

Abstract
Aims
Capmatinib, a mesenchymal–epithelial transition factor tyrosine kinase inhibitor, is metabolized by cytochrome P450 (CYP) 3A4 and aldehyde oxidase. In individuals with hepatic impairment, alterations in hepatobiliary excretion and metabolism could lead to higher capmatinib exposure. We compared the pharmacokinetics of a single oral dose of capmatinib 200 mg administered to participants with varying degrees of hepatic impairment vs. matched controls with normal hepatic function.
Methods
This phase 1, multicentre, open‐label, parallel‐group study enrolled adult participants with normal hepatic function and mild, moderate and severe hepatic impairments. Eligible participants received a single oral dose of 200 mg capmatinib. The pharmacokinetic parameters of capmatinib were analysed and compared across participants with impaired and normal hepatic function.
Results
Of 31 enrolled participants, 29 had an evaluable pharmacokinetic profile: normal (n = 9); mild (n = 6); moderate (n = 8); severe (n = 6). Compared with the normal group, geometric mean (GM) maximum (peak) observed plasma drug concentration after single‐dose administration decreased by 27.6% in the mild group (GM ratio [GMR] = 0.724; 90% confidence interval [CI]: 0.476–1.10), by 17.2% in the moderate group (GMR = 0.828; 90% CI: 0.563–1.22) and remained unchanged in the severe group (GMR = 1.02; 90% CI: 0.669–1.55). Compared with the normal group, GM area under the plasma concentration–time curve from time zero to infinity decreased by 23.3% in the mild group (GMR = 0.767; 90% CI: 0.532–1.11), by 8.6% in the moderate group (GMR = 0.914; 90% CI: 0.652–1.28) and increased by 24% in the severe group (GMR = 1.24; 90% CI: 0.858–1.78).
Conclusion
Mild, moderate and severe hepatic impairment did not have a clinically relevant impact on capmatinib pharmacokinetics. No new safety findings are reported in this study.
Keywords: capmatinib, hepatic impairment, pharmacokinetics
What is already known about this subject
The MET inhibitor capmatinib is predominantly cleared via hepatic metabolism by CYP3A4 and aldehyde oxidase.
Patients with non‐small‐cell lung cancer may have hepatic comorbidities.
In individuals with hepatic impairment, it is unknown whether changes in capmatinib pharmacokinetics would necessitate dose adjustments.
What this study adds
Capmatinib pharmacokinetics were not substantially changed in participants with mild, moderate and severe hepatic impairment vs. those with normal hepatic function.
Single‐dose capmatinib had a manageable safety profile in participants with hepatic impairment.
Capmatinib starting dose adjustment is not required for individuals with mild, moderate or severe hepatic impairment classified according to Child–Pugh score.
1. INTRODUCTION
Dysregulation of the mesenchymal–epithelial transition factor (MET) pathway via MET exon 14 skipping (METex14) mutations, MET gene copy number gain/amplification or MET protein overexpression is a known oncogenic driver that promotes cell–cell detachment and metastasis, epithelial–mesenchymal transition, invasion, angiogenesis, proliferation, and cell survival in many cancers. 1 , 2 , 3 , 4 , 5 , 6 Capmatinib is an orally bioavailable, highly selective and potent (IC50 0.6 nM), ATP‐competitive, small‐molecule MET inhibitor (type 1b). 7 , 8 , 9 , 10 Several clinical studies have demonstrated promising efficacy and a manageable safety profile for capmatinib administered either as a single agent or in combination with other anticancer therapies in various solid tumors. 11 , 12 , 13 , 14 , 15 Capmatinib at a dose of 400 mg oral tablets twice daily (BID) with or without food was granted US Food and Drug Administration (FDA) accelerated approval in May 2020, followed by approval in Japan for the treatment of adult patients with metastatic non‐small‐cell lung cancer (NSCLC) whose tumours harbour METex14 mutations as detected by an FDA‐approved test. 16 The approvals were based on the results from the phase 2, multicohort GEOMETRY mono‐1 study in which capmatinib demonstrated clinically meaningful efficacy and a manageable safety profile in patients with advanced NSCLC harbouring METex14 mutations. 17
Following oral administration, the capmatinib tablet (400 mg BID) was rapidly absorbed, with a median time to maximum observed plasma drug concentration (Tmax) ranging from 1 to 2 hours. 17 The accumulation of capmatinib following repeated administration of 400‐mg BID tablets was low, with a steady state geometric mean (GM) accumulation ratio of 1.4. The effective half‐life (t1/2) was 6.54 hours. 17 M16 (CMN288), the major pharmacologically inactive metabolite of capmatinib in circulation, does not accumulate after multiple dosing. No clinically significant differences have been observed in the pharmacokinetics (PK) of capmatinib based on race (White, Asian, Native American, Black). 16
The PK of capmatinib tablets is linear following a single dose of 200–600 mg in healthy participants. Data from healthy participants indicate that capmatinib is tolerated when administered as a single dose of 600 mg in tablet formulation. Preliminary physiology‐based PK modelling showed up to an ~3‐fold decrease in CL/F in participants with mild, moderate and severe hepatic impairment vs. those with normal hepatic function. Therefore, a single dose of 200 mg capmatinib was considered tolerable in participants with hepatic impairment and was used in this study.
A human absorption, distribution, metabolism and excretion (ADME) study and in vitro phenotyping results showed that capmatinib is cleared via metabolism driven by cytochrome P450 (CYP) 3A4, with a significant contribution from aldehyde oxidase. ADME data suggest that absorbed capmatinib is eliminated from the systemic circulation primarily through liver metabolism and subsequent biliary/faecal excretion. 18 Results from several studies have indicated that the expression, levels and activity of drug‐metabolizing enzymes, including CYP3A4, are altered in liver disease. 19 , 20 , 21 , 22 , 23 , 24 , 25 In individuals with hepatic impairment, alterations in hepatobiliary excretion and metabolism may lead to a higher exposure of capmatinib. Furthermore, hepatic impairment could also alter plasma protein binding (PPB) and consequently trigger changes in unbound concentrations of capmatinib, which is highly bound to plasma protein (96% bound). 16 In individuals with hepatic impairment, it is unknown whether changes in capmatinib PK would necessitate dose adjustments. The aim of this study was to compare the PK of a single oral dose of capmatinib administered to participants with varying degrees of hepatic impairment (by Child–Pugh classification) with that of demographically matched controls with normal hepatic function.
2. METHODS
2.1. Participants
Adult participants (age 18–75 years) with normal or impaired hepatic function were enrolled in this study. Healthy participants were determined by the absence of clinically significant findings in medical history, physical examination, vital signs and electrocardiograms (ECGs), unless consistent with known clinical disease for participants with hepatic impairment. The Child–Pugh classification was used to evaluate and score participants with hepatic impairment. Participants with hepatic impairment were required to have a clinically determined Child–Pugh score, no change in the Child–Pugh hepatic status at least 1 month prior to dosing and confirmed liver disease by at least 1 of the following criteria: history of hepatitis C, histologically by prior liver biopsy showing cirrhosis, clinically by physical examination, laboratory data, liver imaging or endoscopic findings. In addition, participants with hepatic impairment had to meet the following laboratory values: aspartate transaminase ≤5× upper limit of normal (ULN), alanine transaminase ≤5× ULN, total bilirubin ≤3× ULN (≤5× ULN for participants with severe hepatic impairment), calculated creatinine clearance (using Cockcroft–Gault formula) ≥45 mL/min and platelets >50 × 109/L (participants with severe hepatic impairment could be enrolled if platelet count >40 × 109/L).
Key exclusion criteria included the intake of prescription/nonprescription drugs, vitamins, supplements, or herbal remedies, use of proton‐pump inhibitor medications, or the consumption of fruits known to influence the major CYP3A4 drug metabolism pathway of capmatinib within 1 week prior to dosing and until the end of treatment (EOT; day 4). Participants with hepatic impairment could have continued to receive medications required to treat their current disease (unless a CYP3A4 strong and moderate inhibitor). In addition, participants with normal hepatic function who had a positive hepatitis B surface antigen or hepatitis C test result (antibody‐positive participants were allowed if nonviraemic) were excluded. Participants with hepatic impairment with an active grade 3 or 4 hepatic encephalopathy within 4 weeks of study entry, clinical evidence of severe ascites (grade ≥3 as per Common Terminology Criteria for Adverse Events v4.03), or ascites requiring paracentesis within 3 weeks prior to dosing were also excluded from the study.
This clinical study was designed and implemented in accordance with the principles of the Declaration of Helsinki and the Good Clinical Practice guidelines of the International Council for Harmonisation, with applicable local regulations. The study protocol and all amendments were reviewed by the independent ethics committee or institutional review board for each centre. Written informed consent was obtained from all participants before screening.
2.2. Study design
This was a phase 1, multicentre, open‐label, single‐dose, parallel‐group study conducted to evaluate the PK and safety of capmatinib in participants with impaired and normal hepatic function. A single dose of 200 mg capmatinib was administered to all participants in this study. A 2‐stage study design was implemented (Figure 1). In stage 1, participants with mild hepatic impairment (mild group, Child–Pugh A [score 5–6]), moderate hepatic impairment (moderate group, Child–Pugh B [score 7–9]) and normal hepatic function (matched normal/control group) were enrolled. Upon completion of stage 1 (≥6 evaluable participants in the mild and moderate groups and matching control participants in the normal group), an interim analysis was conducted. The study was to be stopped if the estimated GM area under the plasma concentration–time curve (AUC) or maximum (peak) observed plasma drug concentration after single‐dose administration (Cmax) in the mild or moderate hepatic impairment groups was at least 3‐fold higher vs. the normal group (exceeding the established safety margin). If not, stage 2 of the study could be started. In stage 2, participants with severe hepatic impairment (severe group, Child–Pugh C [score 10–15]) were enrolled, along with any additional matching controls (normal group), as needed. The matching criteria applied in stage 1 was implemented.
FIGURE 1.
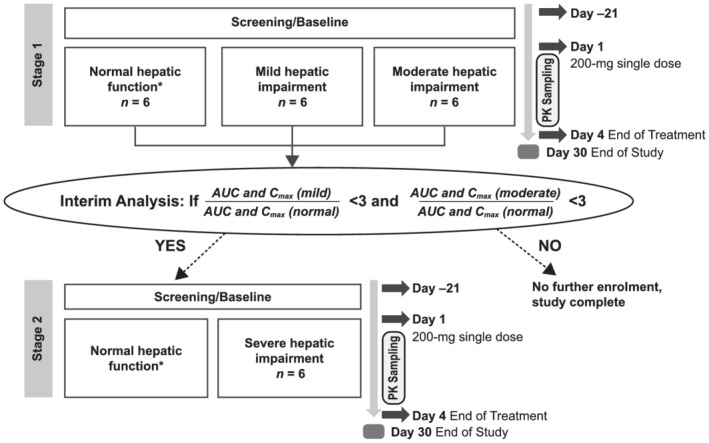
Study design. AUC, area under the plasma concentration–time curve; Cmax, maximum (peak) observed plasma drug concentration after single‐dose administration; PK, pharmacokinetics. *Enrolment of additional participants in the normal group could be required to ensure appropriate matches to the hepatic impairment groups
Enrolment of participants in the normal group was demographically matched to that in the hepatic impairment groups with respect to age (±10 years), body weight (±20%) and sex. Matching participants in the normal group were enrolled after 3 participants from the hepatic impairment groups (mild and moderate groups) were confirmed as evaluable. Sufficient male or female participants were enrolled to ensure that ≥6 evaluable participants in each hepatic impairment group as well as the matching normal group completed the study. Furthermore, enrolment in the normal group remained open until the enrolment in the hepatic impairment groups was complete and the number of matching controls had been achieved for comparison. In the final analysis, PK data from participants in the mild, moderate and severe groups were compared with data from participants in the normal group.
The study consisted of a 21‐day screening/baseline period (day −21 to day −1; Figure 1). Participants who met the eligibility criteria at screening underwent baseline evaluations ~24 hours prior to capmatinib dosing. Baseline safety evaluations were to be available prior to dosing. Participants were confined to the study centre from day −1 to day 4. On day 1, the participants received the study treatment. Between day 1 and day 4, PK samples were collected from participants, and safety assessments were performed. Participants then underwent EOT evaluations on day 4, following which they were discharged. Participants were instructed to contact the study staff during the safety follow‐up period to report adverse events (AEs) and serious AEs (SAEs). Participants were contacted at the end of the study by phone for safety follow‐up 30 days after dosing to collect and record AEs and SAEs that might have occurred following the dose administration and/or to follow‐up on the resolution of ongoing AEs, if applicable. The planned total study duration was ~8 weeks, from screening to the 30‐day safety follow‐up phone call.
2.3. Treatment
All participants had to fast overnight (minimum 10 hours) prior to the administration of capmatinib. On the morning of dosing (day 1), they received a single oral dose of 200 mg capmatinib (2 × 100 mg tablets) taken with ~240 mL of noncarbonated water. Participants continued to fast for at least 4 hours after dosing. Participants could consume water at any time, except for 1 hour before and 1 hour after the administration of capmatinib.
2.4. Study objectives
The primary objective of the study was to compare the PK of a single oral dose of capmatinib 200 mg in participants with impaired and normal hepatic function. Secondary objectives included assessment of safety and PPB of capmatinib in participants with impaired and normal hepatic function. Exploratory objectives included PK assessment of the capmatinib metabolite M16 (CMN288) and PK assessment based on the National Cancer Institute (NCI) Organ Dysfunction Working Group classification of hepatic dysfunction in participants with impaired and normal hepatic function.
2.5. Assessments
The PK parameters of capmatinib were calculated from individual concentration–time profiles by the noncompartmental method using Phoenix WinNonlin v6.4 (Pharsight, Mountain View, CA, USA). For PK evaluations, serial blood samples (4 mL) were collected from a forearm vein at the following time points: predose and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, 48 and 72 hours postdose. Plasma concentrations of capmatinib and its metabolite M16 (CMN288) were measured using a validated liquid chromatography tandem mass spectrometry assay with a lower limit of quantification (LLOQ) of ~1.00 ng/mL (Supplementary Methods). Concentrations below the LLOQ were reported as 0.00 ng/mL, and missing samples were labelled accordingly. All sampling was relative to the administration of capmatinib.
A 7‐mL blood draw was collected at 3 hours (±15 min) postdose on day 1 for PPB assessment of capmatinib using the ultrafiltration method.
Safety assessments included monitoring of haematology, blood chemistry, coagulation parameters, urinalysis, physical examinations, vital signs, weight, performance status, pregnancy tests and cardiac assessments. Haematology, blood chemistry, coagulation parameters and urinalysis assessments were performed at screening, baseline, day 1, day 2 and EOT. Vital signs were measured at screening, baseline, days 1, 2 and 3, and EOT. A complete physical examination, body weight measurements and serum pregnancy test were performed at screening, baseline and EOT. Standard 12‐lead ECGs were performed in triplicate at screening, baseline, dosing visit (day 1) and at EOT. Unscheduled ECGs could also be performed at any time, if clinically indicated. In addition, all AEs and SAEs, with their severity and relationship to capmatinib, were collected.
2.6. Statistical analysis
The full analysis set and safety set included all participants who received at least 1 dose of capmatinib. The PK analysis set (PAS) consisted of all participants who received capmatinib and had an evaluable PK profile.
A formal statistical analysis was performed using a linear model with study group as a fixed effect, which was fitted to the log‐transformed PK parameters (AUClast, AUCinf and Cmax) to assess the effect of hepatic impairment on the PK of a single oral dose of capmatinib. The mild and moderate groups were the test study groups, and the normal group was the reference study group. Point estimates of differences between the study groups and the corresponding 90% confidence intervals (CIs) were calculated and anti‐logged to obtain the point estimates and 90% CIs for the GM ratio (GMR) of the test vs. reference on the original scale. The effect of baseline covariates such as sex, age and weight on PK was investigated as appropriate. Difference in medians between study groups was provided for Tmax. Individual participant values and GMs for AUCinf, AUClast and Cmax were plotted. If the interim analysis results did not show a substantial increase in exposure for either the mild or moderate group (GMR for AUC [AUCinf and AUClast] and Cmax [test: reference] < 3), stage 2 of the study was started. At the completion of stage 2, the above linear model was repeated to compare all hepatic impairment groups (including severe) with the normal group using all data from stages 1 and 2. In the event the study was closed with <6 participants in the severe group, the linear model did not include data from the severe group and the comparison between the severe and normal groups.
The statistical model described for the primary PK analysis was re‐run on the unbound PK parameters (Cmax,u, AUClast,u and AUCinf,u) for capmatinib using PAS. The unbound parameters and concentrations were calculated by multiplying the respective capmatinib parameters and concentrations by the fraction unbound (fu) in the capmatinib concentration data set provided by Bioanalyst. The formulas CL,u/F = (CL/F)/fu and Vz,u/F = (Vz/F)/fu were used for unbound apparent clearance and volume of distribution, respectively.
As an exploratory analysis, descriptive statistics (mean [standard deviation], GM, GM coefficient of variation [CV] %) were presented for PK parameters (except Tmax) by study group for the capmatinib metabolite. Median (minimum–maximum), GM and GM CV% were presented for Tmax.
As an exploratory analysis, the classification of hepatic impairment was done according to the NCI Organ Dysfunction Working Group classification of hepatic dysfunction (NCI, Cancer Therapy Evaluation Program) based on screening visit hepatic parameters. NCI classification was not used for participant assignment. Descriptive statistics (mean [standard deviation], GM, GM CV%) were presented for the primary PK parameters by NCI classification (except Tmax) at final analysis. Median (minimum–maximum), GM and GM CV% were presented for Tmax. A model‐based analysis was performed to assess the effect of hepatic impairment based on NCI classification on capmatinib PK parameters. If any impairment group had <6 participants by NCI classification, the linear model did not include data or comparisons from such groups.
3. RESULTS
The interim PK analysis conducted at the end of stage 1 showed that the GMs of the exposures seen in the mild and moderate groups were <3‐fold higher than those in the normal group. Therefore, stage 2 was conducted as planned.
3.1. Participant disposition
All 31 enrolled participants completed the study and treatment (normal group [n = 10], mild group [n = 7], moderate group [n = 8] and severe group [n = 6]). The full analysis set and safety set included all 31 enrolled participants. PAS included 29 participants; 1 participant in the normal group who did not fast overnight prior to capmatinib dosing and another in the mild group who took prohibited concomitant medication (omeprazole) during treatment were excluded from the PAS.
3.2. Demographic and baseline characteristics
Table 1 summarizes the demographics by study group. The median age of all participants (N = 31) was 55 years (range, 43–64). The majority were male (83.9%) and Caucasian (74.2%); 51.6% had a Hispanic/Latino ethnicity. Demographics were similar across groups. All enrolled participants tested negative for an alcohol test, 4 tested positive for the drug screen test and 12 tested positive for the urine cotinine test. Table 2 provides a summary of the different liver function tests based on the Common Terminology Criteria grades by study group at baseline. The Child–Pugh scores were 5 (4 participants [57.1%]) or 6 (3 participants [42.9%]) in the mild group; 7 (6 participants [75%]), 8 (1 participant [12.5%]), or 9 (1 participant [12.5%]) in the moderate group; and 10 (4 participants [66.7%]), 11 (1 participant [16.7%]) or 12 (1 participant [16.7%]) in the severe group.
TABLE 1.
Demographics by study group (full analysis set)
| Demographic variable | Normal (n = 10) | Mild (n = 7) | Moderate (n = 8) | Severe (n = 6) | All participants (N = 31) |
|---|---|---|---|---|---|
| Median (range) age, y | 54.5 (43.0–62.0) | 57.0 (52.0–62.0) | 56.5 (45.0–64.0) | 49.0 (44.0–60.0) | 55.0 (43.0–64.0) |
| Sex, n (%) | |||||
| Female | 2 (20.0) | 0 | 2 (25.0) | 1 (16.7) | 5 (16.1) |
| Male | 8 (80.0) | 7 (100) | 6 (75.0) | 5 (83.3) | 26 (83.9) |
| Race, n (%) | |||||
| Caucasian | 6 (60.0) | 4 (57.1) | 7 (87.5) | 6 (100) | 23 (74.2) |
| Black | 4 (40.0) | 2 (28.6) | 1 (12.5) | 0 | 7 (22.6) |
| Other | 0 | 1 (14.3) | 0 | 0 | 1 (3.2) |
| Ethnicity, n (%) | |||||
| Hispanic/Latino | 4 (40.0) | 1 (14.3) | 6 (75.0) | 5 (83.3) | 16 (51.6) |
| Other | 6 (60.0) | 6 (85.7) | 2 (25.0) | 1 (16.7) | 15 (48.4) |
| Median (range) weight, kg | 83.6 (56.3–100.5) | 82.4 (69.4–103.9) | 82.2 (61.1–100.5) | 88.7 (60.4–114.9) | 83.7 (56.3–114.9) |
| Median (range) height, cm | 173.8 (150.5–185.0) | 180.0 (170.0–187.0) | 166.6 (158.0–174.1) | 176.5 (155.0–181.0) | 174.0 (150.5–187.0) |
| Median (range) BMI, kg/m2 | 27.9 (21.5–29.4) | 24.2 (20.8–32.1) | 28.3 (22.2–36.8) | 30.2 (23.6–35.6) | 27.4 (20.8–36.8) |
| Median (range) BSA, m2 | 2.0 (1.6–2.3) | 2.1 (1.8–2.3) | 2.0 (1.7–2.2) | 2.1 (1.7–2.4) | 2.0 (1.6–2.4) |
BMI, body mass index; BSA, body surface area.
BMI (kg/m2) = weight (kg)/height (m)2.
BSA (Gehan and George): BSA (m2) = 234.94 × (height [cm]0.422) × (weight [kg]0.515)/10000.
BMI and BSA were calculated using the baseline weight and baseline height.
TABLE 2.
Baseline liver function tests (CTC grade) by study group (safety set)
| Parameter | Normal (n = 10) | Mild (n = 7) | Moderate (n = 8) | Severe (n = 6) |
|---|---|---|---|---|
| Increased ALP (serum), n (%) | ||||
| Grade 0 | 10 (100) | 7 (100) | 7 (87.5) | 5 (83.3) |
| Grade 1 | 0 | 0 | 1 (12.5) | 1 (16.7) |
| Grade 2 | 0 | 0 | 0 | 0 |
| Grade 3 | 0 | 0 | 0 | 0 |
| Grade 4 | 0 | 0 | 0 | 0 |
| Increased bilirubin (total), n (%) | ||||
| Grade 0 | 10 (100) | 7 (100) | 6 (75.0) | 0 |
| Grade 1 | 0 | 0 | 1 (12.5) | 2 (33.3) |
| Grade 2 | 0 | 0 | 1 (12.5) | 4 (66.7) |
| Grade 3 | 0 | 0 | 0 | 0 |
| Grade 4 | 0 | 0 | 0 | 0 |
| Increased SGOT (AST), n (%) | ||||
| Grade 0 | 10 (100) | 3 (42.9) | 3 (37.5) | 0 |
| Grade 1 | 0 | 4 (57.1) | 5 (62.5) | 6 (100) |
| Grade 2 | 0 | 0 | 0 | 0 |
| Grade 3 | 0 | 0 | 0 | 0 |
| Grade 4 | 0 | 0 | 0 | 0 |
| Increased SGPT (ALT), n (%) | ||||
| Grade 0 | 10 (100) | 4 (57.1) | 8 (100) | 5 (83.3) |
| Grade 1 | 0 | 3 (42.9) | 0 | 1 (16.7) |
| Grade 2 | 0 | 0 | 0 | 0 |
| Grade 3 | 0 | 0 | 0 | 0 |
| Grade 4 | 0 | 0 | 0 | 0 |
ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; CTC, Common Terminology Criteria; SGOT, serum glutamic‐oxaloacetic transaminase; SGPT, serum glutamic‐pyruvic transaminase.
Baseline was defined as the last nonmissing value prior to the first dose.
CTC version 4.03 was used.
3.3. PK parameters of capmatinib
The mean plasma concentration–time profiles of capmatinib were largely similar in the normal (n = 9), mild (n = 6) and moderate groups (n = 8) and numerically higher in the severe group (n = 6; Figures 2A and B). The individual participant values and GMs for Cmax, AUClast and AUCinf by study group are presented in Figure 3. Except for 1 participant in the severe group, all individual AUCinf and AUClast values from the 3 hepatic impairment groups were within the ranges of individual values observed from the normal group. Compared with the normal group, GM Cmax decreased by 27.6% in the mild group (GMR = 0.724; 90% CI: 0.476–1.10), by 17.2% in the moderate group (GMR = 0.828; 90% CI: 0.563–1.22) and remained similar in the severe group (GMR = 1.02; 90% CI: 0.669–1.55). Compared with the normal group, GM AUCinf decreased by 23.3% in the mild group (GMR = 0.767; 90% CI: 0.532–1.11), decreased by 8.6% in the moderate group (GMR = 0.914; 90% CI: 0.652–1.28) and increased by 24% in the severe group (GMR = 1.24; 90% CI: 0.858–1.78). Similar results were observed for AUClast. The model‐based adjusted GMs for Cmax, AUClast and AUCinf across groups are provided in Table 3. The interparticipant variability of Cmax, AUCinf and AUClast values, as estimated by GM CV%s (Table 3), was 44.1–56.6, 21.4–54.6 and 20.7–55.2%, respectively, across all groups. Similar Tmax was observed in the normal, mild, moderate and severe groups, with median Tmax ranging from 1.0–1.5 hours (Table 3). Results using sex, age group (<65 and ≥65 y) and weight were similar and followed a similar trend for all comparisons. The effects of the 3 covariates were not clinically relevant across the primary PK variables (Table S1). Compared with the normal group, GM CL/F increased by 30.6 and 9.7% in the mild and moderate groups, respectively, while it decreased by 19.1% in the severe group, consistent with the changes in AUC; GM t½ was shorter by 48.4, 44.5 and 41.5% in the mild, moderate and severe groups, respectively, vs. the normal group. GM Vz/F was lower by 32.9, 39.5 and 53.0% in the mild, moderate and severe groups, respectively, vs. the normal group (Table 3).
FIGURE 2.
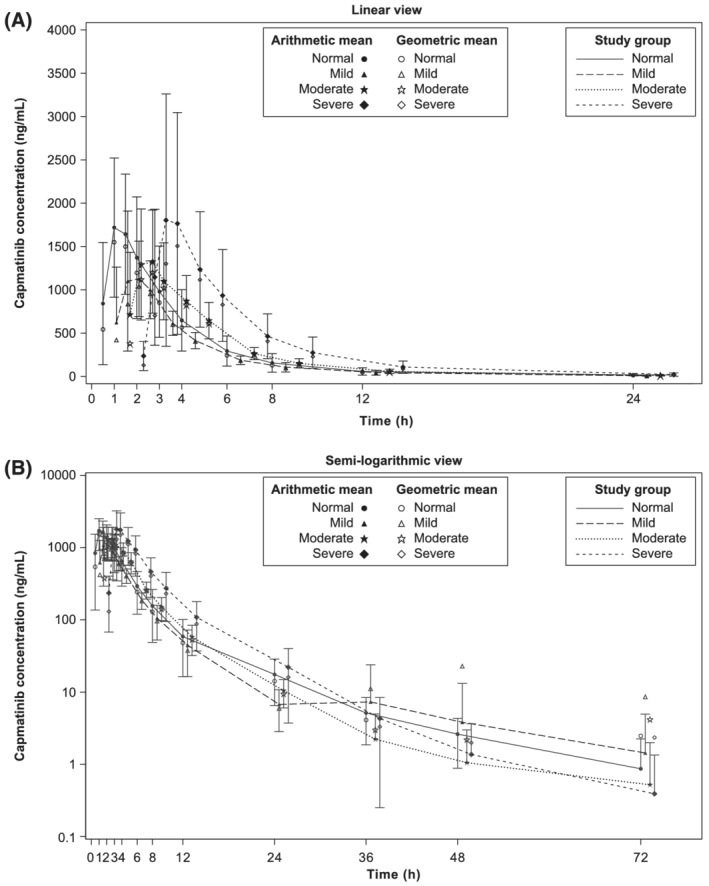
Geometric mean and arithmetic mean (standard deviation) concentration–time profiles for capmatinib by study group in (A) linear view and (B) semi‐logarithmic view (pharmacokinetic analysis set). Zero concentrations at individual time points are excluded from geometric mean computation
FIGURE 3.
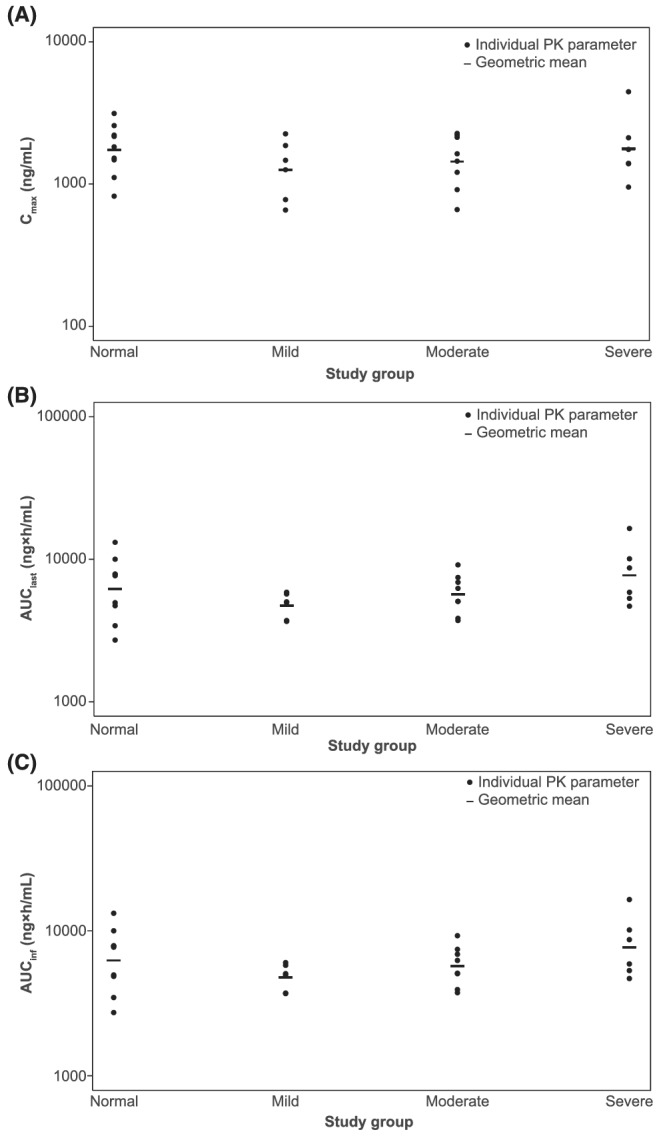
Individual participant values and geometric means for capmatinib PK parameters (A) Cmax, (B) AUClast and (C) AUCinf by study group (pharmacokinetic analysis set). AUC, area under the plasma concentration–time curve; AUCinf, AUC from time zero to infinity; AUClast, AUC from time zero to last measurable concentration sampling time; Cmax, maximum (peak) observed plasma drug concentration after single‐dose administration; PK, pharmacokinetic
TABLE 3.
Summary and statistical analysis of pharmacokinetic parameters for capmatinib by study group (pharmacokinetic analysis set)
| Parameter | Statistics | Normal (n = 9) | Mild (n = 6) | Moderate (n = 8) | Severe (n = 6) |
|---|---|---|---|---|---|
| AUClast (ng × h/mL) | Mean (SD) | 6930 (3350) | 4810 (944) | 5920 (1860) | 8510 (4410) |
| GM | 6190 | 4730 | 5660 | 7710 | |
| CV% GM | 55.2 | 20.7 | 32.5 | 50.1 | |
| AUCinf (ng × h/mL) | Mean (SD) | 6970 (3340) | 4870 (992) | 5960 (1890) | 8530 (4410) |
| GM | 6240 | 4790 | 5700 | 7720 | |
| CV% GM | 54.6 | 21.4 | 32.5 | 50.1 | |
| Cmax (ng/mL) | Mean (SD) | 1870 (732) | 1380 (620) | 1560 (613) | 2020 (1270) |
| GM | 1740 | 1260 | 1440 | 1770 | |
| CV% GM | 44.1 | 51.5 | 47.3 | 56.6 | |
| Tmax (h) | Median (range) | 1.00 (0.500–2.00) | 1.25 (0.500–2.00) | 1.25 (0.500–3.00) | 1.50 (1.00–3.00) |
| GM | N/A | N/A | N/A | N/A | |
| CV% GM | N/A | N/A | N/A | N/A | |
| t½ (h) | Mean (SD) | 12.0 (6.82) | 6.38 (4.88) | 7.62 (8.12) | 6.80 (4.23) |
| GM | 10.4 | 5.37 | 5.77 | 6.08 | |
| CV% GM | 60.3 | 64.2 | 77.0 | 50.1 | |
| CL/F (L/h) | Mean (SD) | 36.1 (19.1) | 42.6 (9.23) | 36.6 (11.4) | 28.2 (11.7) |
| GM | 32.0 | 41.8 | 35.1 | 25.9 | |
| CV% GM | 54.6 | 21.4 | 32.5 | 50.1 | |
| Vz/F (L) | Mean (SD) | 655 (504) | 366 (218) | 340 (227) | 263 (128) |
| GM | 483 | 324 | 292 | 227 | |
| CV% GM | 104.3 | 55.6 | 61.1 | 74.4 |
| Adjusted GMs | GMR (90% CI) | ||||||
|---|---|---|---|---|---|---|---|
| Normal (n = 9) | Mild (n = 6) | Moderate (n = 8) | Severe (n = 6) | Mild/normal | Moderate/normal | Severe/normal | |
| AUCinf (ng × h/mL) | 6240 | 4790 | 5700 | 7720 | 0.767 (0.532–1.11) | 0.914 (0.652–1.28) | 1.24 (0.858–1.78) |
| AUClast (ng × h/mL) | 6190 | 4730 | 5660 | 7710 | 0.764 (0.529–1.10) | 0.915 (0.652–1.28) | 1.24 (0.862–1.80) |
| Cmax (ng/mL) | 1740 | 1260 | 1440 | 1770 | 0.724 (0.476–1.10) | 0.828 (0.563–1.22) | 1.02 (0.669–1.55) |
AUC, area under the plasma concentration–time curve; AUCinf, AUC from time zero to infinity; AUClast, AUC from time zero to last measurable concentration sampling time; CI, confidence interval; CL/F, the apparent total body clearance of drug from the plasma; Cmax, maximum (peak) observed plasma drug concentration after single‐dose administration; CV, coefficient of variation; GM, geometric mean; GMR, geometric mean ratio; SD, standard deviation; t½, elimination half‐life associated with terminal slope (λz) of a semi‐logarithmic concentration–time curve (time); Tmax, time to reach maximum (peak) plasma drug concentration after single‐dose administration (time); Vz/F, apparent volume of distribution during terminal phase.
CV% GM = sqrt (exp [variance for log‐transformed data]‐1) × 100.
Model is a linear model of the log‐transformed PK parameters. Study group was included in the model as a fixed effect. Results were back‐transformed to obtain adjusted GM, GMR and 90% CI.
3.4. PK parameters of unbound capmatinib
The PPB data of capmatinib from 3 hours postdose samples showed that capmatinib was highly bound (96.7%) to human plasma proteins across all participants. The GM unbound fraction of capmatinib across groups is presented in Table S2. The adjusted GMs for Cmax,u, AUClast,u and AUCinf,u across all groups and the GMRs with 90% CIs for all comparisons are presented in Table S3. In the mild group vs. the normal group, GM Cmax,u, AUClast,u and AUCinf,u decreased by 23.9, 19.6 and 19.3%, respectively. In the moderate group, GM Cmax,u, AUClast,u and AUCinf,u were similar to that of the normal group. GM Cmax,u, AUClast,u and AUCinf,u increased by 46, 78 and 78%, respectively, in the severe group vs. the normal group.
3.5. PK of the capmatinib metabolite
A numerically lower GM AUCinf, AUClast, Cmax and t½ of CMN288 was observed in each of the hepatic impairment groups vs. the normal group. GM AUCinf decreased by 52.0, 25.4 and 53.3%; GM AUClast decreased by 52.4%, 25.4 and 53.3%; GM Cmax decreased by 58.1, 41.9 and 74.1%; and GM t1/2 decreased by 31.3, 35.5 and 41.1% in the mild, moderate and severe groups, respectively, vs. the normal group. The GMs and GM CV%s for AUCinf, AUClast, Cmax and t½ across groups are presented in Table 4. The median Tmax was similar in the normal (1.5 h [1.0–2.0 h]), mild (1 h [1.0–2.0 h]) and moderate (1 h [1.0–3.0 h]) groups and numerically higher in the severe group (3 h [1.5–6.0 h]). The metabolite ratio GMs (CV%s) for AUCinf and AUClast were 0.704 (24.1) and 0.702 (24.0) in the normal group, 0.440 (23.8) and 0.437 (24.1) in the mild group, 0.573 (67.7) and 0.573 (68.0) in the moderate group, and 0.266 (32.2) and 0.264 (32.2) in the severe group (Table 4).
TABLE 4.
Summary of pharmacokinetic parameters for M16 (CMN288) by study group (pharmacokinetic analysis set)
| Parameter | Statistics | Normal (n = 9) | Mild (n = 6) | Moderate (n = 8) | Severe (n = 6) |
|---|---|---|---|---|---|
| AUCinf (ng × h/mL) | Mean (SD) | 4820 (1880) | 2300 (691) | 3850 (2490) | 2350 (1200) |
| GM | 4560 | 2190 | 3400 | 2130 | |
| CV% GM | 35.2 | 37.4 | 52.0 | 49.5 | |
| AUClast (ng × h/mL) | Mean (SD) | 4780 (1880) | 2250 (677) | 3830 (2490) | 2330 (1210) |
| GM | 4520 | 2150 | 3370 | 2110 | |
| CV% GM | 35.8 | 37.2 | 52.4 | 50.0 | |
| Cmax (ng/mL) | Mean (SD) | 1050 (328) | 492 (272) | 738 (645) | 287 (130) |
| GM | 1010 | 423 | 587 | 262 | |
| CV% GM | 32.0 | 69.1 | 75.9 | 51.3 | |
| Tmax (h) | Median (range) | 1.50 (1.00–2.00) | 1.00 (1.00–2.00) | 1.00 (1.00–3.00) | 3.00 (1.50–6.00) |
| GM | N/A | N/A | N/A | N/A | |
| CV% GM | N/A | N/A | N/A | N/A | |
| t½ (h) | Mean (SD) | 11.4 (6.92) | 7.86 (5.49) | 7.32 (5.16) | 5.93 (0.907) |
| GM | 9.96 | 6.84 | 6.42 | 5.87 | |
| CV% GM | 57.9 | 55.6 | 50.8 | 14.8 | |
| Metabolite ratio of AUCinf (fold) | Mean (SD) | 0.722 (0.181) | 0.450 (0.0988) | 0.680 (0.443) | 0.276 (0.0802) |
| GM | 0.704 | 0.440 | 0.573 | 0.266 | |
| CV% GM | 24.1 | 23.8 | 67.7 | 32.2 | |
| Metabolite ratio of AUClast (fold) | Mean (SD) | 0.720 (0.180) | 0.447 (0.100) | 0.680 (0.445) | 0.274 (0.0795) |
| GM | 0.702 | 0.437 | 0.573 | 0.264 | |
| CV% GM | 24.0 | 24.1 | 68.0 | 32.2 |
AUC, area under the plasma concentration–time curve; AUCinf, AUC from time zero to infinity; AUClast, AUC from time zero to last measurable concentration sampling time; Cmax, maximum (peak) observed plasma drug concentration after single‐dose administration; CV, coefficient of variation; GM, geometric mean; SD, standard deviation; t½, elimination half‐life associated with terminal slope (λz) of a semilogarithmic concentration–time curve (time); Tmax, time to reach maximum (peak) plasma drug concentration after single‐dose administration (time).
CV% GM = sqrt (exp [variance for log‐transformed data]‐1) × 100.
3.6. PK of capmatinib based on NCI classification
There were differences in the classification of participants when using the NCI and Child–Pugh classification systems. A comparison of the classification of participants with hepatic impairment by NCI and Child–Pugh classification is shown in Table S4. The summary and statistical analysis of PK parameters of capmatinib by NCI classification are shown in Table S5; 14 participants were included in the normal group, 7 in the mild group, 7 in the moderate group and 1 in the severe group. In the mild vs. normal group, GM Cmax, AUClast and AUCinf increased by 35, 27 and 26%. In the moderate vs. normal group, GM Cmax, AUClast and AUCinf increased by 46, 39 and 38% (Table S5). The NCI severe group was not included in the analysis because there was only 1 participant in this group.
3.7. Safety results
Most participants in the mild (7 [100%]), moderate (7 [87.5%]) and severe (4 [66.7%]) groups used some type of concomitant medication or significant nondrug therapy prior to or during the study. The medications most used were furosemide for ascites (7 participants), lisinopril for hypertension (5 participants), clonazepam for anxiety and insomnia (5 participants), and lactulose for hepatic encephalopathy (5 participants).
Overall (N = 31), 6 participants (19.4%) experienced AEs regardless of causality. The most frequently reported AEs were nausea and headache (each reported in 2 participants). Participants in the normal group (n = 10) did not experience any AE. Five participants, 2 each in the mild (n = 7) and moderate (n = 8) groups and 1 in the severe group (n = 6), experienced at least 1 grade 1 AE. One participant in the mild group had grade 2 hypoglycaemia (Table 5). No Grade 3 or 4 AEs were reported.
TABLE 5.
Adverse events regardless of study drug relationship by primary system organ class, preferred term, maximum grade and study group (safety set)
| Primary system organ class | Normal (n = 10) n (%) | Mild (n = 7) n (%) | Moderate (n = 8) n (%) | Severe (n = 6) n (%) | All participants (N = 31) n (%) |
|---|---|---|---|---|---|
| Preferred term | |||||
| Any primary system organ class | 0 | 3 (42.9) | 2 (25.0) | 1 (16.7) | 6 (19.4) |
| Grade 1 | 0 | 2 (28.6) | 2 (25.0) | 1 (16.7) | 5 (16.1) |
| Grade 2 | 0 | 1 (14.3) | 0 | 0 | 1 (3.2) |
| Ear and labyrinth disorders | 0 | 1 (14.3) | 0 | 0 | 1 (3.2) |
| Vertigo | 0 | 1 (14.3) | 0 | 0 | 1 (3.2) |
| Gastrointestinal disorders | 0 | 0 | 2 (25.0) | 1 (16.7) | 3 (9.7) |
| Nausea | 0 | 0 | 2 (25.0) | 0 | 2 (6.5) |
| Diarrhoea | 0 | 0 | 0 | 1 (16.7) | 1 (3.2) |
| Flatulence | 0 | 0 | 0 | 1 (16.7) | 1 (3.2) |
| General disorders and administration site conditions | 0 | 1 (14.3) | 0 | 0 | 1 (3.2) |
| Medical device site dermatitis | 0 | 1 (14.3) | 0 | 0 | 1 (3.2) |
| Metabolism and nutrition disorders | 0 | 1 (14.3) | 0 | 0 | 1 (3.2) |
| Hypoglycaemia | 0 | 1 (14.3) | 0 | 0 | 1 (3.2) |
| Nervous system disorders | 0 | 1 (14.3) | 1 (12.5) | 0 | 2 (6.5) |
| Headache | 0 | 1 (14.3) | 1 (12.5) | 0 | 2 (6.5) |
| Dizziness | 0 | 0 | 1 (12.5) | 0 | 1 (3.2) |
Primary system organ classes are presented alphabetically; preferred terms are sorted within primary system organ class in descending frequency, as reported in the All participants column. A participant with multiple occurrences of an adverse event (AE) under 1 study group is counted only once in the AE category for that study group. A participant with multiple severity ratings for an AE while on a study group is only counted under the maximum rating. A participant with multiple AEs within a primary system organ class is counted only once in the total row at maximum severity grade.
Three participants (9.7%) experienced AEs suspected to be capmatinib related; all AEs suspected to be capmatinib related were of grade 1 severity. In the moderate group, 1 participant had an AE of nausea and 1 had AEs of nausea and headache that were suspected to be drug related. In the severe group, 1 participant had AEs of diarrhoea and flatulence that were suspected to be drug related. No deaths and SAEs were reported during the study treatment and 30‐day follow‐up. Minimal changes in haematological parameters occurred. No clinically relevant changes in chemistry parameters, QTcF or other ECG parameters were observed.
4. DISCUSSION
This phase 1, parallel‐group study was conducted to compare the PK of a single oral dose of capmatinib 200 mg in participants with varying degrees of hepatic impairment vs. a demographically matched cohort with normal hepatic function. Administration of the same dose of capmatinib to all participants allowed for a direct comparison of PK profiles between the impairment groups and the normal group, attributing any observed differences in PK profiles to hepatic impairment (Child–Pugh class A, B or C). As per the NCI Organ Dysfunction Working Group classification of hepatic dysfunction, 1 participant was classified as having severe hepatic impairment. Consequently, the NCI severe group was not included in the PK analysis of capmatinib by NCI classification. This limited the statistical comparison of capmatinib PK parameters by NCI classification between the severe group (n = 1) and the normal group. Prior studies have reported on the effects of varying degrees of hepatic impairment on the PK of anticancer drugs metabolized by CYP3A4. 26 , 27 , 28
A preliminary physiology‐based PK model predicted up to an ~3‐fold decrease in CL/F in all the hepatic impairment groups vs. the normal group. However, the results from this study showed that GM CL/F increased by 30.6 and 9.7% and decreased by 19.1% in the mild, moderate and severe hepatic impairment groups, respectively, vs. the normal group. Consistent with the CL/F results, in participants with mild, moderate and severe hepatic impairment, GM AUCinf decreased by 23.3 and 8.6% and increased by 24%, respectively, vs. those with normal hepatic function. Compared with participants with normal hepatic function, GM Cmax decreased in those with mild and moderate hepatic impairment by 27.6 and 17.2%, respectively, while it remained similar in participants with severe hepatic impairment. The interparticipant variability across all study groups indicated moderate variability for Cmax and low to moderate variability for AUCinf. It should be noted that the individual AUCinf and AUClast values from the 3 hepatic impairment groups were within the ranges of individual values observed from the normal group. Based on the NCI classification, for both the mild and moderate hepatic impairment groups, GM AUCinf, AUClast and Cmax increased by 26–46% vs. the normal group and not decreased as observed from the Child–Pugh classification results. After adjusting for age, sex and body weight, the adjusted GMs of the PK parameters of the various hepatic impairment groups showed similar trends as obtained in the unadjusted primary analysis.
Mild, moderate or severe hepatic impairment did not have a clinically relevant effect on the PPB of capmatinib. Capmatinib was highly bound to plasma proteins (96.7%) across all hepatic groups. Unbound PK parameters largely followed similar trends to that of total capmatinib PK parameters for participants with mild and moderate hepatic impairment. Numerical increases in GM Cmax,u, AUCinf,u and AUClast,u observed in participants with severe hepatic impairment vs. those with normal hepatic function could be related to the higher GM unbound fraction of capmatinib in the severe group (~44.8% higher vs. the normal group).
M16 (CMN288) is a major circulating, pharmacologically inactive metabolite of capmatinib. CMN288 is the metabolic product of aldehyde oxidase. The CYP3A enzyme does not contribute to the formation of CMN288. The exposure of CMN288 was monitored to explore potential impacts of hepatic impairment on the formation of CMN288 through the aldehyde oxidase pathway. Following a single oral dose of capmatinib 200 mg, CMN288 GM AUCinf and AUClast observed in participants from the 3 hepatic impairment groups decreased by 25–53% vs. those with normal hepatic function, indicating reduced metabolism of capmatinib to CMN288 in participants with hepatic impairment.
Therefore, based on the information above, the observed capmatinib PK differences in the mild, moderate and severe hepatic impairment groups vs. normal hepatic function are not considered clinically significant.
The results from this study suggest that capmatinib starting dose adjustment is not required for individuals with mild, moderate, or severe hepatic impairment classified as per the Child–Pugh score. Single‐dose capmatinib was well tolerated and demonstrated a manageable safety profile in participants with mild, moderate and severe hepatic impairment and those with normal hepatic function. No new safety findings emerged from this study.
COMPETING INTERESTS
Thomas C. Marbury is an employee and equity owner of Orlando Clinical Research Center. Xinhui Chen, Xiaoming Cui, Nathalie Pognan, Michelle Quinlan, Shruti Kapoor, Gholamreza Rahmanzadeh and Monica Giovannini are employees of Novartis. Xinhui Chen, Xiaoming Cui, Gholamreza Rahmanzadeh, Nathalie Pognan and Michelle Quinlan own Novartis stock.
CONTRIBUTORS
Xinhui Chen and Thomas C. Marbury contributed to patient accrual, data analysis, data interpretation, manuscript writing and approval. Xiaoming Cui and Monica Giovannini contributed to designing the study, patient accrual, data analysis, data interpretation, manuscript writing and approval. Michelle Quinlan and Gholamreza Rahmanzadeh contributed to data analysis, data interpretation, manuscript writing and approval. Shruti Kapoor and Nathalie Pognan contributed to data interpretation, manuscript writing and approval.
Supporting information
Table S1. Summary of covariate‐adjusted statistical analysis of PK parameters for capmatinib total drug concentration (pharmacokinetic analysis set)
Table S2. Summary of fraction unbound of capmatinib in plasma by study group (pharmacokinetic analysis set)
Table S3. Summary of statistical analysis of PK parameters for capmatinib expressed as unbound drug (pharmacokinetic analysis set)
Table S4. Cross‐tabulation between Child–Pugh and NCI classification (full analysis set)
Table S5. Summary and statistical analysis of PK parameters for capmatinib by NCI classification (pharmacokinetic analysis set)
ACKNOWLEDGEMENTS
The authors thank Gowri Natarajan (Novartis Healthcare Pvt Ltd) for providing medical writing support. This study was funded by Novartis Pharmaceuticals Corporation.
Chen X, Cui X, Pognan N, et al. Pharmacokinetics of capmatinib in participants with hepatic impairment: A phase 1, open‐label, single‐dose, parallel‐group study. Br J Clin Pharmacol. 2022;88(1):91–102. 10.1111/bcp.14929
Principal investigator: The authors confirm that the Principal Investigator for this paper is Dr Thomas C. Marbury and that he had direct clinical responsibility for participants.
Clinical Trial registration: ClinicalTrials.gov NCT02474537.
Funding information Novartis Pharmaceuticals Corporation
DATA AVAILABILITY STATEMENT
Novartis is committed to sharing with qualified external researchers, access to patient‐level data and supporting clinical documents from eligible studies. These requests are reviewed and approved by an independent review panel on the basis of scientific merit. All data provided are anonymized to respect the privacy of participants who have participated in the trial in line with applicable laws and regulations. This trial data availability is according to the criteria and process described on www.clinicalstudydatarequest.com
REFERENCES
- 1. Christensen JG, Burrows J, Salgia R. c‐Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett. 2005;225(1):1‐26. [DOI] [PubMed] [Google Scholar]
- 2. Christensen JG, Schreck R, Burrows J, et al. A selective small molecule inhibitor of c‐Met kinase inhibits c‐Met‐dependent phenotypes in vitro and exhibits cytoreductive antitumor activity in vivo. Cancer Res. 2003;63(21):7345‐7355. [PubMed] [Google Scholar]
- 3. Davis IJ, McFadden AW, Zhang Y, et al. Identification of the receptor tyrosine kinase c‐Met and its ligand, hepatocyte growth factor, as therapeutic targets in clear cell sarcoma. Cancer Res. 2010;70(2):639‐645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Di Renzo MF, Olivero M, Martone T, et al. Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene. 2000;19(12):1547‐1555. [DOI] [PubMed] [Google Scholar]
- 5. Schmidt L, Duh FM, Chen F, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto‐oncogene in papillary renal carcinomas. Nat Genet. 1997;16(1):68‐73. [DOI] [PubMed] [Google Scholar]
- 6. Reungwetwattana T, Liang Y, Zhu V, Ou SI. The race to target MET exon 14 skipping alterations in non‐small cell lung cancer: The why, the how, the who, the unknown, and the inevitable. Lung Cancer. 2017;103:27‐37. [DOI] [PubMed] [Google Scholar]
- 7. Vansteenkiste JF, Van De Kerkhove C, Wauters E, Van Mol P. Capmatinib for the treatment of non‐small cell lung cancer. Expert Rev Anticancer Ther. 2019;19(8):659‐671. [DOI] [PubMed] [Google Scholar]
- 8. Liu X, Wang Q, Yang G, et al. A novel kinase inhibitor, INCB28060, blocks c‐MET‐dependent signaling, neoplastic activities, and cross‐talk with EGFR and HER‐3. Clin Cancer Res. 2011;17(22):7127‐7138. [DOI] [PubMed] [Google Scholar]
- 9. Baltschukat S, Engstler BS, Huang A, et al. Capmatinib (INC280) Is Active Against Models of Non‐Small Cell Lung Cancer and Other Cancer Types with Defined Mechanisms of MET Activation. Clin Cancer Res. 2019;25(10):3164‐3175. [DOI] [PubMed] [Google Scholar]
- 10. Fujino T, Kobayashi Y, Suda K, et al. Sensitivity and Resistance of MET Exon 14 Mutations in Lung Cancer to Eight MET Tyrosine Kinase Inhibitors In Vitro. J Thorac Oncol. 2019;14(10):1753‐1765. [DOI] [PubMed] [Google Scholar]
- 11. Bang YJ, Su WC, Schuler M, et al. Phase 1 study of capmatinib in MET‐positive solid tumor patients: Dose escalation and expansion of selected cohorts. Cancer Sci. 2020;111(2):536‐547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu YL, Zhang L, Kim DW, et al. Phase Ib/II study of capmatinib (INC280) plus gefitinib after failure of epidermal growth factor receptor (EGFR) inhibitor therapy in patients with EGFR‐mutated, MET factor‐dysregulated non‐small‐cell lung cancer. J Clin Oncol. 2018;36(31):3101‐3109. [DOI] [PubMed] [Google Scholar]
- 13. Qin S, Chan SL, Sukeepaisarnjaroen W, et al. A phase II study of the efficacy and safety of the MET inhibitor capmatinib (INC280) in patients with advanced hepatocellular carcinoma. Ther Adv Med Oncol. 2019;11. 1758835919889001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Felip E, Minotti V, Soo R, et al. 1284P MET inhibitor capmatinib plus EGFR tyrosine kinase inhibitor nazartinib for EGFR‐mutant non‐small cell lung cancer. Ann Oncol. 2020;31:S829‐S830. [Google Scholar]
- 15. Schuler M, Berardi R, Lim WT, et al. Molecular correlates of response to capmatinib in advanced non‐small‐cell lung cancer: clinical and biomarker results from a phase I trial. Ann Oncol. 2020;31(6):789‐797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. US Food and Drug Administration . Capmatinib oral tablet prescribing information. 2020; https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213591s000lbl.pdf. Accessed June 16, 2020.
- 17. Wolf J, Seto T, Han JY, et al. Capmatinib in MET Exon 14‐Mutated or MET‐Amplified Non‐Small‐Cell Lung Cancer. N Engl J Med. 2020;383(10):944‐957. [DOI] [PubMed] [Google Scholar]
- 18. Glaenzel U, Jin Y, Hansen R, et al. Absorption, Distribution, Metabolism, and Excretion of Capmatinib (INC280) in Healthy Male Volunteers and In Vitro Aldehyde Oxidase Phenotyping of the Major Metabolite. Drug Metab Dispos. 2020;48(10):873‐885. [DOI] [PubMed] [Google Scholar]
- 19. Verbeeck RK. Pharmacokinetics and dosage adjustment in patients with hepatic dysfunction. Eur J Clin Pharmacol. 2008;64(12):1147‐1161. [DOI] [PubMed] [Google Scholar]
- 20. George J, Liddle C, Murray M, Byth K, Farrell GC. Pre‐translational regulation of cytochrome P450 genes is responsible for disease‐specific changes of individual P450 enzymes among patients with cirrhosis. Biochem Pharmacol. 1995;49(7):873‐881. [DOI] [PubMed] [Google Scholar]
- 21. George J, Murray M, Byth K, Farrell GC. Differential alterations of cytochrome P450 proteins in livers from patients with severe chronic liver disease. Hepatology. 1995;21(1):120‐128. [PubMed] [Google Scholar]
- 22. Guengerich FP, Turvy CG. Comparison of levels of several human microsomal cytochrome P‐450 enzymes and epoxide hydrolase in normal and disease states using immunochemical analysis of surgical liver samples. J Pharmacol Exp Ther. 1991;256(3):1189‐1194. [PubMed] [Google Scholar]
- 23. Ohnishi A, Murakami S, Akizuki S, Mochizuki J, Echizen H, Takagi I. In vivo metabolic activity of CYP2C19 and CYP3A in relation to CYP2C19 genetic polymorphism in chronic liver disease. J Clin Pharmacol. 2005;45(11):1221‐1229. [DOI] [PubMed] [Google Scholar]
- 24. Yang LQ, Li SJ, Cao YF, et al. Different alterations of cytochrome P450 3A4 isoform and its gene expression in livers of patients with chronic liver diseases. World J Gastroenterol. 2003;9(2):359‐363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Elbekai RH, Korashy HM, El‐Kadi AO. The effect of liver cirrhosis on the regulation and expression of drug metabolizing enzymes. Curr Drug Metab. 2004;5(2):157‐167. [DOI] [PubMed] [Google Scholar]
- 26. Morcos PN, Cleary Y, Sturm‐Pellanda C, et al. Effect of Hepatic Impairment on the Pharmacokinetics of Alectinib. J Clin Pharmacol. 2018;58(12):1618‐1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yin OQ, Gallagher N, Tanaka C, et al. Effects of hepatic impairment on the pharmacokinetics of nilotinib: an open‐label, single‐dose, parallel‐group study. Clin Ther. 2009;31(Pt 2):2459‐2469. [DOI] [PubMed] [Google Scholar]
- 28. Abbas R, Chalon S, Leister C, El Gaaloul M, Sonnichsen D. Evaluation of the pharmacokinetics and safety of bosutinib in patients with chronic hepatic impairment and matched healthy subjects. Cancer Chemother Pharmacol. 2013;71(1):123‐132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of covariate‐adjusted statistical analysis of PK parameters for capmatinib total drug concentration (pharmacokinetic analysis set)
Table S2. Summary of fraction unbound of capmatinib in plasma by study group (pharmacokinetic analysis set)
Table S3. Summary of statistical analysis of PK parameters for capmatinib expressed as unbound drug (pharmacokinetic analysis set)
Table S4. Cross‐tabulation between Child–Pugh and NCI classification (full analysis set)
Table S5. Summary and statistical analysis of PK parameters for capmatinib by NCI classification (pharmacokinetic analysis set)
Data Availability Statement
Novartis is committed to sharing with qualified external researchers, access to patient‐level data and supporting clinical documents from eligible studies. These requests are reviewed and approved by an independent review panel on the basis of scientific merit. All data provided are anonymized to respect the privacy of participants who have participated in the trial in line with applicable laws and regulations. This trial data availability is according to the criteria and process described on www.clinicalstudydatarequest.com