

Abstract
Large vertebrates are extremely sensitive to anthropogenic pressure, and their populations are declining fast. The white rhinoceros (Ceratotherium simum) is a paradigmatic case: this African megaherbivore has suffered a remarkable decline in the last 150 years due to human activities. Its subspecies, the northern (NWR) and the southern white rhinoceros (SWR), however, underwent opposite fates: the NWR vanished quickly, while the SWR recovered after the severe decline. Such demographic events are predicted to have an erosive effect at the genomic level, linked to the extirpation of diversity, and increased genetic drift and inbreeding. However, there is little empirical data available to directly reconstruct the subtleties of such processes in light of distinct demographic histories. Therefore, we generated a whole‐genome, temporal data set consisting of 52 resequenced white rhinoceros genomes, representing both subspecies at two time windows: before and during/after the bottleneck. Our data reveal previously unknown population structure within both subspecies, as well as quantifiable genomic erosion. Genome‐wide heterozygosity decreased significantly by 10% in the NWR and 36% in the SWR, and inbreeding coefficients rose significantly by 11% and 39%, respectively. Despite the remarkable loss of genomic diversity and recent inbreeding it suffered, the only surviving subspecies, the SWR, does not show a significant accumulation of genetic load compared to its historical counterpart. Our data provide empirical support for predictions about the genomic consequences of shrinking populations, and our findings have the potential to inform the conservation efforts of the remaining white rhinoceroses.
Keywords: conservation genomics, genomic erosion, northern white rhinoceros, population decline, southern white rhinoceros
1. INTRODUCTION
Earth's biodiversity is experiencing a severe crisis. In the past 100 years, species have gone extinct at rates that are up to 100‐fold higher than conservative estimates of background extinction rates (Ceballos et al., 2015). Deterioration of biodiversity is also pervasively manifested by shrinking population sizes, population extirpations and fragmentation of ranges of distribution in extant species (Ceballos et al., 2017). Alarmingly, these threats to biodiversity can be tightly linked to anthropogenic activities (Ceballos et al., 2015; Otto, 2018; Ripple et al., 2019).
Large vertebrates are particularly sensitive to the impact of anthropogenic pressures, and many wild populations have undergone marked declines (Ripple et al., 2019). Beyond the net loss of individuals and populations, these rapid declines can potentially have dire genetic consequences (Spielman et al., 2004). Theory predicts that populations undergoing dramatic and rapid decreases in size (i.e., bottlenecks) will also lose substantial genetic diversity and subsequently suffer from strong genetic drift and inbreeding depression (Caughley, 1994; Frankham, 2005). All these factors combined can potentially decrease fitness (Reed & Frankham, 2003) and, in the long term, diminish resilience to environmental change (Caughley, 1994).
In an era where genomic‐scale data generation has become increasingly tractable for nonmodel species, and ancient DNA techniques allow us to retrieve such information from even long‐dead biological material, the opportunity now exists to directly observe signals of the demographic history of wild populations (Abascal et al., 2016; Díez‐Del‐Molino et al., 2018; Feng et al., 2019; Robinson et al., 2019; van der Valk et al., 2019). In the case of declining populations, this entails being able to directly detect and measure genomic erosion, that is the suite of genetic symptoms caused by bottlenecks and drift, such as the reduction of genome‐wide heterozygosity, the increase in number and length of runs of homozygosity, and the accumulation of deleterious mutations (Díez‐Del‐Molino et al., 2018).
The white rhinoceros (Ceratotherium simum Burchell, 1817) is an exemplary case of the fate of the megafauna in the Anthropocene. This species is divided into two allopatric populations, also considered subspecies: the northern white rhinoceros (C. s. cottoni, NWR in this text) and the southern white rhinoceros (C. s. simum, SWR in this text) (Harley et al., 2016). Because of their obligate grazing lifestyle, both are bound to sub‐Saharan African grasslands, but their geographical ranges are nonoverlapping (Emslie & Brooks, 1999; Rookmaaker & Antoine, 2012), they are genetically distinct (Harley et al., 2016; Moodley et al., 2018; Tunstall et al., 2018) and they have not come into genetic contact for at least 10,000 years (Tunstall et al., 2018). In the past 150 years they have undergone remarkably different demographic trajectories that share, however, one striking similarity: the occurrence of a dramatic, human‐driven population decline (Emslie & Brooks, 1999; Rookmaaker, 2000; Rookmaaker & Antoine, 2012).
The SWR roamed south of the Zambezi river across present‐day South Africa, Botswana, Namibia, Zimbabwe and Mozambique (Figure 1a), until the expansion of European colonialism into the hinterlands of southern Africa in the 19th century. Habitat clearance and hunting pushed the SWR through a bottleneck, from an estimated size of several hundred thousands to an estimated low of 200 individuals at the turn of the 20th century (Rookmaaker, 2000). Conservation efforts, however, boosted a remarkable recovery of the only surviving population in Kwa‐Zulu, eastern South Africa (Rookmaaker, 2000). There are currently ~18,000 wild individuals (Emslie [IUCN SSC African Rhino Specialist Group], 2020), although they remain threatened due to the relentless poaching for their highly valued horn.
FIGURE 1.
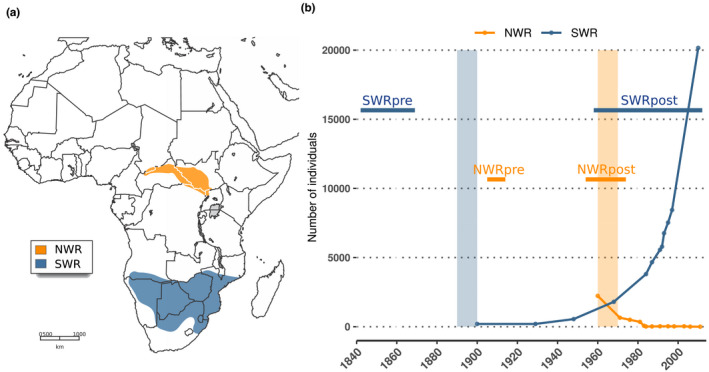
Ranges of distribution and recent demography of the northern and southern white rhinoceros. (a) The historical distribution of the NWR (Ceratotherium simum cottoni) and the SWR (Ceratotherium simum simum) (map adapted from Emslie & Brooks, 1999). (b) Recent demographic histories of the NWR and the SWR according to census size estimates reported in the literature (Emslie [IUCN SSC African Rhino Specialist Group], 2020; Emslie & Brooks, 1999; Rookmaaker, 2000). Absolute values prior to these dates do not exist, but population sizes are known to have been larger from historical records (Emslie [IUCN SSC African Rhino Specialist Group], 2020; Emslie & Brooks, 1999; Rookmaaker, 2000). Vertical shades indicate approximate timing of bottlenecks. Horizontal bars indicate our sampling time windows, which in NWRpre and SWRpre refer to dates of collection, and in NWRpost and SWRpost refer to dates of birth
In contrast, the NWR did not recover from the poaching onslaught that began in the mid‐20th century. Once inhabiting the plains of present‐day South Sudan, northeastern Democratic Republic of the Congo (DRC), Central African Republic and Uganda (Figure 1a), the NWR was rapidly decimated in subsequent poaching bursts boosted by civil and political instability in the area (Emslie, 2008; Emslie & Brooks, 1999; Rookmaaker & Antoine, 2012). Although in 1960 there were still ~2000 NWR in the wild, by 1984, only 350 remained (Emslie & Brooks, 1999). The NWR was declared extinct in the wild in 2011, and is also functionally extinct since only two females remain in captivity (Emslie [IUCN SSC African Rhino Specialist Group], 2020).
A previous genetic study that compared individuals born during or after the population bottleneck, against older samples collected from museums, revealed a loss of diversity at mitochondrial and microsatellite markers as a consequence of these demographic histories (Moodley et al., 2018). In order to expand this assessment to the genome level, and quantify in detail more potential symptoms of genomic erosion (reduced genome‐wide heterozygosity, longer homozygous tracts, or even an accumulation of genetic load), we generated a whole‐genome data set for both NWR and SWR populations, sampled at two time windows each throughout the past ~170 years. With this data set, we investigated (i) the population structure among these pre‐ and post‐bottleneck white rhinoceroses, (ii) the loss of genome‐wide diversity over time, and (iii) whether the surviving SWR have experienced an increase in genetic load.
2. MATERIALS AND METHODS
2.1. Generation of whole‐genome sequence data
Our historical sample collection included keratinous tissue (skin pieces or horn powder) obtained from 30 museum specimens, with collection dates ranging between 1845 and 2010 (see Table S1). Museum sample processing took place in facilities dedicated to ancient DNA work.
The skin pieces were manually cut and then hydrated for 2–3 h at 4°C in 0.5–1 ml of molecular biology grade water. The tissue was then briefly washed with 0.5 ml of a 1% bleach solution, followed by two rinsing steps with molecular biology grade water. Extraction of DNA from skin pieces or horn powder (20–80 mg) was done either with the DNeasy Blood and Tissue Kit (Qiagen) or with the method detailed in Gilbert et al. (2007). In the first case, two modifications were introduced to the manufacturer's guidelines: adding of DTT (dithiothreitol) 1 m to a final concentration of 40 mm to the lysis buffer, and the substitution of the purification columns in the kit by MinElute silica columns (Qiagen) to favour retention of small fragments. In the second approach, lysates were purified using the MinElute PCR Purification Kit (Qiagen), with modifications to retain short DNA fragments based on Dabney et al. (2013): phenol–chloroform separation of the aqueous phase containing the DNA, transfer of this phase to 15 ml of binding buffer, and usage of Zymo‐Spin V Reservoirs coupled to the MinElut® columns to centrifuge this large volume. Concentration of DNA and fragment size distribution in each of the extracts were assessed with a TapeStation 2200 (Agilent).
Sequencing libraries were prepared with 100 ng of extracted DNA, in a maximum volume of 32 µl. For poorly concentrated extracts, 32 µl of the purified lysate was used. We followed the BEST library build protocol (Carøe et al., 2018). The library adapters used were custom‐designed for the BGISEQ 500 Sequencing Platform (Mak et al., 2017). Finished libraries were purified with MinElute columns (Qiagen). Unamplified libraries were characterized in a qPCR (quantitative polymerase chain reaction) to estimate the number of cycles needed to reach the amplification plateau while avoiding excessive PCR cycling (Meyer & Kircher, 2010).
Libraries were amplified in two reactions of 50 µl each, containing 5 U AmpliTaq Gold polymerase (Applied Biosystems), 1× AmpliTaqGold buffer, 2.5 mm MgCl2, 0.4 mg ml–1 bovine serum albumin (BSA), 0.2 mm each dNTP, 0.2 μm BGI forward primer (Mak et al., 2017), 0.2 μm BGI reverse index‐primer (Mak et al., 2017) and 10 µl of library DNA template. PCR products were purified with either MinElute columns (Qiagen), or SPRI beads. DNA concentration was measured with a TapeStation 2200 (Agilent). Amplified libraries that contained >150 ng of DNA were submitted for one lane each of BGISEQ 500 SE100 sequencing.
Regarding the modern samples, we generated new whole‐genome data for nine of them. The sampled material was either the fraction of white cells and platelets from blood samples, or pellets of cell cultures. DNA was extracted with phenol–chloroform separation of aqueous and hydrophobic phases. Sequencing libraries were built using the Illumina TruSeq PCR‐free method on DNA inserts that were 350 bp in average fragment length and following the manufacturer's guidelines. Libraries were then sequenced on an Illumina HiSeq X platform, giving 0.5 lanes per sample in PE150 mode.
2.2. Bioinformatic data processing and quality assessment
Raw data included: 30 newly resequenced genomes from museum specimens, nine newly resequenced genomes from modern samples plus 13 modern genomes that had been previously published (NCBI BioProject PRJNA394025) (Tunstall et al., 2018). Sample names specify first the alpha‐2 code of the country of origin, or the prefix “cap” for individuals born in captivity (the case of six SWR; see Table S1). This is followed by the year of collection for museum samples, and year of birth for modern individuals; a subsequent index number distinguishes samples from the same country and year.
Basic quality summaries of the raw data per sample were obtained with fastqc version 0.11.7 (Andrews, 2010). Then the pipeline paleomix 1.2.13.2 (Schubert et al., 2014) was run for each sample separately. It included: removal of adapters with adapterremoval version 2.2.2 (Schubert et al., 2016), alignment against the GCA_000283155.1 white rhinoceros reference genome (NCBI BioProject PRJNA74583) with the bwa version 0.7.16a backtrack algorithm (Li & Durbin, 2009), duplicate filtering with picard MarkDuplicates (Broad Institute, 2019), and assessment of ancient DNA damage with mapdamage version 2.0.6 (Jónsson et al., 2013). Minimum base quality filtering was set to zero in order to maximize the number of reads aligned.
We performed a number of tests to further evaluate the quality of the data. First, we retrieved the average depth of coverage per sample from the summary file of paleomix. Moreover, we also computed the depth of coverage per scaffold for each sample with samtools version 1.9 (Li et al., 2009) and its option bedcov. We then visualized the distribution of depth of coverage across the 63 biggest autosomal scaffolds for each sample (see Figure S1; see Section 2.3 and Figure S2 for further details on the choice of scaffolds).
Additionally, we calculated error rates per sample with angsd version 0.921 (Korneliussen et al., 2014) and its option ‐doAncError, along with these quality filters: excluding nonprimary, failure and duplicate reads (‐remove_bads 1), discarding reads with multiple best hits (‐uniqueOnly 1), performing Base Alignment Quality calculation (‐baq 1), adjusting base quality for excess mismatches (‐C 50), setting a minimum mapping quality (‐minMapQ 30) and a minimum base quality (‐minQ 20) scores. As an outgroup we used a black rhinoceros resequenced genome that we had among our museum samples, and as a standard of quality we used the modern white rhinoceros sample ZA1969.1 (see Table S1). All error rates were then visualized per sample independently (Figure S3).
To rule out the possibility of cross‐contamination among our samples, we computed allele counts per sample only on the mitochondrial scaffold using the option ‐doCounts and ‐dumpCounts in angsd version 0.921 (Korneliussen et al., 2014). We counted the number of multiallelic mitochondrial sites, and we calculated the fraction of reads supporting the minor allele(s). Multi‐allelic sites at the mitochondrial scaffold are expected to be the result of ancient DNA damage and sequencing error; however, multiallelic sites where the minor allele(s) is represented by a moderate–high fraction of reads would be a sign of probable cross‐contamination. The proportion of multi‐allelic mitochondrial sites varied among samples in our data set, ranging between 7% and 90%. However, the minor allele(s) was supported by <5% of the reads across all multi‐allelic sites in every sample, and by <2% of reads in the majority of multi‐allelic sites (see examples in Figure S4). We therefore excluded the possibility of cross‐contamination events among our samples.
2.3. Variant site finding
We computed genotype likelihoods for the whole set of samples and for several subsets using angsd version 0.921 (Korneliussen et al., 2014) with the GATK genotype likelihood model (‐GL 2). To focus on well‐assembled regions of the reference genome where mapping is accurate and variants can be confidently called, as well as to reduce computing time, we restricted variant site search to scaffolds >13 Mbp in the white rhinoceros reference assembly (Figure S2A). The 66 scaffolds above this cut‐off were probed in an analysis of normalized depth of coverage across female and male samples. Three scaffolds showed half the average depth in samples originating from males, a sign that they belong to the X chromosome (Figure S2B). These scaffolds were therefore discarded from further analyses. The final set included 63 scaffolds, representing 76.17% of the total assembly. For genotype likelihood computation, we also avoided low‐complexity regions of the genome, owing to publicly available information about masked regions in the assembly (masking output file available for download at NCBI BioProject PRJNA74583). In a BED file, we listed all nonmasked regions and provided them to the ‐sites option in angsd. Overall, these nonmasked regions across the 63 longest autosomal scaffolds represented 48.12% of the total assembly (1.19 Gbp).
As input for different statistical analyses, several panels of genotype likelihoods were computed. First, we computed genotype likelihoods per subspecies including all samples (n NWR = 25; n SWR = 27), which were used to detect pairs of closely related individuals (see Section 2.4 for further details). Subsequently, we computed genotype likelihoods for both subspecies together after excluding three samples, each belonging to a closely related pair of individuals (see Section 2.4); this panel included 49 nonclosely related samples. Finally, we also generated a genotype likelihoods panel for each subspecies separately after excluding one of each pair of closely related samples (n NWR =24; n SWR =25) (see Section 2.4). In every case, the minimum number of individuals in which a variant site had to be present (‐minInd) was 95%. Minimum and maximum global depth per site were based on a global depth assessment with angsd (‐doDepth): 200 and 900 for panels combining both subspecies, and 100 and 500 for subspecies‐specific panels. Additionally, the quality filters described in Section 2.2 were also applied in this case: ‐remove_bads 1 ‐uniqueOnly 1 ‐baq 1 ‐C 50 ‐minMapQ 30 ‐minQ 20. As for the output choices, we specified the following: calculate the counts of each of the four possible bases per site (‐doCounts 1), compute genotype likelihoods with the GATK algorithm (‐GL 2), output the genotype likelihoods in nonbinary beagle format (‐doGlf 2), infer major and minor alleles per site from the genotype likelihoods (‐doMajorMinor 1), calculate allele frequencies per site with a fixed major and minor (‐doMaf 1), test for Hardy–Weinberg equilibrium per site (‐doHWE 1), exclude sites whose p‐value in the Hardy–Weinberg test is below the provided value (‐HWE_pval 1e‐2), report statistics per site (‐dosnpstat 1), and exclude variant sites whose p‐value for being a true single nucleotide polymorphism (SNP) is below the given value (‐SNP_pval 1e‐6). Transitions were removed a posteriori with custom‐made code.
These panels of genotype likelihoods were used for the analyses detailed in Sections 2.4 and 2.5. Additionally, we generated genotype calls for each sample individually with gatk version 3.3 (McKenna et al., 2010), via the haplotypecaller (Poplin et al., 2018) and genotypegvcfs tools. These genotype calls were used for the analyses detailed in Section 2.7. During genotyping, sites were filtered by base quality (‐‐min_base_quality_score 20) and mapping quality (‐mmq 30). From the resulting genomic VCF files, indels, mixed variants of SNPs and indels, multinucleotide polymorphisms and other poorly defined variants were removed using vcftools 0.1.16 (Danecek et al., 2011). Subsequently, we omitted sites with a depth of coverage <5×, as well as heterozygous sites where the minor allele was not supported by at least 20% of the reads using custom‐made code.
2.4. Relatedness test
We ran an analysis of relatedness based on a panel of genotype likelihoods per subspecies (n NWR = 25; n SWR = 27) with ngsrelate version 2 (Hanghøj et al., 2019), following the approach described in Waples et al. (2019). We found that one pair of NWR and two pairs of SWR samples showed a relatedness signal (Figure S5), and therefore for analyses of structure (i.e., PCA and admixture), the sample of lowest depth of coverage from each pair was excluded (CD‐un.1, un1856.1, ZA1842.1).
2.5. Principal component and admixture analyses
To run a principal component analysis (PCA), we used pcangsd version 0.973 (Meisner & Albrechtsen, 2018) and a genotype likelihoods panel of transversion sites (n = 897,524 sites) for 49 unrelated samples. The output was a covariance matrix. We then calculated the eigenvectors and eigenvalues using base packages in R version 3.4.4 (R: The R Project for Statistical Computing, 2018).
Using the same set of genotype likelihoods as an input, we ran an assessment of admixture proportions with ngsadmix version 32 (Skotte et al., 2013). For each value of K between two and six, 20 runs of ngsadmix were run. For each value of K, the run with the highest likelihood was chosen for visualization with the software pong (Behr et al., 2016).
2.6. Heterozygosity estimates
To assess individual levels of genome‐wide heterozygosity, we followed the procedure implemented in angsd version 0.921 (Korneliussen et al., 2014), which involves two steps: (i) computing the site allele frequency likelihood of there being zero, one or two alternative alleles based on genotype likelihoods with the angsd option ‐doSaf 1, and (ii) estimating the site frequency spectrum (SFS) from the output of the previous step with realsfs (Korneliussen et al., 2014), by counting the number of homozygous and heterozygous sites across the genome, in the case of a folded, one‐sample SFS.
In the first step, we chose the folded option (‐fold 1), and therefore the ancestral state (‐anc) and the reference (‐ref) parameters were both the white rhinoceros reference assembly. The same quality filters and regions of interest as when computing genotype likelihoods were used in the generation of the SFS (see Section 2.3). Transitions were directly excluded with the option ‐noTrans 1. Sites with depth of coverage <5× were also discarded (option ‐setMinDepth 5) after conducting tests regarding the effect of depth of coverage on heterozygosity estimation (see Relation between depth of coverage and genome‐wide heterozygosity in Appendix S1 and Figure S6).
In the second step, we estimated the SFS in chunks of 1 × 108 covered sites (option ‐nSites) with realsfs (Korneliussen et al., 2014). The resulting estimates of homozygous and heterozygous sites per chunk were summed, and the total count of heterozygous sites was divided by the total number of sites to obtain a genome‐wide estimate of individual heterozygosity at transversions. These estimates were corrected by the sum of the error rates at transversions (see Table S1 and Figure S3), by subtracting the fraction of the heterozygosity estimate owing to errors.
Samples whose average depth of coverage was <5× (n = 4) were excluded from temporal comparisons of genome‐wide heterozygosity, after assessing the effect of depth of coverage on heterozygosity estimation (see Relation between depth of coverage and genome‐wide heterozygosity in Appendix S1, and Figure S6).
2.7. Identification of runs of homozygosity and estimation of inbreeding coefficients
We used bcftools roh (Narasimhan et al., 2016) to identify runs of homozygosity (RoH) from each sample's filtered genotype calls. Individuals whose average depth of coverage was <5× (n = 4), and with any of their error rates exceeding 1% (n = 6; see Figure S3) were excluded from this analysis. Since some samples filled both filtering criteria, eight samples in total were omitted in all RoH and F RoH comparisons (see Table S1). We followed the software manual recommendations for single‐sample RoH detection, and set the allele frequency parameter (‐‐AF‐dflt) to a default of 0.4, since the input was one‐sample VCF files.
From the output of bcftools roh, we retrieved the regions flagged as RoH along the 63 longest autosomal scaffolds, and filtered out RoH of quality <30 phred score, and length <100 kbp (since it is harder to confidently infer short RoH). We calculated the fraction of the total length of the 63 scaffolds considered that was in RoH as a proxy for inbreeding (i.e., F RoH). Additionally, we also calculated average and maximum RoH length per sample.
To assess the performance of this method, we calculated local heterozygosity in sliding windows of 500 kbp with a 100‐kbp slide, also using each sample's filtered genotype calls as input. We visualized the distribution of local estimates of heterozygosity along the longest scaffold per sample together with the location of the RoH identified by bcftools roh, and found a clear overlap (see examples in Figure S7).
To calculate the age of RoH tracts, we used the formula from Thompson (2013): g = 100/2*RoHlength, where g refers to the age of an IBD segment in generations. We followed the approach of van der Valk et al. (2019) where the physical RoH length (in Mbp) is used as a proxy for the length in centimorgans.
2.8. Quantification of loss of genomic diversity
We tested if there was a significant increase or decrease of the following metrics between pre‐ and post‐bottleneck samples (per subspecies): genome‐wide heterozygosity, F RoH, average RoH length and maximum RoH length. We used unpaired, one‐sided Wilcoxon Sum Rank tests via the wilcox.test function in r version 3.4.4 (r: The R Project for Statistical Computing, 2018). This nonparametric test accommodates small and uneven sample sizes between groups.
We then calculated the proportion of increase or decrease of those metrics between groups as median post‐bottleneck value minus median pre‐bottleneck value, divided by the median pre‐bottleneck value. We refer to these as delta estimators (i.e., quantitative, unitless measures of genomic change through time), as proposed by Díez‐Del‐Molino et al. (2018).
2.9. Genetic load assessment
To detect potential functional consequences of genomic erosion, we mapped samples of average depth of coverage >10×, both historical and modern, to the publicly available black rhinoceros reference assembly GCA_013634535.1 (Moodley et al., 2020) (NCBI BioProject PRJNA632573), following the procedure detailed in Section 2.2. Unfortunately, none of the NWRpost samples had high enough depth of coverage to proceed with this analysis, so we evaluated genetic load in the surviving subspecies only, the SWR, based on five historical and 13 modern samples.
We called genotypes per individual with gatk following the approach described in Section 2.3. Regarding the polarization of alleles, we are aware of the impossibility of confirming which allele is ancestral at fixed differences between two species, but for technical reasons we considered “derived” to be the alternative allele among the white rhinoceros samples with respect to the black rhinoceros reference allele.
We annotated the homozygous derived biallelic sites of at least 10× depth of coverage per sample with snpeff version 4.3t (Cingolani et al., 2012) and the available annotation for the black rhinoceros genome (Moodley et al., 2020). Following Kuang et al. (2020), we counted the total number of missense and loss‐of‐function (LoF) homozygous derived sites, and divided those numbers by the total count of synonymous sites. To define LoF sites, we chose the approach of Glicksberg et al. (2019), and considered only sites of high impact under the effect categories “stop_gained,” “frameshift_variant,” “splice_acceptor_variant” and “splice_donor_variant.”
We tested whether the estimates of genetic load differed significantly between pre‐ and post‐bottleneck SWR with two‐sided Wilcoxon Sum Rank tests via the wilcox.test function in r version 3.4.4 (r: The R Project for Statistical Computing, 2018).
3. RESULTS
3.1. A species‐wide, temporal, genomic data set of the white rhinoceros
We generated the most comprehensive white rhinoceros genomic data set to date, consisting of 52 genomes resequenced at an average depth of coverage of 12.4× (see Figure S1 and Table S2 for further details). Of these, 30 were newly generated genomes from historical museum specimens, and 22 originated from modern specimens: 13 were already publicly available (see Tunstall et al., 2018) and nine were newly resequenced (Table S1). These 52 samples represent both NWR (n = 25) and SWR (n = 27) from different geographical locations. Attending to the age of the samples, we sorted them into two time windows for each subspecies (Figure 1a), hereafter referred to as pre‐ and post‐bottleneck, with the suffix pre (e.g., NWRpre) for the former, and post (e.g., NWRpost) for the latter. All pre‐bottleneck samples originated from museum specimens and their date refers to collection date; all modern samples plus four from museum specimens lie in the post‐bottleneck category, and their date refers to the birth of the individual.
Demographic histories and sampling time windows differ between subspecies. For the NWR, a time span of ~70 years is covered, during which a population decline occurred, followed by a rapid collapse (Figure 1b). The SWR, on the other hand, are represented in our study by samples from two time windows at the ends of a period of ~170 years, during which a strong bottleneck was followed by a recovery (Figure 1b).
3.2. Geography and potential sampling gaps underlay the structure in the data set
We ran a PCA on 49 unrelated individuals from both subspecies (see Figure S5) using only transversions to avoid biases due to ancient DNA damage. This revealed a clear separation between NWR and SWR along PC1 (Figure 2a). Moreover, PC2 suggested the existence of structure within each subspecies (Figure 2a).
FIGURE 2.
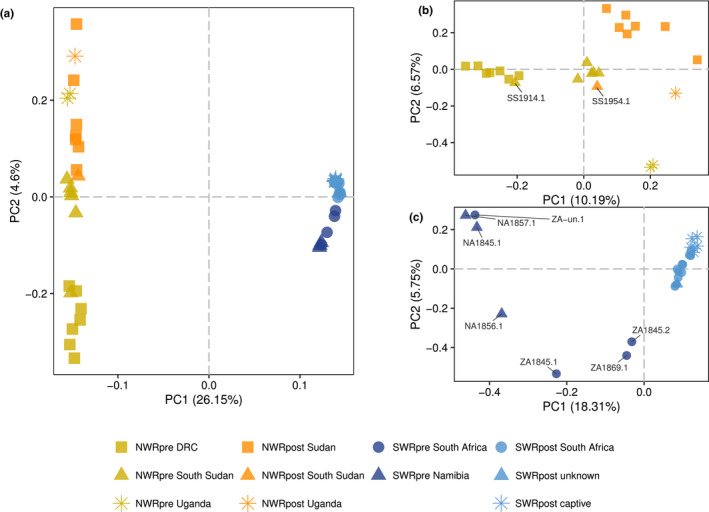
Principal component analysis (PCA) of the genomic variation in northern and southern white rhinoceroses. (a) Visualization of the two first principal components (PCs) of an analysis of 49 unrelated pre‐ and post‐bottleneck NWR and SWR. (b) First two PCs of an analysis of 24 unrelated NWR from both time windows. (c) First two PCs for 25 unrelated SWR, both pre‐ and post‐bottleneck
Therefore, to further untangle subspecies‐specific structure, we ran the same analysis for unrelated NWR and SWR separately (n NWR = 24; n SWR = 25). In the NWR, pre‐bottleneck individuals featured three clusters corresponding to the current DRC (plus an individual from South Sudan, SS1914.1), South Sudan and Uganda (Figure 2b). The post‐bottleneck NWR featured: (i) a group from Sudan (falling relatively close to the historical South Sudan), (ii) an individual from South Sudan (SS1954.1) within the pre‐bottleneck South Sudan, and (iii) one Uganda sample falling in between the pre‐bottleneck Uganda and post‐bottleneck Sudan (Figure 2b).
In the SWR, PC1 clearly separates pre‐ and post‐bottleneck individuals, the former showing structure and the latter clustering tightly together (Figure 2c). Regarding SWRpre, country of origin was known for all but one sample, but metadata regarding the particular locality of origin were not available for three out of seven samples: ZA‐un.1, ZA1869.1 and ZA1845.1. However, it was known that NA1845.1, NA1856.1 and NA1857.1 were individuals from Damaraland, northwestern Namibia, while ZA1845.2 originated in Kwa‐Zulu, eastern South Africa. Two of the Damaraland samples and the Kwa‐Zulu individual separated along both PC1 and PC2 (Figure 2c). Notably, the third Damaraland sample, NA1856.1, did not group together with the other two individuals from this locality. Of the three SWRpre of unknown locality of origin, ZA‐un.1 fell next to two of the Damaraland samples, while ZA1869.1 lay close to the Kwa‐Zulu individual; lastly ZA1845.1 appeared in between the isolated Damaraland sample and the Kwa‐Zulu individual (Figure 2c). All SWRpost fell close to the Kwa‐Zulu sample along PC1 (Figure 2c).
Admixture analysis of 49 unrelated individuals, with values of ancestral components (K) ranging between two and six, showed concordance with the PCA results. The results revealed a clear separation between NWR and SWR at K = 2 (Figure 3), while subsequent values of K separated within‐subspecies clusters. Among the NWRpre, DRC separated first from samples from South Sudan and Uganda, which split next (Figure 3). Individual SS1914.1, as in the PCA, shows affinity for the DRC group, instead of South Sudan. Post‐bottleneck NWR split into a distinct group from Sudan, one South Sudan individual (SS1954.1) showing affinity for the pre‐bottleneck South Sudan, and a Ugandan individual showing affinity for pre‐bottleneck Uganda.
FIGURE 3.
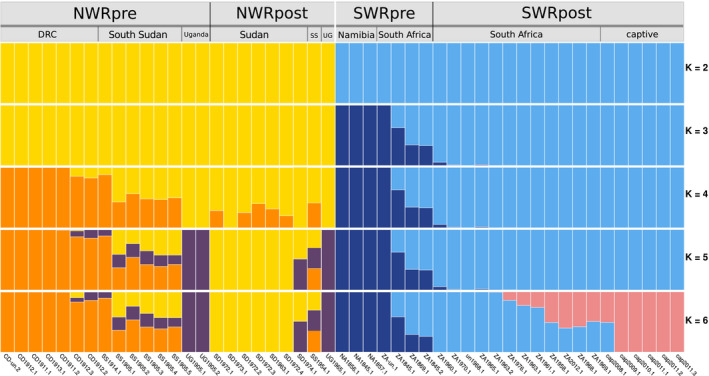
Admixture analysis of pre‐ and post‐bottleneck white rhinoceroses. The analysis included 49 unrelated individuals. The run of highest likelihood of a total of 20 runs is displayed for each value of K
Pre‐bottleneck SWR also displayed structure; a putative western group was featured (the three Namibian and ZA‐un.1), and a putative eastern group of South African individuals (including the Kwa‐Zulu individual ZA1845.2). Post‐bottleneck SWR showed higher affinity for the putative eastern historical group (Figure 3), and at K = 6 the new ancestry component separates captive and some wild SWRpost from the rest (Figure 3).
3.3. Both the NWR and the SWR suffered a loss of genomic diversity over time
To quantify loss of diversity as part of the genomic erosion process within each subspecies, we computed measures of genomic diversity per individual, grouped the individuals by time window and then ran a temporal comparison. First, we assessed if a significant difference existed between temporal groups, and then we calculated the proportional difference between the post‐ and pre‐bottleneck median value of each metric. These intraspecies, quantitative temporal measures of genomic change are referred to as delta estimators (Díez‐Del‐Molino et al., 2018).
Individual genome‐wide heterozygosity decreased significantly between pre‐ and post‐bottleneck individuals in both subspecies (Figure 4a) (W NWR = 106, p NWR = .000846; W SWR = 135, p SWR = .000061). NWRpost showed 10.40% lower heterozygosity than NWRpre, and SWRpost featured a median heterozygosity that was 36.49% lower than SWRpre (Table 1).
FIGURE 4.
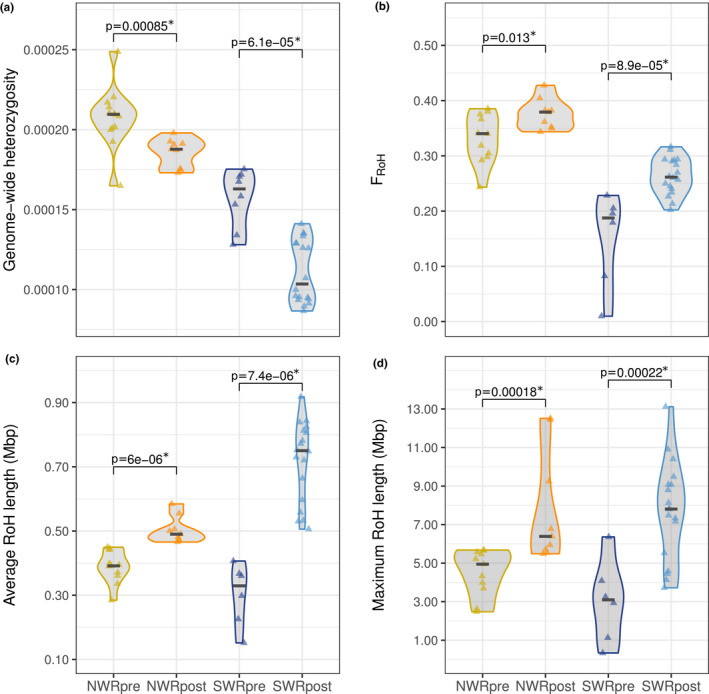
Temporal comparisons of genomic diversity metrics in northern and southern white rhinoceroses. (a) Estimates of individual genome‐wide heterozygosity grouped by subspecies and time window. (b) Estimates of inbreeding as the fraction of the 10 largest autosomal scaffolds within RoH per individual, grouped by subspecies and time window. (c) Average length of RoH, and (d) maximum length of RoH grouped by subspecies and time window. In all four panels, the p‐values above pre‐ and post‐bottleneck groups correspond to one‐sided Wilcoxon Sum Rank tests. Significant p‐values (below a significance threshold of .05) are marked with an asterisk
TABLE 1.
Measures of genomic diversity and delta estimators between pre‐ and post‐bottleneck white rhinoceroses
| Group | Genome‐wide heterozygosity comparisons | F roH, average length of RoH and maximum length of RoH comparisons | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | GW Het | p | Δ GW Het | n | F RoH | p | Δ F RoH | Avg RoH length (bp) | p | Δ Avg RoH length | Max RoH length (bp) | p | Δ Max RoH length | |
| NWRpre | 13 | 0.000210 | .0008463 | −0.1040 | 11 | 0.3403 | .0125300 | 0.1146 | 391,564.0 | .0000060 | 0.2520 | 4,947,425 | .0001786 | 0.2910 |
| NWRpost | 9 | 0.000188 | 9 | 0.3793 | 490,254.0 | 6,387,286 | ||||||||
| SWRpre | 8 | 0.000163 | .0000615 | −0.3649 | 6 | 0.1875 | .0000892 | 0.3942 | 329,295.0 | .0000074 | 1.2782 | 3,095,758 | .0002229 | 1.5219 |
| SWRpost | 18 | 0.000103 | 18 | 0.2615 | 750,207.0 | 7,807,180 | ||||||||
Median values of genome‐wide heterozygosity (GW Het in this table), F RoH, average and maximum length of RoH for each of the four groups. Comparisons between NWRpre and NWRpost, and between SWRpre and SWRpost were performed with one‐sided Wilcoxon Sum Rank tests. Delta estimators (denoted with the Greek letter Δ) were calculated as median post‐bottleneck value minus median pre‐bottleneck value, divided by the median pre‐bottleneck value.
With regard to inbreeding, we calculated F RoH per sample as the fraction of the genome putatively in RoH. We observed a small but significant increase of F RoH between NWRpre and NWRpost (W NWR = 20, p NWR = .01253) of 11.46%. In the SWR, F RoH also increased significantly (W SWR = 4, p SWR = .000089) but by 39.42% (Table 1 and Figure 4b).
The average length of RoH was significantly higher in both NWRpost and SWRpost compared to their historical counterparts (W NWR = 0, p NWR = .000006; W SWR = 0, p SWR = .000007) (Figure 4c). The increase in the NWR amounted to a 25.20% and to a 127.82% in the SWR (Table 1). Moreover, the maximum RoH length was also significantly higher in post‐bottleneck groups (W NWR = 6, p NWR = .000179; W SWR = 6, p SWR = .000223), involving a 29.10% increase in NWR, and a 152.19% rise in the SWR (Table 1 and Figure 4d).
The maximum length of RoH registered in the NWRpre is 5.68 Mbp, while NWRpost individuals harbour RoH of up to 12.51 Mbp (Figure S8). In the SWR, we observed that the longest RoH recorded in the SWRpre group, 6.36 Mbp, is surpassed by most SWRpost that harbour RoH of up to 13.11 Mbp (Figure S8). With the method described in Section 2.7, we calculated that the tracts between 6 and 13 Mbp, which are particularly common among SWRpost, were formed eight to four generations ago (i.e., the shared ancestor of each of these distinct homozygous tracts dates back eight to four generations).
3.4. Genetic load in the SWR did not increase significantly over time
The SWR is the only surviving white rhinoceros subspecies, and according to our results they have suffered an intense loss of genomic diversity. To uncover whether deleterious genetic load might have accumulated in their genomes as another symptom of erosion, we calculated the ratios of missense/synonymous and LoF/synonymous homozygous derived sites across five SWRpre and 13 SWRpost. We did not observe a significant difference in either of the ratios between SWRpre and SWRpost, and the estimates of genetic load ranged more widely among pre‐ than post‐bottleneck individuals (Figure 5).
FIGURE 5.
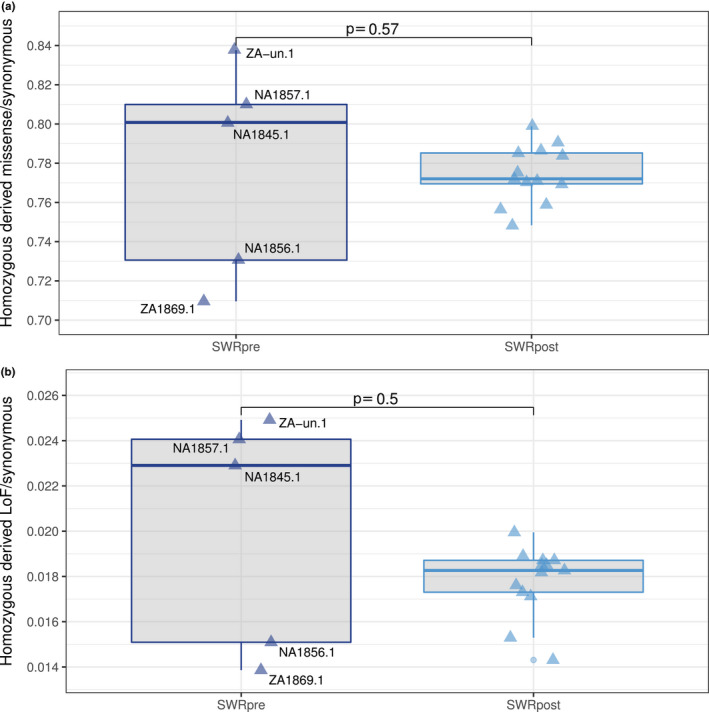
Genetic load through time in the southern white rhinoceros. (a) Distribution of the missense/synonymous ratio at homozygous derived sites in five SWRpre and 13 SWRpost. (b) Distribution of LoF/synonymous ratio at homozygous derived sites in five SWRpre and 13 in SWRpost. In both panels, the p‐values correspond to two‐sided Wilcoxon Sum Rank tests, and the significance threshold is .05
The estimates among SWRpre samples clustered similarly to the pattern previously observed in the PCA (Figure 2c). The sample size was unfortunately too small to assess historical variation in genetic load across geography. However, none of the SWRpost showed a higher estimate of load than the highest among SWRpre (Figure 5).
4. DISCUSSION
In this work, we present a white rhinoceros genomic data set that included 52 individuals from both subspecies, the NWR and SWR, at two different time windows each within the past ~170 years. With this temporal data set we uncovered patterns of population structure, and we assessed within‐subspecies genomic erosion.
Thanks to having historical samples at our disposal, we identified aspects of the population structure of the NWR and the SWR that were previously unknown. We identified three historical NWR clusters, in DRC, South Sudan and Uganda (Figure 2b). The post‐bottleneck NWR in our data set originated mostly from Sudan, which separated as another group, while post‐bottleneck South Sudan NWR aligned with their respective historical population. Post‐bottleneck Uganda did not show as strong an affinity to pre‐bottleneck Uganda (at least in the PCA, Figure 2b). In the SWR, we observed signs of a west–east axis of genomic differentiation among pre‐bottleneck samples (Figures 2c and 3) despite the limited metadata regarding locality of origin and the small sample size. These patterns of structure may be true biological signals of some degree of isolation among populations in historical times, but given the potential gaps in our sampling scheme, and the absence of obvious barriers to gene flow, the clusters we observed within each subspecies might have been part of a more continuous cline caused by isolation by distance over an uninterrupted distribution.
Additionally, the founder effect that gave rise to all extant SWR was detectable in our data set. Kwa‐Zulu, in eastern South Africa, was the only SWR population that survived the extirpations of the late 19th century, and despite suffering a harsh bottleneck, it recovered and became the source of all modern SWR. In our analysis we saw clear affinities between the very homogeneous modern SWR and the historical sample ascribed to Kwa‐Zulu (Figures 2c and 3). The genetic distance between all modern SWR and their presumed historical source propably owes to genetic drift and inbreeding over the last century.
Both the NWR and the SWR experienced some degree of loss of genomic diversity, according to our results, as a consequence of population extirpations and declines. We observed a significant decrease of heterozygosity in both subspecies, and increases in RoH length and inbreeding coefficients measured as F RoH. There were, however, clear differences between the NWR and the SWR in this regard, which might partially owe to the disparity of their respective demographic outcomes, but might be heavily influenced by the difference in time span between the sampling time windows (~70 years in NWR vs. ~170 years in SWR). We therefore discuss these results for each subspecies separately in the following paragraphs.
The NWR disappeared quickly from the wild after the onset of the bottleneck, and some of our NWRpost were born during this declining period. Our results showed a significant loss of heterozygosity (Figure 4a), which would be explained by the progressive extirpation of genomic diversity as the NWR were decimated, but also a small but significant increase of inbreeding coefficients and RoH length (Figure 4b–d and Table 1). This could imply that, in the decades leading up to their extinction, the progressively declining population size might have led to an increase in inbreeding to a certain extent.
In the SWR, both the reduction of heterozygosity and the increase in inbreeding are significant and substantial, as reflected by their delta estimators (Table 1). This agrees with what is known about the recent history of the SWR. Given the strong founder effect they underwent, which implied major extirpation of local diversity, signs of subsequent drift and inbreeding were to be expected. The notable increase of >100% of the average and maximum RoH lengths between pre‐ and post‐bottleneck SWR (Table 1) strongly supports a scenario of intense recent inbreeding.
The SWR is the only white rhinoceros subspecies surviving in the wild, and is subject to conservation efforts. Given the remarkable loss of genomic diversity they suffered, we decided to assess whether modern SWR also carry symptoms of an increased genetic load that could translate into detrimental phenotypic consequences. We observed, however, no overall significant difference in the estimates of genetic load between pre‐ and post‐bottleneck time windows (Figure 5). Despite a potentially structured distribution of genetic load in historical times, the estimates of load of the current SWR remain within the range of values of those in past populations. Importantly, this implies that the recovery of the SWR thus far has occurred without a concomitant accumulation of genetic load. Kuang et al. (2020) presented similar observations where a species of snub‐nosed monkeys, Rhinopithecus roxellana, that suffered strong recent inbreeding did not feature a parallel increase of genetic load. We thus endorse the authors' speculation that, at least in some species, inbreeding might operate at a faster rate than genetic load accumulation (Kuang et al., 2020).
The differing demographic outcomes between the two subspecies make the white rhinoceros an interesting model to study extinction and population management. The NWR was rapidly driven to extinction due to anthropogenic pressure, while the SWR represents a conservation success story. The demographic trajectory of the SWR in the last century proves that white rhinoceroses have the potential to reconstitute thriving populations from a very limited pool of genomic diversity. Additionally, while our results suggest that a careful control of inbreeding would be advisable in the management of the remaining SWR, no evident genetic threats, in the form of heightened genetic load, seem to be haunting them at present.
This does not exclude the possibility that other symptoms of erosion, such as reduced heterozygosity and long homozygous tracts, might pose longer term negative consequences that are yet unknown, particularly in the face of the rapidly changing environments of the Anthropocene. However, state‐of‐the‐art techniques in molecular and reproductive biology, at the service of conservation, might be able to restore (or even increase) some of the rhinoceros genomic diversity lost in the wild, including the potential for creating new NWR populations (Hildebrandt et al., 2018; Saragusty et al., 2016).
Beyond white rhinoceros conservation, this work adds to a growing body of research where temporal data sets provide valuable assessments of threatened species, such as the eastern gorilla (van der Valk et al., 2019), the crested ibis (Feng et al., 2019) or the honeybee (Espregueira Themudo et al., 2020). These studies reveal a pervasive and recent decline in genomic diversity among wild populations, whose consequences might be difficult to foresee. This kind of data set, however, offers the possibility to detect potential genetic threats before they take a toll on the managed populations. Moreover, they could elicit the development of species‐specific protocols to estimate the evolutionary potential of these “eroded” populations.
Species extinctions are often used as a metric of anthropogenic pressure, but population extirpations and census size reductions are more pervasive, precede extinction, and affect both officially threatened and nonthreatened species (Ceballos et al., 2017). As a consequence, snippets of genomic diversity are being erased from the biosphere for good. The viability of a species might not be cancelled in the short term as a result, but its genomic pool will be eroded, which is in itself a biodiversity loss, and potentially entails an added degree of vulnerability to the species in the face of a changing environment. Ultimately, if conservation of biodiversity is entering an era of tailor‐made, population‐specific approaches, temporal data sets hold great power to reveal the hidden consequences (and potential risks) of the human threat to biodiversity.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
AUTHOR CONTRIBUTION
Conceptualization, F.S.B., S.G., J.R.M., M.V.W., M.M., F.G.V., Y.M. and M.T.P.G.; Investigation, F.S.B., M.C., L.D. and O.A.R.; Formal Analysis, F.S.B., S.G., J.R.M., M.V.W., M.M., A.M. and Y.P.; Data Curation, F.S.B., L.D. and O.A.R; Resources, D.C.K., Z.T., T.S.P., G.Z. and T.M.B.; Writing – Original Draft, F.S.B.; Writing –Review & Editing, F.S.B., S.G., J.R.M, M.V.W., M.M., M.C., F.G.V., D.C.K., Z.T., T.S.P., L.D., O.A.R., T.M.B, Y.M. and M.T.P.G; Visualization, F.S.B.; Supervision, Y.M. and M.T.P.G.; Funding Acquisition, M.T.P.G.
Supporting information
Supplementary Material
Table S1
ACKNOWLEDGEMENTS
This work was supported by ERC Consolidator Grant 681396 “Extinction Genomics” to M.T.P.G. and by EMBO Short‐Term Fellowship 7578 to F.S.B. The authors would like to acknowledge support from Science for Life Laboratory, the National Genomics Infrastructure (NGI), Sweden, the Knut and Alice Wallenberg Foundation and UPPMAX for providing assistance in massively parallel DNA sequencing and computational infrastructure. The authors are very grateful to all the museums who provided samples for this study: the American Museum of Natural History, the National Museums Scotland, the Natural History Museum at the National Museum Praha, the Natural History Museum Vienna, the Powell‐Cotton Museum, the Royal Museum for Central Africa Tervuren, and the Swedish Museum of Natural History.
Sánchez‐Barreiro, F. , Gopalakrishnan, S. , Ramos‐Madrigal, J. , Westbury, M. V. , de Manuel, M. , Margaryan, A. , Ciucani, M. M. , Vieira, F. G. , Patramanis, Y. , Kalthoff, D. C. , Timmons, Z. , Sicheritz‐Pontén, T. , Dalén, L. , Ryder, O. A. , Zhang, G. , Marquès‐Bonet, T. , Moodley, Y. , & Gilbert, M. T. P. (2021). Historical population declines prompted significant genomic erosion in the northern and southern white rhinoceros (Ceratotherium simum). Molecular Ecology, 30, 6355–6369. 10.1111/mec.16043
Fátima Sánchez‐Barreiro lead contact.
Yoshan Moodley and M. Thomas P. Gilbert are senior authors.
Contributor Information
Fátima Sánchez‐Barreiro, Email: fatima@palaeome.org.
M. Thomas P. Gilbert, Email: tgilbert@sund.ku.dk.
DATA AVAILABILITY STATEMENT
Of the resequenced genomic data presented in this work, 13 genomes are already available in NCBI BioProject under accession no. PRJNA394025 (Tunstall et al., 2018). Sample‐specific accession codes are listed in Table S1 under accession_code_NCBI. All newly generated data (39 resequenced genomes) are available as FASTQ files at the European Nucleotide Archive (ENA) under Study Primary Accession PRJEB45263. Sample‐specific accession codes are listed in Table S1 under accession_code_ENA. The code used for computational analysis is publicly available at: https://github.com/fasaba/WR_temporal_genomics_code.
REFERENCES
- Abascal, F. , Corvelo, A. , Cruz, F. , Villanueva‐Cañas, J. L. , Vlasova, A. , Marcet‐Houben, M. , Martínez‐Cruz, B. , Cheng, J. Y. , Prieto, P. , Quesada, V. , Quilez, J. , Li, G. , García, F. , Rubio‐Camarillo, M. , Frias, L. , Ribeca, P. , Capella‐Gutiérrez, S. , Rodríguez, J. M. , Câmara, F. , … Godoy, J. A. (2016). Extreme genomic erosion after recurrent demographic bottlenecks in the highly endangered Iberian lynx. Genome Biology, 17(1), 251. 10.1186/s13059-016-1090-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data. Babraham Bioinformatics, Babraham Institute. [Google Scholar]
- Behr, A. A. , Liu, K. Z. , Liu‐Fang, G. , Nakka, P. , & Ramachandran, S. (2016). pong: Fast analysis and visualization of latent clusters in population genetic data. Bioinformatics, 32(18), 2817–2823. 10.1093/bioinformatics/btw327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad Institute (2019). Picard toolkit. Retrieved from https://broadinstitute.github.io/picard/ [Google Scholar]
- Carøe, C. , Gopalakrishnan, S. , Vinner, L. , Mak, S. S. T. , Sinding, M. H. S. , Samaniego, J. A. , Wales, N. , Sicheritz‐Pontén, T. , & Gilbert, M. T. P. (2018). Single‐tube library preparation for degraded DNA. Methods in Ecology and Evolution / British Ecological Society, 9(2), 410–419. 10.1111/2041-210X.12871 [DOI] [Google Scholar]
- Caughley, G. (1994). Directions in conservation biology. The Journal of Animal Ecology, 63(2), 215–244. 10.2307/5542 [DOI] [Google Scholar]
- Ceballos, G. , Ehrlich, P. R. , Barnosky, A. D. , García, A. , Pringle, R. M. , & Palmer, T. M. (2015). Accelerated modern human‐induced species losses: Entering the sixth mass extinction. Science Advances, 1(5), e1400253. 10.1126/sciadv.1400253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceballos, G. , Ehrlich, P. R. , & Dirzo, R. (2017). Biological annihilation via the ongoing sixth mass extinction signaled by vertebrate population losses and declines. Proceedings of the National Academy of Sciences of the United States of America, 114(30), E6089–E6096. 10.1073/pnas.1704949114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani, P. , Platts, A. , Wang, L. L. , Coon, M. , Nguyen, T. , Wang, L. , Land, S. J. , Lu, X. , & Ruden, D. M. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly, 6(2), 80–92. 10.4161/fly.19695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabney, J. , Knapp, M. , Glocke, I. , Gansauge, M.‐T. , Weihmann, A. , Nickel, B. , Valdiosera, C. , Garcia, N. , Paabo, S. , Arsuaga, J.‐L. , & Meyer, M. (2013). Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proceedings of the National Academy of Sciences of the United States of America, 110(39), 15758–15763. 10.1073/pnas.1314445110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C. A. , Banks, E. , DePristo, M. A. , Handsaker, R. E. , Lunter, G. , Marth, G. T. , Sherry, S. T. , McVean, G. , Durbin, R. , & 1000 Genomes Project Analysis Group (2011). The variant call format and VCFtools. Bioinformatics, 27(15), 2156–2158. 10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díez‐Del‐Molino, D. , Sánchez‐Barreiro, F. , Barnes, I. , Gilbert, M. T. P. , & Dalén, L. (2018). Quantifying temporal genomic erosion in endangered species. Trends in Ecology & Evolution, 33(3), 176–185. 10.1016/j.tree.2017.12.002 [DOI] [PubMed] [Google Scholar]
- Emslie, R. (2008). Rhino population sizes and trends. Pachyderm, 44, 88–95. [Google Scholar]
- Emslie, R. (IUCN SSC African Rhino Specialist Group) (2020). IUCN Red List of Threatened Species: White Rhino. IUCN. [Google Scholar]
- Emslie, R. , & Brooks, M. (1999). African rhino: Status survey and conservation action plan (IUCN/SSC African Rhino Specialist Group, Ed.). IUCN. [Google Scholar]
- Espregueira Themudo, G. , Rey‐Iglesia, A. , Robles Tascón, L. , Bruun Jensen, A. , da Fonseca, R. R. , & Campos, P. F. (2020). Declining genetic diversity of European honeybees along the twentieth century. Scientific Reports, 10(1), 10520. 10.1038/s41598-020-67370-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, S. , Fang, Q. I. , Barnett, R. , Li, C. , Han, S. , Kuhlwilm, M. , Zhou, L. , Pan, H. , Deng, Y. , Chen, G. , Gamauf, A. , Woog, F. , Prys‐Jones, R. , Marques‐Bonet, T. , Gilbert, M. T. P. , & Zhang, G. (2019). The genomic footprints of the fall and recovery of the crested ibis. Current Biology, 29, 340–349. 10.1016/j.cub.2018.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankham, R. (2005). Genetics and extinction. Biological Conservation, 126(2), 131–140. 10.1016/j.biocon.2005.05.002 [DOI] [Google Scholar]
- Gilbert, M. T. P. , Haselkorn, T. , Bunce, M. , Sanchez, J. J. , Lucas, S. B. , Jewell, L. D. , Marck, E. V. , & Worobey, M. (2007). The isolation of nucleic acids from fixed, paraffin‐embedded tissues‐which methods are useful when? PLoS One, 2(6), e537. 10.1371/journal.pone.0000537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glicksberg, B. S. , Amadori, L. , Akers, N. K. , Sukhavasi, K. , Franzén, O. , Li, L. I. , Belbin, G. M. , Akers, K. L. , Shameer, K. , Badgeley, M. A. , Johnson, K. W. , Readhead, B. , Darrow, B. J. , Kenny, E. E. , Betsholtz, C. , Ermel, R. , Skogsberg, J. , Ruusalepp, A. , Schadt, E. E. , … Chen, R. (2019). Integrative analysis of loss‐of‐function variants in clinical and genomic data reveals novel genes associated with cardiovascular traits. BMC Medical Genomics, 12(Suppl 6), 108. 10.1186/s12920-019-0542-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanghøj, K. , Moltke, I. , Andersen, P. A. , Manica, A. , & Korneliussen, T. S. (2019). Fast and accurate relatedness estimation from high‐throughput sequencing data in the presence of inbreeding. GigaScience, 8(5), giz034. 10.1093/gigascience/giz034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley, E. H. , de Waal, M. , Murray, S. , & O'Ryan, C. (2016). Comparison of whole mitochondrial genome sequences of northern and southern white rhinoceroses (Ceratotherium simum): The conservation consequences of species definitions. Conservation Genetics, 17(6), 1285–1291. 10.1007/s10592-016-0861-2 [DOI] [Google Scholar]
- Hildebrandt, T. B. , Hermes, R. , Colleoni, S. , Diecke, S. , Holtze, S. , Renfree, M. B. , Stejskal, J. , Hayashi, K. , Drukker, M. , Loi, P. , Göritz, F. , Lazzari, G. , & Galli, C. (2018). Embryos and embryonic stem cells from the white rhinoceros. Nature Communications, 9(1), 2589. 10.1038/s41467-018-04959-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jónsson, H. , Ginolhac, A. , Schubert, M. , Johnson, P. L. F. , & Orlando, L. (2013). mapDamage2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics, 29(13), 1682–1684. 10.1093/bioinformatics/btt193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korneliussen, T. S. , Albrechtsen, A. , & Nielsen, R. (2014). ANGSD: Analysis of next generation sequencing data. BMC Bioinformatics, 15, 356. 10.1186/s12859-014-0356-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang, W. , Hu, J. , Wu, H. , Fen, X. , Dai, Q. , Fu, Q. , Xiao, W. , Frantz, L. , Roos, C. , Nadler, T. , Irwin, D. M. , Zhou, L. , Yang, X. U. , & Yu, L. I. (2020). Genetic diversity, inbreeding level, and genetic load in endangered snub‐nosed monkeys (Rhinopithecus). Frontiers in Genetics, 11, 615926. 10.3389/fgene.2020.615926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25(14), 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. , Durbin, R. , & 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25(16), 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak, S. S. T. , Gopalakrishnan, S. , Carøe, C. , Geng, C. , Liu, S. , Sinding, M.‐H. , Kuderna, L. F. K. , Zhang, W. , Fu, S. , Vieira, F. G. , Germonpré, M. , Bocherens, H. , Fedorov, S. , Petersen, B. , Sicheritz‐Pontén, T. , Marques‐Bonet, T. , Zhang, G. , Jiang, H. , & Gilbert, M. T. P. (2017). Comparative performance of the BGISEQ‐500 vs Illumina HiSeq2500 sequencing platforms for palaeogenomic sequencing. GigaScience, 6(8), 1–13. 10.1093/gigascience/gix049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. , Altshuler, D. , Gabriel, S. , Daly, M. , & DePristo, M. A. (2010). The genome analysis toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20(9), 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisner, J. , & Albrechtsen, A. (2018). Inferring population structure and admixture proportions in low‐depth NGS data. Genetics, 210(2), 719–731. 10.1534/genetics.118.301336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, M. , & Kircher, M. (2010). Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harbor Protocols, 2010(6), db.prot5448. 10.1101/pdb.prot5448 [DOI] [PubMed] [Google Scholar]
- Moodley, Y. , Russo, I.‐R.‐M. , Robovský, J. , Dalton, D. L. , Kotzé, A. , Smith, S. , & Bruford, M. W. (2018). Contrasting evolutionary history, anthropogenic declines and genetic contact in the northern and southern white rhinoceros (Ceratotherium simum). Proceedings. Biological Sciences/The Royal Society, 285(1890), 20181567. 10.1098/rspb.2018.1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moodley, Y. , Westbury, M. V. , Russo, I.‐R. , Gopalakrishnan, S. , Rakotoarivelo, A. , Olsen, R.‐A. , Prost, S. , Tunstall, T. , Ryder, O. A. , Dalén, L. , & Bruford, M. W. (2020). Interspecific gene flow and the evolution of specialization in black and white rhinoceros. Molecular Biology and Evolution, 37(11), 3105–3117. 10.1093/molbev/msaa148 [DOI] [PubMed] [Google Scholar]
- Narasimhan, V. , Danecek, P. , Scally, A. , Xue, Y. , Tyler‐Smith, C. , & Durbin, R. (2016). BCFtools/RoH: A hidden Markov model approach for detecting autozygosity from next‐generation sequencing data. Bioinformatics, 32(11), 1749–1751. 10.1093/bioinformatics/btw044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto, S. P. (2018). Adaptation, speciation and extinction in the Anthropocene. Proceedings. Biological Sciences/The Royal Society, 285(1891), 20182047. 10.1098/rspb.2018.2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poplin, R. , Ruano‐Rubio, V. , DePristo, M. A. , Fennell, T. J. , Carneiro, M. O. , Van der Auwera, G. A. , Kling, D. E. , Gauthier, L. D. , Levy‐Moonshine, A. , Roazen, D. , Shakir, K. , Thibault, J. , Chandran, S. , Whelan, C. , Lek, M. , Gabriel, S. , Daly, M. J. , Neale, B. , MacArthur, D. G. , & Banks, E. (2018). Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv, 2018, 201178. 10.1101/201178 [DOI] [Google Scholar]
- R v3.4.4: The R Project for Statistical Computing (2018). Retrieved from https://www.r‐project.org/
- Reed, D. H. , & Frankham, R. (2003). Correlation between fitness and genetic diversity. Conservation Biology, 17(1), 230–237. 10.1046/j.1523-1739.2003.01236.x [DOI] [Google Scholar]
- Ripple, W. J. , Wolf, C. , Newsome, T. M. , Betts, M. G. , Ceballos, G. , Courchamp, F. , Hayward, M. W. , Valkenburgh, B. , Wallach, A. D. , & Worm, B. (2019). Are we eating the world's megafauna to extinction? Conservation Letters, 358, e12627. 10.1111/conl.12627 [DOI] [Google Scholar]
- Robinson, J. A. , Räikkönen, J. , Vucetich, L. M. , Vucetich, J. A. , Peterson, R. O. , Lohmueller, K. E. , & Wayne, R. K. (2019). Genomic signatures of extensive inbreeding in Isle Royale wolves, a population on the threshold of extinction. Science . Advances, 5(5), eaau0757. 10.1126/sciadv.aau0757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rookmaaker, K. (2000). The alleged population reduction of the southern white rhinoceros (Ceratotherium simum simum) and the successful recovery. Säugetierkundliche Mitteilungen, 45(2), 55–70. [Google Scholar]
- Rookmaaker, K. , & Antoine, P.‐O. (2012). New maps representing the historical and recent distribution of the African species of rhinoceros: Diceros bicornis, Ceratotherium simum and Ceratotherium cottoni . Pachyderm, 52, 91–96. [Google Scholar]
- Saragusty, J. , Diecke, S. , Drukker, M. , Durrant, B. , Friedrich Ben‐Nun, I. , Galli, C. , Göritz, F. , Hayashi, K. , Hermes, R. , Holtze, S. , Johnson, S. , Lazzari, G. , Loi, P. , Loring, J. F. , Okita, K. , Renfree, M. B. , Seet, S. , Voracek, T. , Stejskal, J. , … Hildebrandt, T. B. (2016). Rewinding the process of mammalian extinction. Zoo Biology, 35(4), 280–292. 10.1002/zoo.21284 [DOI] [PubMed] [Google Scholar]
- Schubert, M. , Ermini, L. , Sarkissian, C. D. , Jónsson, H. , Ginolhac, A. , Schaefer, R. , Martin, M. D. , Fernández, R. , Kircher, M. , McCue, M. , Willerslev, E. , & Orlando, L. (2014). Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using PALEOMIX. Nature Protocols, 9(5), 1056–1082. 10.1038/nprot.2014.063 [DOI] [PubMed] [Google Scholar]
- Schubert, M. , Lindgreen, S. , & Orlando, L. (2016). AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Research Notes, 9, 88. 10.1186/s13104-016-1900-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skotte, L. , Korneliussen, T. S. , & Albrechtsen, A. (2013). Estimating individual admixture proportions from next generation sequencing data. Genetics, 195(3), 693–702. 10.1534/genetics.113.154138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielman, D. , Brook, B. W. , & Frankham, R. (2004). Most species are not driven to extinction before genetic factors impact them. Proceedings of the National Academy of Sciences of the United States of America, 101(42), 15261–15264. 10.1073/pnas.0403809101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, E. A. (2013). Identity by descent: Variation in meiosis, across genomes, and in populations. Genetics, 194(2), 301–326. 10.1534/genetics.112.148825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunstall, T. , Kock, R. , Vahala, J. , Diekhans, M. , Fiddes, I. , Armstrong, J. , Paten, B. , Ryder, O. A. , & Steiner, C. C. (2018). Evaluating recovery potential of the northern white rhinoceros from cryopreserved somatic cells. Genome Research, 28(6), 780–788. 10.1101/gr.227603.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Valk, T. , Díez‐Del‐Molino, D. , Marques‐Bonet, T. , Guschanski, K. , & Dalén, L. (2019). Historical genomes reveal the genomic consequences of recent population decline in eastern gorillas. Current Biology, 29(1), 165–170.e6. 10.1016/j.cub.2018.11.055 [DOI] [PubMed] [Google Scholar]
- Waples, R. K. , Albrechtsen, A. , & Moltke, I. (2019). Allele frequency‐free inference of close familial relationships from genotypes or low‐depth sequencing data. Molecular Ecology, 28(1), 35–48. 10.1111/mec.14954 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Table S1
Data Availability Statement
Of the resequenced genomic data presented in this work, 13 genomes are already available in NCBI BioProject under accession no. PRJNA394025 (Tunstall et al., 2018). Sample‐specific accession codes are listed in Table S1 under accession_code_NCBI. All newly generated data (39 resequenced genomes) are available as FASTQ files at the European Nucleotide Archive (ENA) under Study Primary Accession PRJEB45263. Sample‐specific accession codes are listed in Table S1 under accession_code_ENA. The code used for computational analysis is publicly available at: https://github.com/fasaba/WR_temporal_genomics_code.