) CD4 Treg, (v) M-MDSC and (w) PMN-MDSC in tumor. Error bars represent SEM, and the mean values were compared using nonparametric with multiple comparisons (*: P < .05, **: P < .01, ***: P < .001 ****: P < .0001).
Additionally, tumor-infiltrating macrophages in the αCTLA-4/AlloDC combination treatment group had an M1-like phenotype, with higher CD86 (Figure 3h, Supplementary Fig. 6A) and IA-IE expression (Figure 3i, Supplementary Fig. 6B). Moreover, those macrophages also express high iNOS and lower Arginase-I (Figure 3j, Supplementary Fig. S6C-E) indicating features of M1-like functionality. There were also enhanced infiltrating neutrophils and inflammatory monocytes (Inf. mono.) with higher CD86 expression (Figure 3k-l, Supplementary Fig. S6F-G) in the αCTLA-4/AlloDC combination treatment group.
We also observed a general increase in the CD8/CD4 ratio in the αCTLA-4/AlloDC combination group compared to the monotherapy groups (Figure 3m, Supplementary Fig. S7A-F). Notably, both CD8+ and CD4+ tumor-infiltrated T cells exhibited activation features (CXCR6+ and ICOS+) in the combination treatment group (Figure 3n-o, q-r, Supplementary Fig. S7G-H, J-K), with CD8+ T cells additionally exhibiting higher IFN-γ expression (Figure 3p, Supplementary Fig. S7I).
Moreover, CD8+ T-cells from the αCTLA-4/AlloDC combination group did not show any phenotypic signs of exhaustion, while there were slightly more phenotypically exhausted CD4+ T-cells (PD-1+Tim3+LAG3+) in the αCTLA-4/AlloDC combination group (Figure 3s-t). On the other hand, both single and combination groups had less infiltration of CD4+ Tregs, monocytic myeloid-derived suppressor cells (M-MDSC), and polymorphonuclear myeloid-derived suppressor cells (PMN-MDSC) (Figure 3u-w, Supplementary Fig. 8).
Taken together, our data clearly indicate that adding intratumoral administration of AlloDCs to systemic αCTLA-4 therapy enhances intratumoral infiltration of immune cells with anti-cancer features, including host DCs with high antigen-presenting capacity and activation phenotypes; macrophages with M1-like phenotype activated neutrophils and inflammatory monocytes. The combined αCTLA-4/AlloDC treatment also led to an increase in the intratumoral CD8/CD4 ratio with both cell types exhibiting signs of activation and reduced the number of immune-suppressive immune cells, including CD4+ Tregs, M-MDSC, and PMN-MDSC.
Combined αCTLA-4/AlloDC treatment enhances T cell response with a tumor antigen-specific tissue-resident memory phenotype
The increased intratumoral infiltration of CXCR6+ T cells encouraged us to investigate TRM T cells as CXCR6+ T cells are important in positioning these TRM.15 Concordantly, we identified CD49a+CD103+ TRM T cells16 in tumors (Figure 4a-c, Supplementary Fig. S9A), and peripheral CD8 TRM (CD103+CD69+) in draining lymph nodes (Figure 4d, Supplementary Fig. S9C-D) were significantly higher in mice treated with the αCTLA-4/AlloDC combination therapy. DCs in tumor-draining lymph nodes with a tendency for higher Batf3 expression (Supplementary Fig. S10A) and increased cDC1 with high CD86 expression was present in the combination treatment group (Supplementary Fig. S10B-E). These data potentially indicate that TRM was primed by cross-presenting Batf3+ DCs that had migrated from tumor to tumor-draining lymph node.17,18
Figure 4.
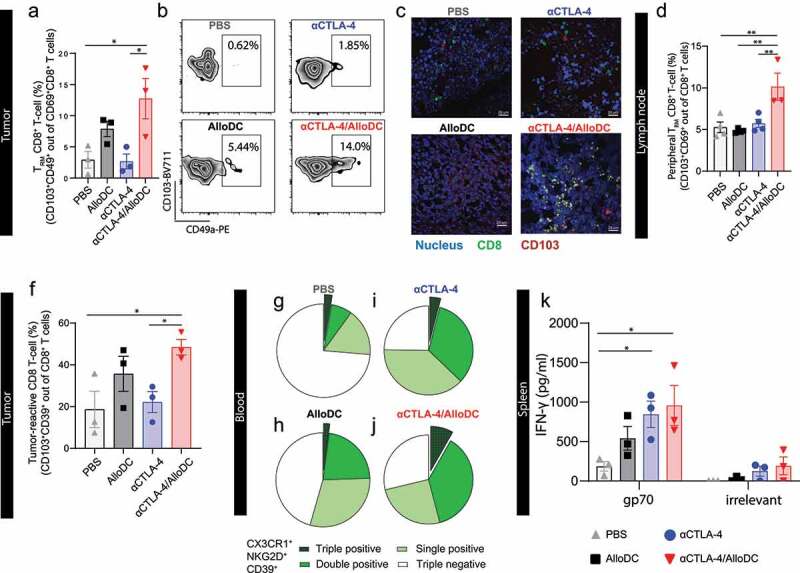
AlloDCs treatment unleashes tumor antigen-specific tissue-resident memory T cells. (a) Percentage of tumor-infiltrating tissue-resident memory (TRM) CD8+ T cells (CD49a+CD103+ out of CD69+CD8+ T cells) in each treatment group, analyzed by flow cytometry. (b) Representative dot plot indicating tumor-infiltrating TRM. (c) Representative fluorescent images of TRM CD8+ T cells in tumor section from different treatment groups. (d) The percentage of peripheral TRM CD8+ T cells in draining lymph nodes. (e) Percentage of potential tumor reacting CD8+ T cells (CD39+CD103+) in tumor samples from different treatment groups. (f-i) Percentage of tumor-matching CD8+ T cells in the blood, showing as triple, double, and single positive of CX3CR1, NKG2D, and CD39 on CD8+ T cells. (j) IFN-γ in vitro culture supernatant of splenocytes, harvested from each treatment group and re-stimulated with either gp70 peptides or a non-relevant peptide (HIV-derived). Error bars represent SEM, and the mean values were compared using nonparametric with multiple comparisons (*: P < .05, **, P < .01).
Furthermore, the frequency of CD8+ T cells with a potential tumor-reactive CD39+CD103+ phenotype19,20 was notably elevated in the combination group (Figure 4f, Supplementary Fig. S9A-B). This was associated with a significantly higher frequency of circulating CD8+ T cells with a “tumor-matching” phenotype (CX3CR1+NKG2D+CD39+)21 in mice in the combination treatment group, compared to mice in the monotherapy groups (Figure 4g-j).
Moreover, CT-26 tumor cells express the endogenous retroviral antigen gp70,22 thus allowing us to investigate endogenous antigen-specific T-cell response. Encouragingly, the splenocytes harvested from combination treatment groups secreted significantly higher IFN-γ (Figure 4k) and had an increasing number of both IFN-γ+CD8+ T cells and IFN-γ+CD4+ T cells (Supplementary Fig. 9E-F) upon stimulation with gp70 peptide, compared to PBS treatment group. These data confirm the establishment of an antigen-specific CD8+ T-cell response. Additionally, the combination treatment may induce a weak un-specific T cell activation since irrelevant peptide stimulation also showed a tendency of increased IFN-γ secretion compared to the PBS treatment group.
Taken together, all the above-mentioned data indicated that combined AlloDC treatment with therapeutic αCTLA-4 treatment enhanced tissue antigen-specific and tissue-resident memory T cells response.
Discussion
Cancer immunotherapy with immune checkpoint inhibitors (ICI), for example, αCTLA-4, has demonstrated long-term effectiveness in the clinic with some patients being cured.23 However, the benefit is still limited to a small fraction of patients,23 which is largely due to the fact that ICI therapy is dependent on the presence of a preexisting basal T cell-mediated anti-tumor response.23–25 Our previous preclinical findings indicate that an adjuvant/immune primer consisting of pro-inflammatory allogeneic dendritic cells (AlloDCs) injected intratumorally could efficiently prime a tumor-specific T cell response and promote tumor infiltration of T cells.7 Our working hypothesis was therefore that combined treatment with AlloDC and αCTLA-4 might improve the anti-tumor efficacy.
The AlloDC approach is aimed at inducing an anti-cancer response via activation and antigen-loading of intratumorally recruited endogenous DC, which subsequently will prime a T cell response.7 This approach thus bypasses the need for antigen-loading of the injected DCs. Since the intratumorally injected DC will serve as a stimulus for an allogeneic rejection process, this will provide a pro-inflammatory milieu potentially reverting the immuno-suppressive tumor microenvironment.7 Therefore, the AlloDCs play multiple roles rather than a simple inflammatory effect. αCTLA-4 was combined with antigen-loaded autologous DCs aiming to block the inhibitory signaling mediated by the injected autologous DCs and augment the T cell priming response.26
Similar to a previous report,27 standard αCTLA-4 therapy did not induce complete tumor regression in mice with subcutaneous CT-26 tumors. Encouragingly, combining AlloDC with αCTLA-4 induced complete tumor regression in 7 out of 10 treated mice. This effect was strictly CD8+ T cell-dependent and all cured mice rejected a subsequent tumor re-challenge. The addition of AlloDC to αCTLA-4 treatment was found to radically affect the tumor microenvironment with drastically altered pathways activation, wherein the most activated pathways were myeloid and lymphoid cell compartment infiltration, antigen presentation, cytokines, and chemokines signaling. We believe the benefit of the addition of AlloDC to αCTLA-4 treatment is based on the enhanced infiltration of different types of immune cells, which generates a pro-inflammatory anti-cancer milieu and further enhanced endogenous T-cell response. Given our previous findings,7,8 the AlloDC plays multi-effect functions in the combination therapy, rather than as a simple inflammatory stimulus. In addition, combination treatment also decreased infiltration of Tregs, M-MDSC, and PMN-MDSC. There was no difference regarding PD1, Tim3, and Lag3 triple-positive CD8+ T cells, but a slightly higher number of triple-positive CD4+ T cells. However, further characterization studies are needed to verify the T cell exhaustion status, including transcription factors analysis (e.g. NFAT, IRF4, TOX, and NR4A)28 and functionality tests. More M1-like macrophages were present in the αCTLA-4/AlloDC group. The αCTLA-4/AlloDC combination treatment also recruited more neutrophils and polarized them into an N1-like phenotype (higher CD86 expression). More importantly, the αCTLA-4/AlloDC combination treatment recruited and activated host DCs for antigen-presentation, which is in line with our previous finding,7 and further synergistically elicited a strong tumor-specific T cell response.
In the combination treatment group, we saw an increased presence of ICOS+CD4+ T cells, which have been reported to be effector cells that produce the Th1 cytokine IFN-γ29,30 and correlated with clinical benefit.30 We also observed an enhanced tumor-infiltration of CXCR6+ T cells in the combination group that was reported to dominate the anti-tumor efficacy of all intratumoral CD8+ T cells.31 This also translated to a higher infiltration and activation of TRM, as CXCR6+ T cells are reported to regulate the translocation of TRM CD8+ T cells.15,31 CD8+ TRM is located in the tumor microenvironments and can be boosted to increase protective immunity against cancer.32 CD49a+CD103+ CD69+ CD8+ TRM cells typically produce Th1-type cytokines, including IFN-γ, TNF, and IL-2 upon stimulation and also have a function in killing tumor cells.32 Higher Batf3 gene expression also indicated the recruitment of Batf3+ DCs, together with an increasing number of cDC1 with higher CD86 expression, in line with the report indicating that activated TRM cells can sustain the cancer immunity cycle via recruitment of Batf3+ DCs.32 However, whether these Batf3+ DCs are lymph node-resident or migratory cDC1 needs to be further analyzed. Finally, we also confirmed that the enhanced T-cell response in the combination therapy group is tumor-antigen specific and reactive, evidenced by the higher presence of potential tumor-reactive CD8+ T cells both in tumor (CD39+CD103+) and in circulation (gated as triple- or double-positive for CX3CR1+, NKG2D+, or CD39+), and their enhanced reactivity against the endogenous antigen (gp70).
In conclusion, we present data showing that the addition of AlloDC turns non-responsive αCTLA-4 therapy into effective treatment. The addition of AlloDC to αCTLA-4 therapy can drastically alter the otherwise immunosuppressive microenvironment, enhancing the infiltration and activation of antigen-presenting DCs which support the effectiveness of tumor-specific CD8+ T cells including TRM cells in the tumors and sustained the cancer immunity cycle to generate long-term protective anti-cancer immunity. Our findings in this preclinical setting warrant further clinical investigation with the corresponding human AlloDC product (ilixadencel) in patients receiving αCTLA-4 therapy, and potentially open new possibilities to combine AlloDC with other checkpoint antibodies.
Supplementary Material
Funding Statement
This work was supported by the Barncancerfonden [PR2020-0167]; Göran Gustafsson Foundation [2013]; Clas Groschinsky Foundation [M19359]; Swedish Research Council [2019-01326]; Swedish Cancer Foundation [190184Pj].
Acknowledgments
We thank the BioVis Platform at Uppsala University for the flow cytometry and imaging service. We also thank Mohanraj Ramachandran (Uppsala University) for designing of flow cytometry panel.
Competing Interests
AK is the founder of Mendus, a company in the field of allogeneic dendritic cell-based immunotherapeutic for cancer. The other authors have no conflicting financial interests.
Contribution
CJ, AI, DY, and AK designed the experiments. CJ, AI, and GF performed the experiments and analyzed the data. CJ and DY wrote the paper and AI, GF, ME, AK and HW revised the paper. All authors read and approved the final version of the manuscript.
Data availability statement
The data that support the findings of this study are available from the corresponding author, [CJ, DY], upon reasonable request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2022.2099642
References
- 1.Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA.. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. 1991;174(3):561–11. doi: 10.1084/jem.174.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107(9):4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pentcheva-Hoang T, Simpson TR, Montalvo-Ortiz W, Allison JP. Cytotoxic T lymphocyte antigen-4 blockade enhances antitumor immunity by stimulating melanoma-specific T-cell motility. Cancer Immunol Res. 2014;2(10):970–980. doi: 10.1158/2326-6066.cir-14-0104. [DOI] [PubMed] [Google Scholar]
- 4.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. New Engl J Medicine. 2010;363(8):711–723. doi: 10.1056/nejmoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. 2016;167(2):397–404.e9. doi: 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crittenden MR, Zebertavage L, Kramer G, Bambina S, Friedman D, Troesch V, Blair T, Baird JR, Alice A, Gough MJ. Tumor cure by radiation therapy and checkpoint inhibitors depends on pre-existing immunity. Sci Rep-uk. 2018;8(1):7012. doi: 10.1038/s41598-018-25482-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fotaki G, Jin C, Kerzeli IK, Ramachandran M, Martikainen MM, Karlsson-Parra A, Yu D, Essand M. Cancer vaccine based on a combination of an infection-enhanced adenoviral vector and pro-inflammatory allogeneic DCs leads to sustained antigen-specific immune responses in three melanoma models. Oncoimmunology. 2018;7(3):e1397250. doi: 10.1080/2162402x.2017.1397250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fotaki G, Jin C, Ramachandran M, Kerzeli IK, Karlsson-Parra A, Yu D, Essand M. Pro-inflammatory allogeneic DCs promote activation of bystander immune cells and thereby license antigen-specific T-cell responses. Oncoimmunology. 2018;7(3):e1395126. doi: 10.1080/2162402x.2017.1395126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karlsson-Parra A, Iskantar A, Jin S, Yu D. Intratumoral injection of allogeneic pro-inflammatory dendritic cells (ilixadencel) in combination with anti-CTLA-4 treatment induce complete tumour responses and anti-tumour immune memory in a mouse tumour model. Ann Oncol. 2020;31:S487. doi: 10.1016/j.annonc.2020.08.683. [DOI] [Google Scholar]
- 10.Karlsson-Parra A, Fröbom R, Berglund E, Berglund D, Nilsson I-L, Linder-Stragliotto C, Suenaert P, Branstrom R. Phase I trial evaluating safety and efficacy of intratumorally administered allogeneic monocyte-derived cells (ilixadencel) in advanced gastrointestinal stromal tumors. J Clin Oncol. 2020;38(5_suppl):15. doi: 10.1200/jco.2020.38.5_suppl.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karlsson-Parra A, Kovacka J, Heimann E, Jorvid M, Zeilemaker S, Longhurst S, Suenaert P. Ilixadencel – an allogeneic cell-based anticancer immune primer for intratumoral administration. Pharmaceut Res. 2018;35(8):156. doi: 10.1007/s11095-018-2438-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity. 2013;39(1):38–48. doi: 10.1016/j.immuni.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fröbom R, Berglund E, Berglund D, Nilsson I-L, Åhlén J, von SK, Linder-Stragliotto C, Suenaert P, Karlsson-Parra A, Bränström R. Phase I trial evaluating safety and efficacy of intratumorally administered inflammatory allogeneic dendritic cells (ilixadencel) in advanced gastrointestinal stromal tumors. Cancer Immunol Immunother. 2020;69(11):2393–2401. doi: 10.1007/s00262-020-02625-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iborra S, Martinez-Lopez M, Khouili SC, Enamorado M, Cueto FJ, Conde-Garrosa R, Fresno CD, Sancho D. Optimal generation of tissue-resident but not circulating memory T cells during viral infection requires crosspriming by DNGR-1(+) dendritic cells. Immunity. 2016;45(4):847–860. doi: 10.1016/j.immuni.2016.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wein AN, McMaster SR, Takamura S, Dunbar PR, Cartwright EK, Hayward SL, McManus DT, Shimaoka T, Ueha S, Tsukui T, et al. CXCR6 regulates localization of tissue-resident memory CD8 T cells to the airways. J Exp Med. 2019;216(12):2748–2762. doi: 10.1084/jem.20181308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Enamorado M, Iborra S, Priego E, Cueto FJ, Quintana JA, Martinez-Cano S, Mejias-Perez E, Esteban M, Melero I, Hidalgo A, et al. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8(+) T cells. Nat Commun. 2017;8(1):16073. doi: 10.1038/ncomms16073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322(5904):1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, Krummel MF. Critical role for CD103(+)/CD141(+) dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell. 2016;30(2):324–336. doi: 10.1016/j.ccell.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corgnac S, Malenica I, Mezquita L, Auclin E, Voilin E, Kacher J, Halse H, Grynszpan L, Signolle N, Dayris T, et al. CD103(+)CD8(+) TRM cells accumulate in tumors of anti-PD-1-responder lung cancer patients and are tumor-reactive lymphocytes enriched with Tc17. Cell Rep Med. 2020;1(7):100127. doi: 10.1016/j.xcrm.2020.100127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de MNF, Goodall CP, Blair TC, Fox BA, McDermott JE, et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun. 2018;9(1):2724. doi: 10.1038/s41467-018-05072-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pauken KE, Shahid O, Lagattuta KA, Mahuron KM, Luber JM, Lowe MM, Huang L, Delaney C, Long JM, Fung ME, et al. Single-cell analyses identify circulating anti-tumor CD8 T cells and markers for their enrichment. J Exp Med. 2021;218(4). doi: 10.1084/jem.20200920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kershaw MH, Hsu C, Mondesire W, Parker LL, Wang G, Overwijk WW, Lapointe R, Yang JC, Wang RF, Restifo NP, et al. Immunization against endogenous retroviral tumor-associated antigens. Cancer Res. 2001;61(21):7920–7924. [PMC free article] [PubMed] [Google Scholar]
- 23.Melero I, Grimaldi AM, Perez-Gracia JL, Ascierto PA. Clinical Development of Immunostimulatory Monoclonal Antibodies and Opportunities for Combination. Clin Cancer Res. 2013;19(5):997–1008. doi: 10.1158/1078-0432.ccr-12-2214. [DOI] [PubMed] [Google Scholar]
- 24.Melero I, Martinez-Forero I, Dubrot J, Suarez N, Palazón A, Chen L. Palettes of vaccines and immunostimulatory monoclonal antibodies for combination. Clin Cancer Res. 2009;15(5):1507–1509. doi: 10.1158/1078-0432.ccr-08-2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber W-J, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515(7528):577–581. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghorbaninezhad F, Asadzadeh Z, Masoumi J, Mokhtarzadeh A, Kazemi T, Aghebati-Maleki L, Shotorbani SS, Shadbad MA, Baghbanzadeh A, Hemmat N, et al. Dendritic cell-based cancer immunotherapy in the era of immune checkpoint inhibitors: from bench to bedside. Life Sci. 2022;297:120466. doi: 10.1016/j.lfs.2022.120466. [DOI] [PubMed] [Google Scholar]
- 27.Du X, Tang F, Liu M, Su J, Zhang Y, Wu W, Devenport M, Lazarski CA, Zhang P, Wang X, et al. A reappraisal of CTLA-4 checkpoint blockade in cancer immunotherapy. Cell Res. 2018;28(4):416–432. doi: 10.1038/s41422-018-0011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seo W, Jerin C, Nishikawa H. Transcriptional regulatory network for the establishment of CD8+ T cell exhaustion. Exp Mol Medicine. 2021;53(2):202–209. doi: 10.1038/s12276-021-00568-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fu T, He Q, Sharma P. The ICOS/ICOSL pathway is required for optimal antitumor responses mediated by anti-CTLA-4 therapy. Cancer Res. 2011;71(16):5445–5454. doi: 10.1158/0008-5472.can-11-1138. [DOI] [PubMed] [Google Scholar]
- 30.Tang DN, Shen Y, Sun J, Wen S, Wolchok JD, Yuan J, Allison JP, Sharma P. Increased frequency of ICOS+ CD4 T cells as a pharmacodynamic biomarker for anti-CTLA-4 therapy. Cancer Immunol Res. 2013;1(4):229–234. doi: 10.1158/2326-6066.cir-13-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang B, Wang Y, Sun X, Deng G, Huang W, Wu X, Gu Y, Tian Z, Fan Z, Xu Q, et al. CXCR6 is required for antitumor efficacy of intratumoral CD8+ T cell. J Immunother Cancer. 2021;9(8):e003100. doi: 10.1136/jitc-2021-003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amsen D, van GK, Hombrink P, van LRAW. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat Immunol. 2018;19(6):538–546. doi: 10.1038/s41590-018-0114-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, [CJ, DY], upon reasonable request.