

Abstract
Phenylketonuria (PKU) is the most common inborn error of metabolism of the liver, and results from mutations of both alleles of the phenylalanine hydroxylase gene (PAH). As such, it is a suitable target for gene therapy via gene delivery with a recombinant adeno‐associated virus (AAV) vector. Here we use the synthetic AAV vector Anc80 via systemic administration to deliver a functional copy of a codon‐optimized human PAH gene, with or without an intron spacer, to the Pah enu2 mouse model of PKU. Dose‐dependent transduction of the liver and expression of PAH mRNA were present with both vectors, resulting in significant and durable reduction of circulating phenylalanine, reaching near control levels in males. Coat color of treated Pah enu2 mice reflected an increase in pigmentation from brown to the black color of control animals, further indicating functional restoration of phenylalanine metabolism and its byproduct melanin. There were no adverse effects associated with administration of AAV up to 5 × 1012 VG/kg, the highest dose tested. Only minor and/or transient variations in some liver enzymes were observed in some of the AAV‐dosed animals which were not associated with pathology findings in the liver. Finally, there was no impact on cell turnover or apoptosis as evaluated by Ki‐67 and TUNEL staining, further supporting the safety of this approach. This study demonstrates the therapeutic potential of AAV Anc80 to safely and durably cure PKU in a mouse model, supporting development for clinical consideration.
Keywords: adeno‐associated virus (AAV) vector, Anc80, gene therapy, inborn error of metabolism, phenylketonuria
Synopsis.
Gene delivery of the human phenylalanine hydroxylase complementary DNA using the synthetic adeno‐associated virus (AAV) serotype Anc80 leads to safe and durable therapeutic cure of phenylketonuria (PKU) in a mouse model of the disease, demonstrating the suitability of AAV vectors for PKU gene therapy.
1. INTRODUCTION
Phenylketonuria (PKU, OMIM #261600)) is an autosomal recessive inborn error of metabolism characterized by a deficiency in phenylalanine hydroxylase (PAH, EC 1.14.16.1), a hepatic enzyme responsible for the conversion of phenylalanine (Phe) to tyrosine (Tyr). PKU is predominantly caused by loss‐of‐function mutations in the PAH gene, leading to disruption of the metabolic pathway of Phe and its over‐accumulation in the blood and central nervous system. Severity is classified into three categories on the basis of blood Phe concentrations: classical PKU (Phe >1200 μmol/L), mild PKU (Phe between 600 and 1200 μmol/L), and mild hyperphenylalaninemia (Phe between 120 and 600 μmol/L). 1 If untreated, PKU is associated with progressive intellectual and behavioral impairment, motor deficits, seizures, microcephaly, and skin and hair hypopigmentation. 2 PKU is typically identified through neonatal screening programs, and the implementation of a Phe‐restricted diet is sufficient to prevent the majority of symptoms. However, high cost of treatment, low compliance owing to unpalatability, and the extant possibility of neuropsychological and neurophysiological complications 3 make alternative treatments to PKU desirable.
One such approach showing positive outcomes is an adeno‐associated virus (AAV)‐based gene therapy targeting a functional recombinant PAH gene to the liver. 4 , 5 , 6 AAV is a nonpathogenic, helper‐dependent virus capable of encoding a transgene cassette of up to 4.8 kb. Upon delivery to the nucleus, the single‐stranded AAV genome is converted to double‐stranded DNA and the vector genome is concatemerized and circularized. 7 In human tissues, AAV vector genomes can persist in nondividing cells in this episomal form, sustaining long‐term expression of the transgene. 8 Studies employing AAV‐mediated PAH gene therapy in mouse models of PKU have achieved near‐complete restoration of liver PAH activity and the reversal of symptoms without dietary limitations in Phe. However, challenges have emerged with evidence of an attenuated long‐term therapeutic response in female mice compared to male mice. 4 , 9
A recent notable advancement in AAV optimization is the use of in silico techniques to identify new synthetic serotypes such as AAV‐Anc80, a predicted AAV ancestor within the lineage of the commonly used AAV serotypes 1, 2, 8, and 9. Synthetic AAV capsid variants developed from Anc80 have compared favorably to naturally occurring AAVs, demonstrating broader transduction profiles, reduced cross‐reactivity, and higher stability and efficiency as a gene transfer vehicle in retina, muscle, and liver. 10 , 11 Furthermore, because it is not a naturally occurring AAV serotype, any restriction in the pool of potential patients due to prior exposure and the presence of serotype‐specific neutralizing antibodies would theoretically be reduced. Anc80's promise as an alternative to natural AAVs marks it as a compelling potential candidate for AAV‐mediated PAH gene therapy. The aim of this study therefore was to evaluate the transduction profile and transgene expression efficiency in the Pah enu2 mouse model of PKU of two distinct Anc80 AAV vectors expressing PAH, under the albumin enhancer and alpha 1 anti‐trypsin (A1AT) promoter, constructed either with or without a minute virus of mice (MVM) intron spacer. 12 , 13
2. MATERIALS AND METHODS
2.1. Animals and animal care
All mice were wild‐type C57BL/6 or homozygous C57BL/6‐Pah enu2/enu2 (referred to as Pah enu2 ) background and were obtained from Jackson Laboratories and bred at Mayo Clinic. Animals were fed standard PicoLab Rodent Diet 20 (LabDiet, St. Louis, Missouri), with 21% protein, 0.91% phenylalanine. Daily observations and weekly body weight assessments were performed by trained animal technical staff with veterinary support from institution veterinarians. Animals were dosed intravenously via orbital plexus at indicated dose levels. Animals were sacrificed using CO2 asphyxiation followed by cervical dislocation in adherence with institutional policies. Blood samples were collected postmortem via the inferior vena cava. Tissues were collected immediately after termination, dabbed to remove excess blood, and weighed for calculation of ratios to terminal body weights.
2.2. Cell lines
Huh7 (Japanese Collection of Research Bioresources Cell Bank: 0403), Hepa1‐6 (AddexBio C0015005 CRL‐1830), and HEK‐293 T (AddexBio T0011002) cells were grown in DMEM (Gibco BRL) supplemented with 10% FBS, 2 mM glutamine, 100 μg/mL streptomycin, 100 U/mL penicillin, and only for HEK‐293T, 110 μg/mL sodium pyruvate.
2.3. AAV vector construct
2.3.1. Cloning and construction of AAV vectors
Synthetic wild‐type (wt) and codon‐optimized (co) sequences of human PAH complementary DNA (cDNA) (NCBI Reference Sequence: NP_000268.1) with and without an internal MVM intron flanked by BspH I and Nru I sites were obtained from ThermoFisher. To generate AAV‐PAH plasmids, the DNA construct for each PAH variant was extracted by EcoR V and Nru I digestion and subcloned into a pAAV2 cloning plasmid previously generated in our laboratory and digested with Pml I, resulting in pAAV‐wtPAH, pAAV‐coPAH, pAAV‐int‐wtPAH, pAAV‐int‐coPAH. These plasmids contain wild‐type AAV2 inverted terminal repeats and have the corresponding PAH ORF downstream of the albumin enhancer and A1AT (AE/A1AT) liver‐specific promoter 12 and a synthetic polyadenylation signal 14 at the 3′ end of the ORF.
2.3.2. Production of AAV vectors
Three vectors were produced, one encoding GFP, one encoding coPAH with the MVM intron in the expression cassette, and one encoding PAH without the MVM intron in the expression cassette.
For each vector, Anc80 AAV vector particles (VPs) were produced in 150‐cm2 flasks containing confluent adherent HEK‐293T cells co‐transfected using linear polyethyleneimine 25 kDa (Polysciences, Warrington, Pennsylvania) with three plasmids: the pAAV (either AAV‐GFP or pAAV‐coPAH or pAAV‐int‐coPAH), pAnc80‐AAP2 (which contain AAV2 rep and Anc80 cap and AAP2 genes) and pΔF6 (which contain adenoviral helper genes) at wt/wt/wt ratio of 1:1:2. After 72 hours, the supernatant was collected and treated with polyethylene glycol solution (PEG8000, 8% vol/vol final concentration) for 48 to 72 hours at 4°C. Supernatant was centrifuged at 1378g for 15 minutes and the pellet was resuspended in lysis buffer (50 mM Tris‐Cl, 150 mM NaCl, 2 mM MgCl2, 0.1% Triton X‐100) and kept at −80°C. Cells containing AAV VPs were collected and treated with lysis buffer and frozen at −80°C. After three cycles of freezing and thawing, VPs obtained from cell supernatants and lysates were purified by ultracentrifugation at 350 000g during 2.5 hours in a 15% to 57% iodioxanol gradient. 15 Finally, the purified virus was concentrated using Amicon Ultra Centrifugal Filters‐Ultracel 100K (Millipore). AAV‐PAH vector titers (viral genomes [VG]/mL) were determined by quantitative PCR (qPCR) with primers specific for A1AT promoter (forward primer: 5′‐TTGCTCCTCCGATAACTGGG‐3′; reverse primer: 5′‐CCCTGTCCTCGTCCGTATTT‐3′). VGs were extracted from DNAase‐treated VPs using the High Pure Viral Nucleic Acid Kit (Roche). The vector titers ranged from 1 × 1013 to 2.5 × 1013 VG/mL.
2.4. In vitro analysis of PAH expression
Huh7 or Hepa1‐6 cells were transfected with pAAV‐PAH plasmids using X‐tremeGENE HP (Roche). Cells were harvested at 48 hours and PAH protein expression was detected by Western blot using a primary rabbit monoclonal antibody specific for PAH (Abcam [EPR12380] ab178430, 1:1000). A goat anti‐rabbit HRP‐conjugated antiserum (Cell Signaling #7074, 1:10 000) was used for detection, and band intensities were quantified utilizing Image Studio Lite software. For AAV infectivity comparison, Huh7 cells were plated and 24 hours later, media was changed to contain 2% FBS and cells were infected with AAV vectors at MOI of 1 × 105 VG/cell. Seventy‐two hours later, cells were harvested and total RNA was isolated from cell lysates using TRIzol Reagent (ThermoFisher), treated with DNase I, Amplification Grade (Invitrogen), and retro‐transcribed into cDNA using M‐MLV reverse‐transcriptase (Invitrogen). qPCR was performed using primers specific for codon‐optimized‐PAH (forward primer: 5′‐ACACATCGAGAGCAGACCCAGC‐3′; reverse primer: 5′‐GCAATGTCGGCGAACTGCTT‐3′). H3F3A was used as normalizing gene (forward primer: 5′‐AAAGCCGCTCGCAAGAGTGCG‐3′; reverse primer: 5′‐ACTTGCCTCCTGCAAAGCAC‐3′.
2.5. Analysis of liver transduction
Small sections of liver (5‐10 mg) were harvested from the right lobe at necropsy and frozen in liquid nitrogen for storage at −80°C until processed for DNA extraction using NucleoSpin Tissue Genomic DNA Purification kit (Macherey‐Nagel, Duren, Germany). The same amount of DNA from each sample was used to quantify VG copies by qPCR using iQ SYBR Green Supermix (BioRad) in a ViiA 7 (ThermoFisher) with primers specific for the A1AT liver promoter (forward, 5′‐TTGCTCCTCCGATAACTGGG‐3′; reverse, 5′‐CCCTGTCCTCGTCCGTATTT‐3′). Mouse glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used as a normalizing gene (forward primer: 5′‐GGATGCAGGGATGATGTTC‐3′; reverse primer: 5′‐TGCACCACCAACTGCTTA′‐3′).
2.6. PAH activity assay
PAH enzyme activity was quantified in crude liver homogenate using a previously established radioactive chromatography assay 16 with correction for protein content measured using a bicinchonic acid microprotein method (Micro BCA Protein Assay Kit, ThermoScientific, 3747 N. Meridian Rd., Rockford, Illinois). The specific liver PAH activities from all experimental animals were normalized to the mean liver PAH activity of saline‐treated wild‐type C57Bl/6 mice.
2.7. Transgene expression analysis
Small sections of liver (5‐10 mg) were harvested from the right lobe at necropsy and frozen in liquid nitrogen for storage at −80°C until processed for total RNA isolation using the Maxwell 16 LEV simplyRNA Tissue Kit (Promega) according to manufacturer's instructions and equal quantities of extracted RNA were reverse transcribed into cDNA using M‐MLV reverse‐transcriptase (Invitrogen). qPCR was performed using iQ SYBR Green Supermix (BioRad) and primers specific for co‐PAH (forward primer: 5′‐ACACATCGAGAGCAGACCCAGC‐3′; reverse primer: 5′‐GCAATGTCGGCGAACTGCTT‐3′) and run on a ViiA 7 (ThermoFisher). Mouse GAPDH was used as a normalizing gene (forward primer: 5′‐GGATGCAGGGATGATGTTC‐3′; reverse primer: 5′‐TGCACCACCAACTGCTTA′‐3′).
2.8. Biochemical analysis
For clinical chemistry analysis, serum was analyzed with the Piccolo Xpress chemistry analyzer (Abaxis, Union City, California) according to the manufacturer's instructions. Phenylalanine values were determined using tandem mass spectrometry and chromatography via Mayo Clinic's internal biochemical PKU test.
2.9. Histology analysis
For histological analysis, tissue samples were fixed in 10% neutral buffered formalin (Protocol, Fisher‐Scientific, Pittsburgh, Pennsylvania) and processed for paraffin embedding and sectioning. For hematoxylin and eosin staining, slides were prepared with standard protocols and evaluated by a board‐certified veterinary pathologist for variations. Ki‐67 immunohistochemistry was performed using a monoclonal anti‐Ki67 primary antibody (MIB‐1; Dako/Agilent, Santa Clara, California) as performed with a Bond III automatic stainer (Leica, Buffalo Grove, Illinois) with a 20‐minute antigen retrieval step using Bond Epitope Retrieval Solution 2 (Leica), and stained with diaminobenzidine (Leica). Ki‐67 and TUNEL quantification was performed by selecting up to three random cross sections per slide manually verified to avoid staining artifacts. Areas were analyzed and quantified using an Aperio ImageScope algorithm that quantifies nuclear staining. Results are reported as percentage of nuclear positivity among cells analyzed.
2.10. Statistical analysis
Numerical data are expressed as mean (±SD). Calculations and statistical analysis were performed using Microsoft Excel 2010, and additional statistical analyses were performed with GraphPad Prism software version 7.03 (San Diego, California). Comparisons between two groups were made using a two‐tailed unpaired t test. One‐way analysis of variance (ANOVA) with multiple comparisons with correction was used for comparison of more than two groups. Two‐way ANOVA with multiple comparisons and Tukey's correction was used to assess statistical significance of data with two variables. Statistical significance was assigned to P‐values <.05.
3. RESULTS
3.1. Relative PAH mRNA expression in vitro allows selection of candidates
The wild‐type (wt) and codon optimized (co) versions of human PAH cDNA, each with and without the MVM intron, were compared for PAH protein expression after transient plasmid transfection of human (HuH7) or murine (Hepa1‐6) hepatocytes in vitro. Forty‐eight hours after transfection cells were harvested and PAH protein expression was analyzed by Western blot (Figure 1A) and band intensities were quantified and normalized to the negative control (cells transfected with pAAV‐GFP, Figure 1B). In both human and mouse cells, the co sequence induced a higher PAH expression than the wt PAH sequence. Further, for both wt and co cDNA and in both cell lines, the presence of the MVM intron resulted in a higher expression of PAH protein.
FIGURE 1.
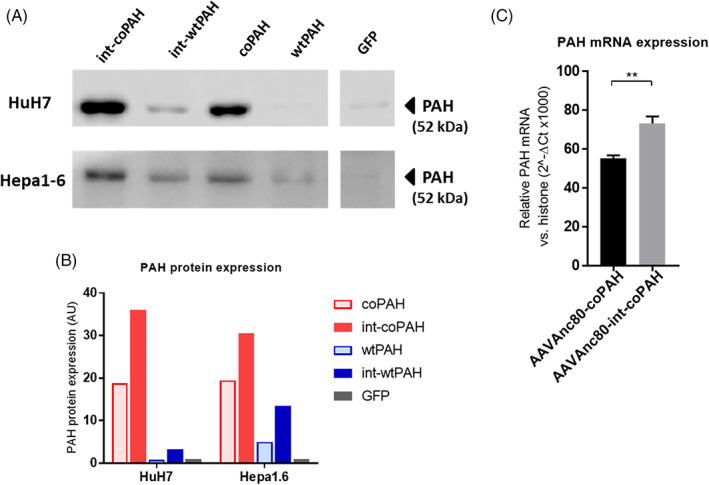
A, Western blot analysis of phenylalanine hydroxylase gene (PAH) expression following plasmid transfection in HuH7 or Hepa1‐6 cell lines with four construct variants of human PAH. B, Relative protein expression levels normalized to a mock control for transfection. Green fluorescent protein (GFP), mock control. C, PAH mRNA levels following transduction with adeno‐associated virus (AAV) with and without the MVM intron spacer in HuH7 cells. Statistical significance was assessed by unpaired t test, **P < .01
Next, Anc80‐coPAH and Anc80‐int‐coPAH vectors were generated containing coPAH or int‐coPAH, respectively, and were used to transduce HuH7 cells. The levels of PAH mRNA were determined 72 hours after transduction (Figure 1C). As with the protein expression following plasmid transfection, AAV Anc80 containing the codon optimized sequence with the internal MVM intron (AAVAnc80‐int‐coPAH) resulted in higher mRNA expression than the AAV Anc80 without intron (AAVAnc80‐coPAH).
3.2. Dose‐dependent GFP expression observed in mice treated with AAV‐Anc80‐GFP
To determine transduction and expression efficiency of AAV‐Anc80‐serotype vectors in Pahenu2 mice, an AAV‐Anc80 vector carrying the GFP reporter gene under the transcriptional control of the AE/A1AT promoter (Anc80‐GFP) was administered at 1.5 × 1012, 5 × 1012, or 1.5 × 1013 VG/kg via intravenous injection (Table 1). As control, wt mice received a dose of 5 × 1012 VG/kg. Animals were evaluated for routine observations, body weight changes, and terminal liver weight after 3 weeks, as well as transduction and transgene expression by the vector. Pahenu2 mice had lower body weights than age‐matched wild‐type mice prior to dosing (Figure 2A), but there were no effects of Anc80‐GFP administration at any dose level on morbidity/mortality, observational data or liver/body weight or ratio (data not shown). All mice survived for the duration of the study.
TABLE 1.
Design of AAV‐GFP mouse study. WT and Pahenu2 mice were assigned to one of six treatment groups with escalating doses of Anc80‐GFP, an adeno‐associated virus vector expressing GFP under the control of the liver‐specific A1AT promoter. Mice were kept on study for 3 weeks after a single administration of vector on day 1. Mice underwent a focused necropsy at the end of the study to evaluate transduction and expression of the vector‐mediated GFP transgene
| Group | Genotype | Article | Dose (VG/kg) | Day 21 (M/F) |
|---|---|---|---|---|
| 1 | Pahenu2 | Anc80‐GFP | 1.5 × 1012 | 3/3 |
| 2 | Pahenu2 | Anc80‐GFP | 5 × 1012 | 3/3 |
| 3 | Pahenu2 | Anc80‐GFP | 1.5 × 1013 | 3/3 |
| 4 | Pahenu2 | Saline | Saline | 2/2 |
| 5 | WT | Anc80‐GFP | 5 × 1012 | 3/3 |
| 6 | WT | Saline | Saline | 2/2 |
Abbreviations: A1AT, alpha‐1 antitrypsin; AAV, adeno‐associated virus; f, female; GFP, green fluorescent protein; m, male; Pahenu2, PKU mouse; VG, vector genomes; WT, wild type.
FIGURE 2.
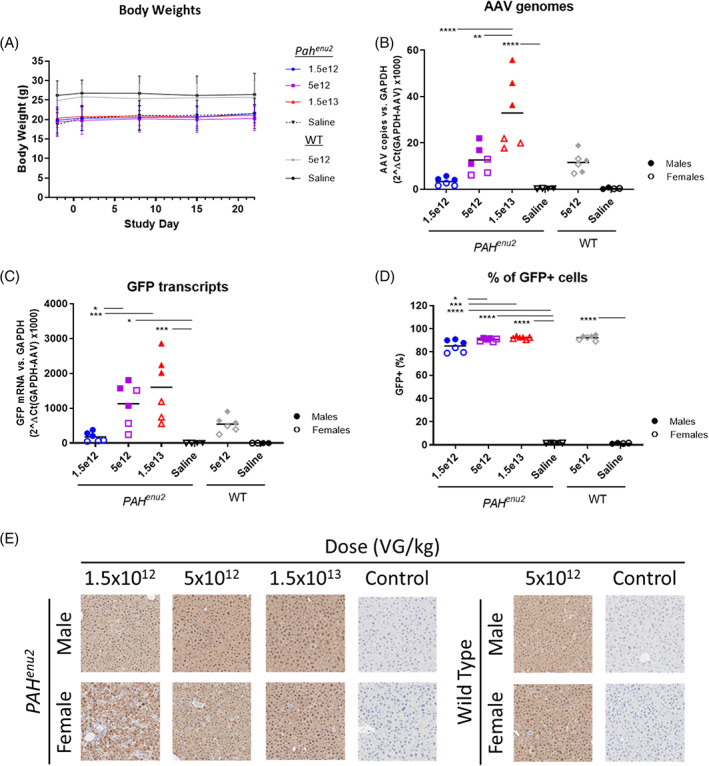
Mice dosed with increasing levels of Anc80‐green fluorescent protein (GFP) showed dose dependent transduction and vector expression. A, There were no dose‐related variations in body weights over the course of the study within genotypes regardless of vector dose administered. Increasing doses of Anc80‐GFP resulted in dose‐dependent increases in, B, adeno‐associated virus (AAV) genomes and C, GFP transcripts present in the livers of Pahenu2 mice. Males showed enhanced responsiveness relative to females in both parameters, with a similar trend present in the wt mice tested at the mid dose of 5 × 1012 VG/kg. D, There was high transduction frequency in the livers at all dose levels tested based on IHC of GFP in representative fixed sections, leaving only incremental gains with increasing doses. E, Immunohistochemistry micrographs of GFP staining in representative sections of livers from male and female mice in each dose group. Dose‐dependent increases in transduction frequency were present in Pahenu2 mice, but were more pronounced in females due to the lower percentage of transduced hepatocytes at the low and mid doses compared to males. Staining in wt mice at the mid dose was similar to that in the Pahenu2 mice, with the same trend toward increased transduction in males. One‐way analysis of variance (ANOVA) with multiple comparisons and Bonferroni correction were used to assess statistical significance, P < .05 (*) .01 (**), .001 (***), or .0001 (****)
Increasing doses of Anc80‐GFP were associated with increasing rates of transduction, GFP transcription, and percentage of GFP‐expressing cells in the liver (Figure 2B‐D). There was no difference between transduction rates at the mid dose group between Pahenu2 mice and wt mice, but there was increased transduction in males compared to females within each dose group, especially at the highest dose (solid vs open symbols in Figure 2B). GFP transcript expression showed a similar trend as AAV transduction; however, Pahenu2 mice showed slightly higher relative expression of GFP mRNA at the same dose level compared to wt mice. Again, a trend supporting more expression in males compared to females within a dose group and genotype was present in GFP mRNA levels (Figure 2C). GFP‐positive cells increased with increasing dose, but the difference between dose groups was minimal (Figure 2D,E). However, the trend for higher percentages in males compared to females was still present.
3.3. AAV‐Anc80 expressing human PAH effectively treats Pah deficiency in knockout mice
Next, AAV‐Anc80 expressing co PAH mRNA with or without the intron were tested for efficacy and safety. Low (5 × 1011 VG/kg) or high (5 × 1012 VG/kg) doses of each AAV vector were administered to 12 to 16‐week‐old wild‐type or Pahenu2 mice via intravenous injection, and animals were evaluated at 4 or 12 weeks (Table 2). Administration of either Anc80‐coPAH or Anc80‐int‐coPAH at 5 × 1012 VG/kg was associated with significant reductions in circulating phenylalanine levels (Figure 3A,B), which were more pronounced in male mice. The reductions were less pronounced at the lower dose of 5 × 1011 VG/kg, but similar over the duration of the study within dose groups, indicating durability of the effect and resulting in near‐normal phenylalanine levels in the high‐dose‐treated Pahenu2 males (Figure 3A). These persistent reductions in phenylalanine resulted in reversion of coat color in the treated Pahenu2 mice, seen as large splotches to complete coverage of blacker fur resembling wild‐type animals that was not present in saline‐treated age‐matched Pahenu2 mice (Figure 3C). Coat color changes were first observed at the high dose with both vectors on Day 22 and in some low dose animals on days 26 to 28.
TABLE 2.
Design of AAV‐PAH mouse study. WT and Pahenu2 mice were assigned to one of six treatment groups with escalating doses of two variants of Anc80‐coPAH, an AAV vector expressing codon optimized PAH either with or without an internal intron under the control of the liver‐specific albumin enhancer and A1AT promoters. Mice were kept on study for 4 or 12 weeks after a single administration of vector on day 1. Mice underwent a full necropsy at the end of the study to demonstrate transduction and expression of the vector‐mediated PAH transgene, as well as routine toxicological evaluation
| Group | Genotype | Article | Dose (VG/kg) | Day 29 (M/F) | Day 85 (M/F) |
|---|---|---|---|---|---|
| 1 | Pahenu2 | Saline | Saline | — | 3/3 |
| 2 | WT | Saline | Saline | — | 3/3 |
| 3 | Pahenu2 | Anc80‐coPAH | 5 × 1011 | 3/3 | 2/2 |
| 4 | Pahenu2 | Anc80‐coPAH | 5 × 1012 | 3/3 | 2/2 |
| 5 | Pahenu2 | Anc80‐int‐coPAH | 5 × 1011 | 3/3 | 2/2 |
| 6 | Pahenu2 | Anc80‐int‐coPAH | 5 × 1012 | 3/3 | 2/2 |
Abbreviations: A1AT, alpha‐1 antitrypsin; AAV, adeno‐associated virus; PAH, phenylalanine hydroxylase gene.
FIGURE 3.
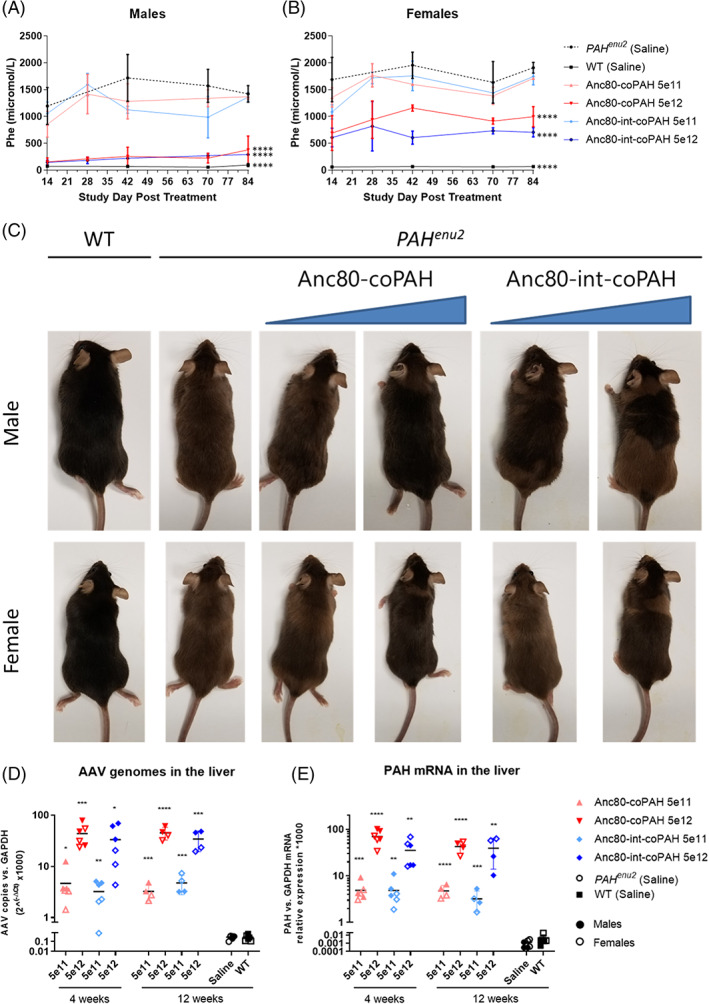
Expression of phenylalanine hydroxylase gene (PAH) corrected the phenylketonuria (PKU) phenotype in Pahenu2 mice. The high dose of 5 × 1012 VG/kg Anc80‐coPAH and Anc80‐int‐coPAH was associated with acute and sustained decreases in circulating phenylalanine levels in males, A, and females, B, which approached wt levels in males with both PAH constructs. Only negligible benefit was indicated at the lower dose of 5 × 1011 VG/kg. C, Wild‐type animals (first column) had much darker coat color than untreated Pahenu2 mice (second column), and restored metabolism of phenylalanine caused a phenotypic switch back to darker coat color in treated male (top row) and female (bottom row) mice which was dose‐dependent. D, Adeno‐associated virus (AAV) genomes in the liver relative to glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) showed similar dose‐dependent increases between the 5 × 1011 VG/kg and 5 × 1012 VG/kg levels. E, PAH mRNA showed corresponding increases between dose levels, with slightly higher transcription with Anc80‐coPAH. In both analyses, results were negligible for wt animals and Pahenu2 mice dosed with saline. One‐way analysis of variance (ANOVA) with multiple comparisons and Dunnett's correction were used to assess statistical significance, P < .05 (*) .01 (**), .001 (***), or .0001 (****)
3.4. AAV‐Anc80‐coPAH administration resulted in stable transduction, transgene expression, and PAH enzymatic activity in mice
Anc80‐coPAH and Anc80‐int‐coPAH transduction in liver was durable, with consistent vector genomes per mouse genome between AAVs at both dose levels (Figure 3D). Average AAV copies were approximately 4 (per 1000 GAPDH copies) at 5 × 1011 VG/kg for both AAVs at both timepoints, but the AAV without intron had slightly higher transduction at 5 × 1012 VG/kg relative to the AAV with intron (approximately 44 vs 34 VG per 1000 GAPDH copies) at both timepoints. WT and Pahenu2 mice treated with saline had only negligible levels.
Stable transduction with either AAV resulted in stable expression of PAH mRNA over the course of the study (Figure 3E). Trends were similar to VG results, whereby both AAVs showed similar PAH mRNA levels at the 5 × 1011 VG/kg dose, but Anc80‐coPAH showed a trend for slightly better transcription than Anc80‐int‐coPAH at the 5 × 1012 VG/kg dose. Again, WT and Pahenu2 mice treated with saline had only negligible levels of PAH mRNA expression. PAH enzyme activity in livers recovered at both 4 and 12 weeks post dose was also significantly increased in most dosed groups relative to the untreated control group, with a trend for higher activity levels in the males (Figure S1). A two‐way ANOVA, with vector and dosage as variables, revealed that dose level indeed had a significant effect on PAH activity (F(3,32) = 6.6, P < .0013) while vector did not (F(1,32 = 0.0, P < .8949). These data indicate that metabolic correction of the phenotype only required as little as 20% of wild‐type PAH activity.
3.5. AAV‐Anc80 vectors were safe at the doses tested in PAH‐deficient mice
Animals were evaluated for routine observations and body weight changes, and blood samples were collected for serum chemistry analysis. Complete necropsy was performed to assess effects of vector administration at 4 and 12 weeks posttreatment. There were no effects of AAV‐Anc80 administration at any dose level on body weights (Figure 4A), morbidity/mortality, observational or liver/body weight ratio data, or postmortem pathology evaluations (Supplemental Tables 1‐5). All mice survived for the duration of the study. Variations in serum chemistry were limited to elevated ALT for Anc80‐int‐coPAH at 5 × 1011 VG/kg after 12 weeks (Figure 4B), which was slightly higher than the untreated Pahenu2 control mice and not present at 5 × 1012 VG/kg. Complete chemistry tables are presented in Supplemental Data (Supplemental Table 5). There were no associated changes in AST, ALP (Figure 4C,D), adverse pathology, or liver histopathology.
FIGURE 4.
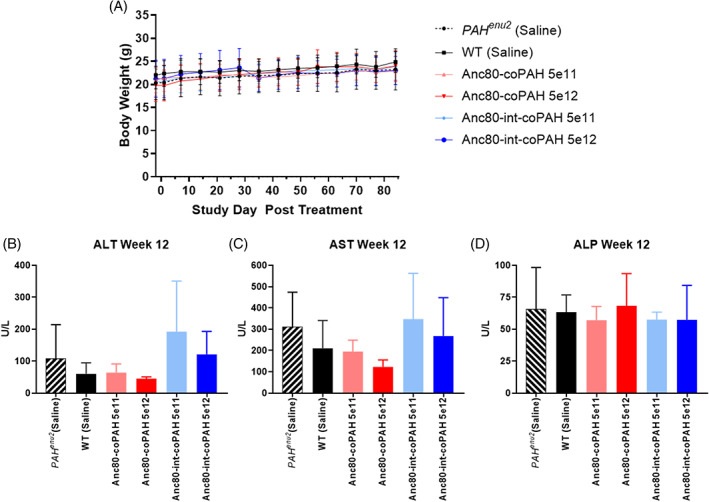
Adeno‐associated virus (AAV) transduction and expression of phenylalanine hydroxylase gene (PAH) was well tolerated in all animals. A, There were no significant variations in body weights over the course of the study within genotypes regardless of vector dose administered. Serum liver enzymes indicated only minor effects on hepatocytes, with Anc80‐int‐coPAH being associated with a minor elevation in, B, ALT at the high dose relative to the untreated Pahenu2 control mice, and no effects of either Anc80 construct on, C, AST, or D, ALP at week 12
Minor changes possibly linked to Anc80‐coPAH or Anc80‐int‐coPAH were limited to a slightly higher prevalence of minimal liver inflammation in some AAV‐dosed mice at both 4 and 12 week necropsy that was not sex‐ or dose‐dependent (Supplemental Tables 3 and 4). This inflammation mostly consisted of randomly scattered mononuclear cells. The small extent of this inflammatory lesion was not interpreted to be adverse.
3.6. AAV‐Anc80‐PAH did not result in increased cell turnover or programmed cell death
There were no effects of either AAV on cell turnover based on Ki67 staining or cell death via apoptosis based on TUNEL staining (Figure 5). Ki‐67 staining was similar in all AAV‐dosed groups (both dose levels) and to control Pahenu2 and WT animals at 1% to 2% of cells. There were also no significant effects of either AAV on frequency of apoptosis based on TUNEL positivity <0.5% of cells. There was a trend for increased TUNEL positivity in untreated Pahenu2 mice compared to wild type, but the overall numbers were very low and not considered biologically relevant.
FIGURE 5.
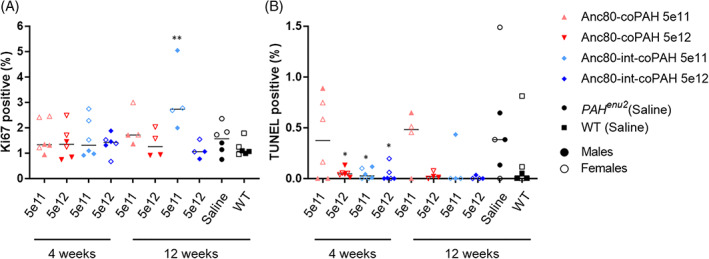
Anc80‐adeno‐associated virus (AAVs) were not associated with changes in cell turnover or apoptosis. A, Percentage of cells that were positive for Ki‐67 staining were similarly low in all groups tested, showing no increased cell turnover resulting from transduction with either AAV. B, Percentage of cells undergoing apoptosis as indicated by TUNEL positivity was also similarly low in all groups tested. In both analyses, results were similar for treated groups, wt animals, and Pahenu2 mice dosed with saline. One‐way analysis of variance (ANOVA) with multiple comparisons and Dunnett's correction were used to assess statistical significance, P < .05 (*) P < .005 (**)
4. DISCUSSION
Single intravenous administration of either Anc80‐coPAH (without intron) or Anc80‐int‐coPAH (with intron) vector was well tolerated at up to 5 × 1012 VG/kg. Administration of both AAVs was associated with marked efficacy, as seen in reduced phenylalanine levels in blood and coat color changes consistent with restored metabolism of phenylalanine. Furthermore, effects were durable through the end of the study after 12 weeks. These findings bode well for confirming the feasibility of a gene therapy for PKU, including demonstration of safety, efficacy, and durability with this AAV platform. Although transduction may be diluted as hepatocytes divide in a younger patient, sufficient transduction to cure PKU is estimated to be around 10% of total liver cells. 17 This threshold would easily be supported by the >80% of liver cell transduction observed with the Anc80‐GFP vector in the current study, even after multiple rounds of cell division in the maturing liver.
However, the response was more prominent in males than females regardless of the Anc80 vector. Differential AAV transduction susceptibility between genders in mice has been previously reported. It appears to be essentially limited to liver transduction and has been suggested to be mediated through an androgen‐dependent pathway. 18 We have previously observed higher transduction and a longer duration of therapeutic effect in males compared to females when treated with an AAV delivering MDR3, a gene whose lack of expression results in progressive familial intrahepatic cholestasis type 3, a different monogenic metabolic disorder of the liver. 19 These sex differences have been determined to be sex hormone related and exist regardless of transgene, promoter or mouse background strain. 18 , 20 , 21 In our study, interestingly the AAV containing the intron had a clearer difference in transduction efficiency between males and females. At the higher dose at both terminal timepoints, the mean AAV genomes (both sexes) were lower for the Anc80‐int‐coPAH than for the intron‐less AAV because all the females had significantly lower transduction (Figure 3D). For the intron‐less AAV, females also were transduced less efficiently although not to the same degree. Despite this, the therapeutic effect lasted the entire duration in females just as well as in males for either vector, even though it was less prominent in females. This indicates that a fully therapeutic treatment could potentially be achieved for female mice simply by increasing the dose. Importantly, there is no evidence to indicate that this phenomenon would also be observed in humans treated with AAV‐mediated gene therapy.
Regardless, Anc80 proved to be a very effective serotype for this monogenic liver disorder, and efficacy was augmented by the use of an intron spacer. Anc80 is a predicted ancestral serotype of current‐day naturally occurring human AAV serotypes 1 to 3 and 6 to 9 that were derived from an in silico process called maximal likelihood of ancestral sequence reconstruction in order to develop an AAV with novel capsid characteristics while maintaining essential functional attributes of known human serotypes, particularly infectivity. 10 Testing of Anc80 AAVs has shown similar transduction efficiency as AAV8 in mouse liver, improved efficiencies in nonhuman primate liver and, importantly, no issues with safety and either no or very low cross‐reactivity with the closest extant human and other primate descendant AAVs. AAV‐Anc80 has also successfully achieved gene transfer to mouse inner ear 22 and retina, 23 as well as human kidney cells. 24 To our knowledge, this is the first demonstration of the use of Anc80 for gene transfer resulting in a phenotypic correction of a metabolic disorder of the liver.
In comparison to the native wt cDNA sequence, codon optimization provides a technical advance that increases the efficiency of expression of the transgene resulting in a higher production of the therapeutic protein. 25 , 26 Several parameters such as mRNA secondary structure, repeat sequences, and guanine‐cytosine nucleotide content dictate the selection of the most efficiently translated codon triplet for each amino acid residue (referred to as codon bias) of the translated protein. Our comparative analysis of the codon optimized sequence with the wt demonstrated substantial expression increase with codon optimization. Although the codon optimization is based on codon translation in human cells, this improvement was also observed in mouse cells in vitro, albeit to a lesser degree than in human cells.
The second technical improvement involves the addition of an intron sequence in between the promoter and the human PAH coding sequence in order to increase transgene expression via multiple mechanisms, including enhancing transcription initiation, transcription elongation, transcription termination, polyadenylation, nuclear export and mRNA stability. Although intron presence in eukaryotic genes provides clear benefit in expression efficiency over intron‐less genes, most introns are much larger than the coding exons so identifying an intronic element small enough to fit within the transgene construct without preventing its packaging within the AAV virus is a challenge. A small intron from MVM of 92 nucleotides in length has shown the most robust potentiation of gene expression from a panel of several small intron sequences. 13 , 27 We incorporated the MVM intron into both the human codon‐optimized and the native wt PAH plasmids and performed a comparative analysis for expression efficiency both in vitro (plasmid transduction of human and mouse hepatocytes) and in vivo (co only). In all cases in vitro, the intron‐containing plasmids or Anc80 vectors showed superior expression compared to the intron‐less sequence. In vivo, the two constructs showed similar transduction and expression, but the one with intron showed a slightly better phenotypic correction.
Effects of administration of these vectors caused no overt sporadic liver enzyme changes and no histological findings attributed to either AAV that were not present in control animals. Importantly, there were no effects of either AAV on liver cell turnover (Ki67 staining) or rates of apoptosis (TUNEL positivity) compared to untreated animals, further supporting the safety of this vector. Furthermore, since AAV would not be replicated during cell division, these results demonstrate that the effectiveness and durability of the gene delivery would not be diluted out by cell turnover induced by transduction.
Based on the similar safety profile and slightly improved efficacy in reducing circulating phenylalanine (particularly in females), these findings favor development of AAV‐Anc80 containing the PAH expression cassette with intron. These data support further development of this and other Anc80‐based AAVs toward consideration of human clinical trials, especially for diseases requiring transduction of a single functional copy of a gene and without localized toxicity at the hepatocyte level. 28
CONFLICT OF INTEREST
N. D. W., L. T. M., A. D., and G. G. A. are employees of Vivet Therapeutics, which funded the study.
AUTHOR CONTRIBUTIONS
Robert A. Kaiser, Nicholas D. Weber, Anne Douar, Rafael Aldabe, Gloria González‐Aseguinolaza, and Joseph B. Lillegard: Designed the study, interpreted data, and wrote the manuscript. Laia Trigueros‐Motos, Kari L. Allen, Michael Martinez, Caitlin J. VanLith, and Lori G. Hillin: Conducted experiments and interpreted data. William Cao: Wrote the manuscript.
ETHICS STATEMENT
This study did not involve human subjects.
ANIMAL CARE AND USE APPROVAL
This study was approved by the Institutional Animal Care and Use Committee of the Mayo Clinic, Rochester, Minnesota.
Supporting information
Appendix S1: Supporting Information
ACKNOWLEDGMENTS
The authors thank Brad Bolon (GEMpath, Longmont, CO) for pathology evaluation. Additionally, the authors would also like to thank Luk Vandenberghe and his laboratory for kindly providing expertise in the Anc80 serotype. This study was funded by Vivet Therapeutics.
Kaiser RA, Weber ND, Trigueros‐Motos L, et al. Use of an adeno‐associated virus serotype Anc80 to provide durable cure of phenylketonuria in a mouse model. J Inherit Metab Dis. 2021;44:1369–1381. 10.1002/jimd.12392
Funding information Vivet Therapeutics
Contributor Information
Rafael Aldabe, Email: raldabe@unav.es.
Joseph B. Lillegard, Email: jlillegard@msn.com.
DATA AVAILABILITY STATEMENT
All data from this study are presented in the manuscript or supplemental information.
REFERENCES
- 1. Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet. 2010;376:1417‐1427. [DOI] [PubMed] [Google Scholar]
- 2. Scriver CR, Waters PJ. Monogenic traits are not simple: lessons from phenylketonuria. Trends Genet. 1999;15:267‐272. [DOI] [PubMed] [Google Scholar]
- 3. Christ SE, Huijbregts SC, de Sonneville LM, White DA. Executive function in early‐treated phenylketonuria: profile and underlying mechanisms. Mol Genet Metab. 2010;99(Suppl 1):S22‐S32. [DOI] [PubMed] [Google Scholar]
- 4. Mochizuki S, Mizukami H, Ogura T, et al. Long‐term correction of hyperphenylalaninemia by AAV‐mediated gene transfer leads to behavioral recovery in phenylketonuria mice. Gene Ther. 2004;11:1081‐1086. [DOI] [PubMed] [Google Scholar]
- 5. Ding Z, Georgiev P, Thony B. Administration‐route and gender‐independent long‐term therapeutic correction of phenylketonuria (PKU) in a mouse model by recombinant adeno‐associated virus 8 pseudotyped vector‐mediated gene transfer. Gene Ther. 2006;13:587‐593. [DOI] [PubMed] [Google Scholar]
- 6. Yagi H, Ogura T, Mizukami H, et al. Complete restoration of phenylalanine oxidation in phenylketonuria mouse by a self‐complementary adeno‐associated virus vector. J Gene Med. 2011;13:114‐122. [DOI] [PubMed] [Google Scholar]
- 7. Nakai H, Yant SR, Storm TA, Fuess S, Meuse L, Kay MA. Extrachromosomal recombinant adeno‐associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J Virol. 2001;75:6969‐6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schnepp BC, Jensen RL, Chen CL, Johnson PR, Clark KR. Characterization of adeno‐associated virus genomes isolated from human tissues. J Virol. 2005;79:14793‐14803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Harding CO, Gillingham MB, Hamman K, et al. Complete correction of hyperphenylalaninemia following liver‐directed, recombinant AAV2/8 vector‐mediated gene therapy in murine phenylketonuria. Gene Ther. 2006;13:457‐462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zinn E, Pacouret S, Khaychuk V, et al. In silico reconstruction of the viral evolutionary lineage yields a potent gene therapy vector. Cell Rep. 2015;12:1056‐1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carvalho JR, Verdelho Machado M. New insights about albumin and liver disease. Ann Hepatol. 2018;17:547‐560. [DOI] [PubMed] [Google Scholar]
- 12. Kramer MG, Barajas M, Razquin N, et al. In vitro and in vivo comparative study of chimeric liver‐specific promoters. Mol Ther. 2003;7:375‐385. [DOI] [PubMed] [Google Scholar]
- 13. Wu Z, Sun J, Zhang T, et al. Optimization of self‐complementary AAV vectors for liver‐directed expression results in sustained correction of hemophilia B at low vector dose. Mol Ther. 2008;16:280‐289. [DOI] [PubMed] [Google Scholar]
- 14. Levitt N, Briggs D, Gil A, Proudfoot NJ. Definition of an efficient synthetic poly(a) site. Gene Dev. 1989;3:1019‐1025. [DOI] [PubMed] [Google Scholar]
- 15. Murillo O, Luqui DM, Gazquez C, et al. Long‐term metabolic correction of Wilson's disease in a murine model by gene therapy. J Hepatol. 2016;64:419‐426. [DOI] [PubMed] [Google Scholar]
- 16. Harding CO, Wild K, Chang D, Messing A, Wolff JA. Metabolic engineering as therapy for inborn errors of metabolism—development of mice with phenylalanine hydroxylase expression in muscle. Gene Ther. 1998;5:677‐683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harding CO, Gibson KM. Therapeutic liver repopulation for phenylketonuria. J Inherit Metab Dis. 2010;33:681‐687. [DOI] [PubMed] [Google Scholar]
- 18. Davidoff AM, Ng CY, Zhou J, Spence Y, Nathwani AC. Sex significantly influences transduction of murine liver by recombinant adeno‐associated viral vectors through an androgen‐dependent pathway. Blood. 2003;102:480‐488. [DOI] [PubMed] [Google Scholar]
- 19. Weber ND, Odriozola L, Martinez‐Garcia J, et al. Gene therapy for progressive familial intrahepatic cholestasis type 3 in a clinically relevant mouse model. Nat Commun. 2019;10:5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Berraondo P, Crettaz J, Ochoa L, et al. Intrahepatic injection of recombinant adeno‐associated virus serotype 2 overcomes gender‐related differences in liver transduction. Hum Gene Ther. 2006;17:601‐610. [DOI] [PubMed] [Google Scholar]
- 21. Paneda A, Vanrell L, Mauleon I, et al. Effect of adeno‐associated virus serotype and genomic structure on liver transduction and biodistribution in mice of both genders. Hum Gene Ther. 2009;20:908‐917. [DOI] [PubMed] [Google Scholar]
- 22. Landegger LD, Pan B, Askew C, et al. A synthetic AAV vector enables safe and efficient gene transfer to the mammalian inner ear. Nat Biotechnol. 2017;35:280‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang L, Xiao R, Andres‐Mateos E, Vandenberghe LH. Single stranded adeno‐associated virus achieves efficient gene transfer to anterior segment in the mouse eye. PLoS One. 2017;12:e0182473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ikeda Y, Sun Z, Ru X, Vandenberghe LH, Humphreys BD. Efficient gene transfer to kidney mesenchymal cells using a synthetic adeno‐associated viral vector. J Am Soc Nephrol. 2018;29:2287‐2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li C, Goudy K, Hirsch M, et al. Cellular immune response to cryptic epitopes during therapeutic gene transfer. Proc Natl Acad Sci U S A. 2009;106:10770‐10774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fischer MD, McClements ME, Martinez‐Fernandez de la Camara C, et al. Codon‐optimized RPGR improves stability and efficacy of AAV8 gene therapy in two mouse models of X‐linked retinitis pigmentosa. Mol Ther. 2017;25:1854‐1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haut DD, Pintel DJ. Intron definition is required for excision of the minute virus of mice small intron and definition of the upstream exon. J Virol. 1998;72:1834‐1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thompson WS, Mondal G, Vanlith CJ, Kaiser RA, Lillegard JB. The future of gene‐targeted therapy for hereditary tyrosinemia type 1 as a lead indication among the inborn errors of metabolism. Expert Opin Orphan Drugs. 2020;8:245‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information
Data Availability Statement
All data from this study are presented in the manuscript or supplemental information.