

Abstract
Risdiplam (Evrysdi) improves motor neuron function in patients with spinal muscular atrophy (SMA) and has been approved for the treatment of patients ≥2 months old. Risdiplam exhibits time‐dependent inhibition of cytochrome P450 (CYP) 3A in vitro. While many pediatric patients receive risdiplam, a drug–drug interaction (DDI) study in pediatric patients with SMA was not feasible. Therefore, a novel physiologically‐based pharmacokinetic (PBPK) model‐based strategy was proposed to extrapolate DDI risk from healthy adults to children with SMA in an iterative manner. A clinical DDI study was performed in healthy adults at relevant risdiplam exposures observed in children. Risdiplam caused an 1.11‐fold increase in the ratio of midazolam area under the curve with and without risdiplam (AUCR)), suggesting an 18‐fold lower in vivo CYP3A inactivation constant compared with the in vitro value. A pediatric PBPK model for risdiplam was validated with independent data and combined with a validated midazolam pediatric PBPK model to extrapolate DDI from adults to pediatric patients with SMA. The impact of selected intestinal and hepatic CYP3A ontogenies on the DDI susceptibility in children relative to adults was investigated. The PBPK analysis suggests that primary CYP3A inhibition by risdiplam occurs in the intestine rather than the liver. The PBPK‐predicted risdiplam CYP3A inhibition risk in pediatric patients with SMA aged 2 months–18 years was negligible (midazolam AUCR of 1.09–1.18) and included in the US prescribing information of risdiplam. Comprehensive evaluation of the sensitivity of predicted CYP3A DDI on selected intestinal and hepatic CYP3A ontogeny functions, together with PBPK model‐based strategy proposed here, aim to guide and facilitate DDI extrapolations in pediatric populations.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ While children can have a different drug–drug interaction (DDI) susceptibility than adults, pediatric DDI studies are often ethically challenging. Physiologically‐based pharmacokinetic (PBPK) modeling can assist in prospective DDI risk evaluation in children.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Can we mechanistically extrapolate the risdiplam time‐dependent inhibition (TDI) of CYP3A studied in healthy adults to children with spinal muscular atrophy using qualified PBPK models?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ The increase in midazolam exposure by risdiplam in healthy adults was <20%. PBPK simulations over ranges of hepatic and intestinal CYP3A ontogenies and TDI parameters indicated a similar DDI propensity in children ≥ 2 months of age, and low likelihood of a clinically relevant CYP3A DDI.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Pediatric PBPK modeling coupled with adequately designed study in adults enables prospective DDI risk assessments in children. Selection of ontogeny models based on sensitivity to enzyme modulation allows for conservative DDI extrapolation to children.
Spinal muscular atrophy (SMA types 1–4) is a severe neuromuscular disease resulting in progressive proximal muscle weakness and paralysis. 1 The underlying cause of this rare disease is an insufficiency in survival motor neuron (SMN) protein due to deletion of and/or loss of function mutation within the SMN1 gene. 2 , 3 Risdiplam (RG7916) distributes into central and peripheral tissues after oral administration and modifies splicing of pre–messenger RNA (mRNA) of the SMN2 gene to increase functional SMN protein in patients with SMA. 4 Type 1 is the most severe form of SMA with an onset of symptoms in infants before the age of 6 months. Infants with this disease are unable to sit unsupported, may require permanent ventilation, and have a life expectancy of 2 years. 5 The clinical trial for risdiplam in infants with Type 1 SMA showed an approximately twofold increase in the SMN protein, an ability to sit without support after 12 months of treatment in 41% of patients, and significant reduction in mortality and need for permanent ventilation. 6 Risdiplam was approved by the US Food and Drug Administration (FDA) in 2020 as the first orally administered drug for SMA treatment, 7 followed by European Medicines Agency in 2021. 8
Risdiplam and its metabolite M1 exhibit time‐dependent inhibition (TDI) of cytochrome P450 (CYP) 3A in vitro in human liver microsomes. 9 Patients with SMA require many medications to treat disease symptoms and complications (e.g., respiratory infections). 1 As these comedications often include various CYP3A substrates, investigation of clinical relevance of potential CYP3A inhibition is important. Although many pediatric patients receive risdiplam, since the typical onset of symptoms is during childhood, 10 a drug–drug interaction (DDI) study without therapeutic need in this highly vulnerable population is considered unethical. Therefore, a DDI study in healthy adults combined with pediatric physiologically‐based pharmacokinetic (PBPK) modeling was prospectively planned to evaluate the CYP3A TDI risk in pediatric patients with SMA.
PBPK modeling in conjunction with in vitro–in vivo extrapolation has been frequently used for prediction of DDIs. 11 , 12 , 13 The mechanistic nature of PBPK modeling enables consideration of age‐dependent differences in physiological parameters 14 and as such has been applied to extrapolate DDI studied in adults to children. 15 , 16 , 17 , 18 , 19 However, wider application of pediatric PBPK modeling in such a context is still lacking. 20 , 21 The hepatic and intestinal CYP3A ontogeny functions are critical physiological information for the prediction of CYP3A‐mediated DDI in children. Several ontogeny models have been proposed based on a combination of surrogate in vitro and in vivo data in children for hepatic CYP3A enzymes, 22 , 23 , 24 , 25 , 26 namely, Salem et al. (“Salem function”) 25 and Upreti and Wahlstrom (“Upreti function”). 26 There is a distinct difference in the predicted hepatic CYP3A activity between these two ontogeny functions, particularly in children ≤ 2 years (Supplementary Material ‐1 ), and thus different susceptibility to hepatic CYP3A modulations can be expected. Recent analysis 17 highlighted the lack of consistency in the selection of hepatic CYP3A ontogeny in pediatric PBPK modeling. In the case of intestinal CYP3A ontogeny, a function derived by Johnson et al. (“Johnson function”) is the only one used. 24 , 27 The fraction metabolized by CYP3A (fmCYP3A) and intestinal availability (FG ) determine the extent of CYP3A victim DDIs. 28 , 29 , 30 , 31 These parameters can be age‐dependent due to the hepatic and intestinal CYP3A ontogenies 24 , 25 , 26 , 27 and/or other age‐related physiological changes (e.g., other contributing enzymes, blood flow, renal and/or biliary excretion 24 , 32 ). Therefore, fmCYP3A and FG in children may differ from adults leading to potentially different sensitivities to DDIs. 17 , 33 , 34
In the current study, the in vivo CYP3A inhibition effect of risdiplam was investigated by a PBPK model‐based approach integrated with a clinical DDI trial to support safe concomitant use of CYP3A substrates in patients with SMA ≥ 2 months old. The strategy consisted of (i) investigation/estimation of the in vivo inactivation constant (k inact) in healthy volunteers at risdiplam exposures relevant to children, and (ii) a mechanistic and comprehensive extrapolation of risdiplam CYP3A DDI from healthy adults to children covering ranges of physiological (intestinal and hepatic CYP3A ontogenies) and drug‐related (in vivo k inact) data. Growth models specific to patients with SMA were developed and implemented in PBPK modeling and DDI extrapolation. In addition, the impact of different hepatic and intestinal CYP3A ontogeny functions on the predicted DDI was theoretically investigated to provide general guidance for selection of ontogeny functions and a PBPK model‐based strategy to extrapolate DDIs from adults to children.
METHODS
Risdiplam PBPK model development
The DDI strategy shown in Figure 1 was applied to risdiplam. A risdiplam PBPK model previously developed for healthy adults 9 was adapted to adult patients with SMA using the Simcyp Simulator version 18. 35 The clearance (CL), consisting of renal CL (CLR, 5%), hepatic intrinsic CL (CLint,H) via CYP3A (20%), and flavin‐containing monooxygenase (FMO)3 (75%), was approximately 30% lower in the SMA population than in healthy adults, based on population pharmacokinetic (PPK) analysis. 35 Therefore, CLR and CLint,H were reduced accordingly. For pediatric patients with SMA, the CLR was scaled by age‐dependent glomerular filtration rate. 24 Multiple ontogeny functions, 25 , 26 , 36 , 37 , 38 have been investigated for scaling risdiplam CLint,H. Ultimately, the Upreti function 26 for both CYP3A and FMO3 enzymes demonstrated the best consistency between the predicted age‐dependent CL/F (apparent clearance) and the post hoc CL/F of the PPK model (Supplementary Material ‐2 ), and this ontogeny was therefore retained in the model. The unbound fraction in plasma was age‐independent. 9 The PBPK models for healthy adults, and pediatric and adult patients with SMA were validated using independent risdiplam pharmacokinetic (PK) data for the respective population. 35 To account for the specific SMA patient demographics and their interindividual variability, growth models of body height and weight were developed (Supplementary Section ‐3 ).
Figure 1.
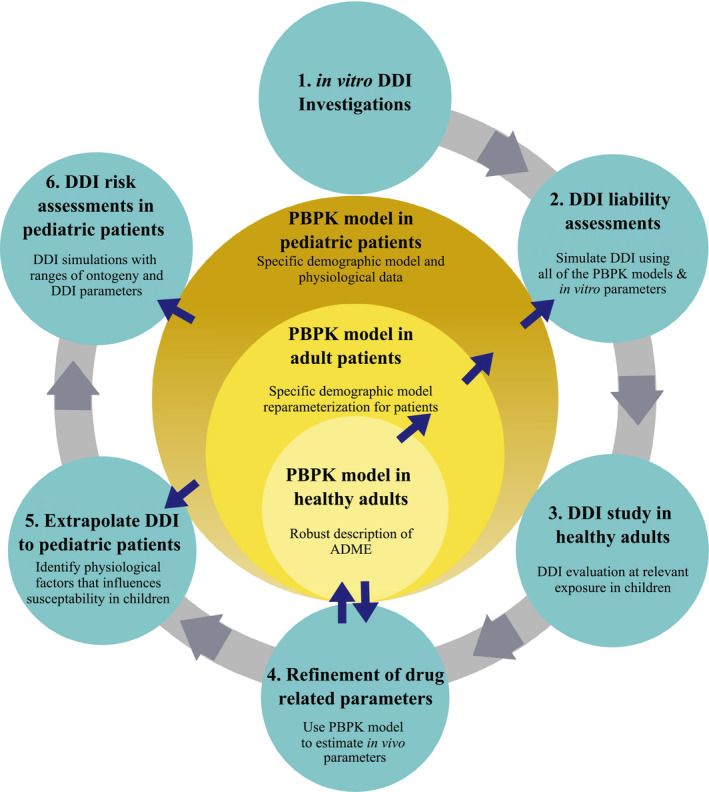
Model‐based CYP3A inhibition risk assessments for pediatric patients with SMA. The arrows for ② indicate the prediction of in vivo CYP3A inhibition of risdiplam using PBPK models for healthy adults and patients with SMA including pediatric populations. The arrows for ④ indicate validation of the risdiplam and midazolam PBPK model for healthy adults using the observations of ③ the clinical DDI study and refinement of the in vivo k inact which was included in the PBPK model. The arrows for ⑤ and ⑥ indicate the extrapolation and DDI risk assessments using the pediatric PBPK model of risdiplam with the in vivo k inact. Different ontogeny functions of CYP3A enzyme predict different susceptibility to CYP3A modulations in children and thus various functions were considered. The risdiplam PBPK model was validated with independent data for each population. 35 The definition of validation used herein is whether the platforms and models accurately predict observations from independent data (i.e., not used during model development). The independent data may have been generated for the same compound/inhibitors (in different studies or subsets of populations) or other compounds with similar properties. ADME, absorption, distribution, metabolism, and elimination; CYP, cytochrome P450; DDI, drug–drug interaction; k inact, inactivation constant; PBPK, physiologically‐based pharmacokinetic; SMA, spinal muscular atrophy.
Risdiplam CYP3A inhibition risk assessments
DDI liability assessments in vitro (Figure 1 (1))
Details of the TDI in vitro study are reported elsewhere. 9 The estimated k inact and inhibition potency (KI ) were 3.9/hour and 13 µM for risdiplam, and 3.8/hour and 13.7 µM for M1, respectively (Supplementary Material ‐4 ).
Initial in silico DDI liability assessments (Figure 1 (2))
The clinical relevance of the in vitro TDI parameters was evaluated by simulating the effect of 14‐day treatment with risdiplam on the oral midazolam PK in virtual healthy adults, and adult and pediatric patients with SMA. This preliminary analysis indicated that the primary CYP3A inhibition site was likely the intestine, and therefore, midazolam was selected as a suitable clinical probe given its FG value (0.55). 39 The preliminary simulations using the default midazolam and the risdiplam PBPK models with the in vitro TDI parameters predicted a 2‐fold and 2.5‐fold increase in midazolam area under the curve (AUC) in adult and pediatric populations, respectively, which were considered clinically relevant and warranted a clinical investigation. 40
Clinical DDI study in healthy adults (Figure 1 (3))
An open‐label trial (NCT03988907) separated into two parts was conducted in 35 healthy adults to investigate safety, tolerability, and PK of risdiplam (Part 1), and to determine its in vivo TDI effect on oral midazolam PK (Part 2). All relevant study documents were approved by the Institutional Review Board and all subjects signed the informed consent prior to enrollment. The study was conducted in full conformance with the principles of the Declaration of Helsinki. Based on the safety and PK evaluation of risdiplam after 5 mg in 8 subjects (Part 1), a risdiplam dose for Part 2 (n = 27) was selected to target a area under the concentration‐time curve over 24 hours (AUC0–24h) of 2000 ng·h/mL, as observed in pediatric patients. Midazolam 2 mg was given 2 days before and 13 days after the initiation of 14‐day risdiplam treatment. In addition to noncompartmental PK analysis, PPK modeling was performed to investigate the effect of risdiplam on midazolam PK. Bioanalysis and further details on the study/analyses performed are in Supplementary Material ‐5 and ‐6 .
Risdiplam PBPK model validation and TDI parameter refinement (Figure 1 (4))
The risdiplam and midazolam PBPK models were evaluated against the data obtained from the DDI study (independent clinical data set). Initial quantitative translation of risdiplam in vitro TDI overestimated the increase in midazolam AUC and maximum concentration (Cmax) by risdiplam. Subsequently, the in vitro k inact value was incrementally refined to match the observations. Ratio of midazolam AUC with and without risdiplam (AUCR) was considered more reliable than Cmax ratio (CmaxR), as this parameter is less dependent on sampling times, and was used primarily for estimation of the in vivo k inact. The in vivo k inact based on the CmaxR was used as a worst‐case scenario in the DDI extrapolation.
Risdiplam PBPK model validation in pediatric patients and TDI extrapolation (Figure 1 (5))
The risdiplam pediatric PBPK model was developed on data collected from 130 subjects (including 94 pediatric patients) and validated using independent data from 289 pediatric patients 35 . Predicted Cmax and AUC of risdiplam were compared with the observations from all 372 pediatric patients with SMA ≥ 2 months old treated with the therapeutic dose of risdiplam (0.2 mg/kg for 2 months to 2 years old, 0.25 mg/kg for patients ≥ 2 years and weighing < 20 kg, or 5 mg once daily for patients ≥ 2 years and weighing ≥ 20 kg 41 ). A detailed evaluation of midazolam pediatric PBPK model (Simcyp version 18) was performed against published clinical data in neonates to 18‐year‐old children (Supplementary Material ‐7 ). Plasma concentrations, systemic and oral CL, and bioavailability (F) of midazolam were examined, with a focus on infants < 2 years due to higher uncertainty in the CYP3A ontogeny functions. The DDI study results in healthy adults were extrapolated to pediatric patients ≥ 2 months (n = 2,000) by simulations using the midazolam and risdiplam PBPK models and the in vivo k inact of risdiplam. The hepatic CYP3A ontogeny functions used for the DDI extrapolation were the Salem 25 and Upreti functions, 26 whereas the Johnson function 24 , 27 was applied for the intestinal CYP3A ontogeny. Simulations were performed following the same DDI study design as in the healthy adults, with the therapeutic risdiplam doses 41 and 0.1 mg/kg oral dose of midazolam. Alteration in the simulated hepatic and intestinal CYP3A activity was assessed. The Qgut model 39 , 42 was applied to predict the effect of risdiplam on FG of midazolam and other CYP3A substrates (Supplementary Material ‐8 ).
Impact of CYP3A ontogeny functions on predicted TDI risk in pediatric patients (Figure 1 (6))
Sensitivity analyses were performed to investigate the general uncertainty of predicted DDIs in children, including (i) an alternative scenario assuming full intestinal CYP3A maturity from birth (“full‐maturity”) to address uncertainty in age‐dependent intestinal drug extraction and (ii) a more conservative risdiplam in vivo k inact based on the midazolam CmaxR to account for a range of clinical observations. In addition, a generalized TDI risk assessment was performed illustrating the impact of choice of ontogeny functions on the predicted CYP3A DDI susceptibility in children considering a theoretical potent CYP3A inhibitor in contrast to the weak CYP3A inhibitor, risdiplam. This analysis was considered of high relevance as current data do not allow an unequivocal selection of hepatic CYP3A ontogeny. The theoretical AUC ratios were calculated using Eq. 1 (Fami et al. 43 ) assuming 95% and 100% inhibition of hepatic and intestinal CYP3A enzymes, respectively, to match reported clinical DDI data of midazolam with potent CYP3A inhibitors. 44 , 45 In this case, I H × k inact/(I H + KI ) ≥ 19‐fold of k deg and the steady‐state Eq. 1 was simplified to Eq. 2. The theoretical AUC and FG ratios were calculated for a virtual pediatric population (0.01–18 years, n = 5,000) using simulated midazolam fmCYP3A and FG obtained for each aforementioned CYP3A ontogeny function.
| (1) |
| (2) |
where k deg,H and kdeg,G are the hepatic and intestinal CYP3A enzyme degradation rate constants, respectively, and I H and I G are hepatic and intestinal inhibitor concentrations, respectively.
[Correction added on 18 September 2021, after first online publication: A typo in the second term of the equation (1), has been corrected in this version.]
RESULTS
Midazolam DDI study in healthy volunteers
Risdiplam treatment for 14 days, alone and coadministered with midazolam, was well tolerated by all subjects. Based on the safety and PK results of Part 1, a risdiplam dose of 8 mg was selected for the DDI assessment (Part 2) which resulted in geometric mean AUC0–24h of 1,730 ng·h/mL (Table 1 ). The ratios of the geometric least‐squares mean of midazolam area under the plasma concentration–time curve from time zero to infinity (AUCinf), area under the plasma concentration‐time curve from time zero to time of last measurable concentration (AUClast), and Cmax in the presence to the absence of risdiplam (90% confidence intervals) were 1.08 (0.93–1.26), 1.11 (1.02–1.20), and 1.16 (1.06–1.28), respectively. The PK parameters of M1 and 1’OH‐midazolam are summarized in Supplementary Material ‐5 . The PPK analysis estimated an 11% increase in the relative bioavailablity (F rel) of midazolam by risdiplam (Supplementary Material ‐6 ), suggestive of a predominant effect on midazolam first‐pass extraction.
Table 1.
Predicted and observed PK parameters of risdiplam after 5 or 8 mg once daily for 14 days
| Parameters | Observed | Predicted with CYP3A inhibition* |
|---|---|---|
| 5 mg | n = 8 | n = 80 |
| Cmax (ng/mL) | 78.6 (23.7%) | 65.2 (40%) |
| AUC0–24h (ng·h/mL) | 1,250 (24.6%) | 1,120 (50%) |
| 8 mg | n = 27 | n = 270 |
| Cmax (ng/mL) | 113 (21.5%) | 102 (36%) |
| AUC0–24h (ng·h/mL) | 1,730 (21.3%) | 1,790 (45%) |
Geometric means (coefficient of variation, CV%) are presented.
*PBPK model prediction. A minimal auto‐inhibition effect is predicted due to the weak inhibition potency, high FG and low fmCYP3A of risdiplam. The geometric mean of predicted risdiplam Cmax and AUC0–24h are all within 0.8–1.25 fold of the observations.
AUC0–24h, area under the concentration‐time curve over 24 hours; Cmax, maximum concentration; CYP, cytochrome P450; FG , intestinal availability; fmCYP3A, fraction metabolized by CYP3A; PBPK, physiologically‐based pharmacokinetic.
Risdiplam PBPK model validation and TDI parameter refinement in healthy volunteers
The PBPK model successfully predicted risdiplam plasma concentration‐time profiles (Figure 2a ) and PK parameters (Table 1 ) after 5 and 8 mg daily for 14 days. Midazolam PK was also well predicted by the default model (Table 2 ). Risdiplam DDI simulations using the in vitro CYP3A kinact (3.9/hour) overpredicted the midazolam AUCR and CmaxR by 91% and 52%, respectively. Although a 10‐fold decrease in k inact recovered the observed CmaxR, the AUCR remained overpredicted (Table 2 ). Consequently, the in vitro k inact was further reduced to 18‐fold to recover the observed AUCR and plasma concentration‐time profiles (Table 2 and Figure 2b ); this in vivo k inact estimate (0.22/hour) was subsequently applied for the extrapolation of TDI to children.
Figure 2.
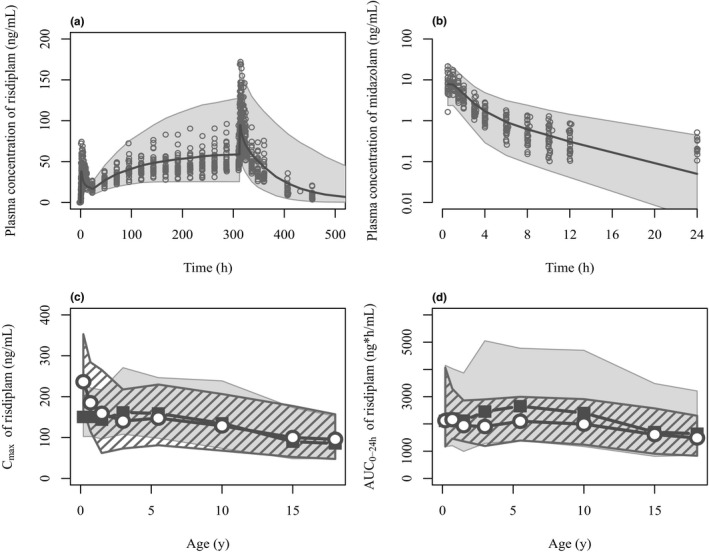
Comparisons of simulated risdiplam and midazolam pharmacokinetics by the PBPK model with the observed data in adults and pediatric population. (a) Risdiplam plasma concentration‐time profiles after oral 8 mg once daily administration for 14 days in 27 healthy subjects. The plot in semi‐log scale as well as the profiles after 5 mg are shown in Supplementary Material ‐5 . (b) Midazolam plasma concentration‐time profiles after 2 mg oral administration in the presence of risdiplam predicted with the refined k inact (1/18 of the in vitro k inact). Sixty percent of the samples at 24 hours after midazolam administration were below the quantification limit (0.1 ng/mL). Observations (circles) and simulations as median (solid line) and 90% prediction interval (shaded area) are shown. Simulated risdiplam (c) Cmax and (d) AUC0–24h values in pediatric patients (gray shade) compared with the observed Cmax and individually estimated AUC0–24h using post hoc PK parameters of population PK model (striped shade). 35 Geometric means of simulated (solid squares) and observed (open circles) values are shown. The observations originate from 372 pediatric patients with SMA aged 2 months–18 years. Geometric means of the simulated risdiplam Cmax and AUC0–24h are within 0.8–1.25 fold of the observations except for Cmax of 2–7 months (0.636) and AUC0–24h of 2–4 years (1.29) and 4–7 years (1.26). AUC0–24h, area under the concentration‐time curve over 24 hours; Cmax, maximum concentration; k inact, inactivation constant; PBPK, physiologically‐based pharmacokinetic.
Table 2.
Observed and predicted midazolam Cmax and AUC ratios
| Cmax a | Cmax ratiob | AUClast a | AUClast ratiob | |
|---|---|---|---|---|
| Observed | ||||
| without risdiplam (n = 27) | 7.65 (48.5%) | n.a | 19.9 (49.0%) | n.a |
| with risdiplam (n = 26) | 8.96 (40.4%) | 1.16 (1.06–1.28) | 22.0 (47.7%) | 1.11 (1.02–1.20) |
| Predictedc | ||||
| without risdiplam | 7.51 (70%) | n.a. | 22.8 (64%) | n.a |
| with risdiplam | ||||
| in vitro k inact = 3.91/hour | 13.2 (67%) | 1.76 (+52%)f | 48.4 (73%) | 2.12 (+91%)f |
| in vivo k inact d = 0.39/hour | 8.72 (70%) | 1.16 (0%)f | 27.0 (66%) | 1.19 (+7.0%)f |
| in vivo k inact e = 0.22/hour | 8.32 (70%) | 1.11 (−4.0%)f | 25.6 (65%) | 1.12 (+1.0%)f |
Observed and predicted midazolam Cmax and AUC ratios using in vitro and estimated in vivo k inact values.
AUClast, area under the plasma concentration‐time curve from time zero to time of last measurable concentration; Cmax, maximum concentration; k inact, inactivation constant; n.a, not applicable.
aGeometric mean (coefficient of variation, CV%). bGeometric least squares means are presented. cPredicted by the PBPK models. d1/10 of the in vitro k inact. e1/18 of the in vitro k inact. f% difference from the corresponding observed values. Number ranges in parentheses are 90% confidence intervals.
PBPK model validation in pediatric patients and TDI extrapolation
The predicted risdiplam Cmax and AUC0–24h in the pediatric patients with SMA after therapeutic doses were consistent with the observed Cmax and AUC0–24h estimated using the post hoc parameters of the PPK model in 372 pediatric patients with SMA ≥ 2 months old (Figure 2c,d ). Plasma concentration–time profiles and CL of midazolam in children (including infants < 2 years) were adequately predicted. While bioavailability of midazolam in children aged 3 days to 12 years was generally well predicted by a combination of Salem and Johnson functions for the hepatic and intestinal CYP3A ontogenies, underpredictions were noted for a combination of Upreti and full‐maturity functions (Supplementary Material ‐7 ). The predicted risdiplam TDI on midazolam PK in pediatric patients ≥ 2 months was consistently low across age range, with a geometric mean midazolam AUCR of 1.09–1.18 (Figure 3a ). The 95th percentiles of the predicted AUCRs were all below 1.4‐fold, comparable to the clinical observations in the healthy adults. Similar trends were seen for the predicted CmaxR (1.08–1.15). The PBPK modeling predicted decreased CYP3A activity by approximately 3% and 20% in the liver and small intestine, respectively, suggesting a predominant risdiplam CYP3A inhibition effect in the intestine and a negligible effect in the liver. Consequently, the predicted midazolam AUCR was almost identical between different hepatic CYP3A ontogeny functions. The analysis also highlighted that the maximal increase in FG (and AUC) cannot exceed 25% even for CYP3A substrates undergoing extensive intestinal extraction due to the limited (20%) reduction in intestinal CYP3A activity by risdiplam (Supplementary Material ‐8 ).
Figure 3.
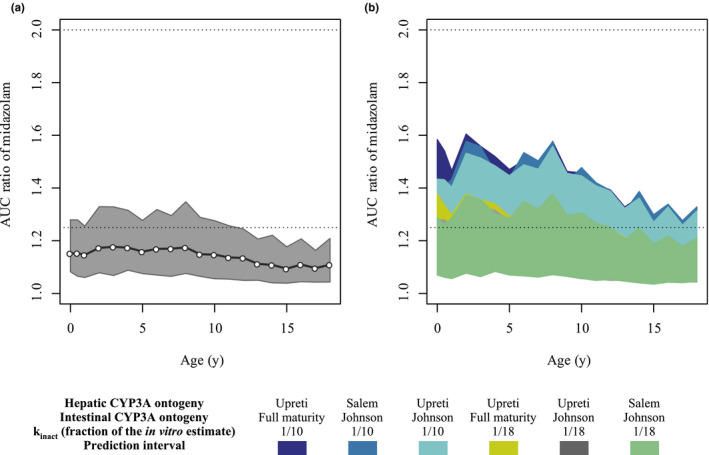
Predicted AUCR of midazolam in the presence of risdiplam in pediatric patients with SMA aged 2 months–18 years. (a) Extrapolation of the DDI using Upreti and Johnson functions for the hepatic and intestinal CYP3A ontogeny, respectively. The open circles and shaded area show geometric means and 5th to 95th percentiles of the predicted midazolam AUCR. The dotted lines indicate the ratios of 1.25 and 2 for weak and moderate inhibition, respectively. (b) Simulations with ranges of ontogeny functions and estimated in vivo k inact values to cover uncertainty in the physiology and drug‐related parameters of the DDI risk assessments. The 5th to 95th percentiles of the predicted AUCR are shown for each scenario. For UGT1A4 of the midazolam model, the steep ontogeny function for UGT1A4 (Supplementary Material ‐9 ) was used. AUCR, ratio of midazolam area under the curve with and without risdiplam; CYP, cytochrome P450; DDI, drug–drug interaction; k inact, inactivation constant; SMA, spinal muscular atrophy; UGT, uridine 5′‐diphospho‐glucuronosyltransferase.
Impact of CYP3A ontogeny functions on predicted TDI risk in pediatric patients
The results of sensitivity analyses performed with combinations of intestinal and hepatic CYP3A ontogeny functions and over a range of in vivo k inact values are shown in Figure 3b . The 95th percentiles of the predicted midazolam AUCR remained significantly below twofold across the age range even with the most conservative scenario, Upreti and full‐maturity functions for the hepatic and intestinal CYP3A ontogenies, respectively, and the k inact based on CmaxR (0.39/hour). The interindividual variability of simulated risdiplam AUC0−24h was larger than observed (Figure 2d ), suggesting that a wider range of individual AUCRs was simulated than is likely to be observed. The extrapolation and sensitivity analyses indicate that clinically significant CYP3A TDI (i.e., more than a twofold increase in midazolam AUC) in pediatric patients ≥2 months old with SMA treated with therapeutic doses of risdiplam is highly unlikely.
Given the lack of sensitivity of risdiplam as a weak CYP3A inhibitor, the impact of hepatic and intestinal CYP3A ontogeny on the DDI predictions in children (aged 0.01–18 years) was evaluated with a theoretical potent CYP3A inhibitor using midazolam as a substrate. The predicted midazolam fmCYP3A and FG (Figure 4a,b , Figures S2–S4 ) and the resulting theoretical midazolam AUC and FG ratios are shown in Figure 4c,d , respectively. The analysis highlighted higher sensitivity to CYP3A modulations when using the Upreti CYP3A ontogeny function, as simulated midazolam AUCRs with a potent inhibitor were significantly higher in children ≤ 2 years relative to the Salem function (Figure 4c ). The simulation also highlighted the importance of ontogeny of other elimination pathways involved which may mature at different rates from CYP3A. Although uridine 5′‐diphospho‐glucuronosyltransferase (UGT)1A4 contributes only 1–2% to midazolam clearance in adults, 46 significantly different AUCR and sensitivity to CYP3A modulation were predicted depending on relative differences in maturation rate between CYP3A and UGT1A4 (Supplementary Material ‐9 ). The simulated midazolam FG ratio was constant across the age range with the Johnson function, whereas increased sensitivity to intestinal CYP3A interaction was shown for children ≤ 2 years with the full‐maturity function (Figure 4d ).
Figure 4.
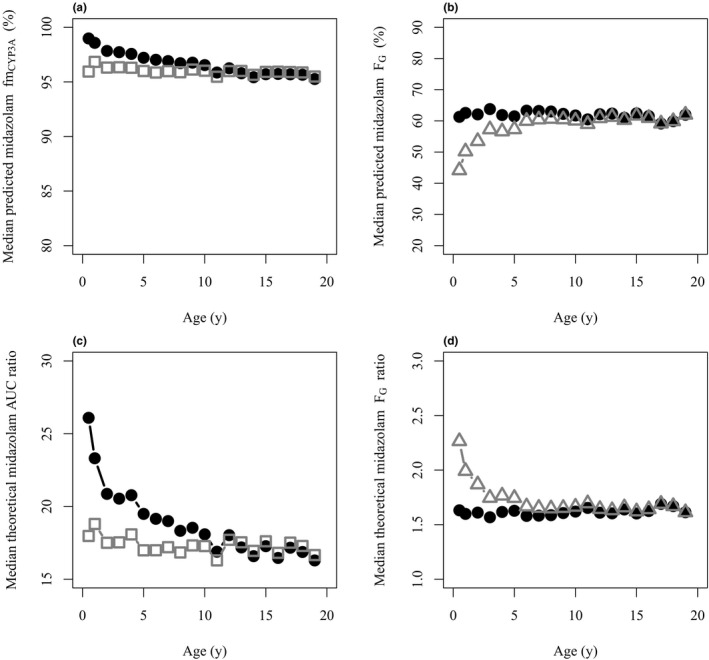
Comparisons of midazolam DDI susceptibility using different CYP3A ontogeny functions to simulate AUC and FG ratios in the presence to the absence of hypothetical potent CYP3A inhibitor in children. (a) Median predicted midazolam fmCYP3A in children using the Salem (open squares) or Upreti (solid circles) functions for the hepatic CYP3A enzyme ontogeny. (b) Median predicted midazolam FG in children using the Johnson (solid circles) or full‐maturity functions (open triangles) for the intestinal CYP3A ontogeny. (c) Median theoretical AUCR of midazolam in the presence to the absence of hypothetical potent CYP3A inhibitor with the hepatic CYP3A enzyme ontogeny according to Salem (open squares) or Upreti (solid circles) are shown. The Johnson function for the intestinal CYP3A ontogeny and the steep ontogeny model for UGT1A4 (Supplementary Material ‐9 ) was used. (d) Median theoretical FG ratios in the presence to the absence of hypothetical potent CYP3A inhibitor with the intestinal CYP3A enzyme ontogeny according to Johnson (solid circles) or full‐maturity functions (open triangles) are shown. All analyses were based on simulations with 5,000 virtual individuals. AUC, area under the curve; AUCR, ratio of midazolam area under the curve with and without the theoretical potent inhibitor; CYP, cytochrome P450; DDI, drug–drug interaction; FG , intestinal availability; fmCYP3A, fraction metabolized by CYP3A; UGT, uridine 5′‐diphospho‐glucuronosyltransferase.
DISCUSSION
A stepwise PBPK model‐based approach (Figure 1 ) was applied for risdiplam with critical qualification of multiple individual aspects to ensure model robustness for prospective prediction of CYP3A TDI risk in pediatric patients with SMA. Important aspects of the strategy include (i) conduct of a clinical DDI evaluation in healthy adults at risdiplam exposures relevant to pediatric patients with SMA, (ii) validation of the pediatric PBPK models of risdiplam (perpetrator) and midazolam (victim drug) with independent data to ensure extrapolative capability, and (iii) identification/assessment of appropriate CYP3A ontogeny functions to determine DDI susceptibility. Each aspect was critically evaluated, and impact of uncertainty was assessed by sensitivity analysis to ensure conservative TDI risk evaluation in the pediatric population (summary of evaluation of all critical inputs in Supplementary Material ‐10 ).
Initial PBPK modeling predicted a twofold or higher increase in midazolam AUC based on the in vitro k inact, in contrast to the clinically inconsequential DDI observed in the healthy adults (midazolam AUCR < 1.25). Overpredictions of in vivo CYP3A TDI using in vitro inhibition microsomal data has been reported previously, in particular for weak to moderate inhibitors. 47 , 48 This trend was also evident here as the in vivo risdiplam k inact that recovered midazolam AUCR/CmaxR was up to 18‐fold lower than the in vitro estimate. The CYP3A inhibition was predicted to occur predominantly in the intestine (20%) and to a negligible extent in the liver (3%). Consistently, the PPK analysis estimated an increase in F rel rather than a decrease in CL/F, indicating an effect of risdiplam on the first‐pass extraction of midazolam. The lack of correlation between risdiplam systemic exposure and fold‐change in midazolam AUC and CL/F (Supplementary Material ‐6 ) further supports this prediction. The estimated increase in midazolam F rel (11%) is consistent with the estimation of the in vivo k inact value based on the observed AUClast ratio (1.11). Although the main metabolite M1 exhibited similar potency to risdiplam in vitro, the intestinal metabolism of risdiplam is negligible 9 and the optimized in vivo k inact would at any rate include contribution from M1 (comparable risdiplam to M1 ratios in adults and children). Taken together, all information is supportive of a negligible risdiplam DDI risk for intravenously administered CYP3A substrates. While a DDI for orally administered CYP3A substrates cannot be completely excluded, it is expected to be negligible for substrates with a lower intestinal extraction than midazolam (i.e., FG > 0.55). Even for substrates undergoing extensive intestinal metabolism, the predicted maximum increase in the absolute bioavailability was up to 25% based on the Q gut model, with a 20% reduction in intestinal CYP3A activity (Supplementary Material ‐8 ). Overall, clinically relevant TDI of CYP3A substrates by risdiplam is not expected.
The risdiplam PBPK model was validated with independent data 35 and a specific growth model for patients with SMA was implemented to allow a more accurate representation of physiological parameters (e.g., organ sizes and blood flow) of the virtual SMA populations. 49 There was a minor difference in PBPK model predictions between the growth model of patients with SMA and the default demographic pediatric model. However, development of a demographic model for a target population represents the best practice if such data are available and it ensures robust prediction of systemic and intestinal concentrations of risdiplam for the DDI extrapolation purposes. The pediatric patients demonstrated higher body weight‐normalized post hoc CL/F than the adolescents and adult patients, 35 indicating an ontogeny function in which enzyme levels per liver weight in children exceed those of adults. Ontogeny of FMO3 enzymes has been mostly investigated in vitro, showing low abundance at birth and monotonic increase with age. 36 , 38 However, in vivo investigation of FMO3 ontogeny is currently limited probably due to lack of clinically sensitive FMO3 substrates. The Upreti function describes higher CYP3A activity per liver weight in children 26 and using it for scaling of both CYP3A and FMO3‐mediated CLint,H produced a highly comparable distribution of CL/F to those of the PPK model (Supplementary Material ‐2 ). PPK modeling elucidated patient’s specific characteristics and age‐dependent process in risdiplam PK. 35 Since risdiplam data in patients with SMA were sparse, this integration of information from PPK modeling was important for subsequent robust PBPK modeling. The validated risdiplam pediatric PBPK model demonstrated accurate prediction of Cmax and AUC0–24h of 372 pediatric patients ≥ 2 months at therapeutic doses (Figure 2 ). These validation steps, together with evaluation of predictive performance of the midazolam pediatric PBPK model, were critical for subsequent model‐based investigation of risdiplam CYP3A TDI risk in pediatric patients with SMA.
Since the predicted CYP3A inhibition by risdiplam was predominantly occured in the intestine, the unbound intestinal concentration and the intestinal CYP3A ontogeny functions were critical for TDI extrapolation to pediatric patients with SMA. The risdiplam PBPK model predicts almost complete absorption and negligible first‐pass effects, and thus a high oral bioavailability. 9 Good prediction of Cmax across age (Figure 2c ) supports adequate prediction of risdiplam absorption and therefore local intestinal concentrations. DDI predictions were performed assuming complete unbound fraction of risdiplam in the enterocytes, ensuring the conservative evaluation of the TDI risk.
Use of the Johnson intestinal CYP3A ontogeny function resulted in a consistent midazolam FG estimate of ~ 0.55–0.60 for children and adults, whereas the full‐maturity function led to lower FG estimates in children ≤ 2 years (Figure 4b ), and thus higher sensitivity to intestinal CYP3A modulations. A combination of the Salem and Johnson functions accurately predicted systemic CL, plasma concentration‐time profiles, and bioavailability of midazolam in children (Supplementary Material ‐7 ). Although FG is not directly observable, these assessments indicate an adequacy of the Johnson function for intestinal CYP3A ontogeny to predict midazolam FG . In contrast, significant underprediction of bioavailability was noted for a combination of the Upreti and full‐maturity functions, suggesting this combination to be a highly conservative and sensitive, but a rather unlikely scenario given the current information. Even with this conservative scenario, the predicted midazolam AUCR (1.10–1.20) and CmaxR (1.08–1.19) in the presence of risdiplam remained < 1.25. This minimal predicted CYP3A‐mediated DDI risk is in line with no safety concerns raised in infants with Type 1 SMA who received various CYP3A substrates (e.g., budenoside, dexamethasone, prednisolone) during the risdiplam treatment (internal data).
As risdiplam is a weak time‐dependent inhibitor, it did not allow complete assessment of the impact of different ontogeny functions on the DDI susceptibility in children. Therefore, a theoretical case of a potent CYP3A inhibitor was explored to illustrate the importance of selection of ontogeny functions on predicted DDI. Simulated midazolam fmCYP3A values in children was >0.9 across the age range and the difference between the hepatic CYP3A ontogeny functions was not very apparent (Figure 4a ). However, the theoretical AUCR using the Upreti function was higher in children ≤ 2 years compared with those with the Salem function which predicted age‐independent AUCR with a potent inhibitor (Figure 4c ). These trends reflect assumption of comparable maturation of CYP3A and UGT1A4 when Salem function was used (Supplementary Material ‐9 , Figure S19 b). In contrast, the Upreti function predicted a more rapid maturation of CYP3A activity compared with UGT1A4, resulting in higher fmCYP3A in children ≤ 2 years. For age ranges when UGT1A4 maturation is advanced relative to CYP3A, the relative contribution of this enzyme increases (Supplementary Material ‐9 and Figure S19 c), resulting in lower sensitivity to CYP3A modulation (Supplementary Material ‐9 and Figure S20 b). Although the contribution of UGT1A4 to hepatic metabolism of midazolam is minor in adults, 46 assumptions of its ontogeny influence the predicted sensitivity to CYP3A‐mediated DDI in children for substrates with fmCYP3A > 0.8, as even minor differences in that range lead to significant differences in predicted DDI with potent CYP3A inhibitors. 50 , 51
In conclusion, a PBPK model‐based extrapolation of risdiplam CYP3A DDI risk was performed in a stepwise manner from healthy adults to the pediatric patients with SMA. Simulations of clinically relevant therapeutic exposures, together with careful design of clinical DDI study in healthy adults and comprehensive evaluation of multiple CYP3A ontogeny functions were performed. As data are not unequivocal on the selection of hepatic CYP3A activity in children, evaluation of both Salem and Upreti ontogenies, together with careful consideration of parallel pathways and intestinal contribution, is recommended for extrapolation of DDIs to pediatric populations within a PBPK framework. This PBPK model–based analysis predicted low DDI propensity of risdiplam with CYP3A substrates in pediatric patients with SMA ≥ 2 months old. The conclusion from the PBPK modeling analysis has been included in the approved prescribing information of risdiplam in the United States. 41
FUNDING
This work has been funded by F. Hoffmann‐La Roche.
CONFLICT OF INTEREST
Y.C., M.G., P.G., A. Günther, K.H., and H.K. are employees of F. Hoffmann‐La Roche. All other authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
Y.C., M.G., and A. Galetin, wrote the manuscript. Y.C., M.G., A. Galetin, and H.K. designed the research. Y.C., M.G., A. Galetin, K.O., L.A., A.Günther, H.K., P.G., and K.H. performed the research. Y.C. and M.G. analyzed the data.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank all the patients with SMA and families who participated in the risdiplam program, the patient groups, and the clinical trial sites and staff for their support. Annie Young for summarizing midazolam DDI study results. Estelle Yau for discussion of growth modeling. Kenichi Umehara, Trevor Johnson, and Karen Rowland Yeo for the discussion on pediatric model validations.
DATA AVAILABILITY STATEMENT
Qualified researchers may request access to individual patient level data through the clinical study data request platform (https://vivli.org/). Further details on Roche’s criteria for eligible studies are available here (https://vivli.org/members/ourmembers/). For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here (https://www.roche.com/research_and_development/who_we_are_how_we_work/research_and_clinical_trials/our_commitment_to_data_sharing.htm).
References
- 1. D'Amico, A. , Mercuri, E. , Tiziano, F.D. & Bertini, E. Spinal muscular atrophy. Orphanet J. Rare Dis. 6, 71 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lefebvre, S. et al. Identification and characterization of a spinal muscular atrophy‐determining gene. Cell 80, 155–165 (1995). [DOI] [PubMed] [Google Scholar]
- 3. Mercuri, E. , Bertini, E. & Iannaccone, S.T. Childhood spinal muscular atrophy: controversies and challenges. Lancet Neurol. 11, 443–452 (2012). [DOI] [PubMed] [Google Scholar]
- 4. Ratni, H. et al. Discovery of risdiplam, a Selective Survival of Motor Neuron‐2 (SMN2) gene splicing modifier for the treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 61, 6501–6517 (2018). [DOI] [PubMed] [Google Scholar]
- 5. Finkel, R.S. et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology 83, 810–817 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baranello, G. et al. Risdiplam in type 1 spinal muscular atrophy. N. Engl. J. Med. 384, 915–923 (2021). [DOI] [PubMed] [Google Scholar]
- 7. US Food and Drug Administration . FDA approves oral treatment for spinal muscular atrophy <https://www.fda.gov/news‐events/press‐announcements/fda‐approves‐oral‐treatment‐spinal‐muscular‐atrophy> (2020). Accessed March 2021.
- 8. European Medicines Agency. Summary of opintion (initial authorisation): Evrysdi (risdiplam) <https://www.ema.europa.eu/en/documents/smop‐initial/chmp‐summary‐positive‐opinion‐evrysdi_en.pdf> (2021). Accessed June 2021.
- 9. Center for Drug Evaluation and Research, US Food and Drug Administration . Application Number: 213535Orig1s000 Clinical Pharmacology Review(s) — risdiplam <https://www.accessdatafdagov/drugsatfda_docs/nda/2020/213535Orig1s000ClinPharmR.pdf> (2020). Accessed March 2021.
- 10. Dangouloff, T. & Servais, L. Clinical evidence supporting early treatment of patients with spinal muscular atrophy: current perspectives. Ther. Clin. Risk Manag. 15, 1153–1161 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grimstein, M. et al. Physiologically based pharmacokinetic modeling in regulatory science: an update from the U.S. Food and Drug Administration's office of clinical pharmacology. J. Pharm. Sci. 108, 21–25 (2019). [DOI] [PubMed] [Google Scholar]
- 12. Guo, Y. et al. Advancing predictions of tissue and intracellular drug concentrations using in vitro, imaging and physiologically based pharmacokinetic modeling approaches. Clin. Pharmacol. Ther. 104, 865–889 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Luzon, E. , Blake, K. , Cole, S. , Nordmark, A. , Versantvoort, C. & Berglund, E.G. Physiologically based pharmacokinetic modeling in regulatory decision‐making at the European Medicines Agency. Clin. Pharmacol. Ther. 102, 98–105 (2017). [DOI] [PubMed] [Google Scholar]
- 14. Salerno, S.N. , Burckart, G.J. , Huang, S.‐M. & Gonzalez, D. Pediatric drug‐drug interaction studies: barriers and opportunities. Clin. Pharmacol. Ther. 105, 1067–1070 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Adiwidjaja, J. , Boddy, A.V. & McLachlan, A.J. Implementation of a physiologically based pharmacokinetic modeling approach to guide optimal dosing regimens for imatinib and potential drug interactions in paediatrics. Front. Pharmacol. 10, 1672 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. US Food and Drug Administration . Application Number: 208684Orig1s000, 208685Orig1s000 clinical pharmacology and biopharmaceutics review(s) — deflazacort <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/208684,208685Orig1s000ClinPharmR.pdf> (2017).
- 17. Lang, J. , Vincent, L. , Chenel, M. , Ogungbenro, K. & Galetin, A. Impact of hepatic CYP3A4 ontogeny functions on drug‐drug interaction risk in pediatric physiologically‐based pharmacokinetic/pharmacodynamic modeling: critical literature review and ivabradine case study. Clin. Pharmacol. Ther. 109, 1618–1630 (2021). [DOI] [PubMed] [Google Scholar]
- 18. Olafuyi, O. , Coleman, M. & Badhan, R.K.S. Development of a paediatric physiologically based pharmacokinetic model to assess the impact of drug‐drug interactions in tuberculosis co‐infected malaria subjects: a case study with artemether‐lumefantrine and the CYP3A4‐inducer rifampicin. Eur. J. Pharm. Sci. 106, 20–33 (2017). [DOI] [PubMed] [Google Scholar]
- 19. Salerno, S.N. et al. Physiologically‐based pharmacokinetic modeling characterizes the CYP3A‐mediated drug‐drug interaction between fluconazole and sildenafil in infants. Clin. Pharmacol. Ther. 109, 253–262 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chapron, B.D. , Chapron, A. & Leeder, J.S. Recent advances in the ontogeny of drug disposition. Br. J. Clin. Pharmacol. (2021) 10.1111/bcp.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang, X. et al. Application of PBPK modeling and simulation for regulatory decision making and its impact on US prescribing information: an update on the 2018–2019 submissions to the US FDA's office of clinical pharmacology. J. Clin. Pharmacol. 60 (suppl. 1), S160–S178 (2020). [DOI] [PubMed] [Google Scholar]
- 22. Björkman, S. Prediction of drug disposition in infants and children by means of physiologically based pharmacokinetic (PBPK) modelling: theophylline and midazolam as model drugs. Br. J. Clin. Pharmacol. 59, 691–704 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Edginton, A.N. & Willmann, S. Physiology‐based versus allometric scaling of clearance in children: an eliminating process based comparison. Paediatr. Perinat. Drug Ther. 7, 146–153 (2006). [Google Scholar]
- 24. Johnson, T.N. , Rostami‐Hodjegan, A. & Tucker, G.T. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin. Pharmacokinet. 45, 931–956 (2006). [DOI] [PubMed] [Google Scholar]
- 25. Salem, F. , Johnson, T.N. , Abduljalil, K. , Tucker, G.T. & Rostami‐Hodjegan, A. A re‐evaluation and validation of ontogeny functions for cytochrome P450 1A2 and 3A4 based on in vivo data . Clin. Pharmacokinet. 53, 625–636 (2014). [DOI] [PubMed] [Google Scholar]
- 26. Upreti, V.V. & Wahlstrom, J.L. Meta‐analysis of hepatic cytochrome P450 ontogeny to underwrite the prediction of pediatric pharmacokinetics using physiologically based pharmacokinetic modeling. J. Clin. Pharmacol. 56, 266–283 (2016). [DOI] [PubMed] [Google Scholar]
- 27. Johnson, T.N. , Tanner, M.S. , Taylor, C.J. & Tucker, G.T. Enterocytic CYP3A4 in a paediatric population: developmental changes and the effect of coeliac disease and cystic fibrosis. Br. J. Clin. Pharmacol. 51, 451–460 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ito, K. , Iwatsubo, T. , Kanamitsu, S. , Ueda, K. , Suzuki, H. & Sugiyama, Y. Prediction of pharmacokinetic alterations caused by drug‐drug interactions: metabolic interaction in the liver. Pharmacol. Rev. 50, 387–412 (1998). [PubMed] [Google Scholar]
- 29. Brown, H.S. , Ito, K. , Galetin, A. & Houston, J.B. Prediction of in vivo drug‐drug interactions from in vitro data: impact of incorporating parallel pathways of drug elimination and inhibitor absorption rate constant. Br. J. Clin. Pharmacol. 60, 508–518 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Galetin, A. , Gertz, M. & Houston, J.B. Potential role of intestinal first‐pass metabolism in the prediction of drug‐drug interactions. Expert. Opin. Drug Metab. Toxicol. 4, 909–922 (2008). [DOI] [PubMed] [Google Scholar]
- 31. Galetin, A. & Houston, J.B. Intestinal and hepatic metabolic activity of five cytochrome P450 enzymes: impact on prediction of first‐pass metabolism. J. Pharmacol. Exp. Ther. 318, 1220–1229 (2006). [DOI] [PubMed] [Google Scholar]
- 32. Johnson, T.N. , Jamei, M. & Rowland‐Yeo, K. How does in vivo biliary elimination of drugs change with age? Evidence from in vitro and clinical data using a systems pharmacology approach. Drug Metab. Dispos. 44, 1090–1098 (2016). [DOI] [PubMed] [Google Scholar]
- 33. Salem, F. , Johnson, T.N. , Barter, Z.E. , Leeder, J.S. & Rostami‐Hodjegan, A. Age related changes in fractional elimination pathways for drugs: assessing the impact of variable ontogeny on metabolic drug‐drug interactions. J. Clin. Pharmacol. 53, 857–865 (2013). [DOI] [PubMed] [Google Scholar]
- 34. Salem, F. , Rostami‐Hodjegan, A. & Johnson, T.N. Do children have the same vulnerability to metabolic drug‐drug interactions as adults? A critical analysis of the literature. J. Clin. Pharmacol. 53, 559–566 (2013). [DOI] [PubMed] [Google Scholar]
- 35. Cleary, Y. et al. Population and physiologically‐based pharmacokinetic modeling of risdiplam in infants, children and adults with spinal muscular atrophy (SMA). ASCPT Annual Meeting 2021, online, March 8–17, 2021. Poster PI‐018.
- 36. Koukouritaki, S.B. , Simpson, P. , Yeung, C.K. , Rettie, A.E. & Hines, R.N. Human hepatic flavin‐containing monooxygenases 1 (FMO1) and 3 (FMO3) developmental expression. Pediatr. Res. 51, 236–243 (2002). [DOI] [PubMed] [Google Scholar]
- 37. Shimizu, M. , Denton, T. , Kozono, M. , Cashman, J.R. , Leeder, J.S. & Yamazaki, H. Developmental variations in metabolic capacity of flavin‐containing mono‐oxygenase 3 in childhood. Br. J. Clin. Pharmacol. 71, 585–591 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xu, M. et al. Genetic and nongenetic factors associated with protein abundance of flavin‐containing monooxygenase 3 in human liver. J. Pharmacol. Exp. Ther. 363, 265–274 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gertz, M. , Harrison, A. , Houston, J.B. & Galetin, A. Prediction of human intestinal first‐pass metabolism of 25 CYP3A substrates from in vitro clearance and permeability data. Drug Metab. Dispos. 38, 1147–1158 (2010). [DOI] [PubMed] [Google Scholar]
- 40. US Food and Drug Administration . Clinical drug interaction studies — cytochrome P450 enzyme‐ and transporter‐mediated drug interactions guidance for industry <https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/clinical‐drug‐interaction‐studies‐cytochrome‐p450‐enzyme‐and‐transporter‐mediated‐drug‐interactions> (2020). Accessed March 2021.
- 41. Evrysdi [highlights of prescribing information]. Genentech, South San Francisco, CA, 2020) <https://wwwaccessdatafdagov/drugsatfda_docs/nda/2020/213535Orig1s000lblpdf> Accessed March 2021.
- 42. Yang, J. , Jamei, M. , Yeo, K.R. , Tucker, G.T. & Rostami‐Hodjegan, A. Prediction of intestinal first‐pass drug metabolism. Curr. Drug Metab. 8, 676–684 (2007). [DOI] [PubMed] [Google Scholar]
- 43. Fahmi, O.A. et al. Comparison of different algorithms for predicting clinical drug‐drug interactions, based on the use of CYP3A4 in vitro data: predictions of compounds as precipitants of interaction. Drug Metab. Dispos. 37, 1658–1666 (2009). [DOI] [PubMed] [Google Scholar]
- 44. Greenblatt, D.J. et al. Inhibition of oral midazolam clearance by boosting doses of ritonavir, and by 4,4‐dimethyl‐benziso‐(2H)‐selenazine (ALT‐2074), an experimental catalytic mimic of glutathione oxidase. Br. J. Clin. Pharmacol. 68, 920–927 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Olkkola, K.T. , Backman, J.T. & Neuvonen, P.J. Midazolam should be avoided in patients receiving the systemic antimycotics ketoconazole or itraconazole. Clin. Pharmacol. Ther. 55, 481–485 (1994). [DOI] [PubMed] [Google Scholar]
- 46. Hyland, R. et al. In vitro and in vivo glucuronidation of midazolam in humans. Br. J. Clin. Pharmacol. 67, 445–454 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen, Y. , Liu, L. , Monshouwer, M. & Fretland, A.J. Determination of time‐dependent inactivation of CYP3A4 in cryopreserved human hepatocytes and assessment of human drug‐drug interactions. Drug Metab. Dispos. 39, 2085–2092 (2011). [DOI] [PubMed] [Google Scholar]
- 48. Mao, J. , Johnson, T.R. , Shen, Z. & Yamazaki, S. Prediction of crizotinib‐midazolam interaction using the Simcyp population‐based simulator: comparison of CYP3A time‐dependent inhibition between human liver microsomes versus hepatocytes. Drug Metab. Dispos. 41, 343–352 (2013). [DOI] [PubMed] [Google Scholar]
- 49. Jamei, M. , Dickinson, G.L. & Rostami‐Hodjegan, A. A framework for assessing inter‐individual variability in pharmacokinetics using virtual human populations and integrating general knowledge of physical chemistry, biology, anatomy, physiology and genetics: a tale of 'bottom‐up' vs 'top‐down' recognition of covariates. Drug Metab. Pharmacokinet. 24, 53–75 (2009). [DOI] [PubMed] [Google Scholar]
- 50. Galetin, A. , Burt, H. , Gibbons, L. & Houston, J.B. Prediction of time‐dependent CYP3A4 drug‐drug interactions: impact of enzyme degradation, parallel elimination pathways, and intestinal inhibition. Drug Metab. Dispos. 34, 166–175 (2006). [DOI] [PubMed] [Google Scholar]
- 51. Obach, R.S. Predicting drug‐drug interactions from in vitro drug metabolism data: challenges and recent advances. Curr. Opin. Drug Discov. Devel. 12, 81–89 (2009). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
Qualified researchers may request access to individual patient level data through the clinical study data request platform (https://vivli.org/). Further details on Roche’s criteria for eligible studies are available here (https://vivli.org/members/ourmembers/). For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here (https://www.roche.com/research_and_development/who_we_are_how_we_work/research_and_clinical_trials/our_commitment_to_data_sharing.htm).